
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHypervalent iodine-mediated β-difluoroalkylboron synthesis via an unusual 1,2-hydrogen shift enabled by boron substitution†
Wen-Xin
Lv
 a,
Yin
Li
a,
Yuan-Hong
Cai
a,
Dong-Hang
Tan
a,
Zhan
Li
a,
Ji-Lin
Li
a,
Qingjiang
Li
a and
Honggen
Wang
*ab
a,
Yin
Li
a,
Yuan-Hong
Cai
a,
Dong-Hang
Tan
a,
Zhan
Li
a,
Ji-Lin
Li
a,
Qingjiang
Li
a and
Honggen
Wang
*ab
aGuangdong Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, 510006, China. E-mail: wanghg3@mail.sysu.edu.cn
bState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmaceutical Sciences, Guangxi Normal University, Guilin 541004, China
First published on 11th February 2022
Abstract
β-Difluoroalkylborons, featuring functionally important CF2 moiety and synthetically valuable boron group, have great synthetic potential while remaining synthetically challenging. Herein we report a hypervalent iodine-mediated oxidative gem-difluorination strategy to realize the construction of gem-difluorinated alkylborons via an unusual 1,2-hydrogen migration event, in which the (N-methyliminodiacetyl) boronate (BMIDA) motif is responsible for the high regio- and chemoselectivity. The protocol provides facile access to a broad range of β-difluoroalkylborons under rather mild conditions. The value of these products was demonstrated by further transformations of the boryl group into other valuable functional groups, providing a wide range of difluorine-containing molecules.
Organofluorine compounds have been widely applied in medicinal chemistry and materials science.1a–d In particular, the gem-difluoro moiety featuring unique steric and electronic properties can act as a chemically inert isostere of a variety of polar functional groups.2a–c Therefore, the construction of gem-difluoro-containing compounds has received considerable attention in recent years. Efficient methods including deoxyfluorination of carbonyl compounds,3a,b photoredox difluorination,4 radical difluorination,5 and cross-coupling reactions with suitable CF2 carriers6a–f are well developed. Alternatively, iodoarene-mediated oxidative difluorination reactions provide valuable access to these motifs by using simple alkenes as starting materials.7a–i Previously, these reactions were generally associated with a 1,2-aryl or 1,2-alkyl migration (Scheme 1a).7a–f Recent developments also allowed the use of heteroatoms as migrating groups, thereby furnishing gem-difluoro compounds equipped with easily transformable functional groups (Scheme 1b). In this regard, Bi and coworkers reported an elegant 1,2-azide migrative gem-difluorination of α-vinyl azides, enabling the synthesis of a broad range of novel β-difluorinated alkyl azides.7g Jacobsen developed an iodoarene-catalyzed synthesis of gem-difluorinated aliphatic bromides featuring 1,2-bromo migration with high enantioselectivity.7h Almost at the same time, research work from our group demonstrated that not only bromo, but also chloro and iodo could serve as viable migrating groups.7i
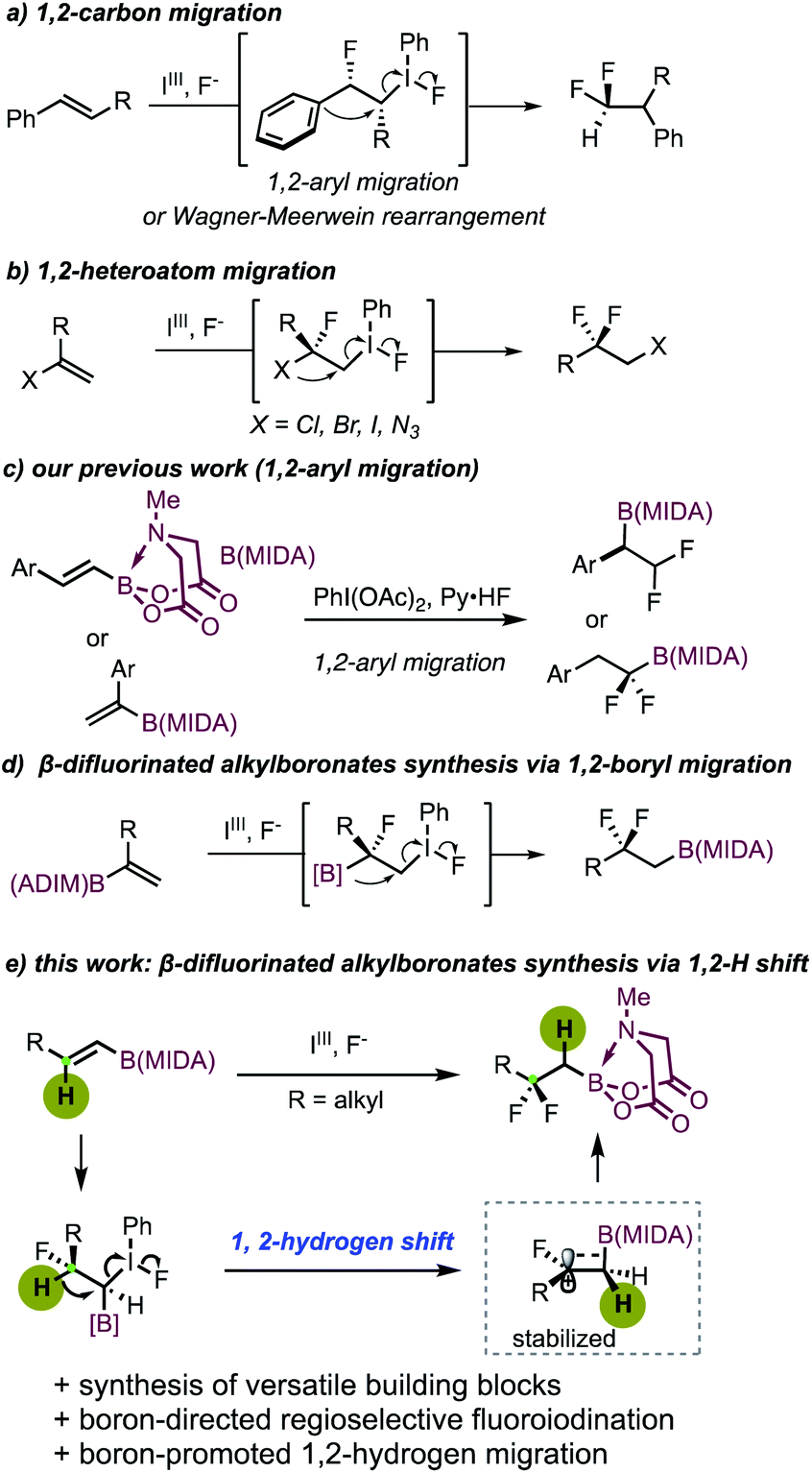 | ||
| Scheme 1 Hypervalent iodine-mediated β-difluoroalkylboron synthesis. | ||
We have been devoted to developing new methodologies for the assembly of boron-containing building blocks by using easily accessible and stable MIDA (N-methyliminodiacetyl) boronates8a–c as starting materials.9a–e Recently, we realized a hypervalent iodine-mediated oxidative difluorination of aryl-substituted alkenyl MIDA boronates.9d Depending on the substitution patterns, the reaction could lead to the synthesis of either α- or β-difluoroalkylborons via 1,2-aryl migration (Scheme 1c). Recently, with alkyl-substituted branched alkenyl MIDA boronates, Szabó and Himo observed an interesting bora-Wagner–Meerwein rearrangement, furnishing β-difluorinated alkylboronates with broader product diversity (Scheme 1d).10 While extending the scope of our previous work,9d we found that the use of linear alkyl-substituted alkenyl MIDA boronates also delivers β-difluoroalkylboron products. Intriguingly, instead of an alkyl- or boryl-migration, an unusual 1,2-hydrogen shift takes place. It should be noted that internal inactivated alkenes typically deliver the 1,2-difluorinated products, with no rearrangement taking place.11a–d Herein, we disclose our detailed study of our second generation of β-difluoroalkylborons synthesis (Scheme 1e). The starting linear 1,2-disubstituted alkyl-substituted alkenyl MIDA boronates, unlike the branched ones,10 could be readily prepared via a two-step sequence consisting of hydroborylation of the terminal alkyne and a subsequent ligand exchange with N-methyliminodiacetic acid. This intriguing 1,2-H shift was found to be closely related to the boron substitution, probably driven thermodynamically by the formation of the β-carbon cation stabilized by a σ(C–B) bond via hyperconjugation.12a–d
To start, we employed benzyl-substituted alkenyl MIDA boronate 1a as a model substrate (Table 1). In accordance with our previous observations,9d the use of F sources such as CsF, AgF and Et3N·HF in association with PhI(OAc)2 (PIDA) as the oxidant and DCM as the solvent led to no reaction (entries 1 to 3). The use of Py·HF (20 equiv) successfully provided β-difluorinated alkylboronate 2a, derived from an unusual 1,2-hydrogen migration, in 39% yield (entry 4). By simply increasing the loading of Py·HF to 40 equivalents, a higher conversion and thus an improved yield of 61% was obtained (entry 5). No further improvement was observed by using a large excess of Py·HF (100 equiv) (entry 6). Other hypervalent iodine oxidants such as PhIO or PIFA were also effective but resulted in reduced yields (entries 7 and 8). A brief survey of other solvents revealed that the original DCM was the optimal one (entries 9 and 10).
|
|
||||
|---|---|---|---|---|
| Entry | F− (equiv) | Oxidant | Solvent | Yield (%) |
| 1 | CsF (2.0) | PIDA | DCM | 0 |
| 2 | AgF (2.0) | PIDA | DCM | 0 |
| 3 | Et3N·HF (40.0) | PIDA | DCM | 0 |
| 4 | Py·HF (20.0) | PIDA | DCM | 39 |
| 5 | Py·HF (40.0) | PIDA | DCM | 61 |
| 6 | Py·HF (100.0) | PIDA | DCM | 55 |
| 7 | Py·HF (40.0) | PIFA | DCM | 52 |
| 8 | Py·HF (40.0) | PhIO | DCM | 26 |
| 9 | Py·HF (40.0) | PIDA | DCE | 49 |
| 10 | Py·HF (40.0) | PIDA | Toluene | 46 |

With the optimized reaction conditions in hand, we set out to investigate the scope and limitation of this gem-difluorination reaction. The reaction of a series of E-type 1,2-disubstituted alkenyl MIDA boronates were first examined. As shown in Scheme 2, the reaction of substrates with primary alkyl (1b, 1e–g), secondary alkyl (1c, 1d), or benzyl (1h–k) groups proceeded efficiently to give the corresponding gem-difluorinated alkylboronates in moderate to good yields. Halides (1i–k, 1m) and cyano (1l) were well tolerated in this reaction. Of note, cyclic alkene 1n is also a viable substrate, affording an interesting gem-difluorinated cyclohexane product (2n).
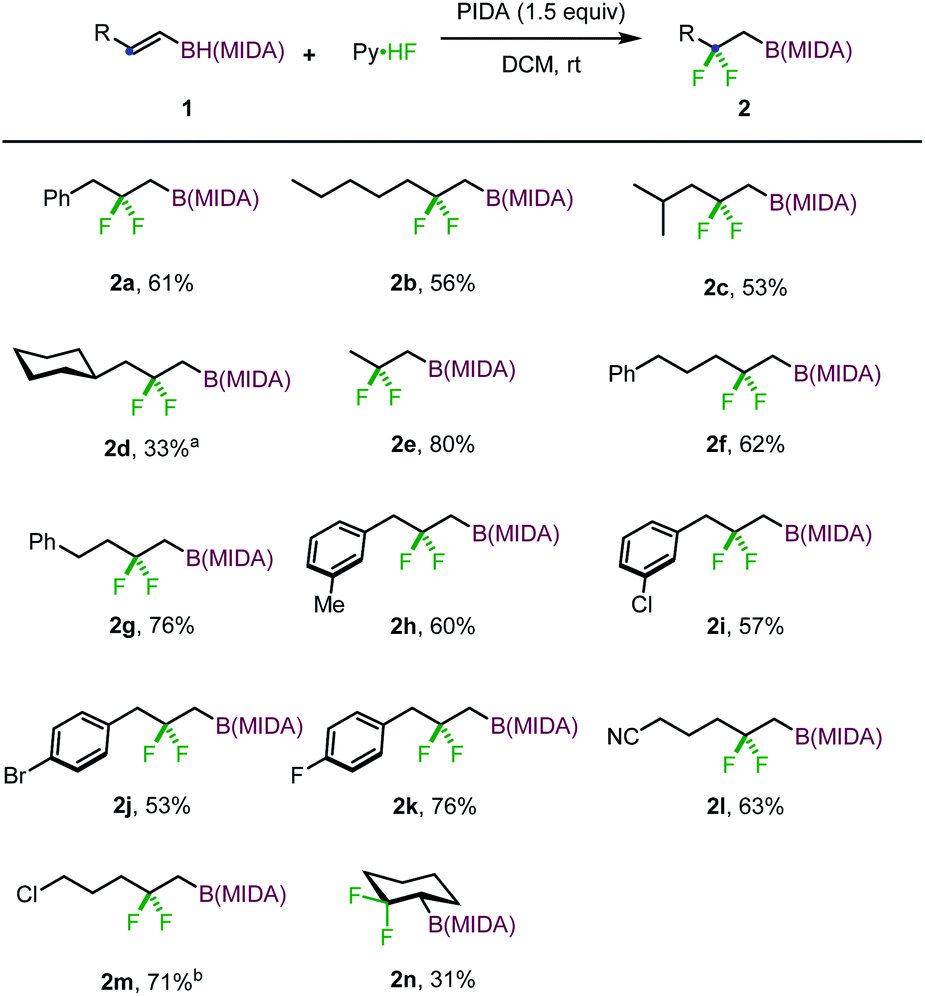 | ||
| Scheme 2 Scope of 1,2-H migratory gem-difluorinations. a 4 h. b PIFA was used. | ||
To define the scope further, the substrates with Z configuration were also employed under the standard reaction conditions (eqn (1) and (2)). The same type of products were isolated with comparable efficiency, suggesting that the reaction outcome is independent of the substrate configuration and substrates with Z configuration also have a profound aptitude of 1,2-hydrogen migration. Nevertheless, the reaction of t-butyl substituted alkenyl MIDA boronate (1p) delivered a normal 1,2-difluorinated alkylboron product (eqn (3)). The 1,2-hydrogen migration was completely suppressed probably due to unfavorable steric perturbation. With an additional alkyl substituent introduced, a 1,2-alkyl migrated product was formed as expected (eqn (4)).
 | (1) |
The gem-difluorination protocol was amenable to gram-scale synthesis of 2a (Scheme 3, 8 mmol scale of 1a, 1.24 g, 50%). To assess the synthetic utility of the resulting β-difluorinated alkylborons, transformations of the C–B bond were carried out (Scheme 3). Ligand exchange of 2a furnished the corresponding pinacol boronic ester 4 without difficulty, which could be ligated with electron-rich aromatics to obtain 5 and 6 in moderate yields. On the other hand, 2a could be oxidized with high efficiency to alcohol 7 using H2O2/NaOH. The hydroxyl group of 7 could then be converted to bromide 8 or triflate 9. Both serve as useful electrophiles that can undergo intermolecular SN2 substitution with diverse nitrogen- (10, 13), oxygen- (14), phosphorus- (11) and sulfur-centered (12) nucleophiles.
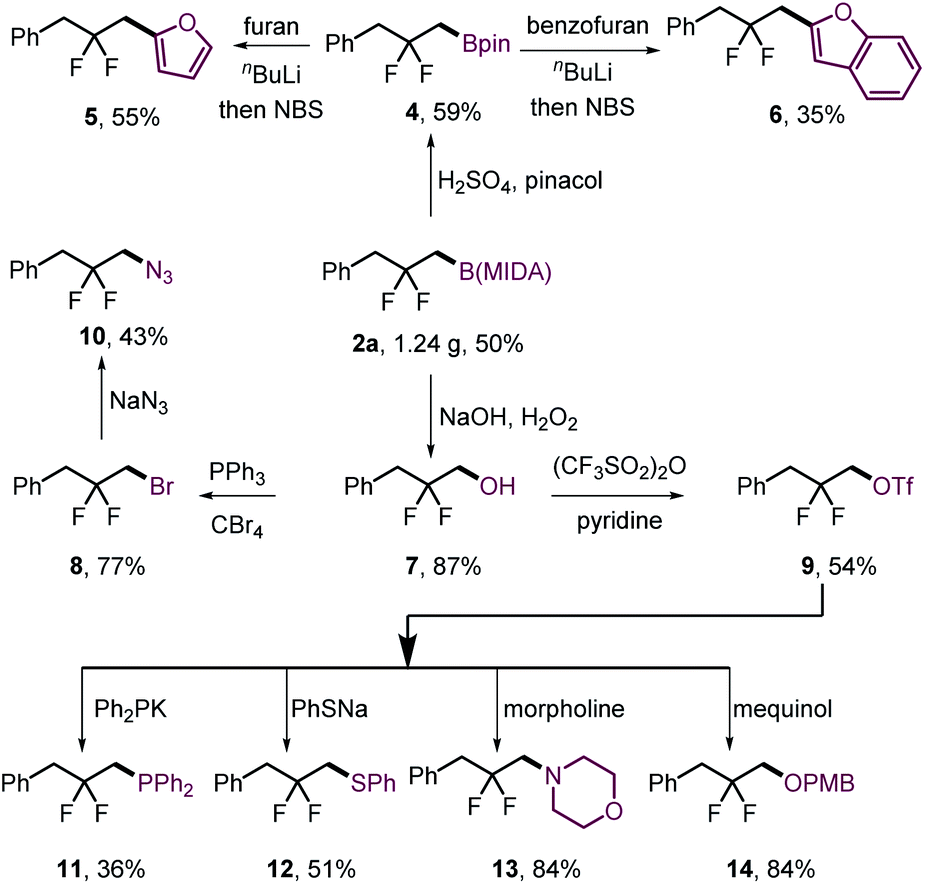 | ||
| Scheme 3 Product derivatizations. PMB = p-methoxyphenyl. | ||
To gain insight into the reaction mechanism, preliminary mechanistic studies were conducted. The reaction employing deuterated alkenyl MIDA boronate [D]-1a efficiently afforded difluorinated product [D]-2a in 72% isolated yield, clearly demonstrating that 1,2-H migration occurred (Scheme 4a). However, when the MIDA boronate moiety was replaced with a methyl group (15), no difluorinated product (derived from 1,2-migration) was detected at all, suggesting an indispensable role of boron for promoting the 1,2-migration event (Scheme 4b). Also, with a Bpin congener of 1a, the reaction led to large decomposition of the starting material, with no desired product being formed (Scheme 4b).
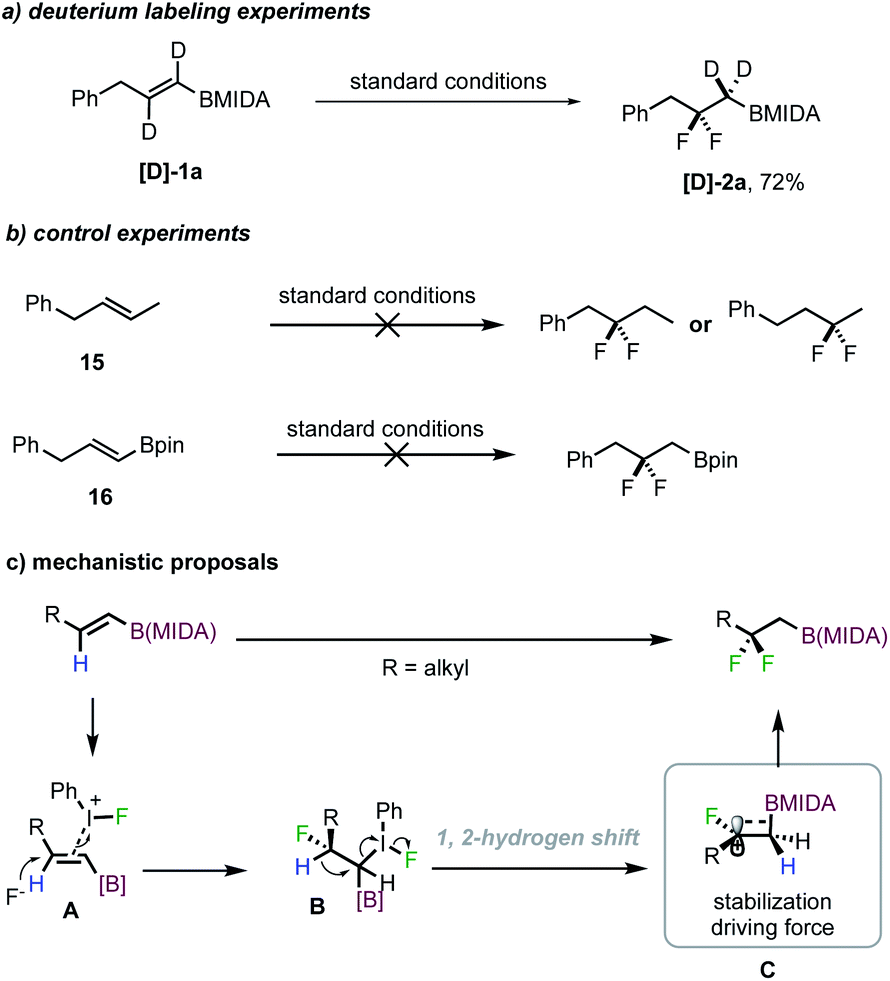 | ||
| Scheme 4 Mechanistic studies and proposals. | ||
Based on the literature precedent and these experiments, a possible reaction mechanism is proposed in Scheme 4c. With linear alkenyl MIDA boronates, the initial coordination of the double bond to an iodium ion triggered a regioselective fluoroiodination to deliver intermediate B. The regioselectivity could arise from an electron-donating inductive effect from boron due to its low electronegativity, consistent with previous observations.13a,b Thereafter, a 1,2-hydrogen shift, rather than the typical direct fluoride substitution of the C–I bond, provides carbon cation C. The formation of a hyperconjugatively stabilized cation is believed to be the driving force for this event.12a–d The trapping of this cation finally forms the product.
In conclusion, we demonstrated herein our second generation of β-difluoroalkylboron synthesis via oxidative difluorination of easily accessible linear 1,2-disubstituted alkenyl MIDA boronates. An unexpected 1,2-hydrogen migration was observed, which was found to be triggered by a MIDA boron substitution. Mild reaction conditions, moderate to good yields and excellent regioselectivity were achieved. The applications of these products allowed the facile preparation of a wide range of gem-difluorinated molecules by further transformations of the boryl group.
Data availability
Data for this work, including experimental procedures, characterization data for all new compounds are provided in the ESI.†Author contributions
W.-X. L. and H. W. conceived the project. W.-X. L., Y. L., Y.-H. C., D.-H. T., Z. L. and J.-L. L. analysed the experimental results. W.-X. L., Q. L. and Y. L. performed the derivatizations and mechanistic studies. H. W. directed the project and composed the manuscript with input from all the authors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22022114, 21971261), the Guangdong Basic and Applied Basic Research Foundation (2020A1515010624), the Fundamental Research Funds for the Central Universities (20ykzd12) and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y093).Notes and references
- References about the development and application of organofluorine compounds, see: (a) D. O'Hagan and D. Harper, J. Fluorine Chem., 1999, 100, 127 CrossRef; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (c) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (d) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed.
- For selected examples about the gem-difluoro moiety, see: (a) C. D. Sessler, M. Rahm, S. Becker, J. M. Goldberg, F. Wang and S. J. Lippard, J. Am. Chem. Soc., 2017, 139, 9325 CrossRef CAS PubMed; (b) D. O'Hagan, Y. Wang, M. Skibinski and A. M. Z. Slawin, Pure Appl. Chem., 2012, 84, 1587 Search PubMed; (c) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed.
- (a) W. J. Middleton, J. Org. Chem., 1975, 40, 574 CrossRef CAS; (b) R. P. Singh and J. M. Shreeve, Synthesis, 2002, 17, 2561–2578 Search PubMed.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS PubMed.
- P. Xu, S. Guo, L. Wang and P. Tang, Angew. Chem., Int. Ed., 2014, 53, 5955 CrossRef CAS PubMed.
- (a) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524 CrossRef CAS PubMed; (b) Z. Feng, Q.-Q. Min, Y.-L. Xiao, B. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1669 CrossRef CAS PubMed; (c) Y. Gu, X. Leng and Q. Shen, Nat. Commun., 2014, 5, 5405 CrossRef CAS PubMed; (d) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909 CrossRef CAS PubMed; (e) G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., Int. Ed., 2012, 51, 12090 CrossRef CAS PubMed; (f) Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95 CrossRef CAS PubMed.
- For selected examples about iodoarene-mediated oxidative difluorination reactions, see: (a) S. M. Banik, J. W. Medley and E. N. Jacobsen, Science, 2016, 353, 51 CrossRef CAS PubMed; (b) F. Scheidt, J. Neufeld, M. Schäfer, C. Thiehoff and R. Gilmour, Org. Lett., 2018, 20, 8073 CrossRef CAS PubMed; (c) B. Zhou, M. K. Haj, E. N. Jacobsen, K. N. Houk and X.-S. Xue, J. Am. Chem. Soc., 2018, 140, 15206 CrossRef CAS PubMed; (d) T. Kitamura, K. Muta and J. Oyamada, J. Org. Chem., 2015, 80, 10431 CrossRef CAS PubMed; (e) N. O. Ilchenko, B. O. A. Tasch and K. J. Szabó, Angew. Chem., Int. Ed., 2014, 53, 12897 CrossRef CAS PubMed; (f) S. Hara, J. Nakahigashi, K. Ishi-i, T. Fukuhara and N. Yoneda, Tetrahedron Lett., 1998, 39, 2589 CrossRef CAS; (g) Y. Ning, P. Sivaguru, G. Zanoni, E. A. Anderson and X. Bi, Chem, 2020, 6, 486 CrossRef CAS; (h) M. D. Levin, J. M. Ovian, J. A. Read, M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 14831 CrossRef CAS PubMed; (i) C. Li, Y. Liao, X. Tan, X. Liu, P. Liu, W.-X. Lv and H. Wang, Sci. China: Chem., 2021, 64, 999 CrossRef CAS.
- (a) T. Mancilla, R. Contreras and B. Wrackmeyer, J. Organomet. Chem., 1986, 307, 1 CrossRef CAS; (b) D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961 CrossRef CAS PubMed; (c) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716 CrossRef CAS PubMed.
- For selected examples of our previous work about the assembly of boron-containing building blocks, see: (a) Y.-F. Zeng, W.-W. Ji, W.-X. Lv, Y. Chen, D.-H. Tan, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 14707 CrossRef CAS PubMed; (b) W.-X. Lv, Y.-F. Zeng, Q. Li, Y. Chen, D.-H. Tan, L. Yang and H. Wang, Angew. Chem., Int. Ed., 2016, 55, 10069 CrossRef CAS PubMed; (c) H. Wang, Y.-F. Zeng, W.-X. Lv and D.-H. Tan, Synlett, 2018, 29, 1415 CrossRef CAS; (d) W.-X. Lv, Q. Li, J.-L. Li, Z. Li, E. Lin, D.-H. Tan, Y.-H. Cai, W.-X. Fan and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 16544 CrossRef CAS PubMed; (e) L. Yang, D.-H. Tan, W.-X. Fan, X.-G. Liu, J.-Q. Wu, Z.-S. Huang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 3454 CrossRef CAS PubMed.
- Q. Wang, M. Biosca, F. Himo and K. J. Szabó, Angew. Chem., Int. Ed., 2021, 60, 26327 CrossRef CAS PubMed.
- For selected examples about difluorination of internal unactivated alkenes, see: (a) I. G. Molnár and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 5004 CrossRef PubMed; (b) S. M. Banik, J. W. Medley and E. N. Jacobsen, J. Am. Chem. Soc., 2016, 138, 5000 CrossRef CAS PubMed; (c) S. V. Kohlhepp and T. Gulder, Chem. Soc. Rev., 2016, 45, 6270 RSC; (d) I. G. Molnár, C. Thiehoff, M. C. Holland and R. Gilmour, ACS Catal., 2016, 6, 7167 CrossRef.
- (a) Z. He and A. K. Yudin, J. Am. Chem. Soc., 2011, 133, 13770 CrossRef CAS PubMed; (b) J. Li and M. D. Burke, J. Am. Chem. Soc., 2011, 133, 13774 CrossRef CAS PubMed; (c) R. K. Shiroodi, O. Koleda and V. Gevorgyan, J. Am. Chem. Soc., 2014, 136, 13146 CrossRef CAS PubMed; (d) C. F. Lee, D. B. Diaz, A. Holownia, S. J. Kaldas, S. K. Liew, G. E. Garrett, T. Dudding and A. K. Yudin, Nat. Chem., 2018, 10, 1062 CrossRef CAS PubMed.
- (a) W.-X. Lv, Z. Li, E. Lin, J.-L. Li, D.-H. Tan, Y.-H. Cai, Q. Li and H. Wang, Chem.–Eur. J., 2019, 25, 4058 CrossRef CAS PubMed; (b) W.-X. Fan, J.-L. Li, W.-X. Lv, L. Yang, Q. Li and H. Wang, Chem. Commun., 2020, 56, 82 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06508d |
| This journal is © The Royal Society of Chemistry 2022 |