
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC(sp3)–H oxygenation via alkoxypalladium(II) species: an update for the mechanism†
Shuaizhong
Zhang‡
,
Jinquan
Zhang‡
 and
Hongbin
Zou
*
and
Hongbin
Zou
*
College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, Zhejiang 310058, P. R. China. E-mail: zouhb@zju.edu.cn
First published on 13th January 2022
Abstract
Pd-catalyzed C(sp3)–H oxygenation has emerged as an attractive strategy for organic synthesis. The most commonly proposed mechanism involves C(sp3)–H activation followed by oxidative addition of an oxygen electrophile to give an alkylpalladium(IV) species and further C(sp3)–O reductive elimination. In the present study of γ-C(sp3)–H acyloxylation of amine derivatives, we show a different mechanism when tert-butyl hydroperoxide (TBHP) is used as an oxidant—namely, a bimetallic oxidative addition-oxo-insertion process. This catalytic model results in an alkoxypalladium(II) intermediate from which acyloxylation and alkoxylation products are formed. Experimental and computational studies, including isolation of the putative post-oxo-insertion alkoxypalladium(II) intermediates, support this mechanistic model. Density functional theory reveals that the classical alkylpalladium(IV) oxidative addition pathway is higher in energy than the bimetallic oxo-insertion pathway. Further kinetic studies revealed second-order dependence on [Pd] and first-order on [TBHP], which is consistent with DFT analysis. This procedure is compatible with a wide range of acids and alcohols for γ-C(sp3)–H oxygenation. Preliminary functional group transformations of the products underscore the great potential of this protocol for structural manipulation.
Introduction
The development of approaches for direct C–O formation of unactivated C(sp3)–H remains a fundamental challenge in synthetic chemistry owing to the high bond dissociation energy of the aliphatic C–H bonds; solving this problem is highly desirable considering the great manipulation potential of organic compounds.1 In the past decade, Pd-catalyzed C(sp3)–H oxygenation has been proven to be a promising way to tackle this challenge through a combination of various oxidants such as PhI(OAc)2,2 K2S2O8,3 and N-fluoropyridinium4 (Scheme 1a). In 2004, Canty's group reported the generation of the 1H NMR-detectable alkylpalladium(IV) intermediate (PdIV(O2CPh)2Me2(L2)) from PdIIMe2(L2) using (PhCO)2O2 as an oxidant.5 This system could deliver the corresponding product with a newly formed C–O bond after decomposition. Following this observation, subsequent Pd-catalyzed C(sp3)–H oxygenation studies continuously proposed that these transformations undergo oxidative addition of an oxygen electrophile to give an alkylpalladium(IV) (C(sp3)–[Pd(IV)(Ln)]–O) species, which further proceeds through C(sp3)–O reductive elimination to give the oxidized products (Scheme 1a). In 2004, the Sanford laboratory demonstrated that PhI(OAc)2 was an effective oxidant that facilitated formation of an alkylpalladium(IV) species affording C(sp3)–H acetoxylation products.6 In addition to the most widely used oxidant PhI(OAc)2, peroxides such as MeCOOOtBu and tert-butyl hydroperoxide (TBHP) also serve as alternative efficient oxidants for C(sp3)–H oxygenation. In 2005, Yu's group pioneered the use of peroxide MeCOOOtBu for oxazoline-directed β-C(sp3)–H acetoxylation, and the alkylpalladium(IV) complex was speculated to be the reaction intermediate.7 Recently, Yu et al. employed TBHP to realize the β-C(sp3)–H lactonization8 or acetoxylation9 of free acid via speculated alkylpalladium(IV) species (Scheme 1b). Despite this progress toward unactivated C(sp3)–H oxygenation, the catalytic mechanism is still unclear owing to the absence of catalytically relevant palladacycle complexes.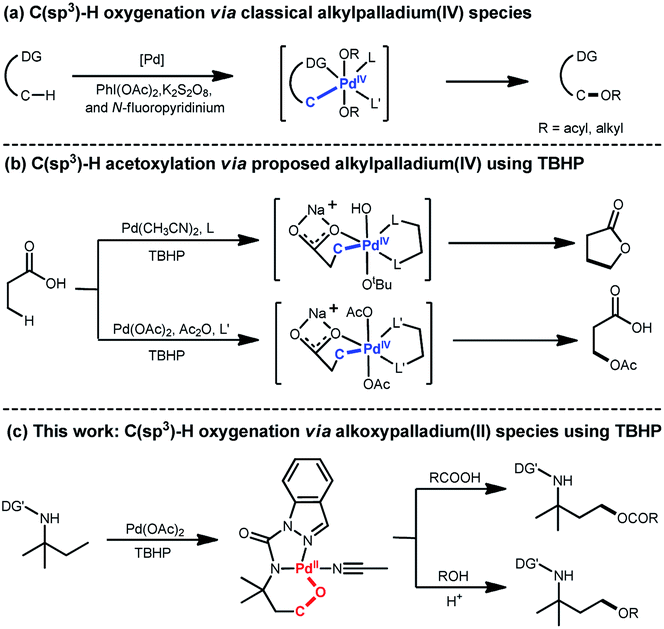 | ||
| Scheme 1 C–H oxygenation with different Pd intermediates. | ||
Herein, we show the Pd-catalyzed γ-C(sp3)–H oxygenation of tert-amylamine (1a) via unexpected alkoxypalladium(II) (C(sp3)–O–[Pd(II)(Ln)]) species using TBHP as the oxidant (Scheme 1c). This interesting finding—together with van Koten's pioneering discovery of C(sp2)–O–[Pd(II)(Ln)] species10 in the presence of TBHP—prompted us to gain insights into this transformation. Experimental and computational studies were hence combined to investigate a plausible reaction mechanism, including the isolation of alkoxypalladium(II) intermediates, density functional theory (DFT) calculations, and further kinetic studies. In addition, a wide range of carboxylic acids, N-protected amino acids, and carboxyl-containing drugs were tested to understand the substrate scope of the unactivated γ-C(sp3)–H acyloxylation as well as the alcohols' scope within γ-C(sp3)–H alkoxylation via the assistance of H+.
Results and discussion
Our initial studies began with the γ-C(sp3)–H acyloxylation of N-(tert-pentyl)-1H-indazole-1-carboxamide (1a) by testing a wide range of reaction parameters, including oxidants, solvents, and temperature (Table 1, refer Tables S1 and S2 in the ESI† for detailed reaction condition screening). Treating 1a with benzoic acid (2a) in the presence of Pd(OAc)2 and N-flouro-2,4,6-trimethylpyridinium tetraflouroborate ([F+]BF4−) in hexafluoroisopropanol (HFIP) at 45 °C for 36 h provided the desired C(sp3)–H acyloxylation product 3a with an isolated yield of 18% (entry 1). Replacing the oxidant with TBHP (5 M in decane) boosted the yield of 3a to 55% (entry 2), while TBHP (70% in water) further increased the yield to 68% (entry 3). Other peroxide oxidants such as cumene hydroperoxide (CHP) and MeCOOOtBu could also drive this reaction with lower yields (entries 4 and 5); TBHP (70% in water) was identified as the choice of oxidant (entry 3). Further solvent screening (entries 6 and 7) showed that entry 7 (acetonitrile solvent) contained the most favorable reaction conditions to afford 3a with an elevated yield of 77%.|
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Solvent | Temp (°C) | Yieldb (%) |
| a For entries 1–7: reaction was conducted with 1a (0.2 mmol), 2a (0.6 mmol), Pd(OAc)2 (0.02 mmol), oxidant (0.6 mmol) and solvent (1 mL). b Isolated yield. | ||||
| 1 | [F+]BF4− | HFIP | 45 | 18 |
| 2 | TBHP (5 M in decane) | HFIP | 45 | 55 |
| 3 | TBHP (70% in water) | HFIP | 45 | 68 |
| 4 | CHP | HFIP | 45 | 23 |
| 5 | MeCOOOtBu | HFIP | 45 | 26 |
| 6 | TBHP (70% in water) | Toluene | 45 | 20 |
| 7 | TBHP (70% in water) | CH 3 CN | 45 | 77 |
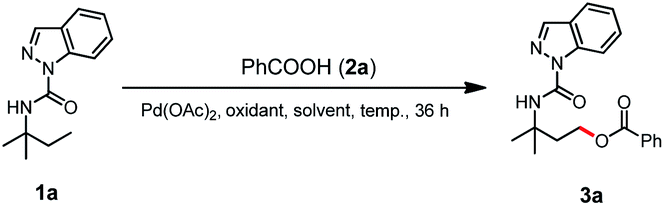
With the optimal γ-C(sp3)–H acyloxylation of 1a established, we next show that simple replacement of benzoic acid with methanol (4a) resulted in the exclusive alkoxylation product 5a with a 24% yield (entry 1, Table 2). Acid additive screening showed that 0.5% acetic acid (AcOH) increased the yield to 39% (entry 2), while 0.5% CF3COOH (TFA) further improved the yield to 48% (entry 3). The reaction temperature was also tested, and the results (entries 3–5) indicated that 60 °C was optimal for 5a formation with a yield of 71%. Further addition of TFA was also tested with a decreased yield of 5a (60%, entry 6). Hence, entry 4 was the best choice for γ-C(sp3)–H alkoxylation of 1a.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Additive | Temp (°C) | Yieldb (%) |
| a For entries 1–6: reaction was conducted with 1a (0.2 mmol), 4a (1.0 mmol), Pd(OAc)2 (0.02 mol), oxidant (0.6 mmol), additive (5 μL) and solvent (1 mL). b Isolated yield. c Additive (10 μL). | ||||
| 1 | CH3CN | — | 45 | 24 |
| 2 | CH3CN | AcOH | 45 | 39 |
| 3 | CH3CN | TFA | 45 | 48 |
| 4 | CH 3 CN | TFA | 60 | 71 |
| 5 | CH3CN | TFA | 80 | 43 |
| 6c | CH3CN | TFA | 45 | 60 |
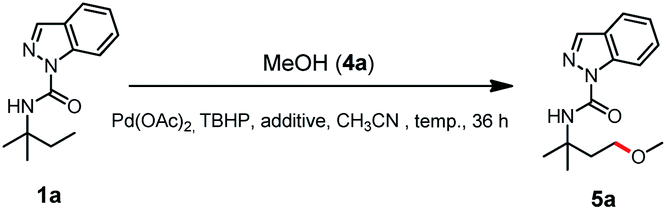
Encouraged by the selective γ-C(sp3)–H acyloxylation and alkoxylation findings, we next performed stoichiometric reactions to investigate the reaction intermediates. Exposing N-(tert-pentyl)-1H-indazole-1-carboxamide (1a) to Pd(OAc)2 in MeCN at room temperature in the presence of TBHP afforded the corresponding mono [5, 6]-fused palladacycle IN17. Replacement of MeCN with AcOH/MeCN (2%, w/w) exclusively resulted in another palladacycle IN14 under otherwise similar reaction conditions (Scheme 2a). Fortunately, the intermediate IN14 was isolated, and its structure was confirmed by X-ray crystallography to be a [5, 6]-fused palladacycle dimer (IN14, CCDC: 2058762†) (Scheme 2a). Intrigued by the discovery of IN14 and IN17, a [5, 5]-fused palladacycle IN6 was also isolated by treating 1a with Pd(OAc)2 in AcOH/MeCN (w/w, 2%) solvent at room temperature.
 | ||
| Scheme 2 Mechanistic studies. | ||
Previously, alkylpalladium(IV) species were reported to be the classical reaction intermediates for C(sp3)–H oxygenation, and a similar mechanism was proposed in Pd-catalyzed β-C(sp3)–H oxygenation using TBHP as the coupling oxidant. Contrary to these general findings, the alkoxypalladium(II) species (IN14 and IN17) were surprisingly found here. This led us to consider three fascinating questions: (1) Are the alkoxypalladium(II) species IN14 and IN17 the real reaction intermediates for this C(sp3)–H oxygenation? (2) Why does this C(sp3)–H oxygenation process prefer the alkoxypalladium(II) species rather than the alkylpalladium(IV) species? (3) What is the possible reaction process to afford the alkoxypalladium(II) species?
Additional experiments were conducted to confirm the role of these isolated intermediates. As expected, a catalytic amount of IN6 was used to replace Pd(OAc)2, thus affording 3a and 5a with 80% and 72% yields, respectively (Scheme 2b). IN6 could be readily transformed to IN14 or IN17 with the assistance of TBHP in AcOH/MeCN (w/w, 2%) or MeCN, respectively. Interestingly, the conversion between IN14 and
IN17 in the corresponding solvent was also observed (Scheme 2a). Similar treatment of 1a with IN14 delivered products 3a and 5a in 84% and 74% yields, respectively (Scheme 2c). Reaction of 1a with IN17 also gave 3a (85%) and 5a (73%) (Scheme 2d). These findings suggest that IN6, IN14, and IN17 are catalytically relevant intermediates in the synthetic process for the desired γ-C(sp3)–H oxygenation products. Direct treatment of IN17 with PhCOOH and MeOH resulted in higher production yields of 3a (88%) and 5a (77%) (Scheme 2e). This result further indicates that this γ-C(sp3)–H oxidative transformation goes through the alkoxypalladium(II) species. Deuterium incorporation experiments of 1a were also investigated, and the results indicate that the C(sp3)–H insertion process is irreversible because no deuterium-labeled 1a was observed (Scheme 2f).
To address questions 2 and 3 more specifically, computational experiments were conducted using DFT calculations via the M062x method11 with Gaussian 16 program suite12 (refer ESI† for computational details). First, the generation of a [5, 5]-fused palladacycle IN6 was studied. This progress starts with the coordination of 1a with Pd(OAc)2 to form a five-membered palladacycle IN3viaTS1 and TS2 with activation free energies of 17.1 and 5.7 kcal mol−1, respectively (Fig. 1). The subsequent γ-C(sp3)–H activation occurs next to obtain the [5, 5]-fused palladacycle IN5viaTS3 and TS4 with activation free energies of 9.0 and 12.6 kcal mol−1, respectively. MeCN was considered to be a coordinating solvent, and the MeCN-coordinated intermediate IN6 was obtained viaTS5 with an energy barrier of 4.9 kcal mol−1. The overall free energy barrier of the C–H cleavage transition state was 20.8 kcal mol−1 (from IN3 to TS4).
 | ||
| Fig. 1 Free energy profiles (kcal mol−1) of C–H activation. | ||
After the generation of the activation intermediate IN6, we next investigated the possibility of alkylpalladium(IV) species (IN8) formation based on previously reported general mechanisms. This assumption is outlined in blue, and the palladium complex IN6 from 1a was used to simplify the computations (Fig. 2). The coordination of TBHP to IN6 results in IN7 with an energy barrier of 6.2 kcal mol−1—this subsequently transforms to the resulting alkylpalladium(IV) intermediate IN8viaTS6 with an activation free energy of 84.6 kcal mol−1. This extremely high free energy barrier indicates that IN8 is not favorable for our reaction, and it may not be relevant in the catalytic cycle.
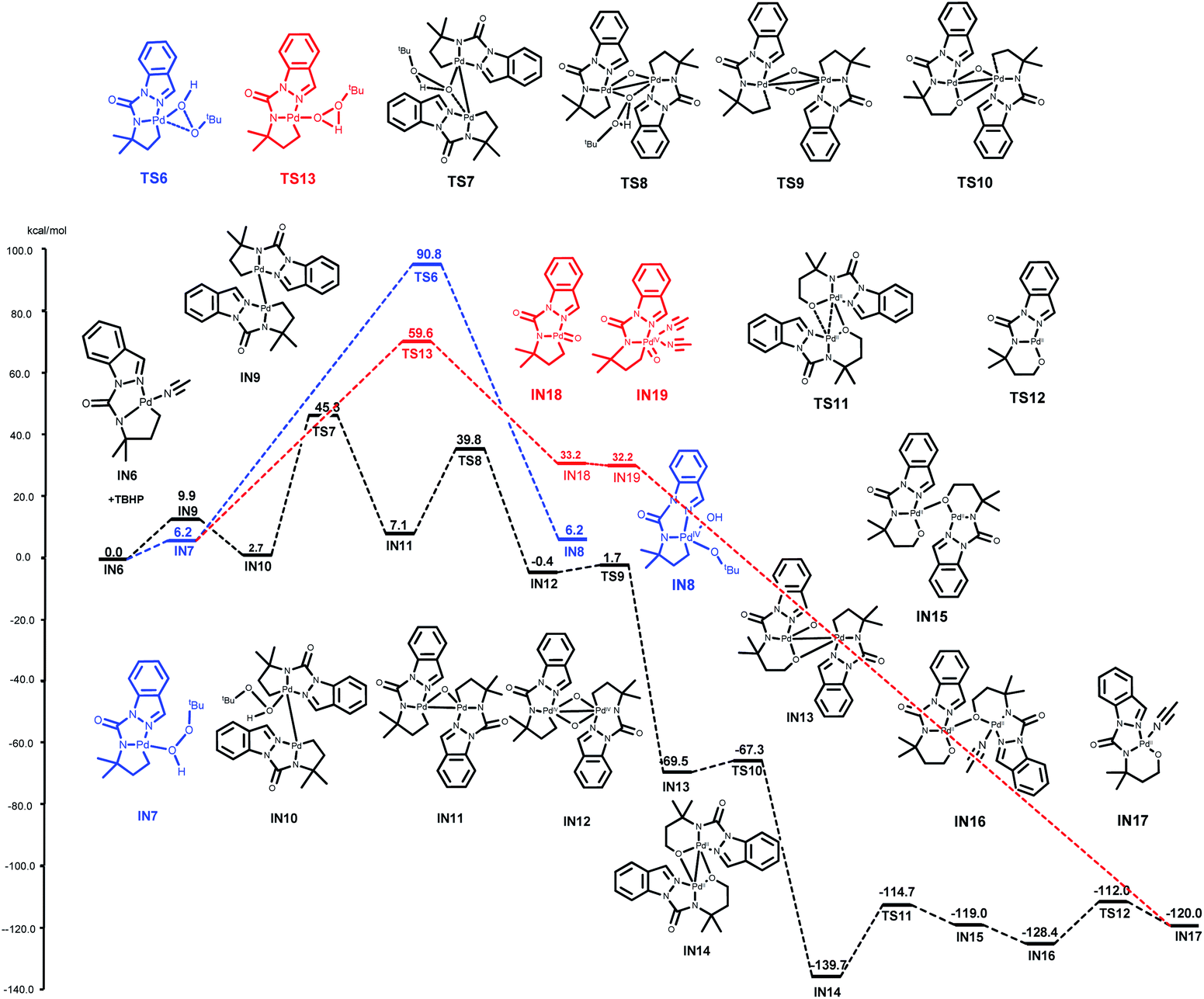 | ||
| Fig. 2 Free energy profiles (kcal mol−1) of oxidative addition. | ||
We then turned our attention to determining a plausible pathway from IN6 to IN17 and two approaches were proposed (black and red lines, Fig. 2). The black line shows the hypothesis that the dimeric palladacycles undergo the entire reaction pathway. As shown by previous reports on bimetallic intermediate formation in C(sp2)–H activation,13 this reaction could also begin with the generation of the dimeric Pd(II) intermediate IN9 from IN6 with an energy barrier of 9.9 kcal mol−1. The coordination of TBHP to IN9 leads to complex IN10. IN10 further undergoes oxidative addition of the O–O bond of TBHP viaTS7 to generate IN11, and IN11 follows the same pathway to obtain Pd(IV) intermediate IN12viaTS8 with activation free energies of 42.6 and 32.7 kcal mol−1, respectively. IN13 is subsequently obtained via C–O bond formation from IN12viaTS9. The desired alkoxypalladium(II) intermediate IN14 is achieved by repeating the same pathway with activation barriers of 2.1 and 2.2 kcal mol−1, respectively. Next, the MeCN-coordinated form intermediate IN16 is formed by the release of weak interaction between Pd and O (viaTS11 and IN15) with the assistance of MeCN and an energy barrier of 25.0 kcal mol−1. Finally, two MeCN-coordinated intermediates IN17 are formed by repeating this process. The overall free energy barrier of this process is 45.3 kcal mol−1 (from IN6 to TS7).
Alternatively, the red line represents a process that involves direct development of the monomer palladacycle complex IN7 to the alkoxypalladium(II) intermediate IN17. IN7 goes through an oxidative reaction to form IN18viaTS13 with an energy barrier of 53.4 kcal mol−1. Subsequent MeCN coordination affords intermediate IN19. However, the overall free energy barrier is 59.6 kcal mol−1 (from IN6 to TS13). The results showed that the bimetallic oxidative addition-oxo-insertion pathway is energetically feasible due to its lower energy barrier than the other two potential processes.
The aforementioned computational calculation results indicated that the oxidative addition progress is likely the turnover-limiting step. As described by Blackmond,14 reaction progress kinetic analysis is a simple, systematic method to obtain a complete picture of a reaction's kinetic profile. 1a and 2aa were used to investigate the dependence of the reaction rate. Reaction rate comparisons between the standard initial condition and higher [1a] or lower [2aa] conditions show zero-order dependence in [1a] or [2aa]. These results indicate that 1a or [2aa] is irrelevant to the rate-determining step of this catalytic cycle. However, lower concentration of [Oxidant] or higher concentration of [Pd] led to a lower or higher reaction rate, respectively, suggesting a positive-order dependence on [Oxidant] and [Pd] (refer ESI Section 6.1†). The dependence of the reaction rates on [Pd] and [TBHP] was also comprehensively investigated (refer ESI Sections 6.2 and 6.3†). A first-order with respect to [TBHP] and a second-order dependence at low [Pd] were observed (Scheme 3), which is consistent with DFT analysis.
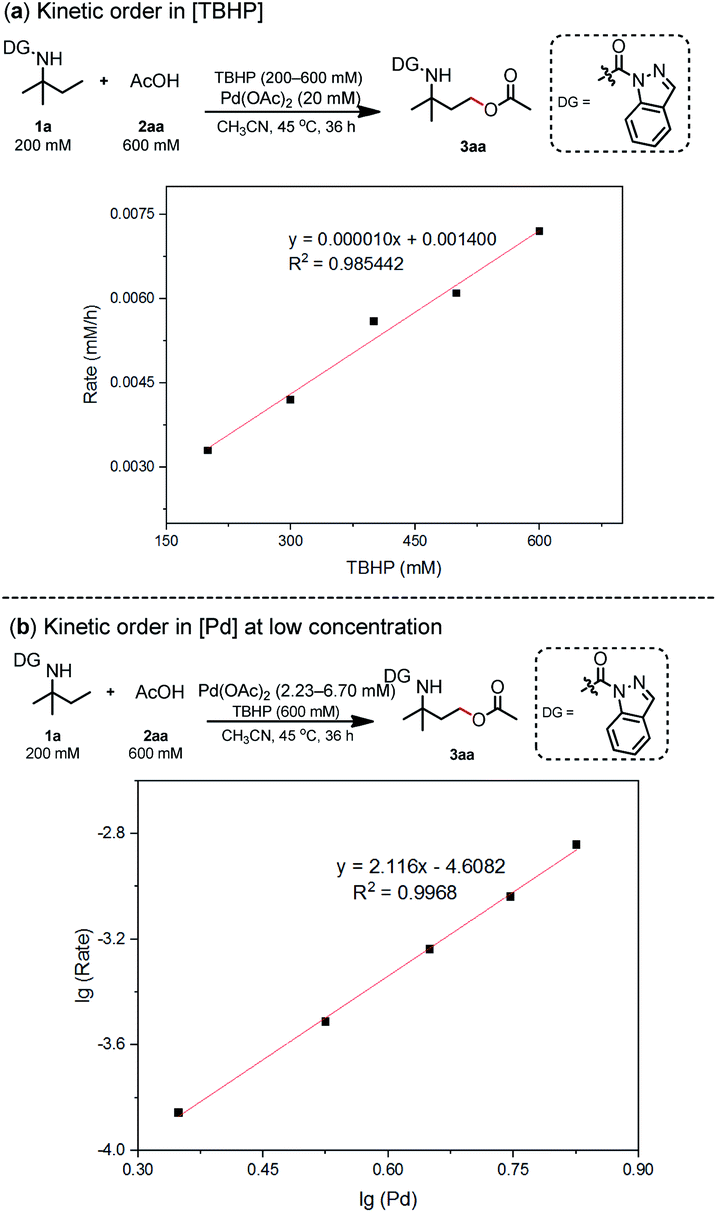 | ||
| Scheme 3 Kinetic studies. | ||
Based on the experimental and computational observations, a plausible C(sp3)–H oxygenation mechanism via the alkoxypalladium(II) species is proposed using 3aa as an example (Scheme 4). The initial coordination of 1a with Pd(OAc)2 and MeCN gives the [5, 5]-fused monomer Pd(II) intermediate IN6. A subsequent dimeric Pd(II) intermediate IN9 is formed by the release of two coordinated MeCN molecules and the following formation of a weak interaction between Pd. Next, the oxidative addition of IN9 by TBHP provides oxenoid oxygen atoms and oxygen insertion into the Pd–C bond gives the dimeric alkoxypalladium(II) intermediate IN14. The MeCN-coordinated monomer alkoxypalladium(II) intermediate IN17 is then promoted by the release of a weak interaction between Pd and O and the formation of the Pd–N (MeCN) bond. Two possible pathways15 from IN17 are proposed to generate the oxygenation product: (1) oxidative addition of IN17 produces LPd(IV)(OAc)2(OR) species and further reductive elimination yields 3aa. However, this pathway was ruled out because we found that direct treatment of IN17 with PhCOOH and MeOH resulted in the acyloxylation and alkoxylation products (Scheme 2e). (2) AcOH is engaged in bonding with the Pd and C center to generate IN20 and subsequent elimination generates IN21, that is redox-neutral at the Pd(II) center. Final proto-depalladation of IN21 in the presence of two additional equivalents of HOAc yields 3aa and regenerates Pd(OAc)2 to continuously drive the catalytic cycle.
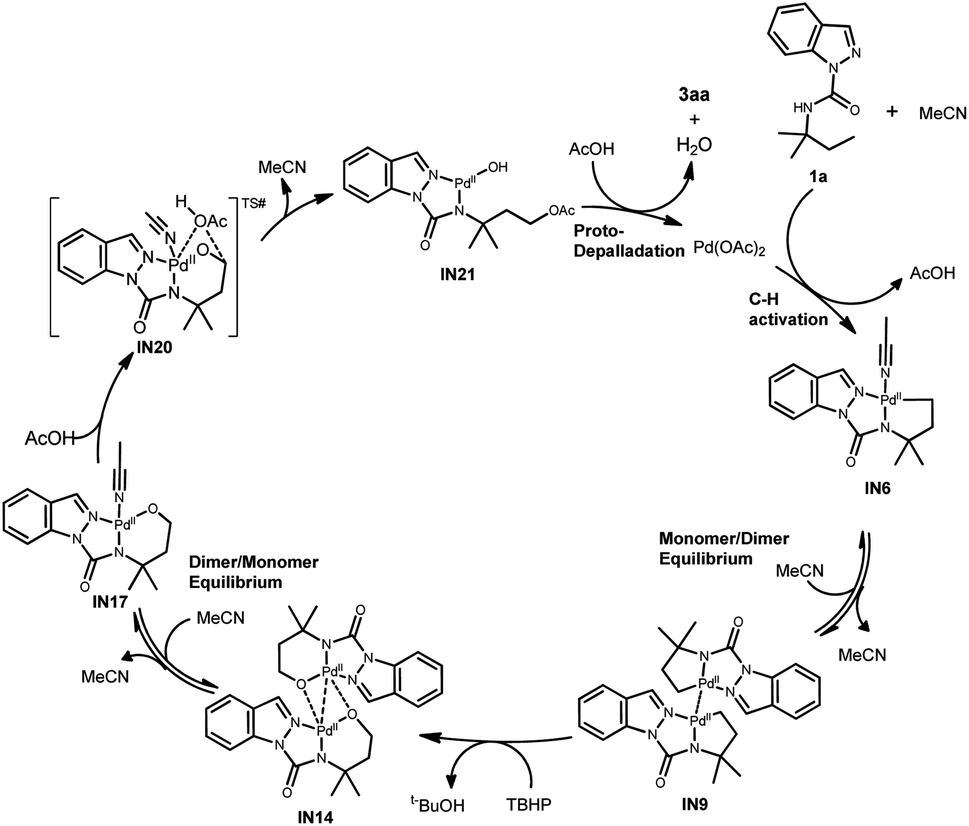 | ||
| Scheme 4 Proposed mechanism. | ||
With the optimized reaction conditions of γ-C(sp3)–H oxygenation in hand (Table 1, entry 7 and Table 2, entry 4), we first investigated the carboxylic acid scope of the acyloxylation reaction (Scheme 5). Various mono-substituted benzoic acids including fluorine, chlorine, bromine, trifluoromethyl, cyano, nitro, methyl, underwent the desired acyloxylation reaction in moderate to excellent isolated yields ranging from 57% to 92% (3a–3n). The results also showed that the electron-withdrawing group (3b–3f) at the para position on the phenyl ring was preferable to those with an electron-donating group (3h, 3i) except for the acetyl group (3g). This observation could also be extended to substituents at the meta or ortho positions in which the electron-withdrawing chloro (3j, 3m) or nitro group (3k) were superior to the electron-donating methyl (3l) or methoxy (3n) group. The same trends were also recorded for the disubstituted benzoic acids—the chloro groups (3o, 3q) had more turnover than their corresponding methyl groups (3p, 3r). Meanwhile, more sterically hindered diphenyl-, naphthyl-, and (methylenedioxy)phenyl-substituted acids gave relatively lower yields (3s–3u). Other aromatic acids containing substituted pyridines, thiophene, and indole were all compatible coupling partners, thus providing products in moderate isolated yields (36–63%, 3v–3y). The substrate scope of aliphatic acids was also investigated (Scheme 5). Primary aliphatic acids including acetic acid (3aa), propionic acid (3ab), isovaleric acid (3ac), neovaleric acid (3ad), and hexanoic acid (3ae) were well tolerated in the C(sp3)–H acyloxylation reaction with good isolated yields (71–81%); formic acid achieved a relatively lower efficiency (3z). Saturated (3af) or unsaturated (3ag) secondary aliphatic acids also successfully led to the products with high efficiency. Aliphatic acids with oxygenated functionalities (3ah, 3ai) or reactive bromine or hydroxyl groups (3aj, 3ak) at the alpha sites were amenable to this transformation, thus affording the targeted products in moderate-to-good isolated yields. Aralkyl group-substituted aliphatic acids could also be efficiently converted to the desired products 3al and 3am with 73% and 75% isolated yields, respectively.
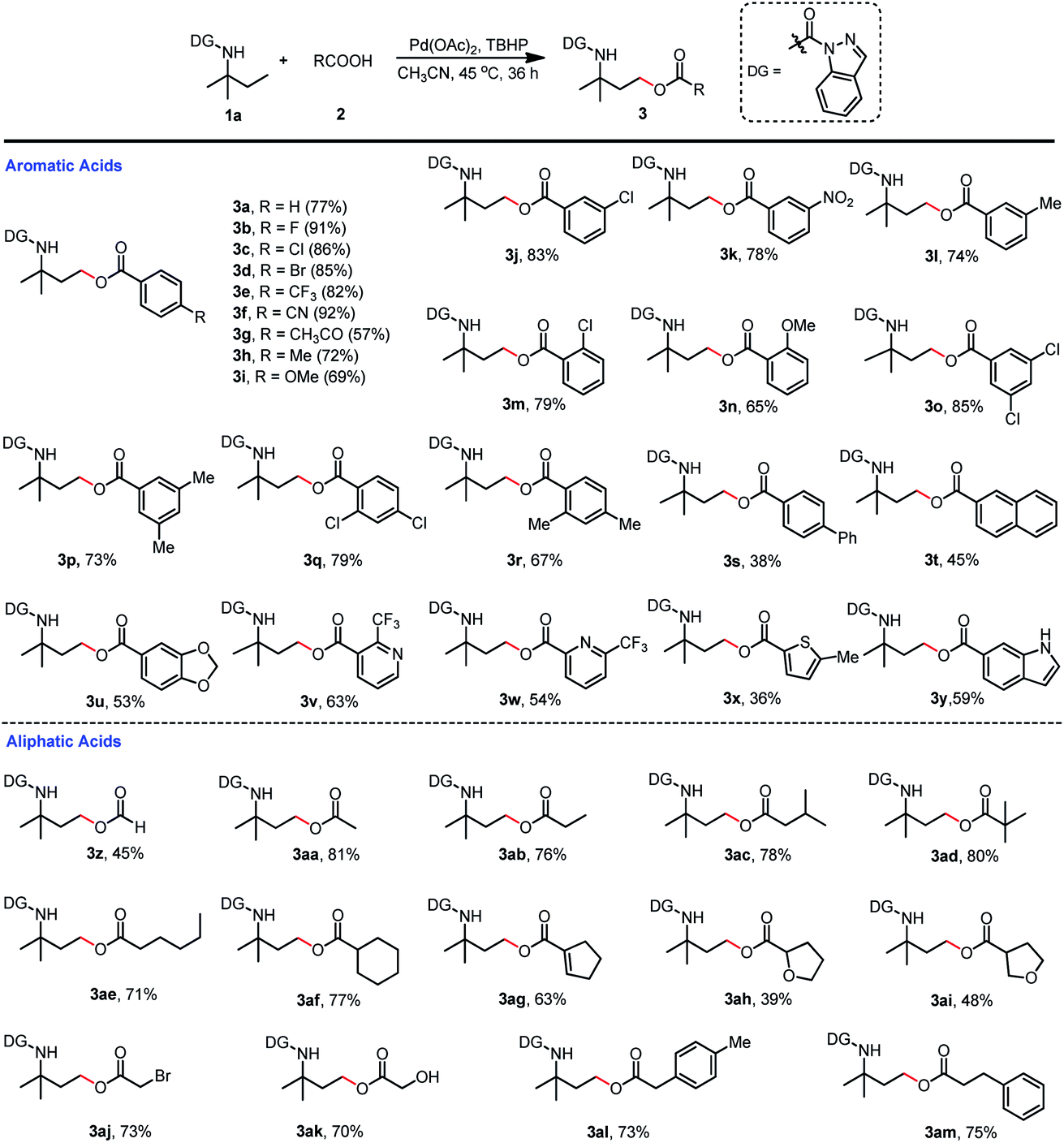 | ||
| Scheme 5 Scope of carboxylic acids for γ-C(sp3)–H acyloxylation. (a) Reaction was conducted with 1a (0.2 mmol), 2 (0.6 mmol), Pd(OAc)2 (0.02 mmol), TBHP (70% in water) (0.6 mmol) and CH3CN (1 mL) at 45 °C for 36 h. (b) Isolated yield. | ||
This protocol could also be well adapted for N-Boc-protected amino acids such as Boc-glycine, Boc-β-alanine, Boc-L-phenylalanine, Boc-L-isoleucine, Boc-D-valine, and Boc-L-threonine to afford the corresponding products (3an–3as) with isolated yields of 61–75% (Scheme 6). Meanwhile, acetyl-L-phenylalanine was also tested to give 3at with a relatively lower yield of 36%, and a higher reaction temperature treatment of Boc-protected tripeptide resulted in the desired product 3au with a slightly lower yield of 33%. These results broadly enhanced the palladium catalyzed γ-C(sp3)–H acyloxylation capabilities and provide a new approach for amino acid derivatization.
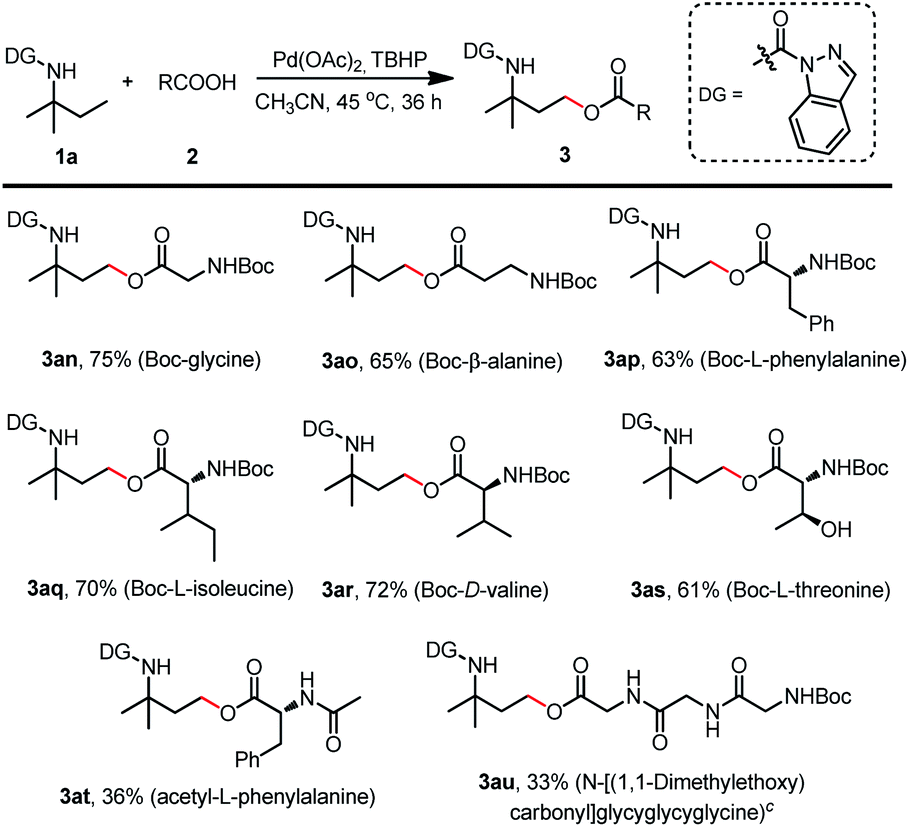 | ||
| Scheme 6 Scope of N-protected amino acids. (a) Reaction was conducted with 1a (0.2 mmol), 2 (0.6 mmol), Pd(OAc)2 (0.02 mmol), TBHP (70% in water) (0.6 mmol) and CH3CN (1 mL) at 45 °C for 36 h. (b) Isolated yield. cReaction was conducted at 60 °C. | ||
Encouraged by these outcomes, we next tested the applicability of this method for the esterification of drugs containing acid moieties (Scheme 7). Probenecid, ibuprofen, flufenamic acid, indomethacin, aspirin, and naproxen resulted in the desired products (3av–3ba) in moderate to good yields (45% to 81%). These findings further demonstrated this protocol's potential to perform late-stage facile functionalization and generate amine-containing drug analogs.
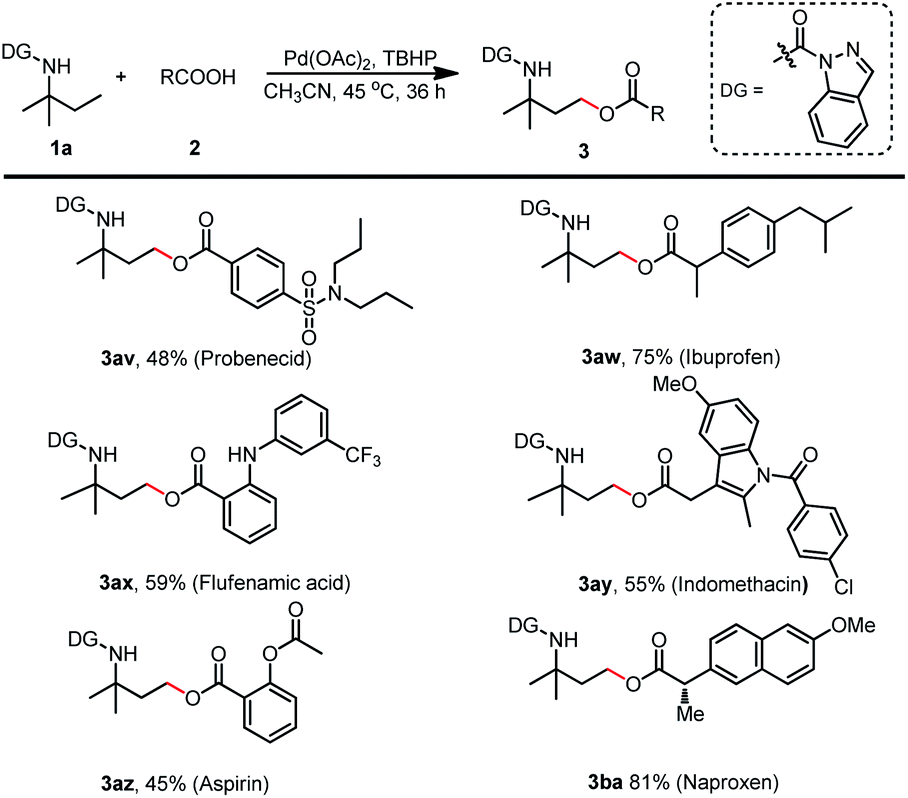 | ||
| Scheme 7 Scope of drugs for esterification. (a) Reaction was conducted with 1a (0.2 mmol), 2 (0.6 mmol), Pd(OAc)2 (0.02 mmol), TBHP (70% in water) (0.6 mmol) and CH3CN (1 mL) at 45 °C for 36 h. (b) Isolated yield. | ||
Schemes 5–7 show that a variety of carboxylic acids were tolerated during the γ-acyloxylation of 1a. With the optimal reaction conditions for 1a γ-alkoxylation in hand (Table 2, entry 4), different alcohols were tested for the substrate scope (Scheme 8). Primary alcohols such as methanol, deuterated methanol, ethanol, n-propanol, n-butanol, n-pentanol, n-octanol, and cyclopentylmethanol successfully afforded the desired alkoxylation products with 45–71% isolated yields (5a–5g). Secondary alcohols such as isopropanol and cyclohexanol were also acceptable for this transformation with moderate isolated yields (5h, 5i).
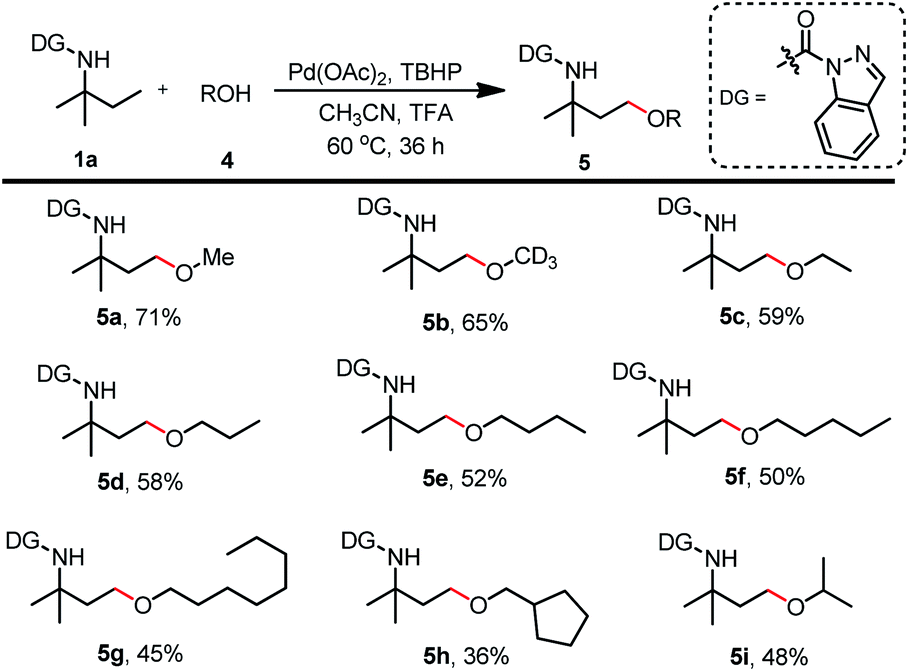 | ||
| Scheme 8 Scope of natural products for C(sp3)–H alkoxylation. (a) Reaction was conducted with 1a (0.2 mmol), 4 (1.0 mmol), Pd(OAc)2 (0.02 mmol), TBHP (70% in water) (0.6 mmol), TFA (5 μL) and CH3CN (1 mL) at 60 °C for 36 h. (b) Isolated yield. | ||
A scaled up (2.0 mmol) oxygenation reaction of 1a was conducted to demonstrate the synthetic utility of this newly developed approach (Scheme 9). Products 3a and 5a were afforded with isolated yields of 54% and 48%, respectively. Increasing the reaction time to 60 h gave a higher yield of 3a (65%) and 5a (54%), thus showing the good efficiency of our protocol (Scheme 9a). Moreover, directing group (DG) deprotection of 3a and 5avia hydrolysis was accompanied by ester bond breakage to deliver a hydroxyl derivative that underwent an in situ one-pot benzoyl protection process to yield the Bz-protected 6 for easy purification and analysis. Hence, removal of DG from 3a led to amino alcohol derivative 6 with 68% yield. Applying the same strategy to 5a resulted in the amino ether derivative 7 with a 72% yield. The partial hydrolysis of the ester bond could also be realized, and product 3h was easily converted into the corresponding primary alcohol 8 with an 89% yield (Scheme 9b). Treatment of 8 with azidotrimethylsilane (TMSN3) and Bi(OTf)3 enabled the formation of 9 with a 68% yield while N-bromosuccinimide (NBS) and dimethylthiourea (DMTU) treatment easily converted the hydroxy group of 8 to a reactive functional bromine group (10) with an 82% yield. Meanwhile, after dealing with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), PPh3, and n-Bu4NCN, the hydroxyl group of 8 could also be transformed to the cyano group (11) with a 54% yield (Scheme 9b). These transformations demonstrate the great potential of the selective C(sp3)–H acyloxylation and alkoxylation for facile access to amine-containing functional molecules.
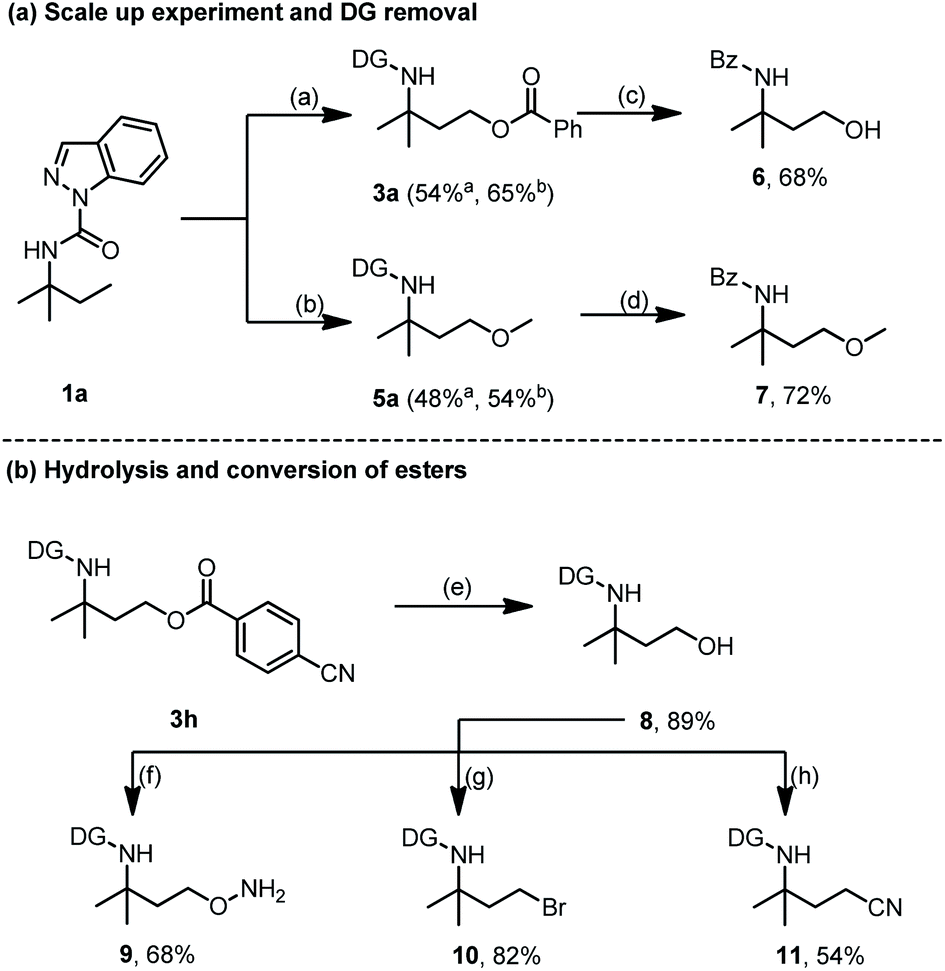 | ||
| Scheme 9 Synthetic applications. Legend: (a) Pd(OAc)2, PhCOOH, TBHP, CH3CN, 45 °C; (b) Pd(OAc)2, MeOH, TFA, TBHP, CH3CN, 60 °C; (c) K2CO3, MeOH, H2O, BzCl, 68%; (d) K2CO3, MeOH, H2O, BzCl, 72%; (e) LiOH, THF, H2O, 89%; (f) TMSN3, Bi(OTf)3, CH3NO2, 68%; (g) NBS, DMTU, CH2Cl2, 82%; (h) DDQ, PPh3, n-Bu4NCN, CH3CN, 54%. aStandard reaction condition. bReaction was conducted for 60 h. | ||
Conclusions
In summary, we developed a general method for γ-C(sp3)–H acyloxylation and alkoxylation of tert-amylamine using 1H-indazole as the directing group and TBHP (70% in water) as an inexpensive oxidant. Unexpectedly, the alkoxypalladium(II) intermediate IN17 was captured, which goes through a SN2 process that is redox-neutral at the Pd(II) center to afford the oxygenation products. To the best of our knowledge, this is the first time that C(sp3)–H oxygenation is realized through a C(sp3)–O–[Pd(II)(Ln)] species in contrast to the classical proposed catalytic pathway with alkylpalladium(IV) species. Experimental and computational experiments were performed to study the reaction mechanism. Two alkoxypalladium(II) intermediates (IN14 and IN17) were isolated and characterized. The DFT calculations showed the preference for the addition-oxo-insertion process with lower energy. The results also revealed that IN17 is generated through a bimetallic oxo-insertion pathway as confirmed by kinetic studies. The results provide an update to the underlying reaction mechanism of palladium-catalyzed oxidative C(sp3)–H oxygenation especially for peroxide-assisted transformations. A variety of acids, N-protected amino acids, acid-containing drugs, and alcohols are all compatible with this reaction. The preliminary simple DG removal and one-step efficient product conversion reaction demonstrate this protocol's potential for structural manipulation to amine-containing diverse functional molecules.Data availability
All experimental and characterization data in this article are available in the ESI. Crystallographic data for compounds IN14 have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under accession number CCDC 2058762.†Author contributions
S. Z. and J. Z. contributed equally. S. Z. and J. Z. conceived the project. S. Z., J. Z. and H. Z. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Key Projects of Natural Science Foundation of Zhejiang Province (LZ21B020001), the National Natural Science Foundation of China (No. 21472170) and the National Key R&D Program of China (No. 2017YFE0102200).Notes and references
- (a) R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2012, 14, 3724–3727 CrossRef CAS PubMed; (b) R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2014, 16, 968–971 CrossRef CAS PubMed; (c) D. Li, Y. Cao and G. Wang, Org. Biomol. Chem., 2015, 13, 6958–6964 RSC; (d) S. Arshadi, A. Banaei, A. Monfared, S. Ebrahimiasl and A. Hosseinian, RSC Adv., 2019, 9, 17101–17118 RSC.
- (a) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391–3394 CrossRef CAS PubMed; (b) S. Zhang, G. He, Y. Zhao, K. Wright, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2012, 134, 7313–7316 CrossRef CAS PubMed; (c) Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991–16994 CrossRef CAS PubMed; (d) X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Petersen and X. Shi, Chem. Sci., 2013, 4, 3712–3716 RSC; (e) Q. Li, S. Zhang, G. He, W. A. Nack and G. Chen, Adv. Synth. Catal., 2014, 356, 1544–1548 CrossRef CAS; (f) Z. Zang, S. Zhao, S. Karnakanti, C. Liu, P. Shao and Y. He, Org. Lett., 2016, 18, 5014–5017 CrossRef CAS PubMed; (g) K. K. Pasunooti, R. Yang, B. Banerjee, T. Yap and C. Liu, Org. Lett., 2016, 18, 2696–2699 CrossRef CAS PubMed; (h) K. Chen, D. Wang, Z. Li, Z. Liu, F. Pan, Y. Zhang and Z. Shi, Org. Chem. Front., 2017, 4, 2097–2101 RSC; (i) Y. Zheng, W. Song, Y. Zhu, B. Wei and L. Xuan, J. Org. Chem., 2018, 83, 2448–2454 CrossRef CAS PubMed; (j) K. K. Ghosh, A. Uttry, A. Koldemir, M. Ong and M. van Gemmeren, Org. Lett., 2019, 21, 7154–7157 CrossRef CAS; (k) C. S. Buettner, D. Willcox, B. G. N. Chappell and M. J. Gaunt, Chem. Sci., 2019, 10, 83–89 RSC.
- (a) L. Zhou and W. Lu, Org. Lett., 2014, 16, 508–511 CrossRef CAS; (b) R. Zhao and W. Lu, Org. Lett., 2017, 19, 1768–1771 CrossRef CAS.
- Y. Chen, Y. Wu, Z. Wang, J. X. Qiao and J. Yu, ACS Catal., 2020, 10, 5657–5662 CrossRef CAS.
- A. J. Canty, M. C. Denney, B. W. Skelton and A. H. White, Organometallics, 2004, 23, 1122–1131 CrossRef CAS.
- L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542–9543 CrossRef CAS.
- R. Giri, J. Liang, J. G. Lei, J. J. Li, D. H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J. Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 7420–7424 CrossRef CAS PubMed.
- Z. Zhuang and J. Yu, Nature, 2020, 577, 656–659 CrossRef CAS PubMed.
- Z. Zhuang, A. N. Herron, Z. Fan and J. Yu, J. Am. Chem. Soc., 2020, 142, 6769–6776 CrossRef CAS PubMed.
- (a) P. L. Alsters, H. T. Teunissen, J. Boersma and G. van Koten, Recl. Trav. Chim. Pays-Bas, 1990, 109, 487–489 CrossRef CAS; (b) P. L. Alsters, H. T. Teunissen, J. Boersma, A. L. Spek and G. van Koten, Organometallics, 1993, 12, 4691–4696 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- (a) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234–11241 CrossRef CAS; (b) D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050–17051 CrossRef CAS PubMed.
- (a) D. G. Blackmond, Angew. Chem., Int. Ed., 2006, 44, 4302–4320 CrossRef PubMed; (b) J. S. Mathew, M. Klussmann, H. Iwamura, F. Valera, A. Futran, E. A. C. Emanuelsson and D. G. Blackmond, J. Org. Chem., 2006, 71, 4711–4722 CrossRef CAS PubMed; (c) P. Ruiz-Castillo, D. G. Blackmond and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 3085–3092 CrossRef CAS PubMed.
- (a) H. Thu, W. Yu and C. Che, J. Am. Chem. Soc., 2006, 128, 9048–9049 CrossRef CAS PubMed; (b) Y. Zhang, B. Shi and J. Yu, Angew. Chem., Int. Ed., 2009, 48, 6097–6100 CrossRef CAS PubMed; (c) B. E. Haines, H. Xu, P. Verma, X. Wang, J. Yu and D. G. Musaev, J. Am. Chem. Soc., 2015, 137, 9022–9031 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, characterization data and NMR spectra. CCDC 2058762 (IN14). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06907a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |