
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuppressing thermal quenching of lead halide perovskite nanocrystals by constructing a wide-bandgap surface layer for achieving thermally stable white light-emitting diodes†
Qinggang
Zhang
ab,
Mengda
He
a,
Qun
Wan
a,
Weilin
Zheng
a,
Mingming
Liu
a,
Congyang
Zhang
 a,
Xinrong
Liao
a,
Wenji
Zhan
a,
Long
Kong
a,
Xiaojun
Guo
b and
Liang
Li
*ac
a,
Xinrong
Liao
a,
Wenji
Zhan
a,
Long
Kong
a,
Xiaojun
Guo
b and
Liang
Li
*ac
aSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: liangli117@sjtu.edu.cn
bDepartment of Electronic Engineering, School of Electronics Information and Electrical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China
cShanghai Engineering Research Center of Solid Waste Treatment and Resource Recovery, Shanghai Jiao Tong University, Shanghai 200240, China
First published on 28th February 2022
Abstract
Lead halide perovskite nanocrystals as promising ultrapure emitters are outstanding candidates for next-generation light-emitting diodes (LEDs) and display applications, but the thermal quenching behavior of light emission has severely hampered their real-world applications. Here, we report an anion passivation strategy to suppress the emission thermal quenching behavior of CsPbBr3 perovskite nanocrystals. By treating with specific anions (such as SO42−, OH−, and F− ions), the corresponding wide-bandgap passivation layers, PbSO4, Pb(OH)2, and PbF2, were obtained. They not only repair the surface defects of CsPbBr3 nanocrystals but also stabilize the phase structure of the inner CsPbBr3 core by constructing a core–shell like structure. The photoluminescence thermal resistance experiments show that the treated sample could preserve 79% of its original emission intensity up to 373 K, far superior to that (17%) of pristine CsPbBr3. Based on the thermally stable CsPbBr3 nanocrystals, we achieved temperature-stable white LED devices with a stable electroluminescence spectrum, color gamut and color coordinates in thermal stress tests (up to 373 K).
Introduction
Lead halide perovskite nanocrystals (NCs) have emerged as outstanding emitters for light emitting diodes (LEDs) and backlight displays owing to their tunable narrow-band emission, high color purity, high defect tolerance, and superior photoluminescence quantum yields (PLQYs).1–4 In the past few years, there have been breakthroughs in the performance of perovskite based LED devices, such as high efficiency, wide color gamut and cost competitiveness.5–7 The rapid development of perovskite LEDs has led to achieving an external quantum efficiency (EQE) of 23.4% at green wavelengths with excellent operating stability (with an EQE of 45.5% based on a hemispherical lens).8 Despite recent rapid progress, one critical bottleneck to the practical applications of perovskite LEDs is the instability of perovskite NCs, which derives from their intrinsically moisture/light/oxygen/heat sensitive ionic structures and low formation energy.9,10 So far, many great efforts have been made to enhance the stability of perovskite NCs against moisture/light/oxygen, and substantial progress has been obtained, including compact barrier matrix encapsulation (SiO2,11 Al2O3,12 TiO2,13 polymeric matrices,14etc.) and surface engineering strategies.15,16 However, another often overlooked problem of perovskite NCs is the severe drop in luminescence intensity with increasing temperature (so-called fluorescence thermal quenching), leading to a very adverse impact on practical display application.17–20 Actually, LED devices inevitably generate heat during operation, and their temperature can easily reach 373 K or even higher,21 which causes the PL emission thermal quenching behaviors of perovskite NCs and leads to color deviation and brightness weakening of the display screen. Therefore, exploring efficient strategies for realizing high PL emission at elevated temperatures and suppressing the thermal quenching of perovskite NCs is very urgent for their practical applications in LEDs.Generally, the thermal quenching of fluorescent materials is usually caused by their surface trap states or thermally activated carrier trapping and phonon-assisted non-radiative decay.18,19,22 In the 1990s, Hines and Guyot-Sionnest proposed a passivation strategy to generate a stable layer with a wider band gap, so as to protect excitations from probing a defective surface.23 Based on this concept, the thermal quenching problems of traditional CdSe quantum dots have been alleviated to a certain extent.22,24 As for perovskite NCs, due to their low trapping activation energy, numerous defects will be inevitably generated with the increase in temperature, which results in a fast drop of PL emission. For example, all-inorganic CsPbBr3 perovskite NCs heated to 373 K have been shown to suffer an over 80% fading of their room-temperature PL emission.18,19,25 To this end, some breakthrough efforts have been proposed to suppress the serious PL emission thermal quenching of perovskite NCs.
Zou et al.17 demonstrated an effective strategy through Mn2+ substitution to effectively increase the formation energy of CsPbX3 (X = Cl, Br, I) NCs, achieving high thermotolerance and anti-thermal quenching of perovskite NCs. Recently, Liu et al.18 proposed a fluoride ion post-synthesis treatment strategy that produces fluorine-rich surfaces with a wider bandgap than the inner CsPbBr3 core, simultaneously greatly improving the trapping activation energy, achieving stable PLQY values of 90% up to 373 K and with no thermal quenching for successive thermal cycles. However, there are few reports on the in-depth mechanism of thermal quenching and corresponding universal strategies, which leads to the lack of regular and technical guidance for the universal suppression of thermal quenching. For classical colloidal perovskite NCs, in the heating process, colloidal perovskite NCs not only produced surface defects but also inevitably caused ion migration and agglomeration due to intrinsic structural instabilities, which makes it difficult to deeply explore the mechanism of thermal quenching and perform follow-up strategies to suppress thermal quenching. Excitingly, our previous work proposed a high-temperature solid-state method to realize the in situ encapsulation and ceramic-like stability of perovskite NCs.26 In this all-solid-state synthesis, perovskite NCs were confined in the internal solid space of silica and isolated from each other, which prevented ion migration and induced particle fusion between NCs during the test with high thermal stress, and therefore, could be outstanding candidates to explore the mechanism of thermal quenching.
In this work, we have designed an anion passivation strategy based on new high temperature solid-state technology that can effectively passivate surface lead defects and suppress PL thermal quenching in CsPbBr3 NCs. The introduced anions (such as SO42−, OH−, and F− ions) can convert the surface lead of CsPbBr3 NCs to wide-bandgap passivation layers with thermodynamical stability. The wide-bandgap passivation layers, such as PbSO4, Pb(OH)2, and PbF2, not only repair the surface defects of CsPbBr3 NCs, but also greatly improve the thermally activated trapping activation energy, and reduce the recombination probability of excitons and thermally activated defects, resulting in highly thermotolerant CsPbBr3 NCs up to 373 K. Meanwhile, thanks to the complete encapsulation of dense SiO2 during the high temperature solid-state reaction, the resulting CsPbBr3 NCs exhibited extraordinary long-term stability in various environments (water, light, HCl solution, and NaOH solution) for up to 3600 hours without obvious PL emission decays. More surprisingly, the corresponding white LED backlight devices can still maintain the stability of the electroluminescence spectrum, color gamut, and color coordinates in thermal stress tests, providing a promising pathway to achieve brightness and color fidelity in real-world backlight displays.
Results and discussion
Thermal resistance performance
To suppress PL thermal quenching in CsPbBr3 NCs, the wide-bandgap passivation layers must chemically and strongly bond to the perovskite surface. The chemically attached coating layers can passivate the surface defects by eliminating surface dangling bonds, and thus enhance the intrinsic luminescence and stability. More importantly, the wide-bandgap passivation layers with higher formation energy have mechanical rigidity, which can stabilize the phase structure of the inner CsPbBr3 NCs and reduce high temperature induced defects. In the experiment, CsPbBr3 NCs were synthesized by a template confined high temperature solid-state method as shown in Fig. S1.† First, mesoporous silica (MCM-41) was selected as the confined template, which can realize the confined growth of high quality CsPbBr3 NCs. Next, perovskite precursors (CsBr and PbBr2), and objective anion salts were first dissolved in water and mixed with mesoporous silica and then dried at 80 °C. The obtained solid powder was ground and calcined at 700 °C for 30 min in an argon atmosphere. Finally, the cooled samples were washed with ultrapure water and dried to obtain the anion-treated CsPbBr3 NCs. In this case, the mole ratio of CsBr and PbBr2 was always kept at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. In order to form a stable passivation layer on the surface of CsPbBr3 NCs, the introduced anions, such as SO42−, OH−, and F− ions, can closely combine with surface lead (Pb2+) to form an insulating layer, corresponding to PbSO4, Pb(OH)2, and PbF2. The surface passivation layer formed strong chemical bonds with the CsPbBr3 NCs and offered much better resistance to many harmful stimuli under continuous heating and high temperature. The compositions of the different samples were evaluated by energy-dispersive spectroscopy (EDS) and the content of perovskite nanocrystals encapsulated into SiO2 was about 18 wt% (Tables S1–S4†). Next, we explored the effects of different anions on the optical properties of CsPbBr3 NCs. In Fig. S2,† the optical absorption and PL spectra of pristine CsPbBr3 and anion-treated CsPbBr3 NCs showed they all almost have identical spectra including the spectral shape and positions. But, the addition of anions effectively enhanced the PLQY of CsPbBr3 NCs. Upon increasing the anion:Pb ratio to 70%, the PLQYs of anion-treated CsPbBr3 NCs all have been effectively improved. Further increasing the anion:Pb ratio to 100% or even higher will lead to a rapid decline in the PLQY, mainly due to the peeling off of the Pb2+ from CsPbBr3 NCs by excessive anions, which means the reduction of the Pb2+ content in CsPbBr3 NCs and results in the transformation of CsPbBr3 into the Cs4PbBr6 phase (Fig. S3†). For this reason, in subsequent thermal resistance tests and structural studies, we focused on the comparison between pristine CsPbBr3 NCs and anion-treated CsPbBr3 NCs with anion:Pb ratio up to 70%.
:1. In order to form a stable passivation layer on the surface of CsPbBr3 NCs, the introduced anions, such as SO42−, OH−, and F− ions, can closely combine with surface lead (Pb2+) to form an insulating layer, corresponding to PbSO4, Pb(OH)2, and PbF2. The surface passivation layer formed strong chemical bonds with the CsPbBr3 NCs and offered much better resistance to many harmful stimuli under continuous heating and high temperature. The compositions of the different samples were evaluated by energy-dispersive spectroscopy (EDS) and the content of perovskite nanocrystals encapsulated into SiO2 was about 18 wt% (Tables S1–S4†). Next, we explored the effects of different anions on the optical properties of CsPbBr3 NCs. In Fig. S2,† the optical absorption and PL spectra of pristine CsPbBr3 and anion-treated CsPbBr3 NCs showed they all almost have identical spectra including the spectral shape and positions. But, the addition of anions effectively enhanced the PLQY of CsPbBr3 NCs. Upon increasing the anion:Pb ratio to 70%, the PLQYs of anion-treated CsPbBr3 NCs all have been effectively improved. Further increasing the anion:Pb ratio to 100% or even higher will lead to a rapid decline in the PLQY, mainly due to the peeling off of the Pb2+ from CsPbBr3 NCs by excessive anions, which means the reduction of the Pb2+ content in CsPbBr3 NCs and results in the transformation of CsPbBr3 into the Cs4PbBr6 phase (Fig. S3†). For this reason, in subsequent thermal resistance tests and structural studies, we focused on the comparison between pristine CsPbBr3 NCs and anion-treated CsPbBr3 NCs with anion:Pb ratio up to 70%.
Next, we evaluated the PL thermal quenching behavior of CsPbBr3 NCs before and after anion (SO42−, OH−, and F− ions) treatment. As shown in Fig. 1, pristine and anion-treated CsPbBr3 NCs settled on the bottom of the glass bottle were placed on a hot plate at T = 293 K, respectively. When heated up to 373 K, the fluorescence of pristine CsPbBr3 NCs decayed rapidly, but the anion-treated CsPbBr3 NCs can still display bright green emission under UV light irradiation (Fig. 1e), indicating their excellent ability to suppress thermal quenching. To further quantitatively analyze the thermal quenching behavior, we monitored the in situ PL spectra of pristine and anion-treated CsPbBr3 NCs during heating ramps from 293 to 373 K, as shown in Fig. 1a–d and S4.† The corresponding integrated PL intensities of the target samples (normalized to the respective value at 293 K) are shown in Fig. 1f. As for pristine CsPbBr3 NCs, upon raising the temperature to 373 K, the PL intensity dropped sharply to ∼17% of their original value at 293 K. Strikingly, the addition of anions (SO42−, OH−, and F− ions) significantly strengthened the thermal resistance of CsPbBr3 NCs, so that the PL intensities of CsPbBr3–OH, CsPbBr3–SO4 and CsPbBr3–F NCs could still be maintained above 40%, 70%, and 79% of their original PL intensities under thermal stress as high as 373 K, respectively (Fig. 1f). Therefore, the strategy for utilizing anions to build thermally stable passivation layers (PbSO4, Pb(OH)2, and PbF2) can realize the remarkable PL thermal resistance of CsPbBr3 NCs and maintain the emission efficiency under high-temperature working conditions. It should be noted that all the passivation reagents are potassium salts, leading to the introduction of K+ ions that may play some roles in anti-thermal quenching. To clarify the influence of K+ ions, KBr was selected to treat CsPbBr3 NCs, and the thermal quenching of CsPbBr3–KBr NCs is shown in Fig. S5.† Upon increasing the temperature to 373 K, the PL intensity of CsPbBr3–KBr NCs dropped steeply to ∼16% of their initial value at 293 K, which was similar to that of the original sample, basically ruling out the contributions of K+ ions in the stabilization process. In this system, anions (SO42−, OH−, and F− ions) played a more decisive role than cations (K+), mainly due to the stable passivation layer derived from anions and surface lead.
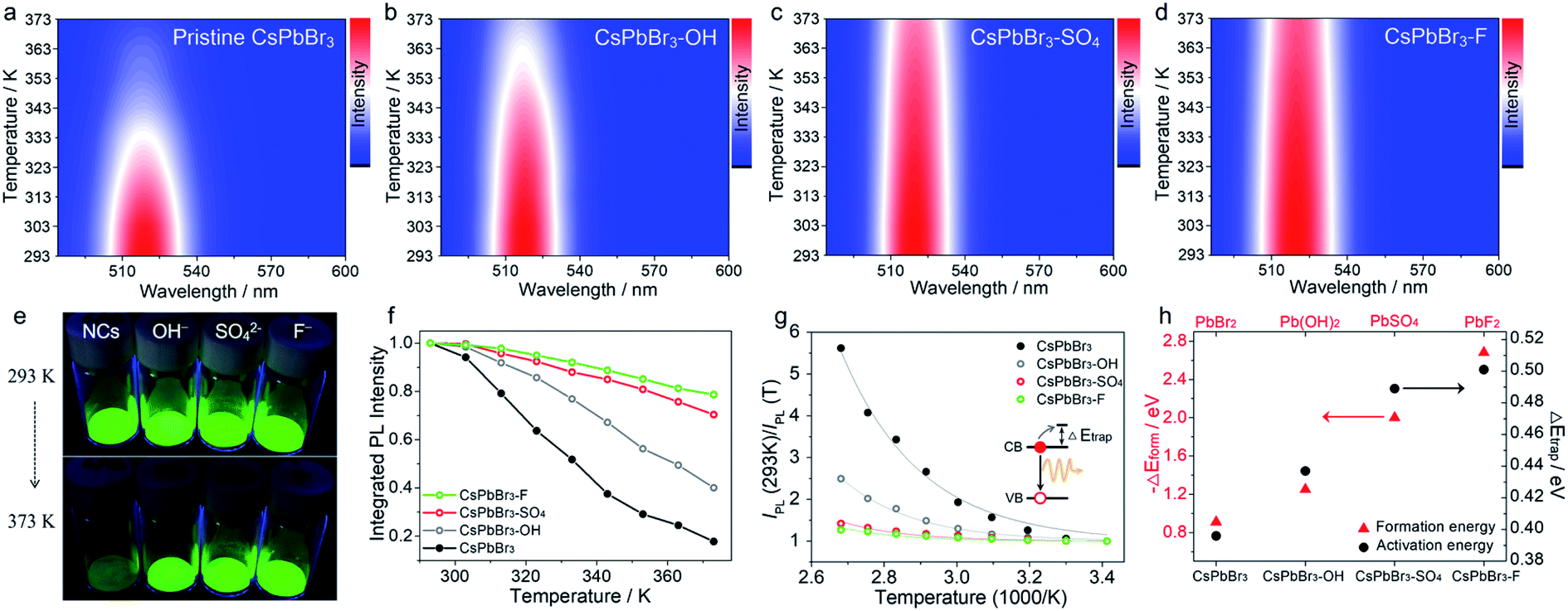 | ||
| Fig. 1 Temperature-dependent PL spectra (excitation wavelength is 450 nm) of pristine CsPbBr3 NCs (a), CsPbBr3–OH NCs (b), CsPbBr3–SO4 NCs (c), and CsPbBr3–F NCs (d) at increasing temperatures from 293 to 373 K. (e) Optical images of pristine and anion-treated CsPbBr3 NCs heated on a hot plate at 293 and 373 K under UV excitation. (f) The integrated PL intensity of pristine and different anion treated CsPbBr3 NCs. (g) Analysis of the PL intensity trend versus 1000/T fitted to a single-trap model. The solid lines are the fitting curves to eqn (1). (h) The trend for formation energy of different lead compounds and ΔEtrap values of different anion-treated CsPbBr3 NCs. | ||
Moreover, we found that there are some correlations between the type of anions and the suppression performance of thermal quenching in NCs. In general, the thermal quenching behavior of NCs or quantum dots is affected by their surface trap states or intrinsic trapping activation energy.24,27 In this work, the suppression effects of the anions on thermal quenching follow a certain trend of F− > SO42− > OH− > Br−. From the viewpoint of chemistry, the four anions have different affinities to Pb2+ (F− > SO42− > OH− > Br−).28,29 Therefore, we speculate that their ability to suppress thermal quenching is determined by the formation energy of the surface lead passivation layers. To further analyze the effect of the different anion treatments, the activation energy for carrier trapping (ΔEtrap) of target CsPbBr3 NCs was estimated by fitting the temperature-dependent integrated PL intensity, through Arrhenius eqn (1):
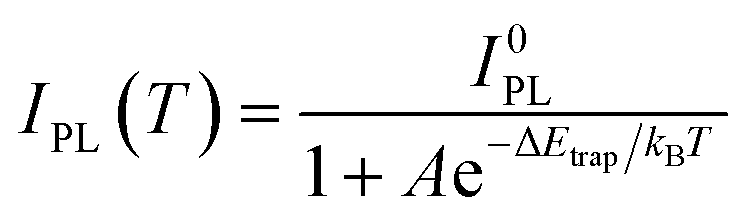 | (1) |
In contrast, the passivation layers with higher formation energy can effectively inhibit ion migration, keep the phase structure stable and reduce the defects of CsPbBr3 NCs at high temperatures, thereby inhibiting non-radiative recombination and improving anti-thermal quenching. Therefore, the trends of ΔEtrap values and formation energy of surface lead layers correlate nearly perfectly with each other (Fig. 1h), further confirming the speculated mechanism of the anion treatment in the excellent thermal resistance of the CsPbBr3 NCs.
Notably, to further demonstrate our concept, the target anion treatments were also applied to colloidal CsPbBr3 NCs without careful optimization (Fig. 2 and S6†). In this case, initial colloidal CsPbBr3 NCs were synthesized by the hot-injection method.18 Then, a certain amount of the anion salt solution was added to the CsPbBr3 toluene solution with stirring for 30 min at room temperature. Finally, the anion-treated colloidal CsPbBr3 NCs solution was obtained via centrifugation. The post treatment resulted in a substantial enhancement of the thermal resistance, demonstrating the universality of the proposed strategy. It should be noted that, in the colloidal NC system, colloidal CsPbBr3–OH NCs cannot be obtained, because the strong base will rapidly destroy the surface ligands of NCs and cause PL quenching. After treatment with SO42− and F− ions, the treated CsPbBr3 NCs showed excellent thermal resistance, as shown in Fig. 2, following a similar trend (F− > SO42− > Br−).
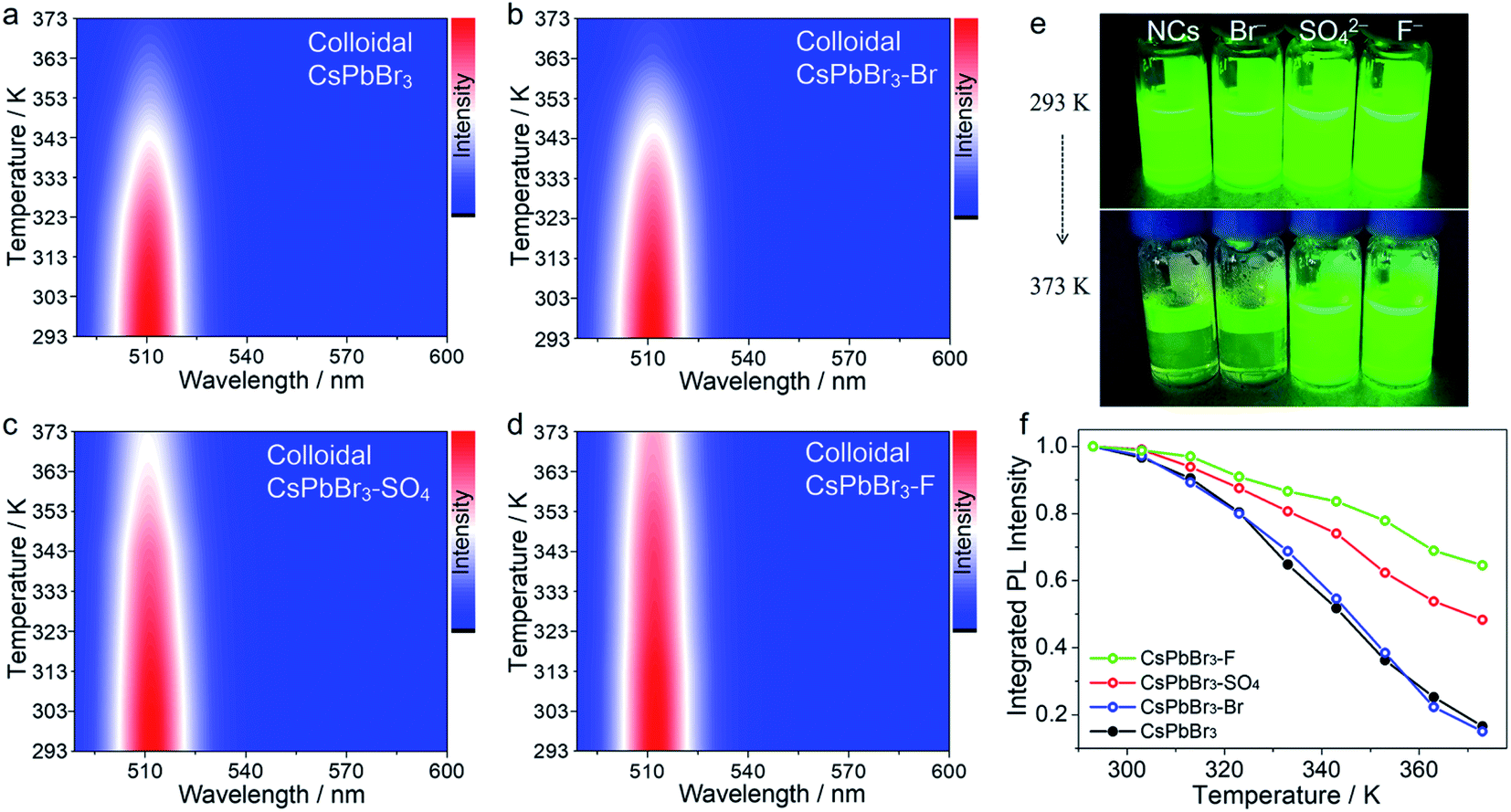 | ||
| Fig. 2 Temperature-dependent PL spectra (excitation wavelength is 450 nm) of colloidal CsPbBr3 NCs (a), colloidal CsPbBr3–KBr NCs (b), colloidal CsPbBr3–SO4 NCs (c), and colloidal CsPbBr3–F NCs (d) at increasing temperatures from 293 to 373 K. (e) Optical images of pristine and anion-treated colloidal CsPbBr3 NCs upon heating on a hot plate at 293 and 373 K under UV excitation. (f) The integrated PL intensity of pristine and different anion treated colloidal CsPbBr3 NCs. | ||
In-depth mechanism insights for suppressing thermal quenching
All the above results showed that the target anions can effectively improve the trapping activation energy and anti-thermal quenching of CsPbBr3 NCs. To understand the mechanism, we took pristine CsPbBr3 NCs and SO42−-treated CsPbBr3 NCs as examples, and performed a series of temperature-dependent in situ characterization studies to confirm the proposed mechanism of surface lead passivation and a wide-bandgap surface layer for suppressing thermal quenching.In situ UV-vis absorption spectra were obtained to monitor photoinduced exciton evolutions of CsPbBr3 NCs at different temperatures from 293 to 373 K (Fig. 3a and b). With the increase in temperature, the absorption band edges of both pristine and SO42−-treated CsPbBr3 NCs remained almost unchanged, but the intensity of their exciton absorption peaks had changed significantly. The stabilities of excitons are determined by external factors such as temperature and carrier concentration.30,31 In this case, high temperature can lead to exciton decomposition and broadening of the exciton absorption peak, resulting in fluorescence decay or even quenching. By comparison, when the temperature increased to 373 K, the exciton absorption peak of the pristine CsPbBr3 NCs became widened significantly, while the SO42−-treated CsPbBr3 NCs still maintained the initial shape of the absorption peak (Fig. 3a and b). To visually observe the evolution of the exciton absorption peak, we treated the absorption spectra of NCs with the second derivative as shown in Fig. 3a and b. Obviously, the decay values of the exciton absorption peak for pristine CsPbBr3 NCs were sharper than that of CsPbBr3–SO4 NCs. We reported the PL intensity of pristine and SO42−-treated CsPbBr3 NCs during five thermal cycles between T = 293 and 373 K, as shown in Fig. S7.† The results showed that the thermal quenching behavior was perfectly reversible, which eliminated the permanent or irreversible destruction of the perovskite structure under thermal stress. Therefore, the observed thermal quenching behavior was purely caused by the surface state of the single CsPbBr3 NCs. Pristine CsPbBr3 NCs have more surface defects, and high temperature will also induce extra defects, such as lattice expansion/distortion, surface ion migration, etc., which will make excitons easier to capture by defects at high temperature, leading to severe PL decay at high temperature. As for the CsPbBr3–SO4 sample, on the one hand, SO42− ion treatment can effectively passivate the surface defects, improve the trapping activation energy, stabilize the structure and inhibit the generation of defects at high temperature; on the other hand, the wide-bandgap PbSO4 passivation layer on the surface of NCs can confine the excitons and remain stable at high temperature, leading to reduced quenching of PL emission.
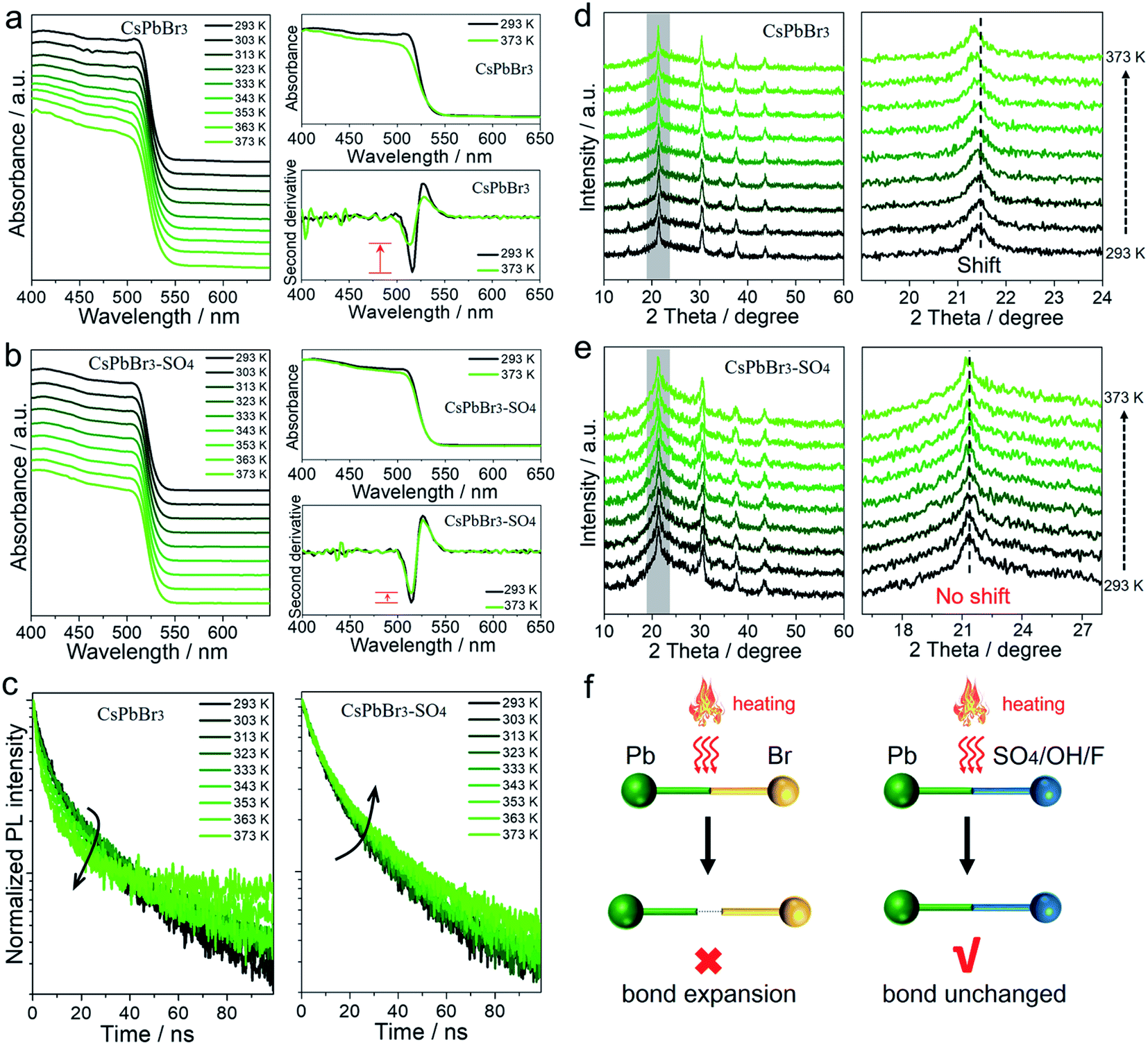 | ||
| Fig. 3 Temperature-dependent in situ characterization studies for pristine and SO42−-treated CsPbBr3 NCs at increasing temperatures from 293 to 373 K. (a and b) In situ UV-vis absorption spectra and treatment of the absorption spectra with the second derivative, (c) in situ PL lifetime decay curves, and (d and e) in situ XRD patterns. (f) Schematic diagram of the suggested mechanism for thermal stability. | ||
In situ PL lifetime decay curves were obtained to evaluate the trap state evolutions during the rise in temperatures (Fig. 3c), and the exponential fitting results are listed in Tables S7–S8.† For pristine CsPbBr3 NCs, with the rise in temperature, the average PL lifetime τave dropped from 25.08 ns to 13.40 ns, implying that carrier trapping was strongly enhanced at elevated temperatures. The accelerated PL kinetics decay originated from the thermally activated escape of carriers to various defects at elevated temperatures, such as halogen vacancies and lead defects, according to previous reports.18,19,24,27 Thanks to SO42− ion treatment, the average PL lifetimes of CsPbBr3–SO4 NCs increased slightly from 24.50 ns to 29.66 ns due to thermally activated delayed fluorescence during the heating process.19,27 Therefore, the SO42− ion passivation strategy can effectively eliminate trap states and reduce the non-radiative recombination path by constructing a PbSO4 passivation layer, which was consistent with the high remaining PL.
In situ XRD was carried out to monitor the crystal structure evolutions of CsPbBr3 NCs before and after SO42− passivation during temperature ramps from 293 to 373 K (Fig. 3d and e). Upon increasing the temperature to 373 K, both pristine and SO42−-treated CsPbBr3 NCs still present diffraction peaks corresponding to the cubic phase of the CsPbBr3 crystal, showing that increasing the temperature up to 373 K will not promote the derivation of other phases from CsPbBr3. After careful analysis, the XRD diffraction peaks of pristine CsPbBr3 NCs shifted toward lower angles with the increase in temperature, meaning that the lattice spacing increased at high temperature, which derived from the bond lengthening and lattice expansion of the CsPbBr3 crystal at high temperature. However, XRD diffraction peaks of CsPbBr3–SO4 NCs remained almost unchanged when the temperature increased from 293 to 373 K. In situ XRD of pure PbSO4 samples during temperature ramps from 293 to 373 K (Fig. S8†) showed that the diffraction peaks of PbSO4 remained almost unchanged when the temperature increased from 293 to 373 K, indicating that lattice expansion can be ignored at 373 K. Therefore, we suspect that the thermally stable PbSO4 passivation layers with higher formation energy can act as a rigid shell to stabilize and suppress the high-temperature induced lattice expansion of inner CsPbBr3 NCs.
Transmission electron microscopy (TEM) images (Fig. 4a and S9–S11†) of pristine CsPbBr3 NCs and CsPbBr3–SO4 NCs showed that CsPbBr3 NCs were uniformly distributed in the collapsed SiO2 system. The scanning electron microscopy (SEM) images of CsPbBr3–SO4 showed that the surfaces of the sample were smooth without impurities through the water washing step (Fig. S12†), which also indicated that the CsPbBr3 NCs were indeed encapsulated inside the SiO2 system. And the average sizes of pristine CsPbBr3 NCs and CsPbBr3–SO4 NCs were 10.5 nm and 12.3 nm, respectively, and the increased size of CsPbBr3–SO4 NCs may be caused by the formation of the PbSO4 layer. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and its corresponding elemental mappings of CsPbBr3–SO4 (Fig. 4a, b and S10†) revealed that the Cs, Pb, Br, and S elements were distributed in CsPbBr3 NCs. Furthermore, we carried out energy-dispersive spectroscopy (EDS) on the samples to accurately determine the location and content of the S element, as shown in Fig. 4c, d and S13–S14.† We selected four points in TEM for EDS analysis, and the results showed that the S element mainly existed on the CsPbBr3 NCs, while there was almost no S element in the blank area (without CsPbBr3 NCs) in the system (Fig. 4c and d), indicating that SO42− ions were indeed adsorbed on CsPbBr3 NCs and tended to combine with Pb2+ to form the PbSO4 passivation layer. The high-resolution TEM (HR-TEM) image and the corresponding fast Fourier transform (FFT) pattern were used to further verify the core–shell like structure. As for pristine CsPbBr3 NCs, the samples showed a regular perovskite lattice, which belonged to the cubic phase (Fig. S9†). Obviously, compared with pristine CsPbBr3 NCs, there were more diffraction points in CsPbBr3–SO4, which belonged to PbSO4 and CsPbBr3 NCs (Fig. 4e, f and S15†). And the HR-TEM images revealed that PbSO4 formed at the edge of the NCs had a periodic lattice, indicating that the PbSO4 layer was tightly coated on the surface of CsPbBr3 NCs to construct a typical core–shell like structure. Therefore, the role of the introduced SO42− ions was to mainly combine with the surface lead of the CsPbBr3 NCs to form a more stable PbSO4 passivation layer,32 as shown in Fig. 4g. After careful comparison, we found that the characteristic XRD peaks of PbSO4 appeared in the system when the doping content of SO42− ions was increased to 70% (Fig. S3†). The Fourier transform infrared (FT-IR) spectra (Fig. 4h and S16†) of the SO42− treated sample showed a characteristic vibration peak at 618 cm−1 ascribed to the tetrahedral structure of SO42− ions, indicating the formation of strong ionic bonds between Pb2+ and SO42− ions;32 and the Raman spectra33 (Fig. 4i) further confirmed that the SO42−-treated CsPbBr3 NCs presented the characteristic peaks of PbSO4 at around 454, 620, and 984 cm−1, which appropriately correspond to the pure PbSO4 standard sample (Fig. S17†). X-ray photoemission spectroscopy (XPS) measurements based on the washed samples further corroborated the perovskite surface composition after SO42− treatment.32 First, the S 2p spectra confirmed the presence of SO42− ions in treated NCs (Fig. S18†), and the Pb 4f spectra revealed noticeable differences between pristine CsPbBr3 NCs and SO42−-treated CsPbBr3 NCs (Fig. 4j). After SO42− treatment, the Pb peaks of CsPbBr3–SO4 NCs shifted toward higher binding energies, indicating the formation of stronger ionic bonding between Pb2+ and SO42− ions. Therefore, the above solid evidence, including HR-TEM, FT-IR, Raman, and XPS spectra, all confirmed that the surfaces of CsPbBr3 NCs were converted to a wide-bandgap PbSO4 passivation layer through the reaction with SO42− ions. Similarly, the introduced F− ions and OH− ions can also form stable passivation layers on the surface of CsPbBr3 NCs (Fig. S19–S20†).
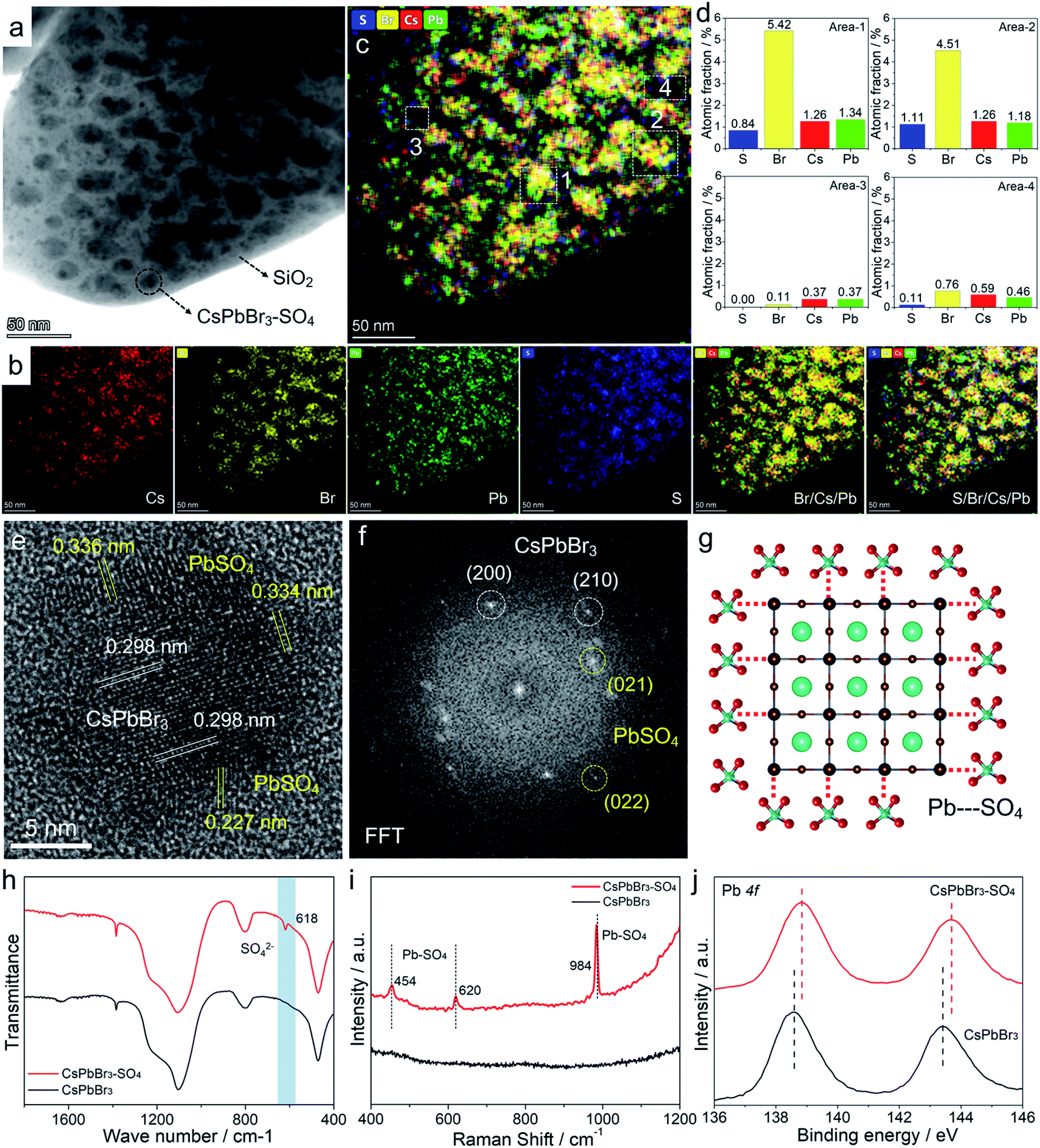 | ||
| Fig. 4 (a and b) TEM image and the corresponding elemental mapping images of the CsPbBr3–SO4 sample. (c and d) Mixed elemental mapping image and the corresponding EDS results of different areas. (e and f) HR-TEM image and the corresponding FFT image of CsPbBr3–SO4 NCs. (g) Schematic diagram of the CsPbBr3–PbSO4 core–shell structure. (h) FT-IR spectra, (i) Raman spectra, and (j) XPS spectra of Pb 4f for pristine and SO42−-treated CsPbBr3 NCs. | ||
Based on the above analysis, we have speculated the mechanism of the anion passivation strategy to suppress thermal quenching, as shown in Fig. 3f. In short, for untreated NCs, high temperature can induce or accelerate surface ion migrations, which produced more surface defects, thereby increasing non-radiative recombination paths and causing PL emission thermal quenching. After being treated by the target anions, the thermally stable passivation layers (PbSO4, Pb(OH)2, and PbF2) can act as a rigid shell to stabilize and suppress the high-temperature induced ion migration of CsPbBr3 NCs, meanwhile effectively repairing surface defects and improving the trapping activation energy, yielding reduced non-radiative recombination paths and suppressed PL emission thermal quenching.
Temperature-stable white light-emitting diodes
It is worth emphasizing that the above experiments and discussions have verified that the anion-treated CsPbBr3 NCs presented excellent high-temperature resistance, which was very considerable for practical display application. For this purpose, we fabricated proof-of-concept white light-emitting diode (WLED) devices with SO42−-treated CsPbBr3 NCs as the active materials, and preliminarily explored their performance at different operating temperatures (Fig. 5 and S21†). The WLED devices were prepared by encapsulating green-emitting CsPbBr3 NCs and commercial red-emitting KSF (K2SiF6:Mn4+) phosphors onto the blue emitting GaN chip (wavelength: 450 nm). As for pristine CsPbBr3 NCs (Fig. 5a and Table S9†), the corresponding WLED devices displayed white light emission at room temperature (293 K), and the CIE color coordinates were optimized at (0.3309, 0.3561). However, when the operating temperature gradually rose to 373 K, the CIE color coordinates shifted significantly from the initial (0.3309, 0.3561) to (0.3756, 0.2469), representing purplish pink luminescence, due to the thermal quenching of the green light components (CsPbBr3 NCs) at high temperatures (Fig. 5b). The WLED devices based on CsPbBr3–SO4 NCs can still display quasi white light emission even at a high temperature of 373 K, corresponding to the color coordinates at (0.3676, 0.3130), as shown in Fig. 5c and Table S10.† And the WLED devices can maintain a stable electroluminescence spectrum in thermal stress tests (Fig. 5d). Furthermore, the color gamut of the WLED devices based on CsPbBr3–SO4 NCs had only a slight attenuation at 373 K, from 130.7% to 128.5% (Fig. S21†), meaning that the corresponding display screen can maintain uniform brightness and color fidelity in the working environment.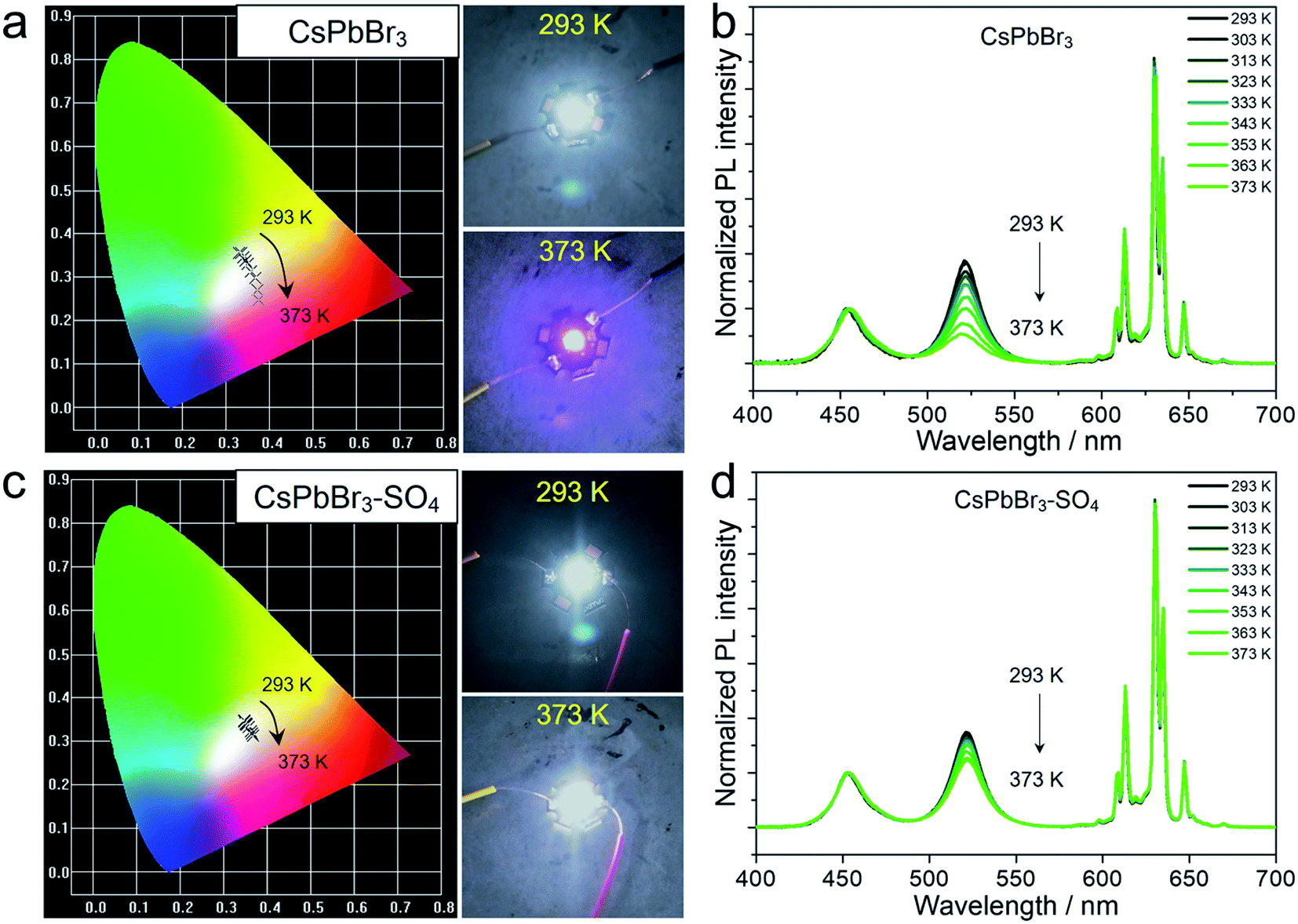 | ||
| Fig. 5 (a and b) The WLED based on pristine CsPbBr3 NCs. (a) The color coordinates of the WLED at increasing temperatures from 293 to 373 K, inset: photographs of the WLED operated at 293 K and 373 K. (b) The emission spectrum of the WLED at increasing temperatures from 293 to 373 K. (c and d) The WLED based on CsPbBr3–SO4 NCs. (c) The color coordinates of the WLED at increasing temperatures from 293 to 373 K, inset: photographs of the WLED operated at 293 K and 373 K. (d) The emission spectrum of the WLED at increasing temperatures from 293 to 373 K. | ||
Meanwhile, aside from the excellent thermal stability, thanks to the complete encapsulation of dense SiO2 during the high-temperature solid-state reaction, the resulting sealed CsPbBr3 NCs exhibited extraordinary long-term chemical and photo stabilities in various environments (water, light, HCl solution, and NaOH solution) for up to 3600 hours without obvious PL emission decays (Fig. S22–S24†).
Conclusions
In summary, we have demonstrated the anion passivation strategy that can convert the surface lead of CsPbBr3 NCs to thermally stable passivation layers to suppress emission thermal quenching behavior and improve the thermal stability of the CsPbBr3 structure. Temperature-dependent in situ characterization studies, including XRD, UV-vis absorption spectra, and PL lifetime, were performed to deeply evaluate the origin of thermal quenching and the mechanism of suppressing thermal quenching by the coating of wide-bandgap passivation layers. We finally confirmed that suppressing the thermal quenching is greatly related to the formation energy of surface lead passivation layers following the order of PbF2 >PbSO4 >Pb(OH)2 > PbBr2. By this anion assisted high-temperature solid-state synthesis, we simultaneously solved the universal chemical and photo stability problems of CsPbBr3 NCs. On the one hand, the dense SiO2 encapsulation successfully protects CsPbBr3 NCs from some factors that can quench photoluminescence, such as moisture, oxygen, and ion migration. On the other hand, by using wide-bandgap passivation layers, the trapping activation energy of CsPbBr3 NCs was significantly enhanced, which allows CsPbBr3 NCs to achieve a thermally stable electroluminescence spectrum, color gamut, and color coordinates at high temperatures. Therefore, we believe that our research results have great guiding significance for the practical application of LED devices or backlight displays in the future.Data availability
All characterization data are available in the ESI.† Raw data are available from the corresponding authors.Author contributions
Q. Z. and L. L. proposed the original research idea. The manuscript was co-written by L. L. and Q. Z. The experiments, including nanocrystal synthesis, thermal quenching experiments, LED encapsulation and the stability test were performed by Q. Z., M. H., Q. W., W. Z., M. L., C. Z., X. L., and W. Z. Q. Z., L. K. and X. G performed the structural characterization. All the authors discussed the results, interpreted the findings, and reviewed the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Key R&D Program (2018YFC1800600); the National Natural Science Foundation of China (NSFC 22175113, 42007125); the China Postdoctoral Science Foundation (2021M702115); the Guangdong Province's 2018-2019 Key R&D Program (2019B010924001); Shanghai Jiao Tong University Scientific and Technological Innovation Funds.References
- T. Chiba, Y. Hayashi, H. Ebe, K. Hoshi, J. Sato, S. Sato, Y.-J. Pu, S. Ohisa and J. Kido, Nat. Photonics, 2018, 12, 681–687 CrossRef CAS.
- H. Tsai, S. Shrestha, R. A. Vilá, W. Huang, C. Liu, C.-H. Hou, H.-H. Huang, X. Wen, M. Li and G. Wiederrecht, Nat. Photonics, 2021, 1–7 Search PubMed.
- C. Zhang, Q. Wan, B. Wang, W. Zheng, M. Liu, Q. Zhang, L. Kong and L. Li, J. Phys. Chem. C, 2019, 123, 26161–26169 CrossRef CAS.
- C. Zhang, S. Wang, X. Li, M. Yuan, L. Turyanska and X. Yang, Adv. Funct. Mater., 2020, 30, 1910582 CrossRef CAS.
- S. Pimputkar, J. S. Speck, S. P. DenBaars and S. Nakamura, Nat. Photonics, 2009, 3, 180–182 CrossRef CAS.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- Q. Zhang, X. Sun, W. Zheng, Q. Wan, M. Liu, X. Liao, T. Hagio, R. Ichino, L. Kong and H. Wang, Chem. Mater., 2021, 33, 3575–3584 CrossRef CAS.
- Y.-H. Kim, S. Kim, A. Kakekhani, J. Park, J. Park, Y.-H. Lee, H. Xu, S. Nagane, R. B. Wexler and D.-H. Kim, Nat. Photonics, 2021, 15, 148–155 CrossRef CAS.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- Q. Zhang, W. Zheng, Q. Wan, M. Liu, X. Feng, L. Kong and L. Li, Adv. Opt. Mater., 2021, 9, 2002130 CrossRef CAS.
- S. Huang, Z. Li, L. Kong, N. Zhu, A. Shan and L. Li, J. Am. Chem. Soc., 2016, 138, 5749–5752 CrossRef CAS PubMed.
- Z. Li, L. Kong, S. Huang and L. Li, Angew. Chem., 2017, 129, 8246–8250 CrossRef.
- Z. J. Li, E. Hofman, J. Li, A. H. Davis, C. H. Tung, L. Z. Wu and W. Zheng, Adv. Funct. Mater., 2018, 28, 1704288 CrossRef.
- Z. Wang, R. Fu, F. Li, H. Xie, P. He, Q. Sha, Z. Tang, N. Wang and H. Zhong, Adv. Funct. Mater., 2021, 31, 2010009 CrossRef CAS.
- G. H. Ahmed, J. Yin, O. M. Bakr and O. F. Mohammed, ACS Energy Lett., 2021, 6, 1340–1357 CrossRef CAS.
- K. Hills-Kimball, H. Yang, T. Cai, J. Wang and O. Chen, Adv. Sci., 2021, 2100214 CrossRef CAS PubMed.
- S. Zou, Y. Liu, J. Li, C. Liu, R. Feng, F. Jiang, Y. Li, J. Song, H. Zeng and M. Hong, J. Am. Chem. Soc., 2017, 139, 11443–11450 CrossRef CAS PubMed.
- M. Liu, Q. Wan, H. Wang, F. Carulli, X. Sun, W. Zheng, L. Kong, Q. Zhang, C. Zhang and Q. Zhang, Nat. Photonics, 2021, 15, 379–385 CrossRef CAS.
- Q. Zhang, Z. Li, M. Liu, L. Kong, W. Zheng, B. Wang and L. Li, J. Phys. Chem. Lett., 2020, 11, 993–999 CrossRef CAS PubMed.
- C. Bi, S. Wang, Q. Li, S. V. Kershaw, J. Tian and A. L. Rogach, J. Phys. Chem. Lett., 2019, 10, 943–952 CrossRef CAS PubMed.
- V. Bachmann, C. Ronda and A. Meijerink, Chem. Mater., 2009, 21, 2077–2084 CrossRef CAS.
- X. Yuan, J. Zheng, R. Zeng, P. Jing, W. Ji, J. Zhao, W. Yang and H. Li, Nanoscale, 2014, 6, 300–307 RSC.
- M. A. Hines and P. Guyot-Sionnest, J. Phys. Chem., 1996, 100, 468–471 CrossRef CAS.
- Y. Zhao, C. Riemersma, F. Pietra, R. Koole, C. de Mello Donegá and A. Meijerink, ACS Nano, 2012, 6, 9058–9067 CrossRef CAS PubMed.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- Q. Zhang, B. Wang, W. Zheng, L. Kong, Q. Wan, C. Zhang, Z. Li, X. Cao, M. Liu and L. Li, Nat. Commun., 2020, 11, 31 CrossRef CAS PubMed.
- B. T. Diroll, G. Nedelcu, M. V. Kovalenko and R. D. Schaller, Adv. Funct. Mater., 2017, 27, 1606750 CrossRef.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder, APL Mater., 2013, 1, 011002 CrossRef.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- D. Lubyshev, P. González-Borrero, E. Marega Jr, E. Petitprez, N. La Scala Jr and P. Basmaji, Appl. Phys. Lett., 1996, 68, 205–207 CrossRef CAS.
- G. Han, B. Chen, L. Zuo, K. Xue, P. Chen, Y. Duan and Y. Zhao, Org. Electron., 2016, 37, 207–212 CrossRef CAS.
- S. Yang, S. Chen, E. Mosconi, Y. Fang, X. Xiao, C. Wang, Y. Zhou, Z. Yu, J. Zhao and Y. Gao, Science, 2019, 365, 473–478 CrossRef CAS PubMed.
- B. Han, A. Xie, Q. Yu, F. Huang, Y. Shen and L. Zhu, Appl. Surf. Sci., 2012, 261, 623–627 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization details and additional figures and tables. See DOI: 10.1039/d1sc06554h |
| This journal is © The Royal Society of Chemistry 2022 |