
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoswitchable architecture transformation of a DNA-hybrid assembly at the microscopic and macroscopic scale†‡
Nadja A.
Simeth§¶
 a,
Paula
de Mendoza§
a,
Victor R. A.
Dubach
b,
Marc C. A.
Stuart
ab,
Julien W.
Smith
a,
Tibor
Kudernac
a,
Wesley R.
Browne
a and
Ben L.
Feringa
*a
a,
Paula
de Mendoza§
a,
Victor R. A.
Dubach
b,
Marc C. A.
Stuart
ab,
Julien W.
Smith
a,
Tibor
Kudernac
a,
Wesley R.
Browne
a and
Ben L.
Feringa
*a
aStratingh Institute for Chemistry, Faculty for Science and Engineering, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
bGroningen Biomolecular Sciences and Biotechnology, Faculty for Science and Engineering, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands
First published on 2nd March 2022
Abstract
Molecular recognition-driven self-assembly employing single-stranded DNA (ssDNA) as a template is a promising approach to access complex architectures from simple building blocks. Oligonucleotide-based nanotechnology and soft-materials benefit from the high information storage density, self-correction, and memory function of DNA. Here we control these beneficial properties with light in a photoresponsive biohybrid hydrogel, adding an extra level of function to the system. An ssDNA template was combined with a complementary photo-responsive unit to reversibly switch between various functional states of the supramolecular assembly using a combination of light and heat. We studied the structural response of the hydrogel at both the microscopic and macroscopic scale using a combination of UV-vis absorption and CD spectroscopy, as well as fluorescence, transmission electron, and atomic force microscopy. The hydrogels grown from these supramolecular self-assembly systems show remarkable shape-memory properties and imprinting shape-behavior while the macroscopic shape of the materials obtained can be further manipulated by irradiation.
Introduction
DNA is known from biology for its high storage information density, its outstanding chemical stability, and its precisely defined supramolecular architecture.1 Indeed, sequence-specific base-pairing allows for readily programmable 2D and 3D structures.2–4 These specific interactions lead to the formation of the thermodynamic product in DNA-based supramolecular assemblies, starting from the meta-stable intermediates formed under kinetic conditions.2 For instance, heat-induced dissociation (melting) of double-stranded DNA (dsDNA) can be reverted by simple cooling of the dsDNA below its melting temperature (Tm).3,5 As a consequence DNA-based materials possess a nano and microscale memory, which results in the thermodynamically favored structure dictated by the base-pair sequence.Due to these unique properties, DNA is particularly versatile in controlling assembly and organization as seen in DNA-based nanotechnology,4,6–15 DNA-origami,8,16–21 hydrogels,22–25 and polymeric materials as a crosslinker.26–29 Alternatively, a single-stranded DNA (ssDNA) can be employed as a readily available and programmable template to construct supramolecular DNA hybrid structures.2 This concept of molecular recognition-driven self-assembly was established by Lehn et al. and can be used in a bottom-up approach to easily obtain complex architectures from simple building blocks.30,31 These assemblies often exhibit distinct properties compared to the individual molecules, owing to the strong intermolecular interactions.2,30,31 The hydrogen bond patterns naturally encoded between the DNA base-pairs direct the assembly of supramolecular fibers,32–34 hydrogels,22–24,32,35 liquid crystals,10,36,37 nanoarrays,38–43 and programmed nanoparticles,44 among others (Fig. 1A).2,45,46
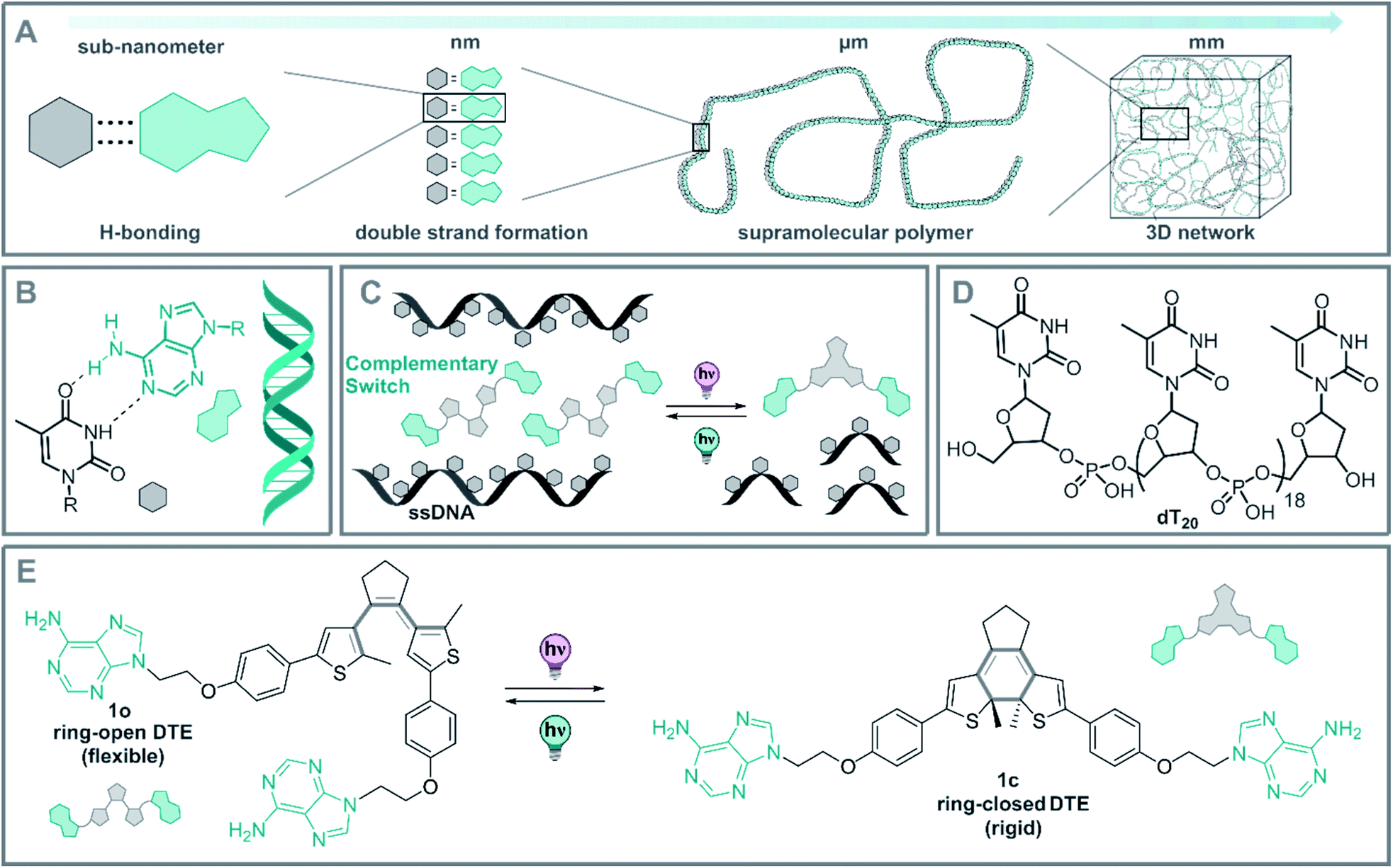 | ||
| Fig. 1 (A) Translation of supramolecular interactions from the molecular to the macroscopic scale. (B) H-base pairing between thymine and adenine and representation of a dsDNA. (C) Design of a DNA-DTE biohybrid system and envisioned interaction between a homo-ssDNA and a photoswitch with complementary base-pairs. (D) Representation of the ssDNA used based on 20 thymine units (dT20). (E) Reversible photoswitching of DTE 1o and 1c through an electrocyclization. | ||
However, the dynamical regulation of these supramolecular architectures and their properties necessitate an external control element to grant an additional level of functionality. Several stimuli have been explored in the context of DNA-structure regulation and DNA hybrid architectures.35,47–51 Within these stimuli, light stands out due the traceless nature of the photon as a reactant, its spatiotemporal precise dosing and the high tunability of its energy and intensity.51 In this way, the structure52–58 and function57,59–63 of DNA and ribonucleic acid (RNA) could be controlled by light. Moreover, controlling these architectures at the molecular level, will allow control of the characteristics of DNA-based supramolecular self-assemblies over various length scales from nano-dimensions up to the macroscopic world (Fig. 1A).
As a part of our ongoing research on photoregulated oligonucleotide structure and function, i.e. photoswitchable DNA hybridization53 and DNA glue,54 we designed a biohybrid system employing the hydrogen-bond driven base-pairing between the nucleic acids adenine and thymine (Fig. 1B) using a short DNA strand and a photoswitch functionalized with the complementary nucleobase as displayed in the conceptional Fig. 1C. For our design, we employed a homo-nucleotide single-strand based on 20 thymine units (dT20, Fig. 1D) and a dithienylethene (DTE) photoswitch64,65 decorated with two adenines (Fig. 1E). This photoactuator can be reversibly isomerized between a more flexible, ring-open (1o) and a rigid, ring-closed isomer (1c). Photoswitches based on such an electrocyclization reaction are P-type photoswitches where both the states are thermally stable even at elevated temperatures.64,66 This beneficial characteristic is crucial to introduce non-invasive photochemical control orthogonal to the thermal melting typical of DNA and DNA hybrids.
Results and discussion
A bis-chloro-functionalized DTE core structure was synthesized according to a procedure published earlier by our group.67 Two bromo ethoxy phenyl groups were installed via Suzuki–Miyaura cross-coupling which were subsequently used to alkylate adenosine to result in DTE 1o (Fig. 1E, see ESI‡ for experimental procedures and characterization).The open isomer obtained is colourless, with an absorption at λmax 267 nm, which tails to ca. 350 nm (EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TE buffer 1:1, see Fig. 2A). This shoulder in the UV-vis absorption spectrum can be used to manifest changes involving DTE in the target system, exploiting the negligible absorbance of DNA at wavelengths longer than 300 nm (see ESI‡). DTE 1o undergoes selective photoisomerization in solution upon irradiation at 300 nm, manifested in an increase in visible absorbance showing distinct isosbestic points (Fig. 2A). The colourless open form was thereby converted into the pink, closed isomer 1c (λmax 521 nm, EtOH:TE buffer 1:1, Fig. 2A) reaching a photostationary state distribution (PSD) of 71:29 C:O at the photostationary state (PSS, determined by 1H NMR spectroscopy in DMSO-D6, see Fig. 2B and ESI‡). Upon subsequent irradiation with visible light (>420 nm) the ring-closed isomer could be converted back quantitatively to the open isomer. The electrocyclization was reversible without fatigue over several cycles, both in the presence and absence of ssDNA (dT20, see insert in Fig. 2A and ESI‡).
:TE buffer 1:1, see Fig. 2A). This shoulder in the UV-vis absorption spectrum can be used to manifest changes involving DTE in the target system, exploiting the negligible absorbance of DNA at wavelengths longer than 300 nm (see ESI‡). DTE 1o undergoes selective photoisomerization in solution upon irradiation at 300 nm, manifested in an increase in visible absorbance showing distinct isosbestic points (Fig. 2A). The colourless open form was thereby converted into the pink, closed isomer 1c (λmax 521 nm, EtOH:TE buffer 1:1, Fig. 2A) reaching a photostationary state distribution (PSD) of 71:29 C:O at the photostationary state (PSS, determined by 1H NMR spectroscopy in DMSO-D6, see Fig. 2B and ESI‡). Upon subsequent irradiation with visible light (>420 nm) the ring-closed isomer could be converted back quantitatively to the open isomer. The electrocyclization was reversible without fatigue over several cycles, both in the presence and absence of ssDNA (dT20, see insert in Fig. 2A and ESI‡).
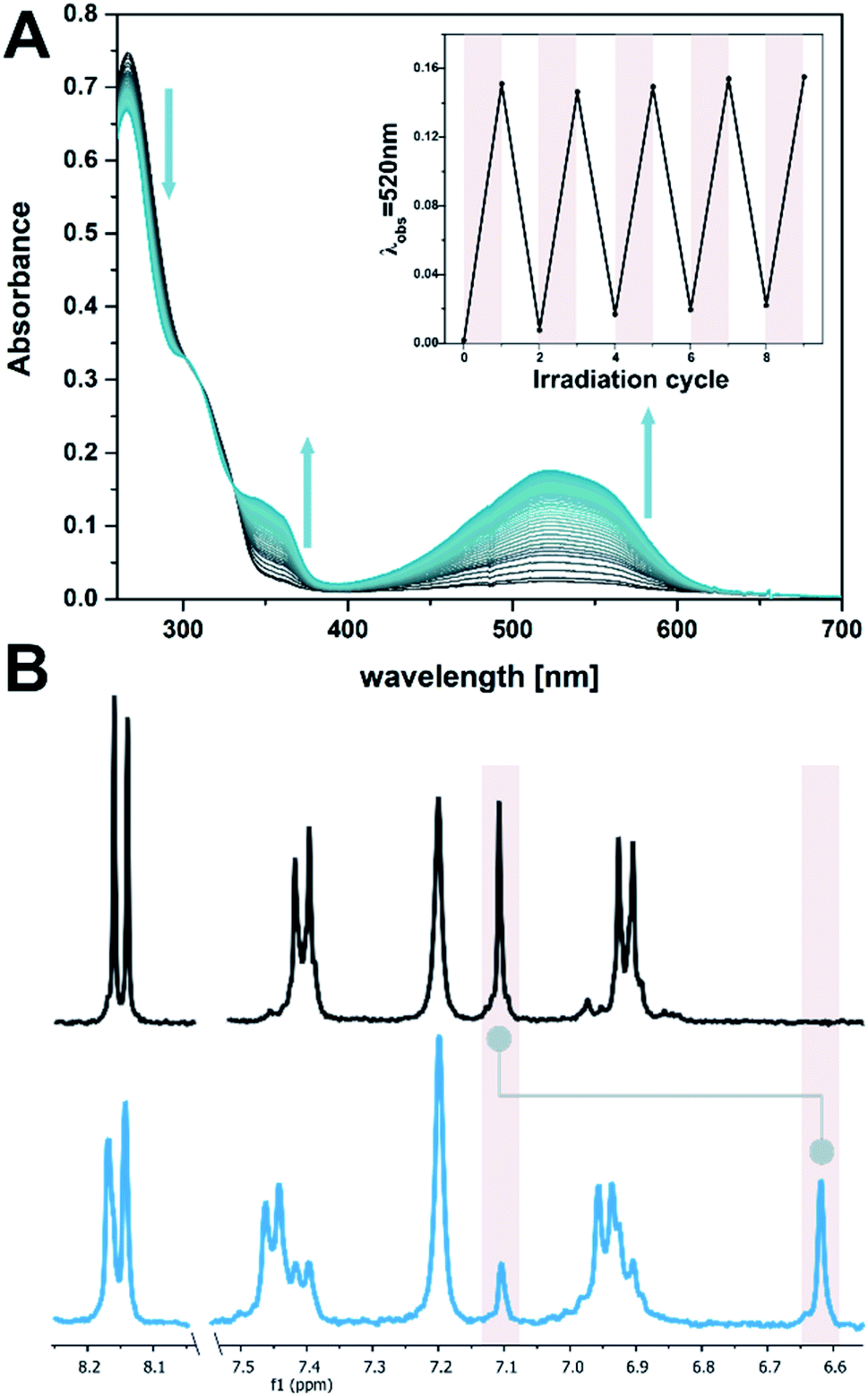 | ||
| Fig. 2 Photochemical isomerization of DTE 1. (A) Time-resolved UV-vis absorption spectrum of 1 (10 μM in EtOH:TE buffer 1:1, with 2 μM dT20) while irradiating at 300 nm (spectra are at 1 s intervals). The insert shows the reversibility of the photochemical electrocyclization over several cycles (alternate irradiation at 300 nm and with visible light, i.e. >420 nm). (B) Aromatic region of the 1H spectrum of DTE 1 in DMSO-D6 (1 mM) before (top) and after (bottom) irradiation at 300 nm. Highlights show the characteristic thiophene singlet at 7.1 ppm (open) or 6.6 ppm (closed isomer), respectively, for which a PSD300 of 71:29 C:O was determined (cf. ESI‡). | ||
dT20 ssDNA and DTE 1o (DTE 1o 0.25 mM and dT20 25 μM, EtOH:TE, 1:1) were mixed followed by a heating–cooling cycle (75 °C → 8 °C → room temperature, for detailed protocols see ESI‡) to facilitate a supramolecular assembly, similar to those used in the study of natural dsDNA hybridization.68,69 The samples were translucent directly after heating, and became cloudy upon cooling (see ESI‡). They were cooled on ice for one hour to favour aggregation. Analysis by circular dichroism (CD) spectroscopy indicated that the structures formed consisted of both ssDNA and DTE, as a negative induced CD (ICD) between 300 nm and 350 nm was observed (Fig. 3A; see Fig. 3B for a representation and images of the samples on the macroscopic scale). This signal can be attributed to the presence of 1o, interacting with ssDNA via H-bonding. Transmission electron microscopy (TEM) confirmed the formation of small aggregates under kinetic conditions (Fig. 3C and D). The process was completed by storing the samples for an additional 18 h at 8 °C and then aging them further at room temperature. The aging process was accompanied by an inversion of the ICD signal at 335 nm (−15 mdeg → +17 mdeg, Fig. 3A and ESI‡) indicating the formation of a thermodynamically favoured assembly over time, a behaviour observed in natural dsDNA also.1 At the macroscopic scale, we could observe that the inversion of the ICD signal was accompanied by the formation of a homogeneous network at ambient conditions (Fig. 3B, central image). Both TEM and atomic force microscopy (AFM) images revealed that this network consisted of regularly twisted ribbons (Fig. 3E and ESI‡). Upon further aging, the network appeared to contract, to eventually form a floating, macroscopic, cylindrical 3D structure in the vial in which the sample was prepared in (Fig. 3B, right image). Analysing the corresponding aged sample with TEM showed that several of the helical fibres had formed larger bundles and are consequently tightly packed (Fig. 3F and ESI‡).
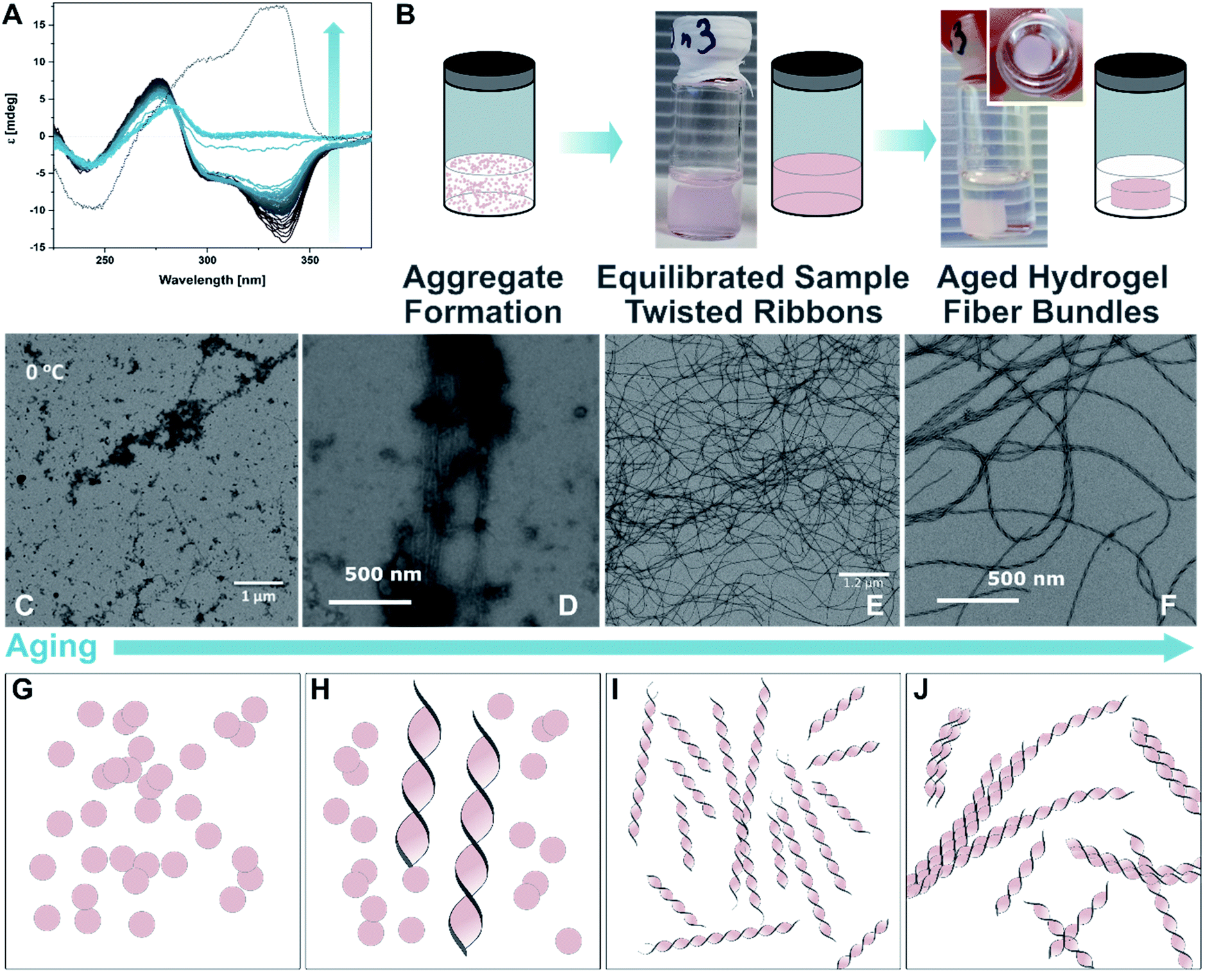 | ||
| Fig. 3 Recognition-driven self-assembly on the nanoscopic, microscopic, and macroscopic scale in the presence of 1o. (A) Aging following by CD spectroscopy. The ICD of 1o in the presence of dT20 (1:1 in EtOH:TE buffer 1:1) inverts over time and is accompanied by the transition from the metastable aggregates to the equilibrated self-assembly (twisted ribbons; grey to blue solid lines: every 60 min over 48 h, dotted line: 4 d). (B) Representation and photographs of the aging process at the macroscopic scale. After the initial formation of aggregates under kinetic conditions (see also ESI‡), the sample is equilibrated at ambient temperature having formed a network, and eventually results in the formation of a shape-resilient hydrogel. (C) and (D) TEM images of 1o and dT20 as metastable aggregates (1:1 in EtOH:TE buffer 1:1, at 0 °C shown in (C) and after 5 min at ambient temperature in (D)), (E) after equilibration as a network of twisted ribbons (1:1 in EtOH:TE buffer 1:1), and (F) as aged sample having formed a hydrogel consisting of twisted ribbons that have formed tight bundles (1:1 in EtOH:TE buffer 1:1, aged at ambient temperature after staining with ethidium bromide; for the corresponding fluorescence microscopy image, see ESI‡). (G–J) Representation of proposed mechanism of the formation of shape-resilient hydrogels based on TEM images (C–F): the aggregates initially formed under kinetic conditions (vial with turbid solution, see ESI‡) slowly start to grow into twisted ribbons at ambient temperature. Over time, the ribbons grow larger forming a network and eventually form tighter bundles after aging. The formation of bundles is probably accompanied by a contraction of the network, expulsion of solvent, and consequently the formation of a hydrogel on the macroscopic scale that has the shape of its vessel imprinted. | ||
Based on these observations, we propose that the initially formed aggregates grow into increasingly longer twisted ribbons once the system is allowed to equilibrate (Fig. 3B, TEM images in Fig. 3C–E, and schemes in Fig. 3G–I). These fibres build a network throughout the vessel that allows formation of a hydrogel. The latter was imaged at different length scales using TEM (vide supra, Fig. 3E and F), AFM (see ESI‡), and fluorescence microscopy after staining the sample with the DNA intercalator ethidium bromide (see ESI‡). After further aging, the twisted ribbons form bundles (TEM image in Fig. 3F, representation in Fig. 3J), that lead to a synchronous contraction of the hydrogel-network under expulsion of solvent. Eventually, a smaller hydrogel was obtained that had the shape of the vial it was grown in imprinted. The formation was completed after ca. 8–10 weeks of aging and resulted in a cylindrical hydrogel with a diameter of ca. 1/3 of the vial (see TOC image and ESI‡). The gel is characterised by an initial storage modulus (G′) of 70 Pa and a loss modulus (G′′) of 10 Pa (details see ESI‡). The gel starts to show inelastic deformation at a strain around 9%, with a final breaking point around 35%. However, repeated measurements of the same sample showed self-healing properties of the gel. Interestingly, G′ increased with every round to ca. 90 Pa after five repetitions, while G′′ stayed constant. Simultaneously, the inelastic regime started earlier, around 4% strain, with the final breaking point around 20% oscillator strain. We assume that by increasing the strain on the gel, additional sites for supramolecular cross-linking between individual fibres become available gradually increasing the stiffness of the gel but making it more prone to breaking. The observed self-healing characteristics of the gel strongly indicates that the key interactions between the individual components are indeed H-bonding interactions that can be disrupted and reformed once stress is released. Besides the molecular self-healing properties, the gel also exhibits unusual shape-resilient properties that are discussed later.
Similar structures did not form when samples with isolated dT20 and 1o were prepared and characterized in the same way. This difference, together with the observed positive ICD signal led us to conclude that the twisted ribbons consist of an assembly of 1o and dT20, as a biohybrid structure based on supramolecular interactions between the two components. TEM imaging showed that the twisted ribbons have a width of 25 nm, a helical pitch of 60–65 nm, and lengths of up to of tens of micrometers (see Fig. 4A). Measurements by AFM confirmed the size of the helical pitch (67 nm, see ESI‡) and showed heights of the twisted ribbons on surface between 5.2–18.7 nm depending on the sample and the position on the twisted ribbon that was chosen for reference (see ESI‡ for further details). The obtained dimensions suggests that the observed structure is consisting of more than one biohybrid double helix (such as envisioned in our design in Fig. 1C). With the calculated diameter of dT20–1o–dT20 being ca. 5 nm (see Fig. 4), the observed ribbons likely consist of ca. five repeating units of dT20–1o–dT20 in x-direction and one unit in z-direction. We hypothesis that the dT20 oligomer serves a crucial role for the growth of the fibre in both x- and y-direction. In y-direction overhangs of different lengths allow for the growths of fibres that are longer than 20 bases. Along the x-axis, free positions around the winded ssDNA could allow for double stand invasion, such as observed, for instance, in peptide nucleic acids,70 or crossovers to neighbouring helices (see schematic drawing in Fig. 4C). Such stabilising crossover threads were previously designed for bundling together several DNA double helices into DNA nanorods.71 We further assume that such interactions could also assist the formation of larger bundles during the aging and shrinking process, that eventually leads to the formation of the observed shape-persistent hydrogel.
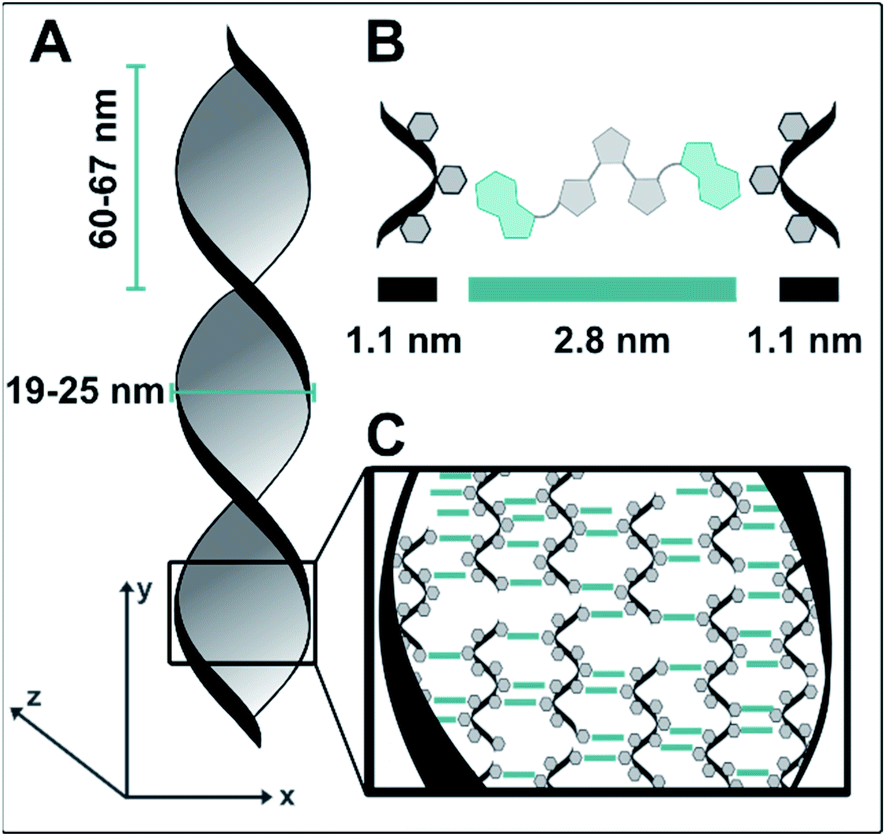 | ||
| Fig. 4 Representation of the twisted ribbons build of 1o and dT20 (1:1 in EtOH:TE buffer) as observed by TEM and AFM. (A) The ribbons are 19–25 nm wide along the x-axis and have a helical pitch of 60–67 nm. In y-direction, they grow several tens of μm, while AFM measurements indicate that they have a thickness of ca. 5 nm along the z-axis. (B) Representation of one unit of dT20–1o–dT20 and its dimension. (C) Structural proposal of the observed ribbons. Based on the dimensions obtained from AFM and TEM, they likely consist of four to five repeating units of dT20–1o–dT20 in x-direction and one unit in z-direction. Interaction between neighbouring helices could be facilitated by double stand invasion or crossovers of some of the dT20 single stands. | ||
We rationalise this structural proposal as the distance along the y-axis at the centre of twisted ribbons is shorter than at the edges. The central structure is a regular helix as displayed in Fig. 1D. However, the supramolecular entities formed at the centre of the ribbon potentially exhibit packing mismatches towards the edges allowing additional molecules to interact and further biohybrid helices to attach. The ultimate width of the ribbons appears to be limited probably due to a delicate balance between stable packing of the available molecules and induced strain upon twisting.
We probed this hypothesis by varying the ratio of the base-pairs between 1o and dT20 to investigate the growth processes of the twisted ribbons further. With sub-stochiometric amounts of DTE 1o, the formation of large fibres was observed (cf. ESI‡). With increasing amounts of switch, the supramolecular structures progressively were wound tighter. At a base pair ratio of ca. 1:1, the fibres showed the highest degree of twisting accompanied by the formation of supercoils, both structural features reminiscent of naturally occurring dsDNA.1 The twisted ribbons, that were observed at ratios around 1:1, appear to be the most stable structure independent of a slight excess of building blocks present and resulted in the formation of hydrogels. However, deviating too much from this ideal ratio gives rise to larger fibres which are not twisted supporting our hypothesis on the structural formation of twisted ribbons. These larger fibres also did not result in the formation of hydrogels.
We studied the formation of the hydrogels consisting of 1o and dT20 in more detail due to their unusual macroscopic properties. Of particular note was the cylindrical shape of the originally obtained structure shows retention of the form of the vial in which the structure grew (vide supra). This hypothesis was tested by preparation of several identical samples which were transferred to vessels of different shape (Fig. 5A and B). A triangular, a cuboid, and a ring-shaped glass vessel were selected in addition to the original cylinder-shaped vial (vide supra). All samples were aged together first at 8 °C and then at ambient temperature in the dark. The initially cloudy solutions showed the formation of homogenous structures throughout the whole vessel after two days aging. Then, the structures slowly contracted, losing contact with the vessel walls analogous to the previously obtained cylindrical shape. The contraction process continued to eventually result in the formation of hydrogels imprinted with the shapes of the containing vessels. Both the triangle, the cuboid, and the ring could be successfully casted. However, the cuboid took ca. 3–6 weeks until a defined, free-floating 3D-structure could be observed, while the ring and the triangle reached the same state already after 1–2 weeks (cf. ESI‡). This indicates that though the differently shaped hydrogels follow the same formation process as the cylindrically shaped gel discussed earlier, the 3D-structure exhibits some sensitivity to complexity of the shape.
 | ||
| Fig. 5 Imprinting distinct shapes to hydrogels; growing of hydrogels of different shape at the microscopic and macroscopic scale. (A) Representation of the formation metastable aggregates (top), a homogeneous network of twisted ribbons (equilibrated, middle), and eventually a tight network of fibre bundles forming a shape-resilient hydrogel (bottom). (B) Photographs of the experimentally observed different states schematically shown in (A) of a cuboid, a ring and a triangular-shaped hydrogel. Note that the cuboid sample was not irradiated, but that ambient light caused a colour-change over time penetrating through the quartz cuvette, while the glass vials filtered larger amounts of UV-light. | ||
Typically, hydrogels that exhibit a well-defined shape are casted following protocols that significantly differ from our approach. For instance, natural or synthetic polymers are dissolved in aqueous media and subsequently crosslinked by UV-light irradiation or other means.72 They can be either casted into their final shape directly or modified afterwards by simply cutting out the desired structure.72,73 These treatments will give the respective hydrogels their permanent shape. Hydrogels that exhibit memory properties of their permanent shape attract considerable attention and are subject of various studies. Upon application of a stimulus, such as pressure,24,74 light,27,29,75etc., they undergo transitions into a metastable structure.72,73,76,77 Applying a counter stimulus, the original permanent shape can be recovered due to a memory code present in the gel. Such memory codes can be, for instance, the entanglement of the polymer chains or the association and dissociation of cross-linking elements based on supramolecular interactions.72,77,78 In particular, the programmable supramolecular interactions of DNA have been studied in this context and could be used to facilitate complex, multi-stimuli responsive behaviour as well as self-healing properties.29,72 For instance, the permanent shape can be converted into a metastable one by irradiation, oxidation, or chelation of cations crucial for the supramolecular interactions. The original shape can be recovered by applying the corresponding counter stimulus.30 Also, polymeric shape-memory hydrogels can be swollen with water to assume their metastable shape and the original shape can be recover upon drying.72,79–81
In sharp contrast, our system memorises the shape it was casted in after having lost contact to the walls of its cast, while still being in the original aqueous solution. As a consequence of a complex shrinking process, that we ascribe to a synchronous formation of fibre bundles throughout the vessel (vide supra). However, the shape-memory we observe here, is fundamentally different from classical shape-memory behaviour, and is to the best of our knowledge unprecedented. Consequently, it will require further studies to be fully understood and to reveal its mechanistic details on the molecular level.
The hydrogels were also shape-resilient when exchanging the solvent to TE buffer or when transferred into a vessel of different shape. For example, a cylindrically shaped gel was transferred into a cuboid vessel, namely a spectroscopy cuvette. During the process, the solvent was exchanged to avoid filter effects caused by excess photoswitch in solution. We irradiated the same sample at 300 nm and observed a colour change to purple and further shrinking of the hydrogel, while the cylindrical shape was largely retained. Macroscopically, the gel appeared to be more fragile, as small pieces detached. However, TEM showed intact twisted ribbons and a network density comparable to a non-irradiated sample that followed the same aging procedure. Also, G′ and G′′ did not alter significantly and the gel kept its self-healing properties (more details see ESI‡). In contrast, when the sample was irradiated with UV light and subjected to another heating–cooling cycle before aging, no gel formation was observed. We found that the closed isomer 1c formed only small aggregates with DNA confirmed by an ICD in the visible region (Fig. 6A). TEM imaging showed that the sample also contained short, twisted ribbons (25 nm width, 60–65 nm helical pitch, average length 1–1.5 micrometers, cf.Fig. 6B and ESI‡) that we ascribe to remaining 1o in the mixture due to incomplete photoisomerization (PSD300 of 71:29 C:O, vide supra).
 | ||
| Fig. 6 Analysis of 1c. (A) Irradiation of the equilibrated self-assembled structures (black line) at 300 nm and accompanied O → C electrocyclization leads to loss of the ICD (grey line). A subsequent heating–cooling cycle results in a new interaction between DNA and 1c as indicated by a different ICD (blue line). (B) TEM images of 1c and dT20 equilibrated (1:1 in EtOH:TE buffer 1:1 after irradiation to the PSS300). | ||
We tested the propensity of the hydrogel to be further manipulated by photochemical stimuli (cf. setting in Fig. 7A). We imprinted a ring shape into a hydrogel containing 1o and dT20 (1:1 in EtOH:TE buffer 1:1) following our standard protocol (heating, cooling, equilibration at room temperature and aging). Then, we ruptured one side mechanically to obtain a sheet (Fig. 7B). The central area of the hydrogel-sheet was irradiated at 300 nm by placing an LED in front of the vial (Fig. 7A). Similar to the irradiated cylindrical sample discussed above, irradiation with light locally induced shrinking of the hydrogel, formation of a hole, and eventually, photochemical tearing at the irradiation site (see Fig. 7C and ESI‡). Upon illumination, the colour of the gel changed from practically colourless to pink, which indicates O → C photoisomerization of the switch. However, TEM images of irradiated hydrogels show structurally intact twisted ribbons (vide supra and ESI‡) and do not allow to correlate photoswitching at the molecular level with the observed macroscopic effects. Also, shrinking and tearing due to local heating through the LED can be excluded as the same experiment employing a 700 nm or a 526 nm LED, respectively, left the hydrogel unaltered. Consequently, absorption of light of a suitable wavelength, such as provided by the 300 nm LED, by 1o appears crucial to locally manipulate the shape of the hydrogel either through photoswitching or photothermal effects mediated by the dye.
 | ||
| Fig. 7 Photochemical tearing of a sheet-shaped hydrogel. (A) Irradiation setting consisting of a 300 nm LED and a glass vial containing the previously casted hydrogel-sheet. (B) Schematic representation of the experiment displaying the initially casted ring, which was mechanically teared to form a sheet and then irradiated at the central part of the sheet. The irradiation induced initially local shrinking and led eventually to photochemically induced tearing of the sheet into two fragments. (C) Photographs of the experiment summarised in (B) showing the vial containing the hydrogel-sheet from the side and the bottom at different time-points of irradiation (pictures were taken ca. every hour). The initially local shrinking leads to the formation of a hole and finally to a tearing of the sheet. Also, the colour change accompanied with O → C photoisomerization can be seen. | ||
In contrast to irradiation, heating to ca. 80 °C led to melting and dissolution of the shape-memory hydrogels and loss of the 3D structure (see ESI‡), probably breaking the hydrogen-base pair interactions of the supramolecular ensemble. We employed this property to recover the originally assembly, which showed the formation of metastable aggregates, by subsequent cooling of the sample. In a second aging process, the shape-memory hydrogels could be recovered (see ESI‡). Similarly, samples containing 1c were irradiated with visible light to induce the photochemical C → O reaction. A subsequent heating–cooling–aging cycle allowed us to recover the originally obtained hydrogels (see ESI‡). The full cycle could also be followed by CD spectroscopy and TEM (see ESI‡) confirming that the biohybrid system goes in the second cycle through the same steps as it did in the first one demonstrating the full reversibility and recyclability of our supramolecular biohybrid ensemble.
Conclusion
In conclusion, we have designed a molecular recognition-driven hydrogel in which ssDNA initially acts as a template for the chiral self-assembly of a molecular photoswitch. The meta-stable aggregates of 1o and dT20 obtained under kinetic conditions eventually formed an increasingly tight network of twisted ribbons as the equilibrated product, driven by the self-correction properties of the template. The thus obtained hydrogel showed self-healing properties on the molecular scale and a fascinating memory of the vessel shape it grew in, eventually forming a miniaturised 3D copy of the cast by the increased association of the supramolecular biohybrid fibres. A comparable observation of shape memory in hydrogels is to the best of our knowledge unprecedented and detailed future studies are needed to elucidate all mechanistic aspects and the general applicability of this fascinating phenomenon.Irradiation with UV light induced local shrinking of the gel, which could be employed to manipulate the originally casted shape photochemically in a spatially controlled manner. Treatment with visible light followed by a heating–cooling cycle allowed to recover the initial kinetic product. The full cycle can be followed using a combination of UV-vis and CD spectroscopy as well as fluorescence microscopy, AFM and TEM. The photoregulation and modulation of the architecture of this recognition-driven biohybrid self-assembly on the microscopic and macroscopic scale represents a promising step towards precise control of the shape and properties of smart materials and structural organization in hydrogels, potentially impacting broad subjects like controlled cargo-release, DNA origami and soft robotics.
Data availability
ESI† containing synthetic procedures, analytical data, such as NMR spectra, further microscopy images, data supporting the rheology measurements, photographs of the gelation experiments over time.Author contributions
NAS, PM, and BLF designed the study. NAS and PM synthesized the compounds and performed UV-vis spectroscopy. NAS, PM, and WRB performed CD spectroscopy. VD and MCAS performed microscopy. JWS and TK performed rheology. NAS and PM wrote the paper. BLF supervised the work. All authors discussed and commented on the manuscript. NAS and BLF acquired funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ms Dhanya Babu for her advice and help regarding the fluorescence microscopy and Dr Anouk Lubbe, Dr Stefano Crespi, and Dr Wojciech Danowski for fruitful discussion, and Pavlo Gordiichuk and Xiaoyan Zhang for their help with initial microscopy measurements. We gratefully acknowledge generous support from the Humboldt Foundation (Feodor-Lynen Fellowship to NAS), the Horizon 2020 Framework Program (ERC Advanced Investigator Grant No. 694345 to BLF), and the Ministry of Education, Culture and Science of the Netherlands (Gravitation Program No. 024.001.035 to BLF).References
- A. J. F. Griffiths, W. M. Gelbart, J. H. Miller and R. C. Lewontin, in Modern Genetic Analysis, W. H. Freeman, New York, 1999 Search PubMed.
- M. Surin and S. Ulrich, ChemistryOpen, 2020, 9, 480–498 CrossRef CAS PubMed.
- T. LaBean, in 2006 International Electron Devices Meeting, IEEE, 2006, pp. 1–3 Search PubMed.
- Z.-G. Wang and B. Ding, Adv. Mater., 2013, 25, 3905–3914 CrossRef CAS PubMed.
- A. Vologodskii and M. D. Frank-Kamenetskii, Phys. Life Rev., 2018, 25, 1–21 CrossRef PubMed.
- H. Li, J. D. Carter and T. H. LaBean, Mater. Today, 2009, 12, 24–32 CrossRef CAS.
- B. Lu, S. Vecchioni, Y. P. Ohayon, R. Sha, K. Woloszyn, B. Yang, C. Mao and N. C. Seeman, ACS Nano, 2021, 15, 16788–16793 CrossRef CAS PubMed.
- K. Tapio and I. Bald, Multifunct. Mater., 2020, 3, 032001 CrossRef CAS.
- D. Ghosh, L. P. Datta and T. Govindaraju, Beilstein J. Nanotechnol., 2020, 11, 124–140 CrossRef CAS PubMed.
- R. Wang, G. Zhang and H. Liu, Curr. Opin. Colloid Interface Sci., 2018, 38, 88–99 CrossRef CAS.
- J. Chen and N. C. Seeman, Nature, 1991, 350, 631–633 CrossRef CAS.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2018, 3, 17068 CrossRef CAS.
- W. Wang, C. Chen, S. Vecchioni, T. Zhang, C. Wu, Y. P. Ohayon, R. Sha, N. C. Seeman and B. Wei, Angew. Chem., Int. Ed., 2021, 133, 25985–25990 CrossRef.
- N. C. Seeman, Biochemistry, 2003, 42, 7259–7269 CrossRef CAS PubMed.
- N. C. Seeman, Annu. Rev. Biochem., 2010, 79, 65–87 CrossRef CAS PubMed.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- P. Wang, T. A. Meyer, V. Pan, P. K. Dutta and Y. Ke, Chem, 2017, 2, 359–382 CAS.
- S. Dey, C. Fan, K. V. Gothelf, J. Li, C. Lin, L. Liu, N. Liu, M. A. D. Nijenhuis, B. Saccà, F. C. Simmel, H. Yan and P. Zhan, Nat. Rev. Methods Primers, 2021, 1, 13 CrossRef CAS.
- A. Gopinath, C. Thachuk, A. Mitskovets, H. A. Atwater, D. Kirkpatrick and P. W. K. Rothemund, Science, 2021, 371, eabd6179 CrossRef CAS PubMed.
- P. W. K. Rothemund, in Nanotechnology: Science and Computation, Springer-Verlag, Berlin/Heidelberg, 2006, pp. 3–21 Search PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- J. Shi, Z. Shi, Y. Dong, F. Wu and D. Liu, ACS Appl. Bio Mater., 2020, 3, 2827–2837 CrossRef CAS PubMed.
- J. Gačanin, C. V. Synatschke and T. Weil, Adv. Funct. Mater., 2020, 30, 1906253 CrossRef.
- J. Bush, C.-H. Hu and R. Veneziano, Appl. Sci., 2021, 11, 1885 CrossRef CAS.
- L. Yue, S. Wang, V. Wulf and I. Willner, Nat. Commun., 2019, 10, 4774 CrossRef PubMed.
- A. Buchberger, H. Saini, K. R. Eliato, A. Zare, R. Merkley, Y. Xu, J. Bernal, R. Ros, M. Nikkhah and N. Stephanopoulos, ChemBioChem, 2021, 22, 1755–1760 CrossRef CAS.
- F. Huang, M. Chen, Z. Zhou, R. Duan, F. Xia and I. Willner, Nat. Commun., 2021, 12, 2364 CrossRef CAS PubMed.
- X. Zhang, J.-H. Wang, D. Tan, Q. Li, M. Li, Z. Gong, C. Tang, Z. Liu, M.-Q. Dong and X. Lei, Anal. Chem., 2018, 90, 1195–1201 CrossRef CAS PubMed.
- Z. Li, G. Davidson-Rozenfeld, M. Vázquez-González, M. Fadeev, J. Zhang, H. Tian and I. Willner, J. Am. Chem. Soc., 2018, 140, 17691–17701 CrossRef CAS PubMed.
- J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
- J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10423–10424 CrossRef CAS PubMed.
- R. Iwaura, K. Yoshida, M. Masuda, M. Ohnishi-Kameyama, M. Yoshida and T. Shimizu, Angew. Chem., Int. Ed., 2003, 42, 1009–1012 CrossRef CAS PubMed.
- R. Iwaura, F. J. M. Hoeben, M. Masuda, A. P. H. J. Schenning, E. W. Meijer and T. Shimizu, J. Am. Chem. Soc., 2006, 128, 13298–13304 CrossRef CAS PubMed.
- R. Iwaura, Soft Matter, 2017, 13, 8293–8299 RSC.
- W. Guo, C.-H. Lu, R. Orbach, F. Wang, X.-J. Qi, A. Cecconello, D. Seliktar and I. Willner, Adv. Mater., 2015, 27, 73–78 CrossRef CAS PubMed.
- T. Bellini, G. Zanchetta, T. P. Fraccia, R. Cerbino, E. Tsai, G. P. Smith, M. J. Moran, D. M. Walba and N. A. Clark, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1110–1115 CrossRef CAS PubMed.
- G. Zanchetta, Liq. Cryst. Today, 2009, 18, 40–49 CrossRef CAS.
- L.-A. Fendt, I. Bouamaied, S. Thöni, N. Amiot and E. Stulz, J. Am. Chem. Soc., 2007, 129, 15319–15329 CrossRef CAS.
- P. G. A. Janssen, S. Jabbari-Farouji, M. Surin, X. Vila, J. C. Gielen, T. F. A. de Greef, M. R. J. Vos, P. H. H. Bomans, N. A. J. M. Sommerdijk, P. C. M. Christianen, P. Leclère, R. Lazzaroni, P. van der Schoot, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 1222–1231 CrossRef CAS PubMed.
- M. V. Ishutkina, A. R. Berry, R. Hussain, O. G. Khelevina, G. Siligardi and E. Stulz, Eur. J. Org. Chem., 2018, 2018, 5054–5059 CrossRef CAS PubMed.
- A. Pérez-Romero, A. Domínguez-Martín, S. Galli, N. Santamaría-Díaz, O. Palacios, J. A. Dobado, M. Nyman and M. A. Galindo, Angew. Chem., Int. Ed., 2021, 60, 10089–10094 CrossRef PubMed.
- S. Wang, M. C. Forster, K. Xue, F. Ehlers, B. Pang, L. B. Andreas, P. Vana and K. Zhang, Angew. Chem., Int. Ed., 2021, 60, 9712–9718 CrossRef CAS PubMed.
- H. Kashida and H. Asanuma, Phys. Chem. Chem. Phys., 2012, 14, 7196 RSC.
- G. Yao, J. Li, Q. Li, X. Chen, X. Liu, F. Wang, Z. Qu, Z. Ge, R. P. Narayanan, D. Williams, H. Pei, X. Zuo, L. Wang, H. Yan, B. L. Feringa and C. Fan, Nat. Mater., 2020, 19, 781–788 CrossRef CAS PubMed.
- P. G. A. Janssen, J. Vandenbergh, J. L. J. van Dongen, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2007, 129, 6078–6079 CrossRef CAS.
- R. Iwaura, Chem.–Eur. J., 2019, 25, 2281–2287 CrossRef CAS PubMed.
- J. Rodriguez, J. Mosquera, S. Learte-Aymamí, M. E. Vázquez and J. L. Mascareñas, Acc. Chem. Res., 2020, 53, 2286–2298 CrossRef CAS PubMed.
- F. Wang, X. Liu and I. Willner, Angew. Chem., Int. Ed., 2015, 54, 1098–1129 CrossRef CAS PubMed.
- A. Cecconello, L. V. Besteiro, A. O. Govorov and I. Willner, Nat. Rev. Mater., 2017, 2, 17039 CrossRef CAS.
- O. I. Wilner and I. Willner, Chem. Rev., 2012, 112, 2528–2556 CrossRef CAS.
- A. S. Lubbe, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2017, 46, 1052–1079 RSC.
- C. Dohno and K. Nakatani, Chem. Soc. Rev., 2011, 40, 5718–5729 RSC.
- A. S. Lubbe, Q. Liu, S. J. Smith, J. W. de Vries, J. C. M. Kistemaker, A. H. de Vries, I. Faustino, Z. Meng, W. Szymanski, A. Herrmann and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 5069–5076 CrossRef CAS PubMed.
- N. A. Simeth, S. Kobayashi, P. Kobauri, S. Crespi, W. Szymanski, K. Nakatani, C. Dohno and B. L. Feringa, Chem. Sci., 2021, 12, 9207–9220 RSC.
- Y. Kamiya and H. Asanuma, Acc. Chem. Res., 2014, 47, 1663–1672 CrossRef CAS PubMed.
- H. Asanuma, T. Ishikawa, Y. Yamano, K. Murayama and X. Liang, ChemPhotoChem, 2019, 3, 418–424 CrossRef CAS.
- H. Asanuma, X. Liang, H. Nishioka, D. Matsunaga, M. Liu and M. Komiyama, Nat. Protoc., 2007, 2, 203–212 CrossRef CAS PubMed.
- X. Liang, T. Mochizuki and H. Asanuma, Small, 2009, 5, 1761–1768 CrossRef CAS PubMed.
- M. Zhou, X. Liang, T. Mochizuki and H. Asanuma, Angew. Chem., Int. Ed., 2010, 49, 2167–2170 CrossRef CAS PubMed.
- P. K. Jain, V. Ramanan, A. G. Schepers, N. S. Dalvie, A. Panda, H. E. Fleming and S. N. Bhatia, Angew. Chem., Int. Ed., 2016, 55, 12440–12444 CrossRef CAS PubMed.
- E. V. Moroz-Omori, D. Satyapertiwi, M.-C. Ramel, H. Høgset, I. K. Sunyovszki, Z. Liu, J. P. Wojciechowski, Y. Zhang, C. L. Grigsby, L. Brito, L. Bugeon, M. J. Dallman and M. M. Stevens, ACS Cent. Sci., 2020, 6, 695–703 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, F. Xie, J. Lin and L. Xu, Chem. Sci., 2020, 11, 11478–11484 RSC.
- W. Zhou, W. Brown, A. Bardhan, M. Delaney, A. S. Ilk, R. R. Rauen, S. I. Kahn, M. Tsang and A. Deiters, Angew. Chem., Int. Ed., 2020, 59, 8998–9003 CrossRef CAS PubMed.
- Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- L. N. Lucas, J. J. D. de Jong, J. H. van Esch, R. M. Kellogg and B. L. Feringa, Eur. J. Org. Chem., 2003, 2003, 155–166 CrossRef.
- Y.-H. Shim, P. B. Arimondo, A. Laigle, A. Garbesi and S. Lavielle, Org. Biomol. Chem., 2004, 2, 915–921 RSC.
- Y. Chai, M. Munde, A. Kumar, L. Mickelson, S. Lin, N. H. Campbell, M. Banerjee, S. Akay, Z. Liu, A. A. Farahat, R. Nhili, S. Depauw, M.-H. David-Cordonnier, S. Neidle, W. D. Wilson and D. W. Boykin, ChemBioChem, 2014, 15, 68–79 CrossRef CAS PubMed.
- A. Porcheddu and G. Giacomelli, Curr. Med. Chem., 2005, 12, 2561–2599 CrossRef CAS PubMed.
- R. Wang, W. Liu and N. C. Seeman, Chem. Biol., 2009, 16, 862–867 CrossRef CAS PubMed.
- J. M. Korde and B. Kandasubramanian, Chem. Eng. J., 2020, 379, 122430 CrossRef CAS.
- J. Shang, X. Le, J. Zhang, T. Chen and P. Theato, Polym. Chem., 2019, 10, 1036–1055 RSC.
- A. T. Neffe, C. Löwenberg, K. K. Julich-Gruner, M. Behl and A. Lendlein, Int. J. Mol. Sci., 2021, 22, 5892 CrossRef CAS PubMed.
- C. Li, A. Iscen, L. C. Palmer, G. C. Schatz and S. I. Stupp, J. Am. Chem. Soc., 2020, 142, 8447–8453 CrossRef CAS PubMed.
- Q. Bian, L. Fu and H. Li, Nat. Commun., 2022, 13, 137 CrossRef CAS PubMed.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- J. L. Zhang, W. M. Huang, G. Gao, J. Fu, Y. Zhou, A. V. Salvekar, S. S. Venkatraman, Y. S. Wong, K. H. Tay and W. R. Birch, Eur. Polym. J., 2014, 58, 41–51 CrossRef CAS.
- R. Zamani Alavijeh, P. Shokrollahi and J. Barzin, J. Mater. Chem. B, 2017, 5, 2302–2314 RSC.
- B. Maiti, A. Abramov, L. Franco, J. Puiggalí, H. Enshaei, C. Alemán and D. D. Díaz, Adv. Funct. Mater., 2020, 30, 2001683 CrossRef CAS.
Footnotes |
| † Dedicated in memory of Ned Seeman, a pioneer in DNA-nanotechnology. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06490h |
| § These authors contributed equally. |
| ¶ Current address: Institute for Organic and Biomolecular Chemistry, University of Goettingen, Tammanstr. 2, 37077 Goettingen, Germany. |
| This journal is © The Royal Society of Chemistry 2022 |