
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn engineered third electrostatic constriction of aerolysin to manipulate heterogeneously charged peptide transport†
Hongyan
Niu
a,
Meng-Ying
Li
ab,
Yi-Lun
Ying
 *ab and
Yi-Tao
Long
a
*ab and
Yi-Tao
Long
a
aState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China
bChemistry and Biomedicine Innovation Center, Nanjing University, Nanjing 210023, P. R. China. E-mail: yilunying@nju.edu.cn
First published on 3rd February 2022
Abstract
Reading the primary sequence directly using nanopores remains challenging due to the complex building blocks of 20 proteinogenic amino acids and the corresponding sophisticated structures. Compared to the uniformly negatively charged polynucleotides, biological nanopores hardly provide effective ionic current responses to all heterogeneously charged peptides under nearly physiological pH conditions. Herein, we precisely design a N226Q/S228K mutant aerolysin which creates a new electrostatic constriction named R3 in-between two natural sensing regions for controlling the capture and translocation of heterogeneously charged peptides. At nearly physiological pH, the decoration of positive charges at this constriction gives a large velocity of electroosmotic flow (EOF), leading to a maximum 8-fold increase in frequency for the heterogeneously charged peptides with the net charge from +1 to −3. Even the duration time of the negatively charged peptide Aβ35-25D4 in N226Q/S228K AeL also rises from 0.07 ± 0.01 ms to 0.63 ± 0.01 ms after introducing the third electrostatic constriction. Therefore, the N226Q/S228K aerolysin nanopore with three electrostatic constrictions realizes the dual goals of both capturing and decelerating heterogeneously charged peptides without labelling, even for the folded peptides.
Introduction
Proteins play an important role in multiple physiological processes. Given the success of nanopore single-molecule DNA sequencing,1–3 more studies are focused on single protein sensing and sequencing with biological nanopores.4–7 Currently, two potential strategies have been proposed for nanopore protein sequencing. One method is to adopt a protein fingerprinting approach like the shotgun proteomics used in mass spectrometry.8 The other strategy employs DNA motor enzymes to ratchet a peptide–oligonucleotide conjugate into the nanopore.6,7,9 The former method requires the digestion of the protein. Then, all peptide fragments should be captured and recognized by a nanopore. But compared to the uniformly negatively charged polynucleotides, the driving electrophoretic force (EPF) for heterogeneously charged peptides (amino acids can be neutral, positively, or negatively charged) is not always efficient. The latter protein sequencing idea needs a highly sensitive nanopore interface for prolonging the duration of short peptide segments in the sensing region in order to achieve the effective discrimination of a single amino acid. However, due to the complex physicochemical properties and small volumes of 20 proteinogenic amino acids (e.g., charge, hydrophilicity, etc.), nanopore protein sequencing is still challenging because of the low resolution for a single amino acid. Even so, various types of biological nanopores have been engineered for studying peptide translocation, analysing peptide interactions and controlling single protein/peptide capture and translocation through non-covalent interactions.10–15 Wild-type aerolysin (WT AeL) has shown an excellent ability to discriminate amino acids from short peptides with the help of a charged polyarginine carrier, which holds promise for peptide sequencing. But WT AeL hardly provides effective ionic current responses to all heterogeneously charged peptides.15 Some previous studies exploited electroosmotic flow (EOF) capturing of molecules in α-hemolysin, Frac, aerolysin, etc., which is independent of the carrying charges in peptides.16–19 EOF can compete or cooperate with EPF acting on the threading molecule to enhance capture possibility.20 Studies have shown that the strength and direction of the EOF depend on the charge distribution and shape of a nanopore, and are related to its ion selectivity.21–24 Therefore, a low pH buffer (<4) was used to protonate residues at the nanopore lumen, leading to an enhanced EOF.16,21 But the extremely low pH conditions would not only induce instability of the detection system (biological nanopores and lipid bilayer) but also change the structures of the protein, causing relatively low reproducibility. To enhance the EOF at a mild physiological pH (pH = 7–8), the most practical strategy is to design the single-molecule sensing interface of biological nanopores by using mutagenesis. However, if the net driving force (resultant of EOF and EPF) is stronger than the repelling force from the electrostatic barrier inside the nanopore confinement, the translocation of peptides is accelerated. The transient residence time may be beyond the bandwidth of the current amplifier, which would barely be recorded.25 If the electrostatic repelling force is too strong than the net driving force, the peptide may not overcome the energy barrier for a successful translocation. Therefore, the design of the nanopore sensing interface should not only enhance but also compromise the driving force and repelling force for all heterogeneously charged peptide sensing. Some efforts have been made to achieve this goal. By introducing extra positive charged lysine into the nanopore, a T232K mutant AeL achieves neutral peptide (Tau306-316) sensing.26 Results show that the capture frequency increased compared with WT AeL. But the neutral peptide bumped out from the pore opening due to the strong repelling force, leading to a short duration below 1 ms. Herein, we design a third electrostatic constriction AeL that works together with the two pre-existing sensitive regions to control the capture and translocation of heterogeneously charged peptides. A new electrostatic constricted region named R3 is created by introducing a positively charged amino acid in-between two natural constrictions. The EOF of N226Q/S228K AeL has a nearly 2-fold increase compared to that of WT AeL due to the enhanced anion selectivity. All the heterogeneously charged peptides (net charge (NC) from +1 to −3) in the experiments could thread and successfully translocate through a N226Q/S228K AeL at a nearly physiological pH. The duration time of the heterogeneously charged peptides was prolonged about 7 to 72 times with this strategy. Therefore, our N226Q/S228K AeL nanopore realizes the dual goals of both capturing and decelerating heterogeneously charged peptide translocation for nanopore sensing without labelling.Results and discussion
Design a new constriction region (R3) in AeL
Usually, the introduction of charged residues inside a nanopore lumen could regulate the EOF and the electrostatic barrier for controlling the transport of molecules. The EOF of WT AeL directs from cis-to-trans side under positive voltages, which could facilitate the cis-side capture of peptides. According to our previous studies, the R1 constricted region (T218 ∼ D222 and T274 ∼ S278) at the entrance of AeL determines the selectivity of the nanopore.27–31 The introduction of charged residues at R1 achieved a charge selective AeL sensor.32To meet the dual goals of both effective capturing and prolonging the translocation time of heterogeneously charged peptides, we chose two mutant sites in-between the two constricted regions of R1 and R2, which were the N226 site and S228 site, respectively. Asparagine(N)-226 was replaced by a glutamine (Q) residue with a bulky volume (denoted as N226Q) to build a third constricted region (named R3) below the R1 region (Fig. 1a). Then, a positively charged lysine residue was introduced at the 228 site (denoted as S228K). This design aimed to enhance the EOF for peptide capture while forming an electrostatic trap for decelerating the peptide translocation speed. The molecular dynamics (MD) simulations support that the diameter of N226Q/S228K AeL shrinks from approximately 1.2 nm to 0.8 nm in the designed R3 region, compared with WT AeL (Fig. 1a and S4†). The electrostatic potential rises higher at R3, indicating a stronger electrostatic field and a stronger force on the charged residue of the transported peptide.
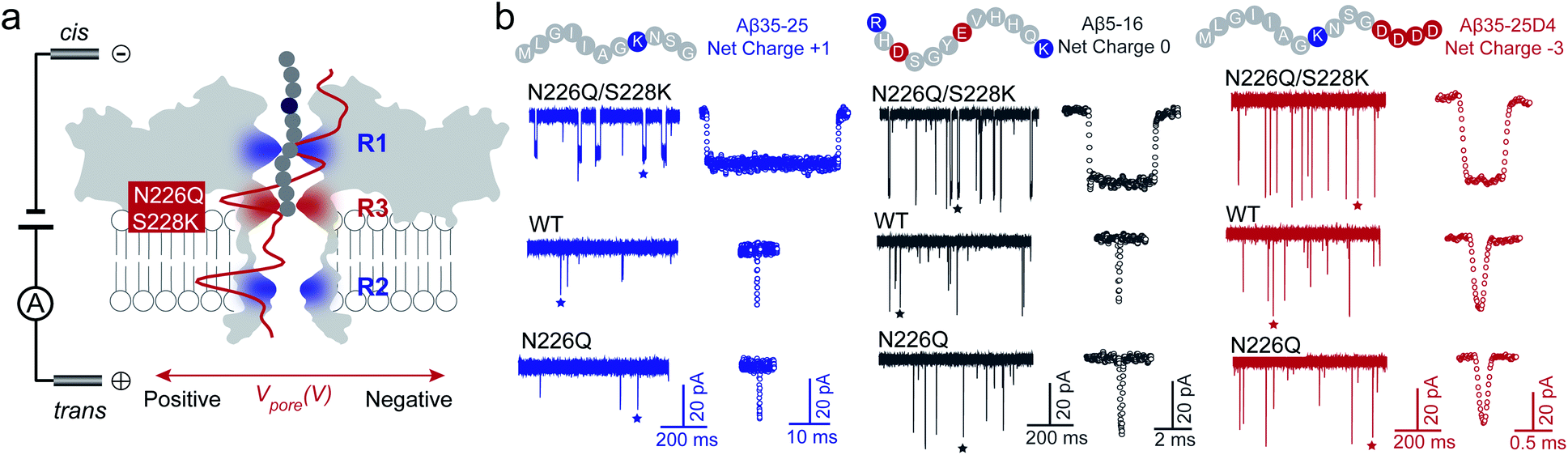 | ||
| Fig. 1 Building a new constricted region of AeL for heterogeneously charged peptide sensing. (a) Three electrostatic constricted regions of N226Q/S228K AeL for peptide sensing. The red (N226Q/S228K) band represents the new third electrostatic constricted region (R3). The other two electrostatic constrictions (R1: R220 and R2: K238) are also marked here (blue band). An all-atom model of the full-length aerolysin nanopore system was developed using the program NAMD6 and visualized using the program VMD. The red line is the electrostatic potential distribution along the N226Q/S228K AeL at +120 mV. (b) The typical current traces of heterogeneously charged peptides are obtained with N226Q/S228K AeL, WT AeL and N226Q AeL, respectively. From left to right: Aβ35-25, Aβ5-16, and Aβ35-25D4. The peptides were added to the cis chamber. All the nanopore experiments were performed in 1.0 M KCl, 10.0 mM Tris, and 1.0 mM EDTA at pH 8.0 under +120 mV bias. The final concentrations of the peptides were all 40.0 μM. Experimental details and procedures of molecular dynamics could be found in the ESI (ESI Methods and Fig. S3†). | ||
Next, we quantified the strength of EOF in the mutant N226Q/S228K AeL. The ion selectivity of AeL was calculated according to the reversal potential (Vm) by using the Goldman–Hodgkin–Katz equation.33 The Vm was measured by using asymmetric KCl concentrations on either side of the nanopore (ESI Methods and Fig. S3†). The calculated results reveal that the ratios of cation to anion permeability (PK+/PCl−) decrease from 0.52 (WT AeL) to 0.17 (N226Q/S228K AeL) after the mutation (ESI Methods†). Moreover, PK+/PCl− of N226Q AeL is comparable to that of WT AeL, which is 0.54. The calculated permeability ratio reveals that mutant N226Q/S228K AeL is more favorable for Cl− transport. These results demonstrate that the design of the R3 constricted region is feasible to modulate the ion permeability of AeL. Then the velocity of EOF is estimated34 and the results show that the electroosmotic velocity of N226Q/S228K AeL is about 2 times larger than that of WT AeL (detailed calculations are shown in ESI Methods†). These findings further support our design that charges at the R3 constriction could provide considerable EOF for cis-side capturing.
Driving and slowing the translocation of heterogeneously charged peptides
As shown in Fig. 1, three heterogeneously charged peptides were chosen as model peptides to examine the sensing ability of N226Q/S228K AeL. They are the fragments of amyloid-β peptides, which deposit in the brain with the progression of Alzheimer's disease.35 The chemical properties of the peptides are shown in Table S1.† Peptides were added to the cis side of AeL. The pH of the electrolyte solution in the cis chamber was 8.0, which could aid in maintaining the stability of biological nanopores, membranes and peptides. By using WT AeL, all heterogeneously charged peptides produced short blockades (Fig. 1b). For the positively charged Aβ35-25 (sequence: MLGIIAGKNSG, net charge (NC) +1), the direction of EPF is the trans-to-cis side, which is against cis-side capturing (Fig. 2a). Due to the insufficient EOF, positively charged peptides could hardly enter the WT AeL. With the larger strength of EOF, the sensing of Aβ35-25 (NC +1) with N226Q/S228K AeL gives an increase of 3 times the event frequency compared with WT AeL. Here, the event frequency, which is defined as the reciprocal of the interval time between two close blockades, is adopted as an indicator of peptide capturing. Moreover, electrostatic repulsion at the S228K site and the non-covalent interactions between the peptide and AeL in R3 slow the translocation of Aβ35-25. The duration of Aβ35-25 (NC +1) with N226Q/S228K AeL increases to 14.44 ± 2.12 ms at +120 mV, nearly 72 times larger than with WT AeL (Fig. 2a). As for the neutral peptide Aβ5-16 (sequence: RHDSGYEVHHQK, NC 0), the EOF also acts as the dominant driving force. Therefore, the frequency of Aβ5-16 with N226Q/S228K AeL shows a nearly 3-fold increase compared with WT AeL at +120 mV. The neutral peptide Aβ5-16 contains two negatively charged amino acids and two positively charged amino acids which experience electrostatic interactions with R3. Consequently, the duration of Aβ5-16 is prolonged to 3.57 ± 0.34 ms inside N226Q/S228K AeL, which is nearly 8 times larger than that of WT AeL (Fig. 2b). Moreover, N226Q/S228K also exhibits the capability of negatively charged peptide sensing. When positive voltage is applied, the direction of EPF for Aβ35-25D4 (sequence: MLGIIAGKNSGDDDD, NC −3) is cis-to-trans which is in accordance with EOF. The combination of EOF and EPF would increase the capture rate but accelerate the translocation speed for negatively charged peptides. The event frequency of Aβ35-25D4 (NC −3) with N226Q/S228K AeL is about 3 times higher than that with WT AeL. By virtue of the electrostatic trap and interaction at R3, under such a strong driving force, Aβ35-25D4 (NC −3) still exhibits a slow translocation with a duration time of 0.63 ± 0.01 ms (Fig. 2c). Note that N226Q AeL could not effectively trap the heterogeneously charged peptides, leading to low event frequencies and fast durations. Overall, the results demonstrate that the R3 in N226Q/S228K AeL allows for easy and long-time trapping of heterogeneously charged peptides before the translocation.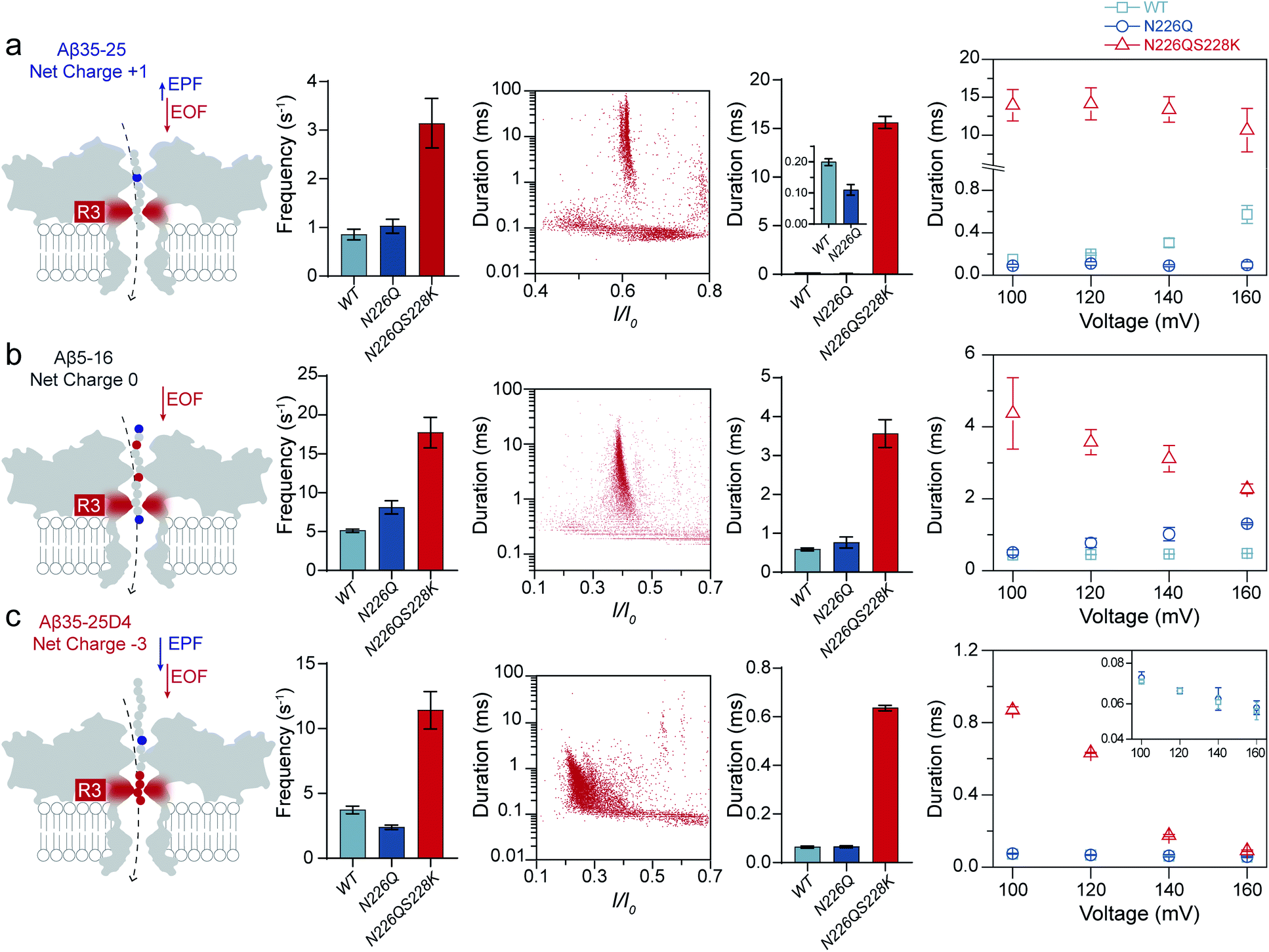 | ||
| Fig. 2 Sensing of heterogeneously charged peptides with N226Q/S228K AeL at pH 8.0 (a) Aβ35-25; (b) Aβ5-16; (c) Aβ35-25D4. From left to right: schematic representation of the EPF and EOF modulating the motion of heterogeneously charged peptides; the capture frequencies with various mutant AeLs at +120 mV; scatter plots showing the relationship between current blockade (I/I0) and duration; the durations with various mutant AeLs at +120 mV; effects of the applied voltages on the durations. | ||
To achieve nanopore peptide sequencing, the peptide should translocate through the nanopore without retracting back. Ideally, as peptides pass through the nanopore sensitive domain, all the sequence information of peptides can be read in single amino acid resolution. For N226Q/S228K AeL, the durations of Aβ35-25D4 and Aβ5-16 have negative correlations with voltages (+100 mV to +160 mV), which suggests the successful translocation (Fig. 2b and c). The duration time for Aβ35-25 is divided into two regimes (Fig. 2a). At low applied voltages (<+120 mV), the duration time increases; while at high voltages (>+120 mV), the duration time decreases. Below the threshold voltage, the net force of EOF and EPF is not able to overcome the high energy barrier for Aβ35-25 to translocate through the nanopore. When the applied voltage is higher than +120 mV, the peptide could overcome the energy barrier and the elevated voltage could accelerate the translocation speed. Regarding WT AeL and N226Q AeL, the durations of Aβ35-25 and Aβ5-16 stay constant or show positive correlations with voltages. Hence, the energy barrier in the R3 region is well tuned for decelerating the peptide translocation. The frequencies of heterogeneously charged peptides at different voltages with N226Q/S228K AeL are also shown in Table 1. The frequencies of all peptides have positive correlations with voltages. And the frequencies of Aβ5-16 are higher than those of Aβ35-25D4 under the same voltage, which may be caused by the length difference. Meanwhile, the numerous negatively charged residues at the cis entrance of AeL could also obstruct the entrance of the long biopolymer with rich negatively changes.36
| Contentsa | Aβ35-25D4 | Aβ5-16 | Aβ35-25 | Aβ25-35 |
|---|---|---|---|---|
| a The unit of frequency is s−1. And the standard deviations are also shown here. | ||||
| +100 mV | 6.35 ± 0.32 | 9.54 ± 0.64 | 1.80 ± 0.17 | 1.50 ± 0.28 |
| +120 mV | 11.75 ± 1.53 | 17.78 ± 1.92 | 3.13 ± 0.51 | 2.65 ± 0.21 |
| +140 mV | 17.56 ± 0.62 | 27.78 ± 4.81 | 4.70 ± 0.56 | 4.60 ± 0.28 |
| +160 mV | 18.92 ± 1.25 | 44.44 ± 9.62 | 7.70 ± 1.40 | 7.40 ± 0.42 |
Capturing and threading of a β-sheet peptide
Previous studies have shown that the bulky conformation of folded peptides could hinder their translocation probability.37 To evaluate the general sensing ability of N226Q/S228K AeL, a β-sheet peptide Aβ25-35 (sequence: GSNKGAIIGLM, NC +1), the reverse sequence analogue of Aβ35-25 (NC +1, random coil), was also driven into the nanopore at applied voltages (Fig. 3a). The diameter of the entrance of N226Q/S228K AeL is ∼2 nm which is suitable for accommodation of the β-sheet (∼2 nm). But the diameters of all three constricted regions are nearly 1 nm, which could give a high entropy barrier for bulky β-sheet peptide translocation. As a result, using WT AeL and N226Q AeL, Aβ25-35 produced only bumping events with a shorter duration (<0.50 ms) (Fig. 3c). However, employing N226Q/S228K AeL, the frequency and duration time of Aβ25-35 exhibit a ∼7-fold and ∼23-fold increase, respectively (Fig. 3a). The voltage-dependent duration of Aβ25-35 displays a threshold at +120 mV (Fig. S5†), which has a similar trend to that of Aβ35-25. Therefore, the folded peptide could also be driven into the wide entrance of N226Q/S228K AeL due to enhanced EOF, and then it undergoes a series of conformational changes at R1–R3 before its final translocation into linear form.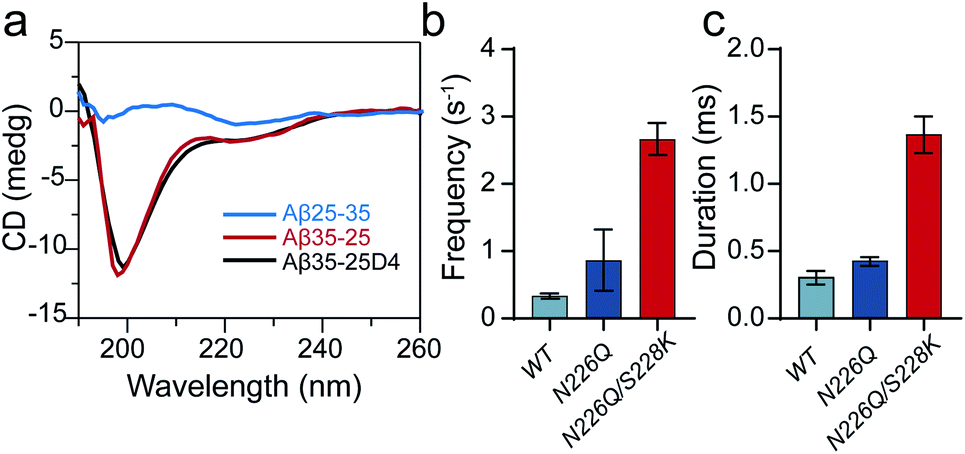 | ||
| Fig. 3 Sensing a β-sheet peptide Aβ25-35 with N226Q/S228K AeL. (a) The circular dichroism spectra of Aβ25-35 (blue), Aβ35-25 (red) and Aβ35-25D4 (black). The frequencies (b) and durations (c) of Aβ25-35 (NC +1) at +120 mV with various AeL mutants. | ||
Conclusions
In this study, a new third-electrostatic-constriction (R3) is created in the middle of the AeL nanopore by site-directed mutagenesis. The diameter of R3 shrinks from ∼1.2 nm to ∼0.8 nm. The decoration of positively charged amino acids in this confined space gives a large velocity of EOF in N226Q/S228K AeL, leading to an at least 3 times increase in capturing frequency for all heterogeneously charged peptides (NC +1 to −3). Moreover, the R3 region provides a dramatically electrostatic potential fluctuation and a higher possibility for strong non-covalent interactions so as to prolong the duration time of peptides. Even for the highly negatively charged Aβ35-25D4 (NC −3) with the largest driving force, N226Q/S228K AeL prolongs the duration to approximately 9 times. More importantly, folded Aβ25-35 (NC +1) could also be captured and completely threaded through N226Q/S228K AeL. These results suggest that N226Q/S228K AeL could be applied in the identification of folded peptides. The design strategy is practical without labelling or exposing the peptides and nanopores to extreme conditions (i.e., low pH). And with this engineering principle, further studies will focus on regulating the multiple constriction regions for peptide sensing. Our findings would be helpful to address the challenge of heterogeneously charged polypeptide capturing and prolonging, which could facilitate the achievement of the ultimate goal for nanopore protein sequencing.Data availability
The datasets generated during and/or analysed during the current study are not publicly available due to copyright policy and funding policy but are available from the authors on reasonable request.Author contributions
H. Y. N., Y. L. Y. and Y. T. L. conceived the idea. H. Y. N. performed the experiments. M. Y. L. made the MD simulation for AeLs. H. Y. N., Y. L. Y., and Y. T. L. analyzed the data. H. Y. N. and Y. L. Y. wrote the paper. Y. L. Y. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21834001, 21922405 and 22027806). Y. L. Y. acknowledges the sponsor from the National Ten Thousand Talent Program for young top-notch talent. We would like to thank Dr Xue-Yuan Wu for the preparation of WT Aerolysin and Miss Minyue Shen for data processing.Notes and references
- I. M. Derrington, T. Z. Butler, M. D. Collins, E. Manrao, M. Pavlenok, M. Niederweis and J. H. Gundlach, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16060–16065 CrossRef PubMed.
- A. H. Laszlo, I. M. Derrington, B. C. Ross, H. Brinkerhoff, A. Adey, I. C. Nova, J. M. Craig, K. W. Langford, J. M. Samson, R. Daza, K. Doering, J. Shendure and J. H. Gundlach, Nat. Biotechnol., 2014, 32, 829–833 CrossRef CAS PubMed.
- M. Drndić, Nat. Nanotechnol., 2014, 9, 743 CrossRef PubMed.
- H. Ouldali, K. Sarthak, T. Ensslen, F. Piguet, P. Manivet, J. Pelta, J. C. Behrends, A. Aksimentiev and A. Oukhaled, Nat. Biotechnol., 2020, 38, 176–181 CrossRef CAS PubMed.
- B. Yuan, S. Li, Y.-L. Ying and Y.-T. Long, Analyst, 2020, 145, 1179–1183 RSC.
- S. Yan, J. Zhang, Y. Wang, W. Guo, S. Zhang, Y. Liu, J. Cao, Y. Wang, L. Wang and F. Ma, Nano Lett., 2021, 21, 6703–6710 CrossRef CAS PubMed.
- H. Brinkerhoff, A. S. Kang, J. Liu, A. Aksimentiev and C. Dekker, Science, 2021, 374, 1509–1513 CrossRef CAS PubMed.
- F. L. R. Lucas, R. C. A. Versloot, L. Yakovlieva, M. T. Walvoort and G. Maglia, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- Z. Chen, Z. Wang, Y. Xu, X. Zhang, B. Tian and J. Bai, Chem. Sci., 2021, 12, 15750–15756 RSC.
- Q. Zhao, D. A. Jayawardhana, D. Wang and X. Guan, J. Phys. Chem. B, 2009, 113, 3572–3578 CrossRef CAS PubMed.
- A. J. Wolfe, M. M. Mohammad, S. Cheley, H. Bayley and L. Movileanu, J. Am. Chem. Soc., 2007, 129, 14034–14041 CrossRef CAS PubMed.
- X. Chen, Y. Zhang, P. Arora and X. Guan, Anal. Chem., 2021, 93, 10974–10981 CrossRef CAS PubMed.
- M. M. Mohammad, S. Prakash, A. Matouschek and L. Movileanu, J. Am. Chem. Soc., 2008, 130, 4081–4088 CrossRef CAS PubMed.
- S. Li, X.-Y. Wu, M.-Y. Li, S.-C. Liu, Y.-L. Ying and Y.-T. Long, Small Methods, 2020, 4, 2000014 CrossRef CAS.
- S. Li, C. Cao, J. Yang and Y.-T. Long, ChemElectroChem, 2019, 6, 126–129 CrossRef CAS.
- A. Asandei, I. Schiopu, M. Chinappi, C. H. Seo, Y. Park and T. Luchian, ACS Appl. Mater. Interfaces, 2016, 8, 13166–13179 CrossRef CAS PubMed.
- S. P. Bhamidimarri, J. D. Prajapati, B. van den Berg, M. Winterhalter and U. Kleinekathöfer, Biophys. J., 2016, 110, 600–611 CrossRef CAS PubMed.
- L.-Q. Gu, S. Cheley and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15498–15503 CrossRef CAS PubMed.
- A. E. Chavis, K. T. Brady, G. A. Hatmaker, C. E. Angevine, N. Kothalawala, A. Dass, J. W. Robertson and J. E. Reiner, ACS Sens., 2017, 2, 1319–1328 CrossRef CAS PubMed.
- M. Boukhet, F. Piguet, H. Ouldali, M. Pastoriza-Gallego, J. Pelta and A. Oukhaled, Nanoscale, 2016, 8, 18352–18359 RSC.
- G. Huang, K. Willems, M. Soskine, C. Wloka and G. Maglia, Nat. Commun., 2017, 8, 1–11 CrossRef CAS PubMed.
- G. Huang, K. Willems, M. Bartelds, P. van Dorpe, M. Soskine and G. Maglia, Nano Lett., 2020, 20, 3819–3827 CrossRef CAS PubMed.
- G. Di Muccio, B. M. Della Rocca and M. Chinappi, ArXiv Prepr, ArXiv210403390, 2021.
- Y. Wang, K. Tian, X. Du, R.-C. Shi and L.-Q. Gu, Anal. Chem., 2017, 89, 13039–13043 CrossRef CAS PubMed.
- Z. Gu, H. Wang, Y.-L. Ying and Y.-T. Long, Sci. Bull., 2017, 62, 1245–1250 CrossRef CAS.
- M.-Z. Huo, Z.-L. Hu, Y.-L. Ying and Y.-T. Long, Proteomics, 2021, 2100041 CrossRef PubMed.
- Y.-Q. Wang, M.-Y. Li, H. Qiu, C. Cao, M.-B. Wang, X.-Y. Wu, J. Huang, Y.-L. Ying and Y.-T. Long, Anal. Chem., 2018, 90, 7790–7794 CrossRef CAS PubMed.
- C. Cao, M.-Y. Li, N. Cirauqui, Y.-Q. Wang, M. Dal Peraro, H. Tian and Y.-T. Long, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- M.-Y. Li, Y.-L. Ying, J. Yu, S.-C. Liu, Y.-Q. Wang, S. Li and Y.-T. Long, JACS Au, 2021, 1, 967–976 CrossRef CAS PubMed.
- J.-G. Li, M.-Y. Li, X.-Y. Li, X.-Y. Wu, Y.-L. Ying and Y.-T. Long, Langmuir, 2022, 38(3), 1188–1193 CrossRef PubMed.
- M.-Y. Li, Y.-L. Ying, S. Li, Y.-Q. Wang, X.-Y. Wu and Y.-T. Long, ACS Nano, 2020, 14, 12571–12578 CrossRef CAS PubMed.
- Y.-Q. Wang, C. Cao, Y.-L. Ying, S. Li, M.-B. Wang, J. Huang and Y.-T. Long, ACS Sens., 2018, 3, 779–783 CrossRef CAS PubMed.
- W. F. Pickard, Math. Biosci., 1976, 30, 99–111 CrossRef CAS.
- F. Piguet, F. Discala, M.-F. Breton, J. Pelta, L. Bacri and A. Oukhaled, J. Phys. Chem. Lett., 2014, 5, 4362–4367 CrossRef CAS PubMed.
- A. Hatami, S. Monjazeb, S. Milton and C. G. Glabe, J. Biol. Chem., 2017, 292, 3172–3185 CrossRef CAS PubMed.
- Z.-L. Hu, M.-Y. Li, S.-C. Liu, Y.-L. Ying and Y.-T. Long, Chem. Sci., 2019, 10, 354–358 RSC.
- Y.-X. Hu, Y.-L. Ying, Z. Gu, C. Cao, B.-Y. Yan, H.-F. Wang and Y.-T. Long, Chem. Commun., 2016, 52, 5542–5545 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06459b |
| This journal is © The Royal Society of Chemistry 2022 |