
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePt(II)-coordinated tricomponent self-assemblies of tetrapyridyl porphyrin and dicarboxylate ligands: are they 3D prisms or 2D bow-ties?†
Paola A.
Benavides
 a,
Monica A.
Gordillo
a,
Ashok
Yadav
a,
M. Andrey
Joaqui-Joaqui
b and
Sourav
Saha
*a
a,
Monica A.
Gordillo
a,
Ashok
Yadav
a,
M. Andrey
Joaqui-Joaqui
b and
Sourav
Saha
*a
aDepartment of Chemistry, Clemson University, Clemson, South Carolina 29634, USA. E-mail: souravs@clemson.edu
bDepartment of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA
First published on 9th March 2022
Abstract
Thermodynamically favored simultaneous coordination of Pt(II) corners with aza- and carboxylate ligands yields tricomponent coordination complexes with sophisticated structures and functions, which require careful structural characterization to paint accurate depiction of their structure–function relationships. Previous reports claimed that heteroleptic coordination of cis-(Et3P)2PtII with tetrapyridyl porphyrins (M′TPP, M′ = Zn or H2) and dicarboxylate ligands (XDC) yielded 3D tetragonal prisms containing two horizontal M′TPP faces and four vertical XDC pillars connected by eight Pt(II) corners, even though such structures were not supported by their 1H NMR data. Through extensive X-ray crystallographic and NMR studies, herein, we demonstrate that self-assembly of cis-(Et3P)2PtII, M′TPP, and four different XDC linkers having varied lengths and rigidities actually yields bow-tie (![[X triangles, left, right]](https://www.rsc.org/images/entities/char_22c8.gif) )-shaped 2D [{cis-(Et3P)2Pt}4(M′TPP) (XDC)2]4+ complexes featuring a M′TPP core and two parallel XDC linkers connected by four heteroleptic PtII corners instead of 3D prisms. This happened because (i) irrespective of their length (∼7–11 Å) and rigidity, the XDC linkers intramolecularly bridged two adjacent pyridyl-N atoms of a M′TPP core via PtII corners instead of connecting two cofacial M′TPP ligands and (ii) bow-tie complexes are entropically favored over prisms. The electron-rich ZnTPP core of a representative bow-tie complex selectively formed a charge-transfer complex with highly π-acidic 1,4,5,8,9,12-hexaazatriphenylene-2,3,6,7,10,11-heaxacarbonitrile but not with a π-donor such as pyrene. Thus, this work not only produced novel M′TPP-based bow-tie complexes and demonstrated their selective π-acid recognition capability, but also underscored the importance of proper structural characterization of supramolecular assemblies to ensure accurate depiction of their structure–property relationships.
)-shaped 2D [{cis-(Et3P)2Pt}4(M′TPP) (XDC)2]4+ complexes featuring a M′TPP core and two parallel XDC linkers connected by four heteroleptic PtII corners instead of 3D prisms. This happened because (i) irrespective of their length (∼7–11 Å) and rigidity, the XDC linkers intramolecularly bridged two adjacent pyridyl-N atoms of a M′TPP core via PtII corners instead of connecting two cofacial M′TPP ligands and (ii) bow-tie complexes are entropically favored over prisms. The electron-rich ZnTPP core of a representative bow-tie complex selectively formed a charge-transfer complex with highly π-acidic 1,4,5,8,9,12-hexaazatriphenylene-2,3,6,7,10,11-heaxacarbonitrile but not with a π-donor such as pyrene. Thus, this work not only produced novel M′TPP-based bow-tie complexes and demonstrated their selective π-acid recognition capability, but also underscored the importance of proper structural characterization of supramolecular assemblies to ensure accurate depiction of their structure–property relationships.
Introduction
Owing to the dynamic, directional, and self-selecting/rectifying nature of metal–ligand coordination bonds, metal-driven self-assembly processes have emerged as one of the most attractive and powerful tools of supramolecular chemistry, yielding myriads of supramolecular coordination complexes (SCCs) ranging from discrete metallacycles1–10 and cages11–23 to extended coordination polymers and metal–organic frameworks24–26 over several decades. To obtain the target SCCs and to avoid statistical mixtures of different possibilities, only one rigid organic ligand is combined with a metal ion at an appropriate stoichiometry, which usually yields bicomponent coordination complexes. However, the resulting bicomponent SCCs often lack the structural and functional diversity and tunability needed for various advanced applications. Expanding the scope of coordination-driven self-assembly strategies, recently researchers have discovered26–56 that cis-capped Pt(II) and Pd(II) corners simultaneously bind a carboxylate and a pyridyl ligand, which preferentially yield thermodynamically favored heteroleptic Pt(N,O) complexes instead of two different homoleptic complexes.29–34,47–53 Furthermore, when two different homoleptic PtII(COO−)2 and PtII(pyridyl)2 complexes were mixed together at an appropriate stoichiometry, they spontaneously reorganized into thermodynamically more stable heteroleptic Pt(N,O) complexes.30,32,33 These revelations paved the way for metal-driven self-assembly of tricomponent metallacycles and cages containing two complementary ligands that could further diversify their structures, compositions, properties, and functions.While it is fairly straightforward to assemble 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:4 tricomponent rectangles30,32,41 having two parallel dicarboxylate and two parallel dipyridyl arms connected by four heteroligated PtII(N,O) corners, the formation of 2:4:8 tricomponent tetragonal prisms featuring two cofacial tetratopic (tetrapyridyl or tetracarboxylate) ligands and four complementary ditopic (dicarboxylate or dipyridyl) linkers requires the latter to intermolecularly connect the tips of two separate cofacial tetratopic ligands via eight heteroleptic Pt(N,O) corners30,32,35,36 instead of intramolecularly bridging two adjacent tips of the same tetratopic ligand. If a ditopic linker intramolecularly bridges two adjacent binding sites of the tetratopic ligand via shared Pt(N,O) corners, then entropically more favored 1:2:4 bow-tie () complexes featuring one tetratopic core and two ditopic linkers connected by four heteroleptic corners would be formed instead of 2:4:8 tetragonal prisms containing two cofacial tetratopic faces connected by four ditopic linkers via eight heteroleptic corners.54 Nevertheless, previous reports have claimed30,33,34 that Pt(II)-driven tricomponent self-assembly of tetrapyridyl porphyrin (M′TPP, M′ = Zn- or H2) and various aromatic and aliphatic dicarboxylate (XDC) linkers having varied lengths and rigidities yielded tetragonal prisms [{cis-(Et3P)2Pt}8(M′TPP)2(XDC)4]8+ featuring two parallel M′TTP faces and four XDC pillars connected by eight heteroligated PtII(N,O) corners. The resulting complexes preserved the photophysical properties of M′TPP chromophores33 and displayed promising applications in cancer photodynamic therapy39 and guest encapsulation.55 Encouraged by these literature reports,30,33,34,39,56 we attempted to construct bi-chromophoric tetragonal prisms consisting of two M′TPP faces and four dicarboxylate linkers having complementary redox- and optically active aromatic cores, such as naphthalene- and perylene diimides that could support ligand-to-ligand photoinduced electron and/or energy transfer events. Surprisingly, none of our attempts to construct tetragonal prisms having two parallel M′TPP faces and four XDC linkers based on the reported protocols was successful despite the fact that the lengths of our XDC linkers were much longer than the distances between the two adjacent pyridyl-N atoms of free M′TPP ligands (dN–N/free = 10.9 Å), which precluded the possibility of intramolecular bridging of two adjacent pyridyl groups via heteroligated PtII(N,O) corners.
:2:4 tricomponent rectangles30,32,41 having two parallel dicarboxylate and two parallel dipyridyl arms connected by four heteroligated PtII(N,O) corners, the formation of 2:4:8 tricomponent tetragonal prisms featuring two cofacial tetratopic (tetrapyridyl or tetracarboxylate) ligands and four complementary ditopic (dicarboxylate or dipyridyl) linkers requires the latter to intermolecularly connect the tips of two separate cofacial tetratopic ligands via eight heteroleptic Pt(N,O) corners30,32,35,36 instead of intramolecularly bridging two adjacent tips of the same tetratopic ligand. If a ditopic linker intramolecularly bridges two adjacent binding sites of the tetratopic ligand via shared Pt(N,O) corners, then entropically more favored 1:2:4 bow-tie () complexes featuring one tetratopic core and two ditopic linkers connected by four heteroleptic corners would be formed instead of 2:4:8 tetragonal prisms containing two cofacial tetratopic faces connected by four ditopic linkers via eight heteroleptic corners.54 Nevertheless, previous reports have claimed30,33,34 that Pt(II)-driven tricomponent self-assembly of tetrapyridyl porphyrin (M′TPP, M′ = Zn- or H2) and various aromatic and aliphatic dicarboxylate (XDC) linkers having varied lengths and rigidities yielded tetragonal prisms [{cis-(Et3P)2Pt}8(M′TPP)2(XDC)4]8+ featuring two parallel M′TTP faces and four XDC pillars connected by eight heteroligated PtII(N,O) corners. The resulting complexes preserved the photophysical properties of M′TPP chromophores33 and displayed promising applications in cancer photodynamic therapy39 and guest encapsulation.55 Encouraged by these literature reports,30,33,34,39,56 we attempted to construct bi-chromophoric tetragonal prisms consisting of two M′TPP faces and four dicarboxylate linkers having complementary redox- and optically active aromatic cores, such as naphthalene- and perylene diimides that could support ligand-to-ligand photoinduced electron and/or energy transfer events. Surprisingly, none of our attempts to construct tetragonal prisms having two parallel M′TPP faces and four XDC linkers based on the reported protocols was successful despite the fact that the lengths of our XDC linkers were much longer than the distances between the two adjacent pyridyl-N atoms of free M′TPP ligands (dN–N/free = 10.9 Å), which precluded the possibility of intramolecular bridging of two adjacent pyridyl groups via heteroligated PtII(N,O) corners.
Prompted by these unexpected outcomes, we took a closer look at the reported 1H NMR spectra of the putative M′TPP-based tetragonal prisms30,33,34,56 and recognized that all of them actually displayed two distinct singlets (1:1 ratio) for pyrrole protons—one set of four pyrrole protons was significantly more shielded than the other four (in contrast, all eight pyrrole protons of free M′TPP ligands are chemically equivalent and show one singlet). These NMR signatures revealed that the pyrrole rings of M′TPP ligands in these complexes were no longer chemically equivalent, i.e., they resided in two completely different environments. However, in the proposed tetragonal prisms, all pyrrole rings of M′TPP should have enjoyed the same chemical environment and displayed a characteristic singlet peak.47–49,53,55 Thus, the reported 1H NMR signals were not consistent with the proposed tetragonal prism structures and indicated the formation of 2D bow-tie () structures, a possibility that was previously overlooked. In bow-tie () complexes, the two opposite pyrrole rings of M′TPP would be located inside two isosceles triangles formed by two parallel XDC linkers and therefore shielded accordingly, while the other two opposite pyrrole rings would remain exposed and not shielded by the XDC linkers. These inconsistencies prompted us to carefully examine whether or not the Pt(II)-driven self-assembly processes of M′TPP and XDC ligands indeed produce tricomponent prisms or yield an entirely different architecture, namely bow-ties having the same ratio (4:1:2) of the three components.
Herein, we report the self-assembly and in-depth characterization of eight novel bow-tie complexes [{cis-(Et3P)2Pt}4(M′TPP)(XDC)2]·4(TfO) (Scheme 1) composed of M′TPP ligands (M′ = Zn and H2) and four XDC linkers, namely 1,6-hexane-, 4,4′-biphenyl-, 1,4-benzene-, and 2,6-naphthalene- dicarboxylates (HDC, BPDC, BDC, and NDC), having different lengths and rigidities. The most direct and compelling evidence of bow-tie architectures of M′TPP-based tricomponent SCCs came from their first ever single-crystal X-ray structures, which were fully consistent with their solution phase 1H, 31P, and 2D (COSY, ROESY, and DOSY) NMR spectra as well as the ESI-MS data. The energy-minimized structures of these complexes were also in good agreement with the experimental results. These mutually corroborating results unequivocally demonstrated that each complex was composed of an M′TPP core and two parallel XDC linkers connected by four heteroligated (Et3P)2PtII(N,O) corners. Interestingly, the formation of M′TPP-based tricomponent bow-tie complexes instead of 3D prisms is also consistent with recent reports54,57–59 describing the formation of similar ‘triangular dicycles’ based on other tetratopic cores and ditopic linkers. Furthermore, we demonstrate the ability of an electron rich ZnTPP-based bow-tie complex to form a π-donor/acceptor (D/A) charge-transfer (CT) complex with highly electron deficient 1,4,5,8,9,12-hexaazatriphenylene-2,3,6,7,10,11-heaxacarbonitrile (HATHCN) but not with another π-donor such as pyrene.
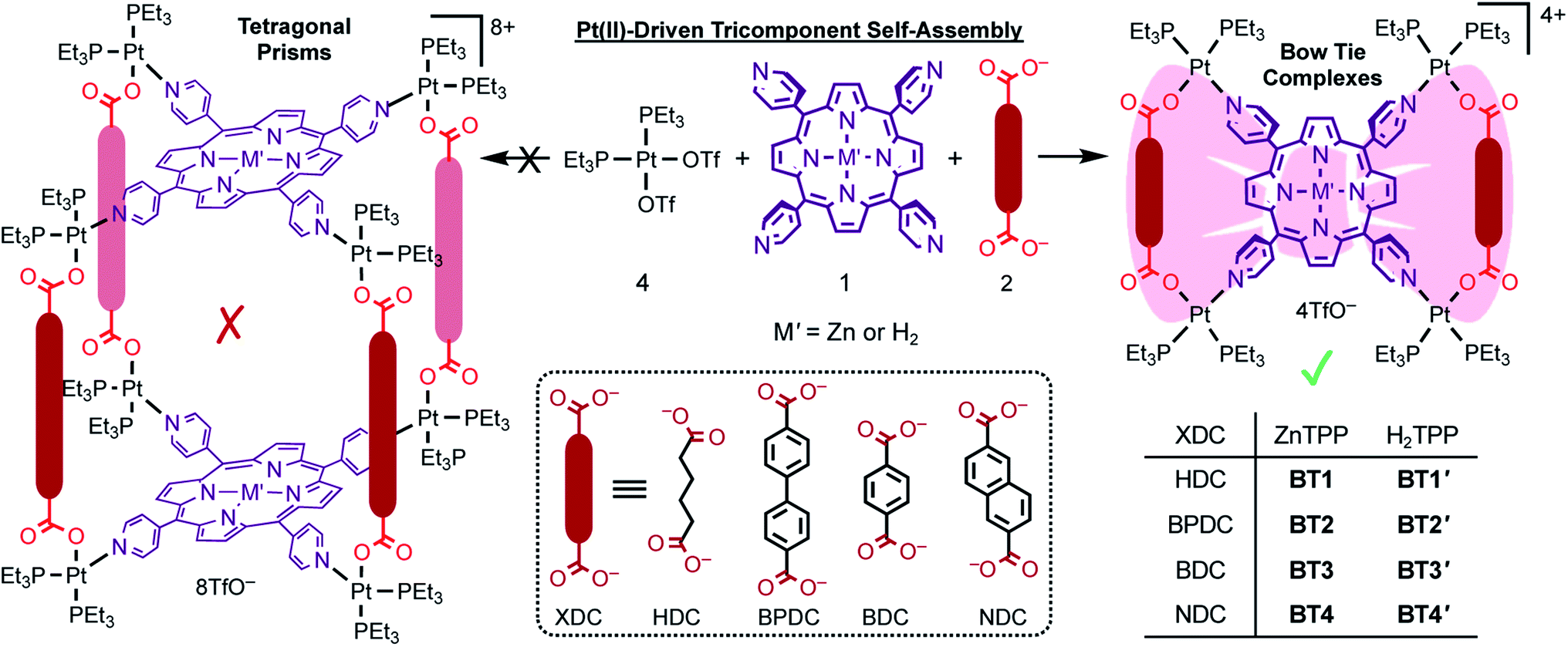 | ||
| Scheme 1 Pt(II)-driven self-assembly of M′TPP ligands (M′ = Zn or H2) and four different XDC linkers (HDC, BPDC, BDC, and NDC) yielded novel bow-tie complexes [{cis-(Et3P)2Pt}4(M′TPP)(XDC)2]·4(TfO) (BT1–BT4 and BT1′–BT4′). No tetragonal prism was formed irrespective of the length and rigidity of the XDC linkers. | ||
Results and discussion
Heteroleptic coordination-driven self-assembly of M′TPP-based tricomponent bow-ties (![[X triangles, left, right]](https://www.rsc.org/images/entities/h3_char_22c8.gif) )
)
To determine whether the heteroleptic coordination of cis-(Et3P)2PtII corners with tetratopic M′TPP ligands and ditopic XDC linkers yield tetragonal 3D prisms containing two cofacial M′TPP ligands and four XDC linkers held together by eight Pt(II) corners30,33,34 or 2D bow-ties containing a M′TPP core and two parallel XDC linkers connected by four shared Pt(II) corners, herein, we employed four XDC ligands—HDC, BPDC, BDC, and NDC—having different lengths, rigidities, and electron densities. For consistency, we applied the same self-assembly conditions reported in the literature,30,33,34 namely (i) 4:1:2 stoichiometry of cis-(Et3P)2Pt(TfO), M′TPP, and XDC; (ii) solvent mixtures (1:1:1 CH2Cl2/MeCN/MeNO2 or 4:1 Me2CO/H2O) that adequately solubilized all components; (iii) temperature (∼60 °C), and (iv) reaction time (∼18 h) that were used to synthesize the proposed prisms. Our comprehensive 1D and 2D NMR, ESI-MS, single-crystal X-ray, and computational studies unequivocally demonstrated that regardless of their rigidity and lengths (∼7–11 Å), all four XDC linkers formed bow-tie complexes BT1–BT4 and BT1′–BT4′ (Scheme 1, Fig. 1) while no 3D prism was identified. Notably, the HDC and BDC linkers were also previously employed to assemble M′TPP-based tricomponent SCCs,30,33,34,39 and the reported NMR spectra of the resulting complexes were the same as those of our unequivocally characterized bow-tie structures.
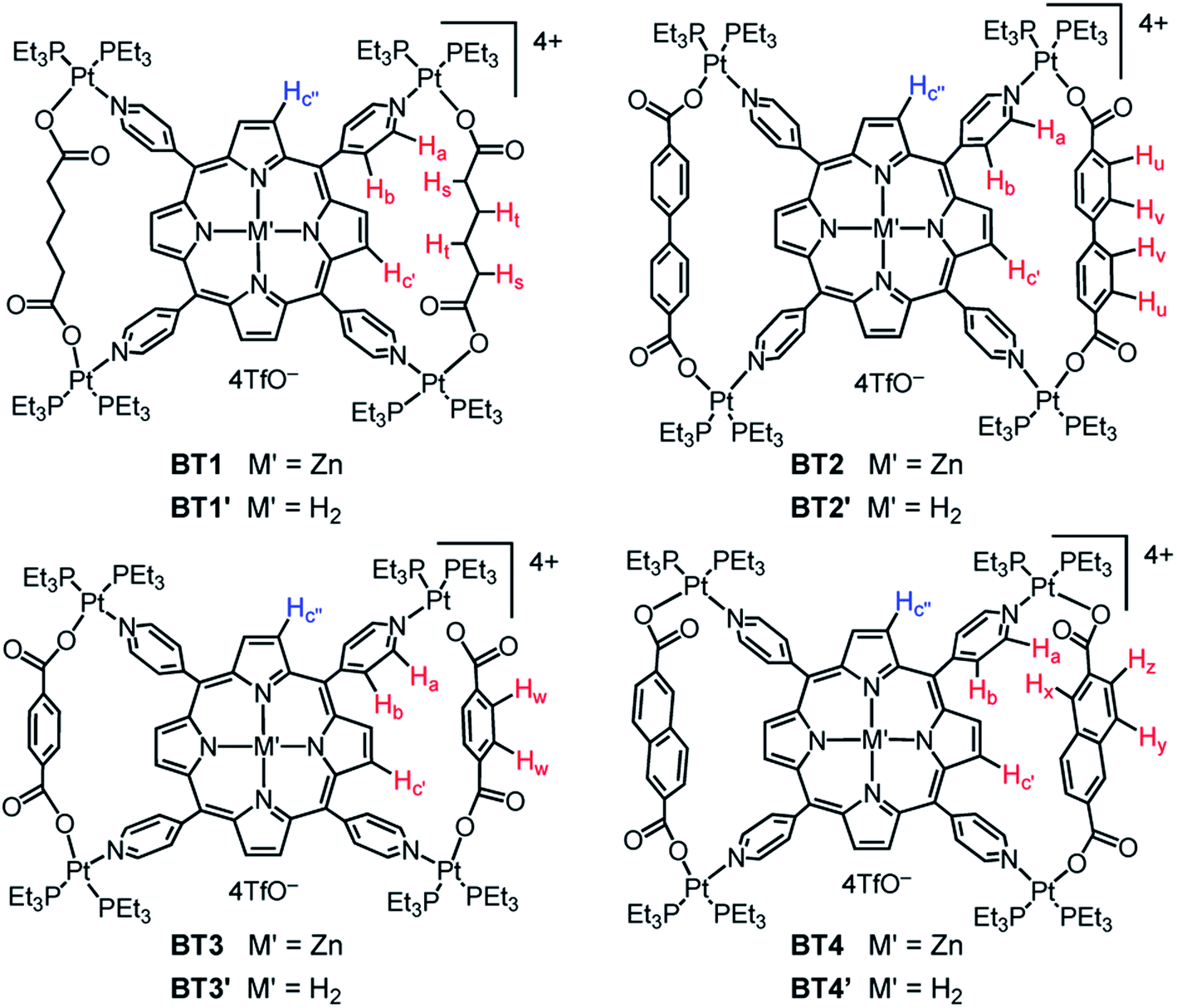 | ||
| Fig. 1 The chemical structures of bow-tie complexes. | ||
Single-crystal structures of bow-tie complexes
The single-crystal X-ray analysis of these tricomponent SCCs presented the most direct and conclusive evidence of their bow-tie structures consisting of a M′TPP core and two parallel XDC linkers held together by four heteroleptic (Et3P)2Pt(N,O) corners (Fig. 2). Notably, this was the first time the crystal structures of any M′TPP-based tricomponent SCCs could be determined. The crystals were obtained from the respective NMR solutions (acetone-d6) via either slow evaporation or vapor diffusion of CH2Cl2 or Et2O, assuring that the solid-state crystal structures and solution NMR data belonged to the same materials. The bow-tie complexes based on rigid aromatic BPDC, BDC, and NDC linkers crystallized easily, but those containing flexible aliphatic HDC linkers did not. The structural features of all bow-tie complexes (Table 1) were fully consistent with their respective NMR and ESI-MS data, confirming that the same species were present both in solutions and solid crystals. The crystal structures of these bow-tie complexes shined light on why the M′TPP core displayed two distinct 1H NMR signals for the enclosed Hc′ and exposed Hc′′ protons (Fig. 1) and why only Hc′ protons were shielded by and ROE-coupled to XDC linkers but Hc′′ protons were not.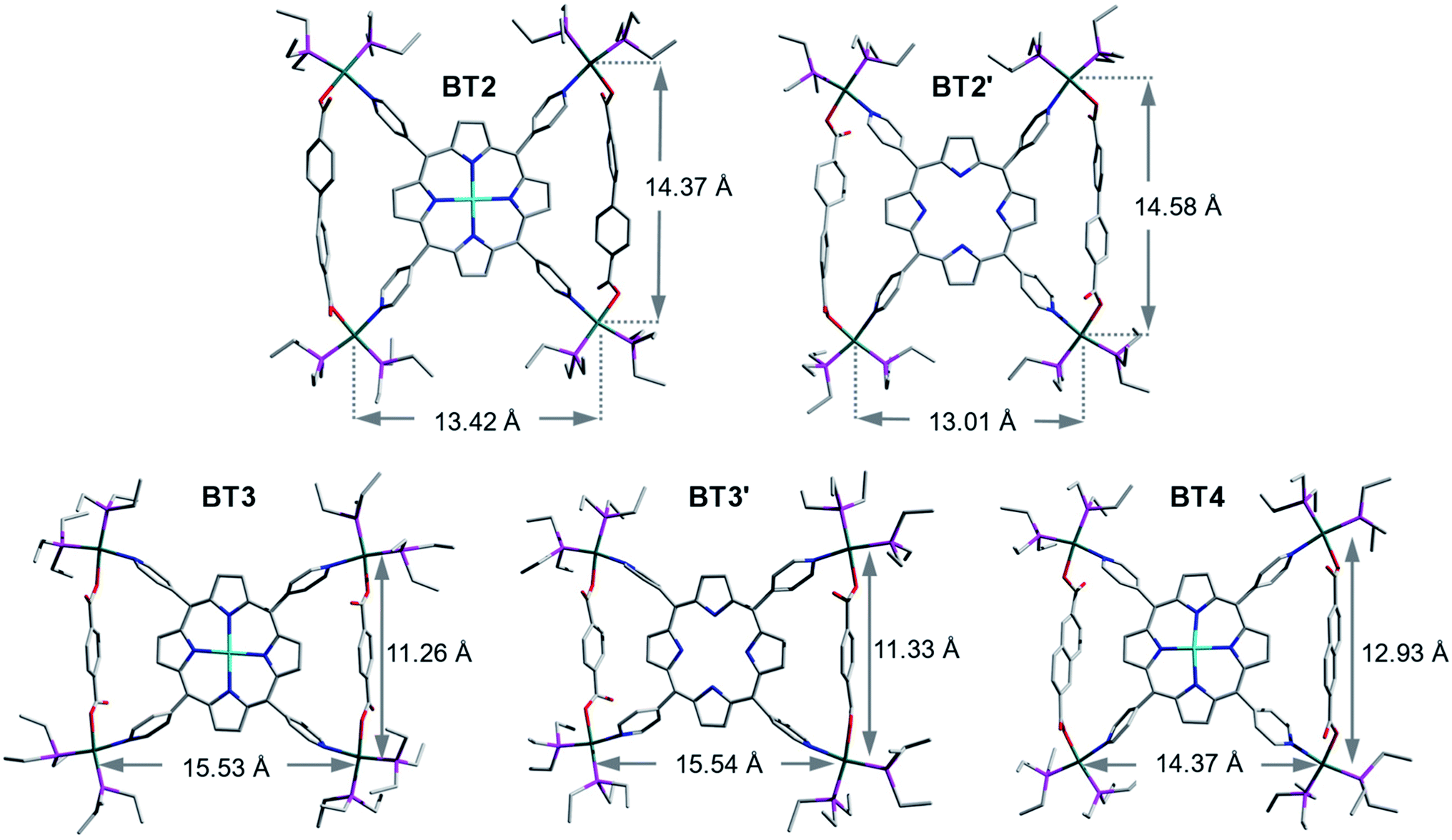 | ||
| Fig. 2 Single-crystal structures of BT2, BT2′, BT3, BT3′ and BT4 bow-tie complexes. Atom legends: green: Pt, cyan: Zn, pink: P, red: O, blue: N, and grey: C. The H-atoms and TfO− anions were omitted for clarity. | ||
| BT1 calc.* | BT2 | BT2′ | BT3 (calc.*) | BT3′ (calc.*) | BT4 | |
|---|---|---|---|---|---|---|
| Space group | — |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c | C2/c | I41/a | P21/c |
| d Pt–Pt/int (Å) | 11.55* | 14.37 | 14.58 | 11.26 (11.33*) | 11.33 (11.33*) | 12.93 |
| d Pt–Pt/ext (Å) | 15.41* | 13.42 | 13.01 | 15.53 (15.59*) | 15.54 (15.56*) | 14.37 |
| d N–N/int (Å) | 10.19* | 11.38 | 11.52 | 10.09 (10.12*) | 10.07 (10.20*) | 10.83 |
| d N–N/ext (Å) | 11.63* | 10.37 | 10.18 | 11.39 (11.69*) | 11.61 (11.68*) | 10.80 |
| d Hc′–XDC-center (Å) | 2.84* | 3.37 | 3.11 | 3.18 (3.50*) | 3.05 (3.44*) | 2.95 |
| d Hc′–pyridine-center (Å) | 2.96* | 3.41 | 3.51 | 3.02 (2.98*) | 2.96 (3.01*) | 3.19 |
| d Hc′′–pyridine-center (Å) | 3.37* | 3.08 | 3.00 | 3.41 (3.47*) | 3.47 (3.41*) | 3.26 |
| ∠N–Pt–O (°) | 96*, 98* | 82, 85 | 82, 83 | 78, 83 (93*) | 80, 83 (93*) | 82, 82 |
| ∠(NPy–center–NPy)int (°) | 82* | 95 | 97 | 83 (81*) | 82 (82*) | 88 |
| ∠(NPy–center–NPy)ext (°) | 98* | 85 | 83 | 97 (99*) | 98 (98*) | 89 |
| θ porphyrin/pyridyl-dh (°) | 82*, 83* | 76, 77 | 64, 73 | 86, 88 (84*) | 76, 80 (85*) | 65, 76 |
| θ pyrrole/XDC-dh (°) | — | — | — | 80 (90*) | 85 (90*) | 87 |
The bow-tie ()-shaped BT2 and BT2′ complexes (Fig. 2: [{(Et3P)2Pt}4(M′TPP)(BPDC)2]4+, M′ = Zn or H2) crystallized in the P and C2/c space groups, respectively. The ∠N–Pt–O angles of slightly distorted square-planar Pt(II) corners ranged between ca. 82–85° (two diagonally opposite angles were the same). In BT2 and BT2′, the distances between two BPDC-bridged Pt(II) corners (dPt–Pt/int) were ca. 14.5 and 14.6 Å, respectively, whereas those between two adjacent Pt(II) corners not bridged by BPDC (dPt–Pt/ext) were 13.4 and 13.0 Å, respectively. This happened because the long BPDC linker (lBPDC = 11.2 Å)60 positioned the two bridged Pt(II) corners farther away from each other, which in turn shortened the dPt–Pt/ext. Consequently, the distances between the two adjacent pyridyl-N atoms coordinated to two BPDC-bridged Pt(II) corners, i.e., the pyridyl-N atoms that belonged to the same isosceles triangle, were also noticeably longer (dN–N/int ≈ 11.4 and Å) than the distance between two adjacent pyridyl-N atoms that were not part of the same triangle (dN–N/ext ≈ 10.4 Å). These N–N distances in BT2 and BT2′ deviated from the uniform distance between two adjacent pyridyl-N atoms of free M′TPP ligands (dN–N/free ≈ 10.9 Å).61,62 Furthermore, in BT2, the angles of projection between two adjacent pyridyl rings that belonged to the same triangle (∠(NPy–center–NPy)int) expanded to 95° (97° in BT2′), while the angle between two adjacent pyridyl rings that were not part of the same triangle (∠(NPy–center–NPy)ext) contracted to 85° (83° in BT2′). Thus, these angles deviated by 3–5° from the angles of projection (∼90°) between the adjacent pyridyl rings of free M′TPP in order to incorporate long BPDC linkers within the isosceles triangles of bow-tie structures. The dihedral angles between the porphyrin core and the pyridyl arms were 76° in BT2 (73° in BT2′). The enclosed pyrrole rings of M′TPP and the twisted BPDC core (torsion angle between two phenyl rings ≈ 40°) located across the bay were not coplanar but formed large dihedral angles. The average distances from the enclosed Hc′ pyrrole protons located inside the triangles to the center of the closest phenyl ring of the BPDC linker (dHc′–XDC) was ca. 3.4 Å in BT2 (3.1 Å in BT2′) and to the center of the nearest pyridyl ring (dHc′–pyridine-center) was 3.4 Å in BT2 (3.5 Å in BT2′). Both distances were the longest among the bow-tie complexes presented here.
Although the entire single-crystal structures of BT2 and BT2′ complexes, including their ancillary Et3P ligands on the Pt(II) corners, were well-resolved, only the basic bow-tie skeletons of BT3, BT3′, and BT4 complexes consisting of the M′TPP core, two parallel BDC and NDC linkers, and four Pt(II) corners were fully resolved, but the fluxional CH3CH2-groups and TfO− anions were not, which caused large R-values. Nevertheless, since the ancillary ligands and counterions were not integral parts of the bow-tie structures, the poor resolution of these highly disordered components had little effect on key structural features that influenced their respective NMR spectra.
The BDC-based bow-tie complexes BT3 and BT3′ (Fig. 2: [{(Et3P)2Pt}4(M′TPP)(BDC)2]4+, M′ = Zn or H2) crystallized in C2/c and I41/a space groups, respectively. The ∠N–Pt–O angles at distorted square-planar Pt(II) corners ranged between ca. 78–83°. The distances between two adjacent Pt(II) corners bridged by short BDC linkers (lBDC = 6.9 Å)60 were ca. 11.3 Å (dPt–Pt/int), whereas the distances between two adjacent Pt(II) corners not bridged by BDC (dPt–Pt/ext) were ca. 15.5 Å. Thus, the short BDC linkers placed the two bridged Pt(II) corners closer to each other while increasing the distances between the two non-bridged Pt(II) corners. Consequently, the distances between the pyridyl-N atoms that belonged to the same triangle were also shorter (dN–N/int ≈ 10.1 Å) than those between two pyridyl-N atoms that were not part of the same triangle (dN–N/ext ≈ 11.5 Å). Consequently, the projection angles between two adjacent pyridyl rings belonging to the same triangle (∠(NPy–center–NPy)int) shrunk to ca. 82°, while the angles between two adjacent pyridyl rings not belonging to the same triangle (∠(NPy–center–NPy)ext) expanded to ca. 98° (ca. 8° deviations from the ideal ∼90° angle in free M′TPP) in order to accommodate short BDC linkers within the triangles. The dihedral angles between the enclosed pyrrole rings of M′TPP and the BDC core located across the bay were 80–85°, i.e., they were almost orthogonal to each other. The enclosed Hc′ pyrrole protons were projected toward the center of the BDC ring (dHc′–XDC ≈ 3.1 Å) and the adjacent pyridyl rings, which were pulled closer (dHc′–pyridine-center ≈ 3 Å) by the BDC linker.
The NDC-based bow-tie complex BT4 [{(Et3P)2Pt}4(ZnTPP)(NDC)2]4+ (Fig. 2) possessed the P21/c space group. All four ∠N–Pt–O angles of distorted square-planar Pt(II) corners were ca. 82°. The distances between two adjacent NDC-bridged Pt(II) corners (dPt–Pt/int = 12.9 Å) were slightly shorter than those between two adjacent Pt(II) corners not bridged by a NDC linker (dPt–Pt/ext = 14.4 Å). However, the distances between the N-atoms of two adjacent pyridyl rings belonging to the same triangle (dN–N/int) and those between two adjacent pyridyl-N atoms that were not part of the same triangle (dN–N/ext) were almost same (ca. 10.8 Å) and close to the uniform distances between two adjacent pyridyl-N atoms of the free ZnTPP ligand (dN–N/free ∼ 10.9 Å). As a result, the angles between two adjacent pyridyl rings belonging to the same triangle (∠(NPy–center–NPy)int) and those between two adjacent pyridyl rings not belonging to the same triangle (∠(NPy–center–NPy)ext) in BT4 were also close to ideal ∼90° angle between adjacent pyridyl rings found in the free ZnTPP ligand. Thus, the intermediate length of the NDC linker (lNDC = 9.2 Å)60 caused the least distortion of the ZnTPP ligand in order to form the isosceles triangles. In BT4, the ZnTPP and the NDC planes were nearly orthogonal to each other (dihedral angles = 87°). The enclosed Hc′ pyrrole protons located inside the triangles were projected toward the NDC core across the bay (dHc′–XDC ≈ 3 Å) and to the center of the adjacent pyridyl rings (dHc′–pyridine-center ≈ 3.2 Å).
Energy optimized structures
Since the crystal structures of BT1 or BT1′ complexes containing flexible HDC linker were not available, we calculated their energy minimized structures using Gaussian 09 software (Fig. 3). To verify the accuracy of these calculated structures, we also calculated the energy minimized structures of BDC-based bow-tie complexes, which were in good agreement with their actual crystal structures (Table 1). In BT1 and BT1′ complexes, the ∠N–Pt–O angles at the distorted square-planar Pt(II) corners were ca. 96–98°. The two adjacent HDC-bridged Pt(II) corners (dPt–Pt/int) were located ca. 11.6 Å apart, whereas the two adjacent Pt(II) corners not bridged by HDC (dPt–Pt/ext) were located ca. 15.4 Å apart. The bridged Pt–Pt distances in BT1 were comparable to those found in BT3, suggesting that HDC and BDC have similar lengths. Like BDC, the short HDC linkers also brought the two bridged Pt(II) corners closer to each other, expanding the distances between the two adjacent non-bridged Pt(II) corners. Consequently, the angles between two adjacent pyridyl rings of M′TPP that belonged to the same triangle (∠(NPy–center–NPy)int) shrunk to ∼82°, while the angles between the two adjacent pyridyl rings that were not part of the same triangle (∠(NPy–center–NPy)ext) expanded to ∼98° in order to accommodate short HDC linkers within the isosceles triangles of these bow-tie structures. The distances from the enclosed Hc′ pyrrole protons to the center of the nearest pyridyl ring (dHc′–pyridine-center) were ca. 2.96 Å and to the Ht protons of the HDC linker located across the bay were ca. 2.8 Å. The calculated structures of these complexes were consistent with their respective NMR spectra.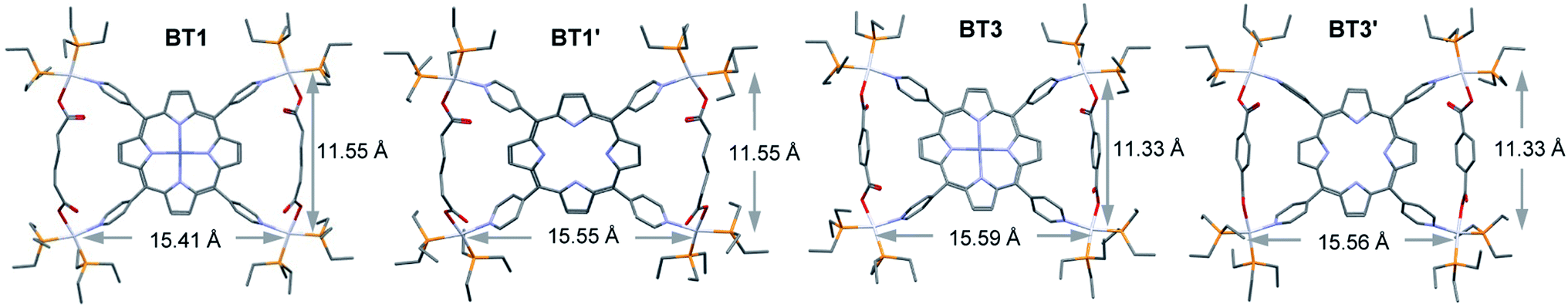 | ||
| Fig. 3 The optimized structures of BT1, BT1′, BT3, and BT3′ complexes calculated by the PM6 method. | ||
Thus, the single-crystal and energy-minimized structures of M′TPP-based tricomponent SCCs demonstrated that regardless of the length (within the range of 6.9–11.2 Å) and rigidity of the XDC linkers, the Pt(II)-driven self-assembly of M′TPP and XDC ligands yielded bow-tie complexes instead of 3D prisms. This happened because the pyridyl arms of M′TPP ligands deviated from their original projections in order to incorporate the XDC linkers within the isosceles triangles of bow-tie structures.
31P NMR spectroscopy
Simultaneous coordination of a pyridyl group of M′TPP and a carboxylate group of XDC with cis-(Et3P)2PtII was also evident from the 31P NMR spectra of the resulting SCCs (Fig. 4 and S1†). While cis-(Et3P)2Pt(TfO)2 displayed a characteristic singlet at 11.14 ppm indicating that both Pt(II)-coordinated P atoms were chemically equivalent, the resulting BT1–BT4 and BT1′–BT4′ complexes displayed two distinct doublets—one at ca. 0–2 ppm that corresponded to the P-nucleus trans- to the pyridyl-N atom and another at 6–7 ppm (2JP–P ≈ 20–21 Hz) corresponding to the P-nucleus trans- to the carboxylate O-atom—indicating that the two ancillary Et3P ligands were not chemically equivalent.30,33,34 The fact that no other 31P NMR signal was observed further confirmed the exclusively heteroleptic coordination of two different ligands with the Pt(II) corners and ruled out the formation of any homoleptic complex. However, the 31P NMR spectra shed little light on the actual structures and compositions of the resulting tricomponent SCCs, i.e., whether they were 3D prisms or 2D bow-ties, as both structures would feature the same heteroleptic cis-(Et3P)2PtII(N,O) corners.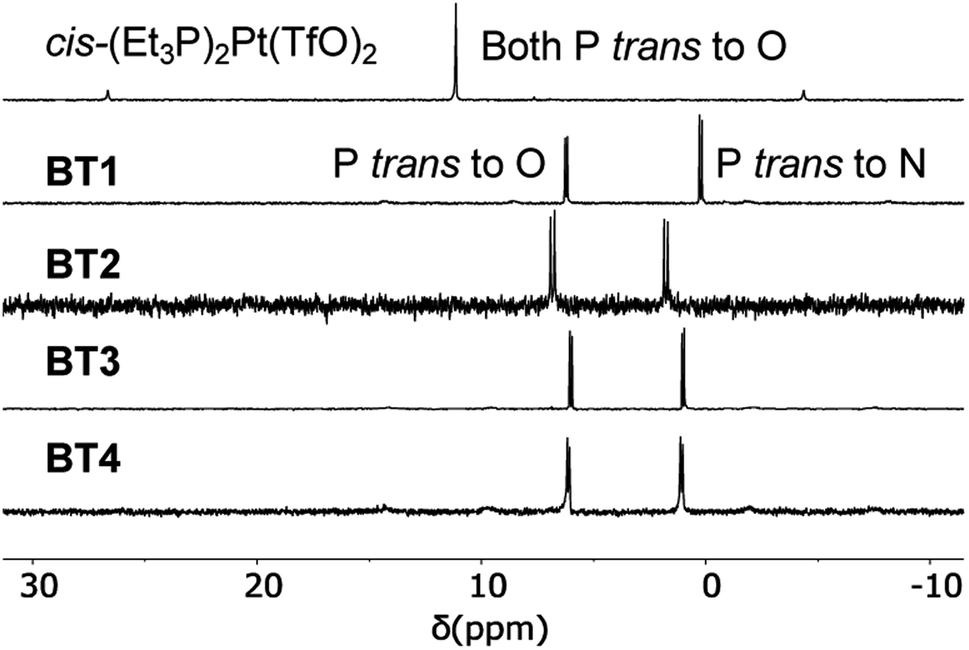 | ||
| Fig. 4 Partial 31P NMR spectra (122 MHz, acetone-d6) of the cis-(Et3P)2Pt(TfO)2 and BT1–BT4 complexes. | ||
1H NMR spectroscopy
The 1H NMR spectra of the resulting complexes (Fig. 5, S2 and S3†) also presented telltale signs of their bow-tie structures and ruled out tetragonal prism formation. In comparison to the D4h-symmetric free ZnTPP ligand, which displayed two doublets at 8.58 and 7.95 ppm corresponding to its Ha and Hb pyridyl protons (8 each), respectively, and a sharp singlet at 8.61 ppm for all eight chemically equivalent Hc pyrrole protons (Fig. 5), the BT1–BT4 complexes not only displayed a significant downfield shift of Ha and Hb pyridyl protons due to Pt(II)-coordination, but most tellingly, two distinct singlets with a 1:1 integral ratio for Hc′ and Hc′′ (pyrrole) protons indicating that the pyrrole rings of ZnTPP were no longer chemically equivalent. The H2TPP-based BT1′–BT4′ complexes (Fig. S2†), and all previously reported M′TPP-based tricomponent SCCs,30,33,34 also displayed two distinct singlets for the pyrrole protons, which indicated that the pyrrole rings of M′TPP resided in two different environments, a telltale sign of bow-tie complexes, not prisms.
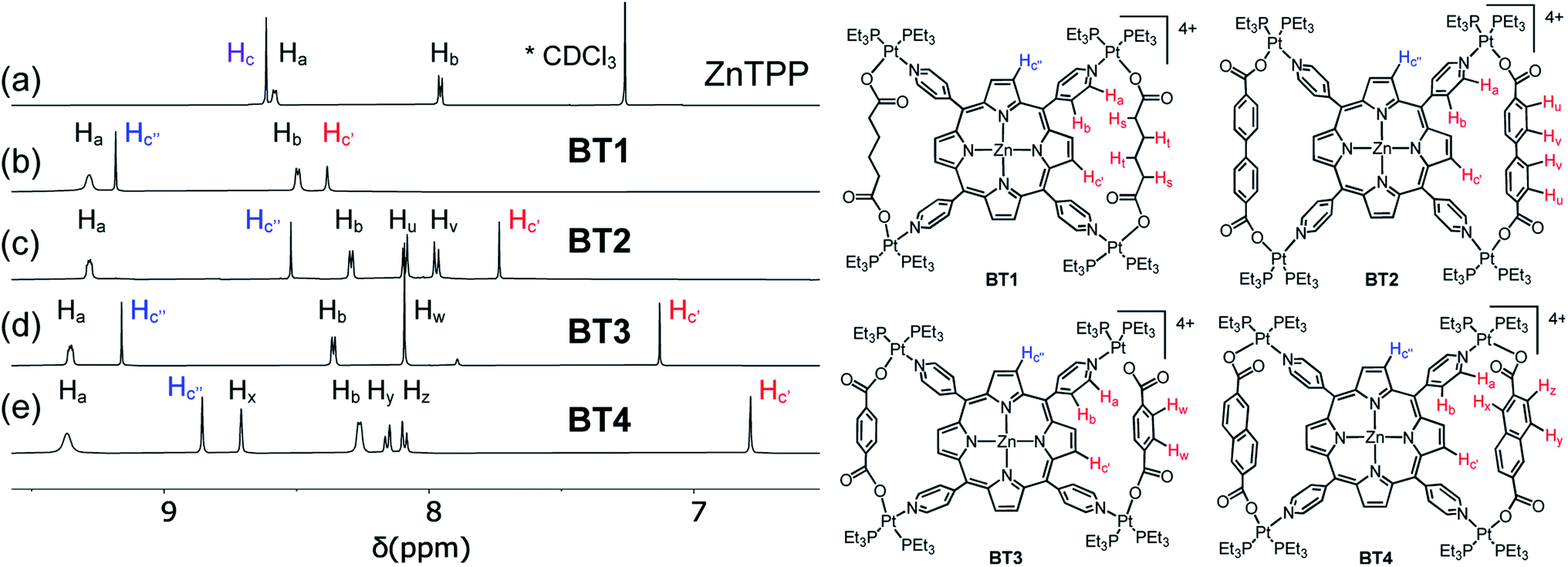 | ||
| Fig. 5 Partial 1H NMR spectra (500 MHz) of (a) ZnTPP, (b) BT1, (c) BT2, (d) BT3, and (e) BT4. The enclosed Hc′ pyrrole protons (highlighted in red) located inside the isosceles triangles of bow-tie structures were shielded by adjacent XDC linkers, whereas the exposed Hc′′ pyrrole protons (highlighted in blue) were not. | ||
The singlet peaks corresponding to four Hc′ pyrrole protons of the BT1–BT4 complexes appeared at 8.38, 7.73, 7.13, and 6.79 ppm, respectively, which were significantly up-field shifted from the Hc signal (8.61 ppm) of free ZnTPP, while the singlets corresponding to four Hc′′ pyrrole protons appeared at 9.18, 8.52, 9.16, and 8.86 ppm, respectively. Thus, the up-field shifts of Hc′ signals were directly correlated to the increasing electron cloud, i.e., the shielding effect of XDC linkers (HDC < BPDC < BDC < NDC), suggesting that these pyrrole protons were located inside the isosceles triangles formed by two parallel XDC linkers, whereas the Hc′′ pyrrole protons were not. The aliphatic HDC linkers in BT1 exerted the least shielding effect, causing the smallest up-field shift of the enclosed Hc′ pyrrole protons located across the bay, followed by the BPDC linkers having two twisted phenyl rings that were not properly aligned with the enclosed Hc′ pyrrole protons of BT2, as seen from its single-crystal structure (vide supra). Having the largest π-cloud and the maximum shielding effect among the four XDC linkers employed here, the NDC linkers in BT4 caused the largest up-field shift of enclosed Hc′ protons, followed by the BDC linkers in BT3. Although the exposed Hc′′ pyrrole protons were not shielded by distant XDC linkers, their chemical shifts were affected by the length of the XDC linkers, which controlled their distances from the adjacent pyridyl rings of ZnTPP. This was further evident from their crystal structures and certain structural features summarized in Table 1. For instance, the shorter HDC and BDC linkers (l ≈ 7 Å) placed the bridged Pt(II) corners closer to each other, which in turn, pulled the pyridyl rings of the ZnTPP ligand away from the exposed Hc′′ pyrrole protons of the BT1 and BT3 complexes. As a result, the shielding effect of pyridyl rings on Hc′′ pyrrole protons in BT1 and BT3 was diminished, and they appeared at more downfield positions (ca. 9.2 ppm) than the Hc protons of the free ZnTPP ligand. On the other hand, the longest BPDC linkers (lBPDC = 11.2 Å) in BT2 held the bridged Pt(II) corners farther away from each other, which in turn, pushed the pyridyl rings of ZnTPP closer to the exposed Hc′′ pyrrole protons making them more shielded and up-field shifted than the Hc protons of free ZnTPP. Having an intermediate length, the NDC linkers (lNDC = 9.2 Å) in BT4 pulled the pyridyl rings of ZnTPP slightly away from exposed Hc′′ protons, causing a slight downfield shift. The exact same trends were observed for H2TPP-based BT1′–BT4′ complexes (Fig. S2†).
Thus, the splitting of pyrrole protons of M′TPP ligands into two chemically nonequivalent and noncoupled Hc′ and Hc′′ protons in the tricomponent SCCs and the variable shielding of the former by XDC linkers were the unmistakable signs of bow-tie complexes where two opposite pyrrole rings bearing four Hc′ protons were located inside the isosceles triangles formed by two parallel XDC linkers while the other two pyrrole rings bearing four Hc′′ protons remained exposed. It is worth noting that previously reported M′TPP-based tricomponent SCCs, including those containing HDC and BDC ligands,32,33 essentially displayed the same 1H NMR characteristics as those displayed by our BT1–BT4 and BT1′–BT4′ complexes, i.e., two distinct singlets for Hc′ and Hc′′ pyrrole protons, which are the characteristic signs of bow-tie structures. If the resulting tricomponent SCCs were indeed tetragonal prisms having two cofacial M′TPP planes connected by four XDC linkers via heteroleptic (Et3P)2PtII(N,O) corners,30,33,34,56 then all sixteen pyrrole protons of M′TPP faces should have remained chemically equivalent and displayed one singlet peak instead of splitting into two chemically non-equivalent Hc′ and Hc′′ protons with two distinct singlets, as observed in other porphyrin-based prisms.47–49,53,55 However, that was not observed for any Pt(II)/M′TPP/XDC-based tricomponent SCCs,30,33,34,56 which ruled out the prism formation.
1H–1H COSY NMR spectroscopy
The COSY NMR spectra of these M′TTP-based tricomponent SCCs (Fig. S4†) provided further insights into their actual structures by revealing the coupling between the adjacent (α- and β-) protons. Conspicuously missing from the COSY NMR spectra of all these SCCs were any α/β-coupling between the Hc′ and Hc′′ protons of the M′TPP ligand, which further indicated that these two chemically non-equivalent protons did not belong to the same pyrrole ring but to two separate pyrrole rings located in different environments. This scenario was possible only in bow-tie structures where two opposite pyrrole rings carrying the Hc′ protons were located inside the triangles, whereas the other two pyrrole rings bearing Hc′′ protons remained exposed. If these tricomponent SCCs were indeed tetragonal prisms containing two cofacial M′TPP panels linked by four XDC linkers, then either all the pyrrole protons of M′TPP should have remained chemically equivalent instead of splitting into distinct Hc′ and Hc′′ protons, or each pyrrole ring would have carried one Hc′ and one Hc′′ protons involved in α/β-coupling (in case of twisted prisms).63 The absence of such couplings ruled out the prism formation.ROESY NMR spectroscopy
Another piece of powerful evidence of bow-tie complex formation was found in the ROESY NMR spectra (Fig. 6 and S5†), which revealed long-range coupling between the enclosed Hc′ pyrrole protons of M′TPP and the XDC protons located across the bay. For example, the enclosed Hc′ pyrrole protons were coupled with (i) the Ht protons of HDC in BT1 and BT1′, (ii) the Hu and Hv protons of BPDC in BT2 and BT2′, (iii) the Hw protons of BDC in BT3 and BT3′, and (iv) the Hx, Hy, and Hz protons of NDC in BT4 and BT4′, indicating that the enclosed pyrrole rings were located in close proximity to the XDC linkers. In addition, the enclosed Hc′ protons were also coupled to the adjacent Hb pyridyl protons of M′TPP ligands. However, no such ROE-coupling between the exposed Hc′′ pyrrole protons and the distant XDC protons were observed (the Hc′′ protons were only coupled to Hb protons of the adjacent pyridyl ring in some cases), further verifying that the enclosed Hc′ and exposed Hc′′ pyrrole protons were located in two different chemical environments. This scenario is possible only in bow-tie structures, not in prisms.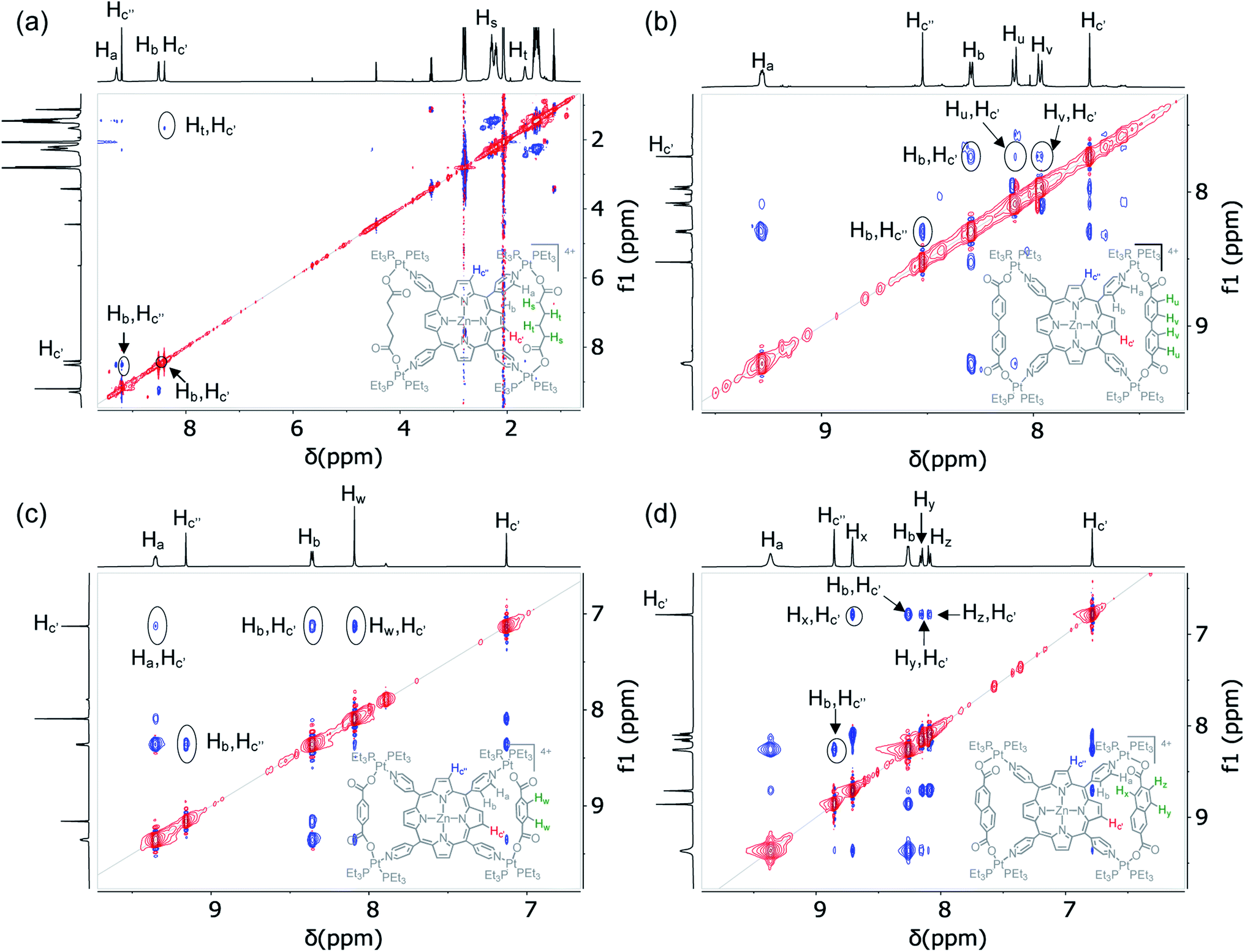 | ||
| Fig. 6 Partial 1H–1H ROESY NMR spectra (500 MHz, acetone-d6) of (a) BT1, (b) BT2, (c) BT3, and (d) BT4 show that the enclosed Hc′ pyrrole protons of these bow-tie complexes located inside the isosceles triangles are through-space coupled with the protons of adjacent XDC linkers but the exposed Hc′′ pyrrole protons are not coupled with the distant XDC protons. | ||
DOSY NMR spectroscopy
Diffusion NMR experiments (acetone-d6) showed that (Fig. S6†) irrespective of the length of XDC linkers (l ≈ 7–11 Å), the BT2, BT3, and BT4 complexes possessed similar diffusion coefficients (D = 5.40 × 10−10, 5.63 × 10−10, and 5.67 × 10−10 m2 s−1, respectively) and hydrodynamic radii (rH = 12.64, 12.11, and 12.04 Å, respectively). Notably, the hydrodynamic radii of these complexes were roughly two-third of a legitimate porphyrin-based tetragonal prism (rH = 17.54 Å),47 and they were not affected by the length of XDC linkers (the longer linkers should have yielded larger cages but did not affect the overall size of 2D bow-tie complexes), which further ruled out the alleged cage formation.ESI-MS analysis
The ESI-MS analysis revealed (Fig. S7†) the characteristic m/z peaks of the [M-2TfO]2+ species of bow-tie complexes. The ZnTPP-based BT1–BT4 and H2TPP-based BT2′, BT3′, and BT4′ displayed the respective [M-2TfO]2+ peaks at m/z = 1497.09, 1593.58, 1517.01, 1567.05, 1561.50, 1485.52, and 1535.60, but no peak corresponding to any tetragonal prisms. Interestingly, the previously reported30 ESI-MS profile of a tricomponent SCC featuring (Et3P)2PtII, H2TPP, and BDC revealed a prominent m/z peak at 1481.38, which possibly represented the [M-2PF6−]2+ species of the bow-tie complex [{(Et3P)2Pt}4(H2TPP)(BDC)2]4+·4(PF6−). Although previous reports have assigned certain m/z peaks to [{(Et3P)2Pt}8(M′TPP)2(XDC)4]8+·8(X−) prisms,30,33,34,56,64 in light of our extensive X-ray crystallographic and NMR analyses, those peaks could be attributed to dimers of [{(Et3P)2Pt}4(M′TPP) (XDC)2]4+·4(X−) bow-tie complexes (after the loss of certain counterions) instead of any prisms. Thus, in light of the discovery of M′TPP-based tricomponent bow-tie complexes and a better understanding of why the corresponding tetragonal prisms were not formed, it appears that careful 2D NMR and X-ray crystallographic studies are vital for accurate structural characterization of similar tricomponent SCCs.Molecular recognition via π-donor/acceptor charge transfer interaction
Having assembled and accurately characterized M′TPP-based novel tricomponent bow-tie complexes, we turned our attention to explore their molecular recognition capabilities. We hypothesized that the electron-rich M′TPP core of bow-tie complexes will selectively recognize electron deficient π-systems through π-donor/acceptor CT interaction but not other π-donors. To test this hypothesis, we employed BT4 ([{(Et3P)2Pt}4(ZnTPP)(NDC)2]4+) as a host, which contains the least strained ZnTPP ligand (i.e., ∠(NPy–Zn–NPy)int ≈ ∠(NPy–Zn–NPy)ext ≈ 88–89°) because of the intermediate length of the rigid NDC linker, and highly electron-deficient HATHCN and electron-rich pyrene compounds as potential guests.The BT4·HATHCN complex formation was evident from the 1H NMR titration experiment (Fig. 7a). The characteristic Hc′ and Hc′′ (pyrrole) protons of the ZnTPP core gradually shifted up-field with the increasing amount of HATHCN (it has no proton), revealing that the ZnTPP core was shielded by cofacially aligned HATHCN. The greater up-field shift and shielding of Hc′′ signal (Δδ = 0.15 ppm) than Hc′ signal (Δδ = 0.06 ppm) suggests that HATHCN is positioned more above the exposed pyrrole rings than sterically crowded (by NDC) enclosed pyrrole rings. The protons on the pyridyl and NDC rings, which are aligned almost perpendicularly to the Zn-porphyrin and HATHCN cores, shifted downfield, indicating that they were not shielded by HATHCN. The formation constant of the BT4·HATHCN complex (Ka = 2.5 × 103 M−1, 3:7 CD2Cl2/CD3NO2, 25 °C) calculated from the 1H NMR titration data (Fig. S8†)65 is comparable to that of similar CT complexes of other electron-rich Zn-porphyrin and π-acidic HAT-derivatives.66 In contrast, during 1H NMR titration of BT4 with pyrene (Fig. 7b), none of their signals shifted, indicating the lack of any meaningful interaction between the two electron-rich species. The 31P NMR spectrum of BT4 remained unchanged in the presence of HATHCN (Fig. S9†), confirming that it did not interfere with the heteroleptic coordination of (Et3P)2Pt2+ corners with the ZnTPP and NDC ligands. The ESI-MS analysis also revealed the [M-TfO]3+ peak (m/z = 1123.36) of the 1:1 BT4·HATHCN complex (Fig. S10†).
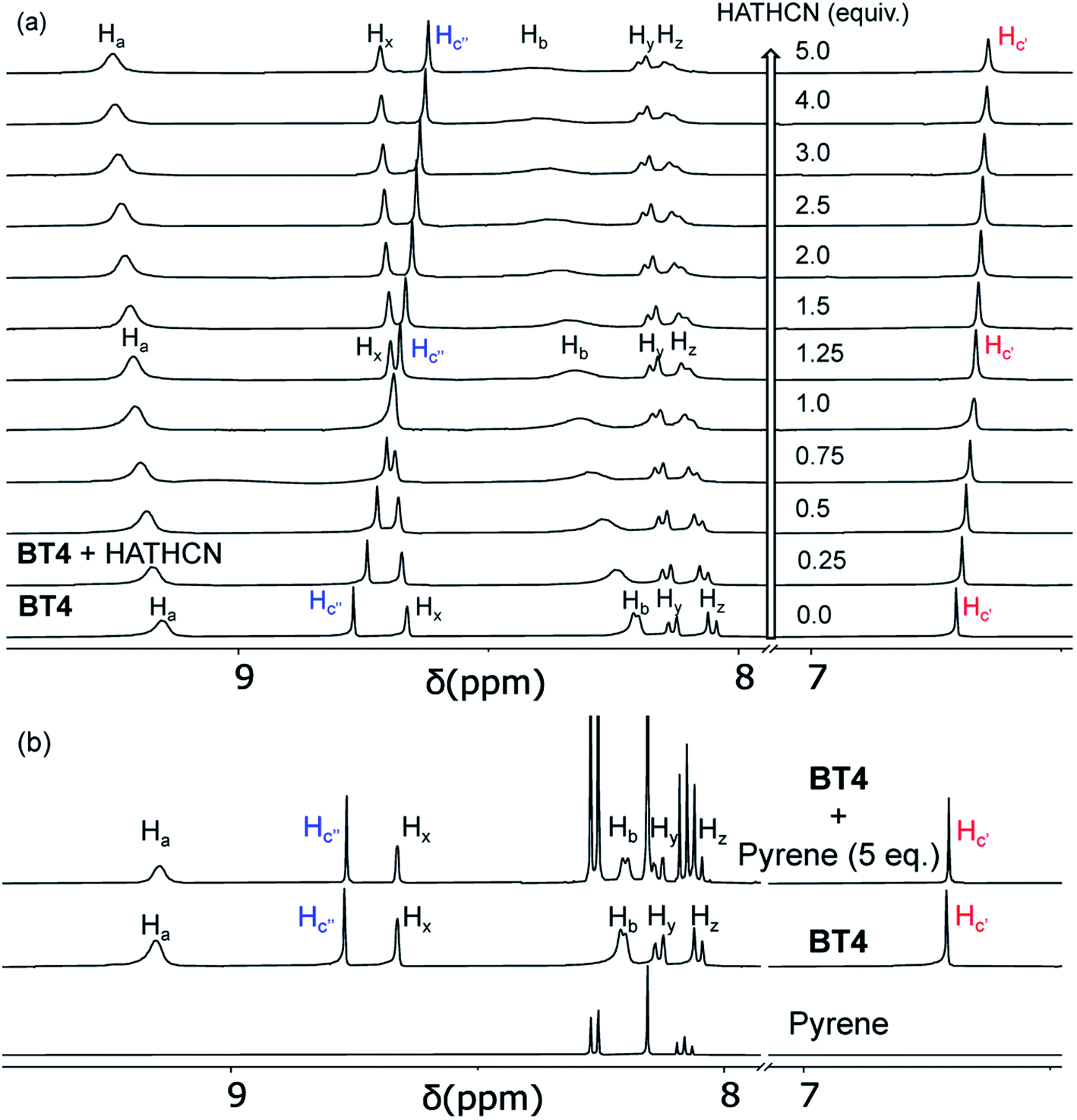 | ||
| Fig. 7 The 1H NMR titration data (500 MHz, 3:7 CD2Cl2/CD3NO2) of BT4 with (a) HATHCN and (b) pyrene show gradual up-field shift, i.e., shielding of Hc′ and Hc′′n (pyrrole) signals of the ZnTPP core by the former but no such change with the latter. | ||
The UV-vis spectrum BT4·HATHCN complex displayed (Fig. 8a) a noticeable decrease in Soret and Q bands intensities (compared to the free BT4 spectrum) with a concomitant appearance of a characteristic broad CT band (650–870 nm) centered at ∼725 nm, indicating ZnTPP/HATHCN π-D/A CT interaction. In addition, in the BT4·HATHCN CT complex, the Q bands of ZnTPP (560 and 602 nm) and the longest wavelength absorption of HATHCN (322 nm), which correspond to S0 → S1 transitions, were noticeably blue-shifted (∼4–6 nm) compared to the free species, a characteristic sign of face-to-face π-D/A interaction between these two complementary π-systems.67 In contrast, no UV-vis absorption change of BT4 was observed in the presence of pyrene (Fig. 8b), indicating the lack of any meaningful electronic interaction between these two electron rich species.
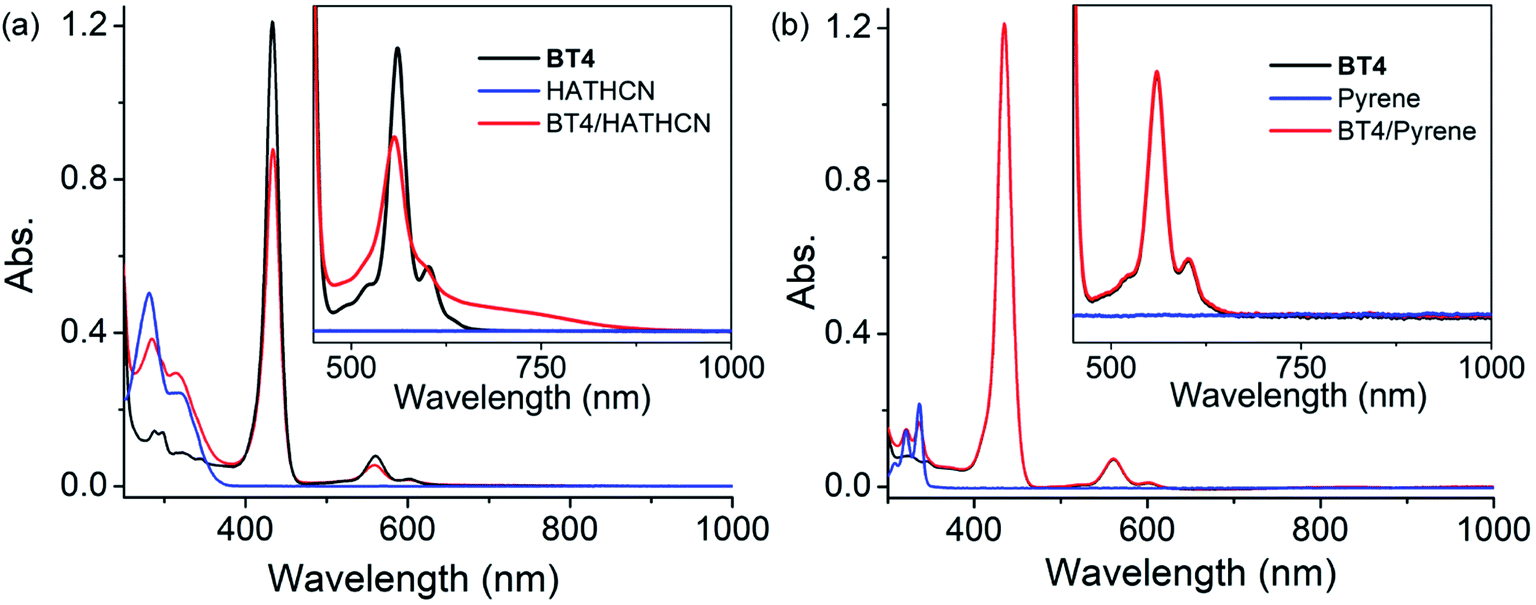 | ||
| Fig. 8 The UV-vis spectra of BT4 (in CH2Cl2) in the presence of (a) HATHCN and (b) pyrene. Insets: the amplified 475–1000 nm regions show the appearance of the CT band with HATHCN but not with pyrene. | ||
The cyclic voltammetry analysis (Fig. S11†) showed that in the BT4·HATHCN complex, the first oxidation (anodic) peak of the ZnTPP core shifted by +40 mV—from +1.14 V for free BT4 to 1.18 V for the BT4·HATHCN CT complex (vs. Ag/AgCl in 0.1 M Bu4N·TfO/CH2Cl2 solution)—suggesting that the CT interaction between the electron-rich ZnTPP core and highly π-acidic HATHCN (LUMO: −4.8 eV)68 made the oxidation of the π-donor harder in the complex. In contrast, no significant shift of the first oxidation (anodic) peak of the ZnTPP core was observed in the presence of electron-rich pyrene due to the lack of such interaction.
Conclusions
In summary, we have demonstrated that Pt(II)-driven social self-assembly of a tetratopic M′TPP ligand and ditopic XDC linkers having different lengths (6.9–11.2 Å) and rigidities yielded novel 2D bow-tie complexes [{(Et3P)2Pt}4(M′TPP)(XDC)2]·(4TfO) featuring a M′TPP core and two parallel XDC linkers that were held together by four heteroligated PtII(N,O) corners. The most direct and compelling evidence of bow-tie complexes came from the SXRD data, which were fully consistent with their NMR characteristics. The 31P NMR spectra of the resulting tricomponent SCCs revealed the formation of heteroleptic PtII(N,O) corners bearing one carboxylate and one pyridyl group, while the 1H and 2D NMR studies presented telltale signs of bow-tie structures by revealing that the two opposite pyrrole rings carrying the more shielded Hc′ protons were located inside two isosceles triangles formed by two parallel XDC linkers, while the other two pyrrole rings bearing the less shielded Hc′′ protons remained exposed. The pyridyl arms of M′TPP deviated from their original projections in order to accommodate different XDC linkers having different lengths (∼7–11 Å) and rigidities into isosceles triangles. This led to the formation of 2D bow-tie complexes, which were entropically more favored over 3D tetragonal prisms. Thus, these comprehensive studies not only unveiled novel M′TPP-based bow-tie complexes, but also ruled out the corresponding prism formation. These revelations underscore the importance of careful multiprobe characterization and careful data analyses to ensure accurate identification of structures and compositions of SCCs, which is key to proper depiction of their structure–property/function relationships because without accurate characterization in the first place, we run the risk of misassigning the properties and functions to nonexistent species, as it happened evidently in the case of illusive prisms despite having all the advanced characterization tools and techniques at our disposal. In addition to assembling and accurately characterizing novel bow-tie-shaped tricomponent coordination complexes based on M′TPP ligands, we have demonstrated that these electron-rich species can bind highly π-acidic HATHCN through strong π-D/A CT interaction, but do not interact with π-donors. While the focus of the foregoing studies was to determine the accurate structures and compositions of M′TPP-based tricomponent SCCs and to demonstrate their molecular recognition capability involving π-donor/acceptor interaction, in light of these new revelations it appears that some fascinating properties and functions, such as photodynamic cancer therapy that were previously attributed to M′TPP-based prisms, actually belonged to the corresponding bow-tie complexes. Further studies of potential applications of these bow-tie complexes, such as light-harvesting and energy transduction systems are underway in our laboratory.Data availability
All relevant experimental and computational data associated with this work have been reported above and in the ESI.†Author contributions
PAB and MAG conducted most of the experiments, analyzed data, prepared all figures, and helped draft and edit the manuscript; AY helped refine the crystal structures; AJJ performed all ESI-MS analyses; and SS supervised the project, verified data analysis, edited and reviewed the manuscript, and secured funding. All authors read the manuscript and agreed with the content.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Science Foundation (NSF award no. CHE-1660329 and DMR-1809092) and Clemson University. We also acknowledge the NSF-MRI grant CHE-1725919 for the 500 MHz NMR instrument used for our studies. We thank Dr Colin McMillen for solving the crystal structures of the bow-tie complexes. We also thank the handling editor and the reviewers for strongly encouraging us to emphasize the importance of proper structural characterization of complex supramolecular assemblies for accurate depiction of their structure–function relationships and to highlight how rigorous structural analyses presented here led to rectification of previously mischaracterized tetragonal prisms as bow-tie complexes for the long-term benefit of supramolecular chemistry community.References
- M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645 CrossRef CAS.
- P. J. Stang and D. H. Cao, J. Am. Chem. Soc., 1994, 116, 4981 CrossRef CAS.
- P. J. Stang, B. Olenyuk, J. Fan and A. M. Arif, Organometallics, 1996, 15, 904 CrossRef CAS.
- P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502 CrossRef CAS.
- A. J. Lees and S.-S. Sun, Inorg. Chem., 2001, 40, 3154 CrossRef PubMed.
- F. Würthner, C.-C. You and C. R. Saha-Möller, Chem. Soc. Rev., 2004, 33, 133 RSC.
- N. C. Gianneschi, M. S. Masar III and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825 CrossRef CAS PubMed.
- B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554 CrossRef CAS PubMed.
- H. T. Chifotides, I. D. Giles and K. R. Dunbar, J. Am. Chem. Soc., 2013, 135, 3039 CrossRef CAS PubMed.
- M. A. Gordillo, P. A. Benavides and S. Saha, Cryst. Growth Des., 2019, 19, 6017 CrossRef CAS.
- K. Kumazawa, K. Biradha, T. Kusukawa, T. Okano and M. Fujita, Angew. Chem., Int. Ed., 2003, 42, 3909 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 112, 734 CrossRef PubMed.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed.
- S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419 RSC.
- S. Fujii, T. Tada, Y. Komoto, T. Osuga, T. Murase, M. Fujita and M. Kiguchi, J. Am. Chem. Soc., 2015, 137, 5939 CrossRef CAS PubMed.
- W. M. Bloch and G. H. Clever, Chem. Commun., 2017, 53, 8506 RSC.
- T. K. Ronson, W. Meng and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 9698 CrossRef CAS PubMed.
- W. Brenner, T. K. Ronson and T. K. Nitschke, J. Am. Chem. Soc., 2017, 139, 75 CrossRef CAS PubMed.
- S. Saha, I. Regeni and G. H. Clever, Coord. Chem. Rev., 2018, 374, 1 CrossRef CAS.
- S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052 CrossRef CAS PubMed.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991 RSC.
- D. Bardhan and D. Chand, Chem.–Eur. J., 2019, 25, 12241 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed.
- W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed.
- K. W. Chi, C. Addicott, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2004, 126, 16569 CrossRef CAS PubMed.
- M. Wang, Y.-R. Zheng, K. Ghosh and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 6282 CrossRef CAS PubMed.
- Y. R. Zheng, W. J. Lan, M. Wang, T. R. Cook and P. J. Stang, J. Am. Chem. Soc., 2011, 133, 17045 CrossRef CAS PubMed.
- Y.-R. Zheng, Z. Zhao, M. Wang, K. Ghosh, J. B. Pollock, T. R. Cook and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 16873 CrossRef CAS PubMed.
- M. Wang, Y.-R. Zheng, T. R. Cook and P. J. Stang, Inorg. Chem., 2011, 50, 6107 CrossRef CAS PubMed.
- J. B. Pollock, T. R. Cook, G. L. Schneider and P. J. Stang, Chem.–Asian J., 2013, 8, 2423 CrossRef CAS PubMed.
- Y. Shi, I. Sánchez-Molina, C. Cao, T. R. Cook and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9390 CrossRef CAS PubMed.
- Y. Ye, T. R. Cook, S. P. Wang, J. Wu, S. Li and P. J. Stang, J. Am. Chem. Soc., 2015, 137, 11896 CrossRef CAS PubMed.
- Y. Sun, C. Chen, J. Liu, L. Liu, W. Tuo, H. Zhu, S. Lu, X. Li and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 17903 CrossRef CAS PubMed.
- G. Yu, T. R. Cook, Y. Li, X. Yan, D. Wu, L. Shao, J. Shen, G. Tang, F. Huang, X. Chen and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13720 CrossRef CAS PubMed.
- Z. Li, X. Yan, F. Huang, H. Sepehrpour and P. J. Stang, Org. Lett., 2017, 19, 5728 CrossRef CAS PubMed.
- M. Zhang, M. L. Saha, M. Wang, Z. Zhou, B. Song, C. Lu, X. Yan, X. Li, F. Huang, S. Yin and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 5067 CrossRef CAS PubMed.
- G. Yu, S. Yu, M. L. Saha, J. Zhou, T. R. Cook, B. C. Yung, J. Chen, Z. Mao, F. Zhang, Z. Zhou, Y. Liu, L. Shao, S. Wang, C. Gao, F. Huang, P. J. Stang and X. Chen, Nat. Commun., 2018, 9, 4335 CrossRef PubMed.
- Y. Sun, Y. Yao, H. Wang, W. Fu, C. Chen, M. L. Saha, M. Zhang, S. Datta, Z. Zhou, H. Yu, X. Li and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 12819 CrossRef CAS PubMed.
- X. Chang, Z. Zhou, C. Shang, G. Wang, Z. Wang, Y. Qi, Z.-Y. Li, H. Wang, L. Cao, X. Li, Y. Fang and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 1757 CrossRef CAS PubMed.
- X. Chang, Z. Zhou, C. Shang, G. Wang, Z. Wang, Y. Qi, Z. Y. Li, H. Wang, L. Cao, X. Li, Y. Fang and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 1757 CrossRef CAS PubMed.
- Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889 RSC.
- S. Ghosh and P. S. Mukherjee, Inorg. Chem., 2009, 48, 2605 CrossRef CAS PubMed.
- A. K. Bar, G. Mostafa and P. S. Mukherjee, Inorg. Chem., 2010, 49, 7647 CrossRef CAS PubMed.
- D. Samanta, S. Shanmugaraju, S. A. Joshi, Y. P. Patil, M. Nethaji and P. S. Mukherjee, Chem. Commun., 2012, 48, 2298 RSC.
- C. García-Simón, M. Garcia-Borràs, L. Gómez, I. Garcia-Bosch, S. Osuna, M. Swart, J. M. Luis, C. Rovira, M. Almedia, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Chem.–Eur. J., 2013, 19, 1445 CrossRef PubMed.
- C. García-Simón, M. Garcia-Borràs, L. Gómez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 5557 CrossRef PubMed.
- C. Colomban, G. Szalóki, M. Allain, L. Gómez, S. Goeb, M. Sallé, M. Costas and X. Ribas, Chem.–Eur. J., 2017, 23, 3016 CrossRef CAS PubMed.
- C. Colomban, V. Martin-Diaconescu, T. Parella, S. Goeb, C. García-Simón, J. Lloret-Fillol, M. Costas and X. Ribas, Inorg. Chem., 2018, 57, 3529 CrossRef CAS PubMed.
- C. Fuertes-Espinosa, A. Gómez-Torres, R. Morales-Martínez, A. Rodríguez-Fortea, C. García-Simón, F. Gándara, I. Imaz, J. Juanhuix, D. Maspoch, J. M. Poblet, L. Echegoyen and X. Ribas, Angew. Chem., Int. Ed., 2018, 57, 11294 CrossRef CAS PubMed.
- C. Colomban, C. Fuertes-Espinosa, S. Goeb, M. Sallé, M. Costas, L. Blancafort and X. Ribas, Chem.–Eur. J., 2018, 24, 4371 CrossRef CAS PubMed.
- C. García-Simón, A. Monferrer, M. Garcia-Borràs, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Chem. Commun., 2019, 55, 798 RSC.
- V. Vajpayee, S. Bivaud, S. Goeb, V. Croué, M. Allain, B. V. Popp, A. Garci, B. Therrien and M. Sallé, Organometallics, 2014, 33, 1651 CrossRef CAS.
- E. Ubasart, O. Borodin, C. Fuertes-Espinosa, Y. Xu, C. García-Simón, L. Gómez, J. Juanhuix, F. Gándara, I. Imaz, D. Maspoch, M. Delius and X. Ribas, Nat. Chem., 2021, 13, 420 CrossRef CAS PubMed.
- G. Yu, Y. Ye, Z. Tong, J. Yang, Z. Li, B. Hua, L. Shao and S. Li, Macromol. Rapid Commun., 2016, 37, 1540 CrossRef CAS PubMed.
- C. Li, H. Nian, Y. Dong, Y. Li, B. Zhang and L. Cao, Inorg. Chem., 2020, 59, 5720 Search PubMed.
- Z. Yang, Y. Wang, X. Liu, R. T. Vanderlinden, R. Ni, X. Li and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 13689 CrossRef CAS PubMed.
- Y. Hou, Z. Zhang, S. Lu, J. Yuan, Q. Zhu, W. P. Chen, S. Ling, X. Li, Y.-Z. Zheng, K. Zhu and M. Zhang, J. Am. Chem. Soc., 2020, 142, 18763 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed.
- S. Lipstman and I. Goldberg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, 371 CrossRef PubMed.
- L. R. Dinelli, G. Von Poelhsitz, E. E. Castellano, J. Ellena, S. E. Galembeck and A. A. Batista, Inorg. Chem., 2009, 48, 4692 CrossRef CAS PubMed.
- A. N. Oldacre, M. R. Crawley, A. E. Friedman and T. R. Cook, Chem.–Eur. J., 2018, 24, 10984 CrossRef CAS PubMed.
- Z. Zhao, Z. Zhang, H. Wang, X. Li and M. Zhang, Isr. J. Chem., 2019, 59, 299 CrossRef CAS PubMed.
- (a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC; (b) www.supramolecular.org .
- T. Aoki, H. Sakai, K. Ohkibo, T. Sakanoue, T. Takenobu, S. Fukizumi and T. Hasobe, Chem. Sci., 2015, 6, 1498 RSC.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
- G. Aragay, A. Frontera, V. Lloveras, J. Vidal-Gancedo and P. Ballester, J. Am. Chem. Soc., 2013, 135, 2620 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2081606–2081610. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06533e |
| This journal is © The Royal Society of Chemistry 2022 |