
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diboramacrocycles: reversible borole dimerisation–dissociation systems†
Sonja
Fuchs
ab,
Arumugam
Jayaraman
 ab,
Ivo
Krummenacher
ab,
Laura
Haley
ab,
Marta
Baštovanović
ab,
Maximilian
Fest
ab,
Krzysztof
Radacki
ab,
Holger
Helten
ab and
Holger
Braunschweig
*ab
ab,
Ivo
Krummenacher
ab,
Laura
Haley
ab,
Marta
Baštovanović
ab,
Maximilian
Fest
ab,
Krzysztof
Radacki
ab,
Holger
Helten
ab and
Holger
Braunschweig
*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 4th February 2022
Abstract
We report that the outcome of the tin–boron exchange reaction of a mixed thiophene-benzo-fused stannole with aryldibromoboranes is associated with the steric bulk of the aryl substituent of the borane reagent, leading to either boroles or large diboracycles as products. NMR spectroscopic studies indicate that the two products can reversibly interconvert in solution, and mechanistic density functional theory (DFT) calculations reveal boroles to be intermediates in the formation of the diboracyclic products. The addition of Lewis bases to the diboracycles leads to the corresponding borole adducts, demonstrating that they react as “masked” boroles. Additionally, the reaction of the title compounds with a series of organic azides affords complex heteropropellanes, formally 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 borole-azide adducts, that deviate from the usual BN aromatic compounds formed via nitrogen atom insertion into the boroles.
:1 borole-azide adducts, that deviate from the usual BN aromatic compounds formed via nitrogen atom insertion into the boroles.
Introduction
The class of five-membered boroles continues to be of great interest beyond their antiaromatic π-electron conjugation.1 Due to a range of additional attributes, such as their high Lewis acidity, electron-accepting ability, and chromophoric properties, boroles are a promising platform for diverse applications ranging from reagents in chemical synthesis to electronic materials.2,3Intense research during the past decade has demonstrated that the properties of boroles are highly sensitive to the nature of the substituents surrounding the five-membered ring. Typically highly reactive in nature, air and moisture stable derivatives could be synthesised by incorporating the bulky 2,4,6-tris(trifluoromethyl)phenyl group on the boron atom, while the steric bulk of the remaining substituents suppresses dimerisation via Diels–Alder cycloadditions.4 Likewise, the extension of the borole π system by fusion of aromatic groups can be used to modulate key properties.3 For example, dibenzannulated boroles, also known as 9-borafluorenes,3b display reduced antiaromaticity, whereas dithiophene-fused boroles possess increased antiaromaticity that even surpasses the antiaromaticity of their non-fused derivatives.5 The greater antiaromaticity and consequently lower stability of the doubly thiophene-fused derivatives is also reflected in their preparation.5 A systematic study by the group of He recently revealed that the success of the classic tin–boron exchange reaction is correlated with the degree of antiaromaticity of the boroles (Scheme 1).6 While the reaction of doubly benzo-fused and mixed thiophene-benzo-fused stannole precursors with dichloro(phenyl)borane led to the desired boroles (I and II), the tin–boron exchange with the doubly thiophene-fused precursor led instead to a diboracyclic structure (III), where the unfavourable cyclic four-electron delocalisation is avoided.6 Such macrocyclic ring structures, in which the boron atoms are doubly biaryl-bridged, have been observed by Piers7 and also postulated by Wagner as intermediates in dimerisation pathways of 9-H-9-borafluorene.8 Although boroles have not been previously thought to be playing a role in tin–boron exchange reactions where large diboracycles are formed,6 they might thus represent viable intermediates.
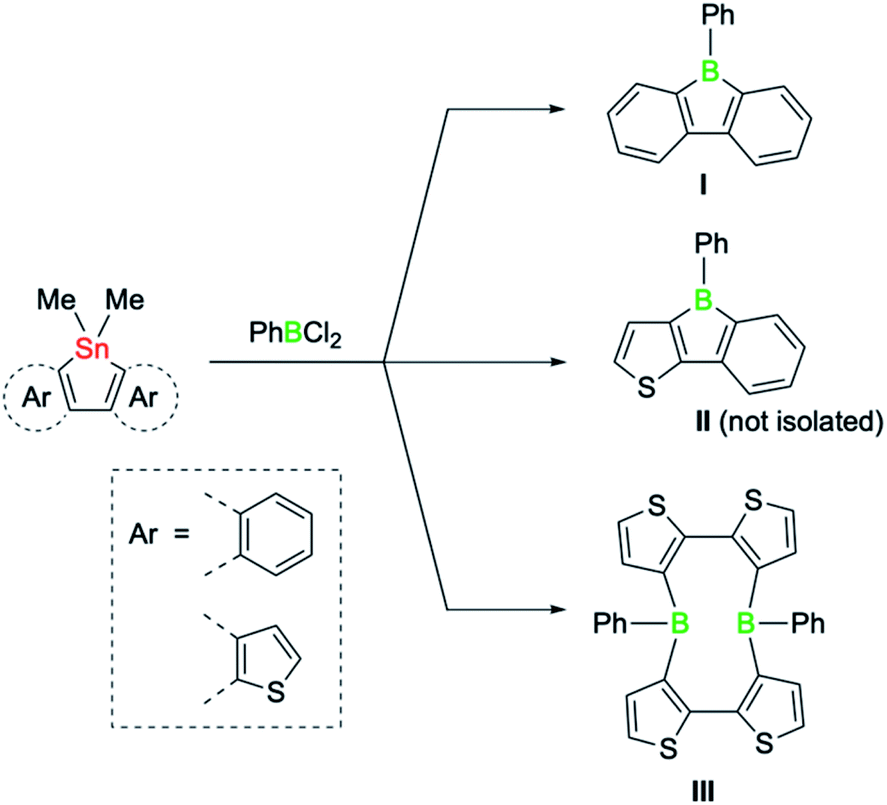 | ||
| Scheme 1 Products of the tin–boron exchange reaction of fused stannoles with PhBCl2, according to He et al.6 | ||
In this contribution, we present experimental insights into how steric factors influence borole and diboramacrocycle formation, respectively, in the tin–boron exchange reaction of a mixed thiophene-benzo-fused stannole with a series of aryldibromoboranes. Computational studies identifying the mechanistic steps of the transformation clarify the relationship between the two products and establish boroles as intermediates in the formation of the large diboracyclic ring systems. In view of the poorly studied influence of fused thiophene rings on the reactivity of boroles, we further canvassed the reactivity of a derivative towards a series of organic azides. In contrast to its dibenzo analog, where the reaction yields BN-phenanthrenes by ring expansion of the central borole unit,9 the reaction proceeds by a distinct mechanism to structurally complex heteropropellanes.
Results and discussion
The tin–boron exchange reaction of the mixed thiophene-benzo-fused stannole 16 with different aryldibromoboranes10 produced either borole (2c) or diboracyclic products (3a, b; Scheme 2, top). Selective formation of the borole 2c was observed only with the sterically most hindered dibromo(mesityl)borane, whereas employing the less-hindered phenyl- and 2,4-xylyldibromoboranes led to the diboracycles 3a and 3b in 79% and 37% isolated yield, respectively. The products were obtained as red (2c) or yellow solids (3a, b) and were characterised by single-crystal X-ray diffraction analysis (see ESI† for details). The structural parameters of the borole 2c, and the diboracycle 3a (shown in Fig. 1) and 3b, consisting of a ten-membered ring structure, are comparable to experimental data for related systems.5–7,11 The large heterocycles 3a and 3b, with the composition of formal borole dimers, adopt a boat-like conformation with transannular boron–boron interactions of 2.779(4)/2.790(4) Å (3a) and 3.291(4) Å (3b).6,7 In particular, the short boron–boron distance in 3a suggests a significant overlap of the vacant pz orbitals of the neighbouring boron centres.12 This is also seen in the LUMO of 3a, as shown in Fig. 1.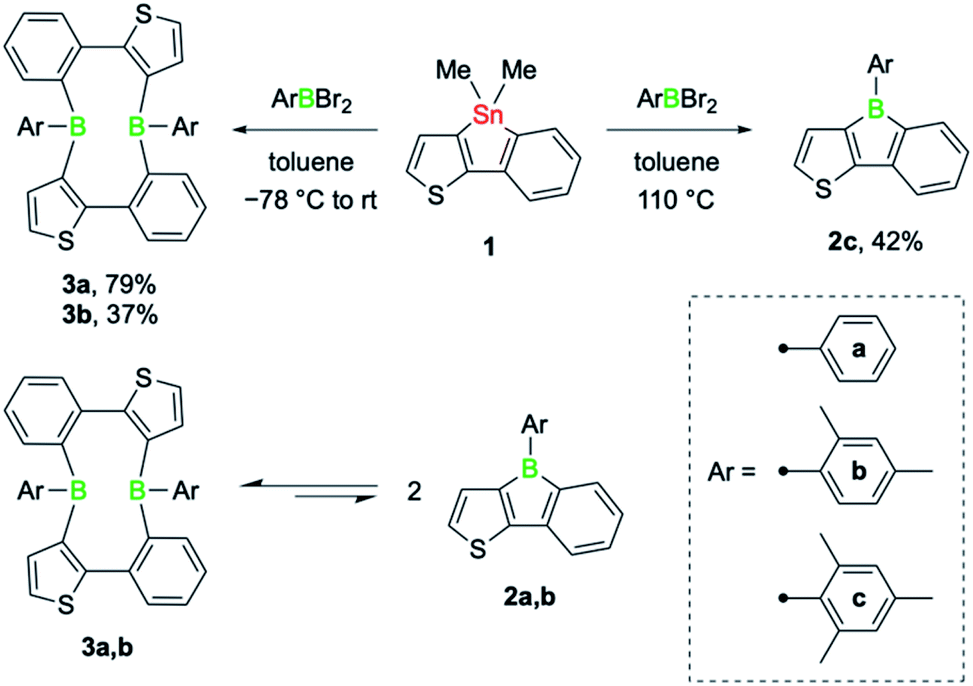 | ||
| Scheme 2 Isolated products of the tin–boron exchange reaction between stannole 1 and various aryldibromoboranes (top), and dynamic equilibrium between the cyclic dimers 3a, b and the corresponding annulated boroles 2a, b in solution (bottom). | ||
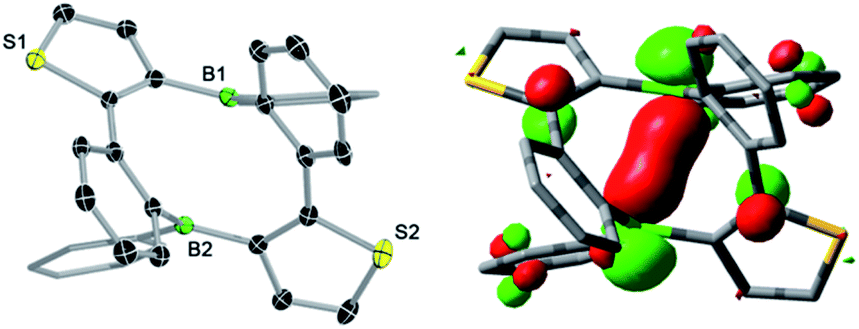 | ||
| Fig. 1 Molecular structure of 3a with thermal ellipsoids at 50% probability (left) and LUMO of 3a (isovalue = 0.05, right). | ||
Solution 1H and 11B NMR spectroscopy at room temperature revealed that 3a and 3b dissociate readily to the monomeric boroles 2a and 2b, respectively (Scheme 2, bottom). While 3b exhibits only very broad resonances in the 1H and 11B NMR spectra, distinct 11B NMR signals for the monomer (2a: δ(11B) = 60.0 ppm) and dimer (3a: δ(11B) = 64.0 ppm) can be observed upon dissolution of 3a in toluene-d8. On heating, the two peaks become better resolved, clearly indicating the presence of a monomer–dimer equilibrium in solution. Although readily observable by NMR spectroscopy, borole 2a was not isolable.6 The higher yield of phenyl-substituted dimer 3a relative to that of the xylyl derivative 3b suggests that a sterically less hindered boron environment favours dimer formation. Indeed, computations performed at the B3LYP-D3BJ/6-311g++(d,p)/SCRF(SMD = benzene)//B3LYP-D3BJ/6-31g(d,p) level of theory showed that thermodynamically, dimer formation is favoured in all cases, and that the relative stability decreases with increasing steric hindrance of the B-aryl substituent (phenyl, ΔG = −7.1 kcal mol−1; xylyl, ΔG = −5.7 kcal mol−1; mesityl, ΔG = −1.3 kcal mol−1). Although thermodynamically slightly favoured, dimer formation in the case of the mesityl derivative might be more challenging due to steric hindrance and we have not observed any dimer formation experimentally.
Borole 2c is characterised by a lowest-energy absorption at 457 nm in hexane, which is slightly hypsochromically shifted compared to its π-extended benzothiophene-substituted analog (λmax = 474 nm).5b Like many other fused borole derivatives, 2c was found to be fluorescent.5a,13 It exhibits a broad emission with a peak maximum around 590 nm and a fluorescence lifetime of τ = 11.5 ns (see Fig. S4 in the ESI† for details).
We then investigated the reactivity of diboracycle 3a toward adduct formation with Lewis bases. Addition of two equivalents of pyridine and tri(p-tolyl)phosphine to 3a led to complete consumption of 3a and formation of the borole adducts 2a-pyr and 2a-P, respectively (Scheme 3). The 11B NMR spectroscopic signals of both adducts appear as relatively sharp singlets at 1.1 ppm (2a-pyr) and −10.0 ppm (2a-P), respectively. Their structures were unambiguously confirmed by single-crystal X-ray crystallography (see Scheme 3 for the structure of 2a-pyr). The structural parameters, including the boron–heteroatom bonds and the geometry around the boron atoms, are consistent with other structurally characterised pyridine- and phosphine-borole adducts.14,15 The reactivity of 3a towards the Lewis base pyridine differs from that of the corresponding doubly thiophene-fused derivative, for which the bis(pyridine) adduct was observed under comparable conditions.6 The selective formation of the borole-pyridine adduct 2a-pyr from 3a implies that diboracycle 3a rapidly dissociates to borole 2a before reacting with pyridine. It is thus able to serve as a source for the borole monomer, in analogy to the Diels–Alder dimers of boroles.16
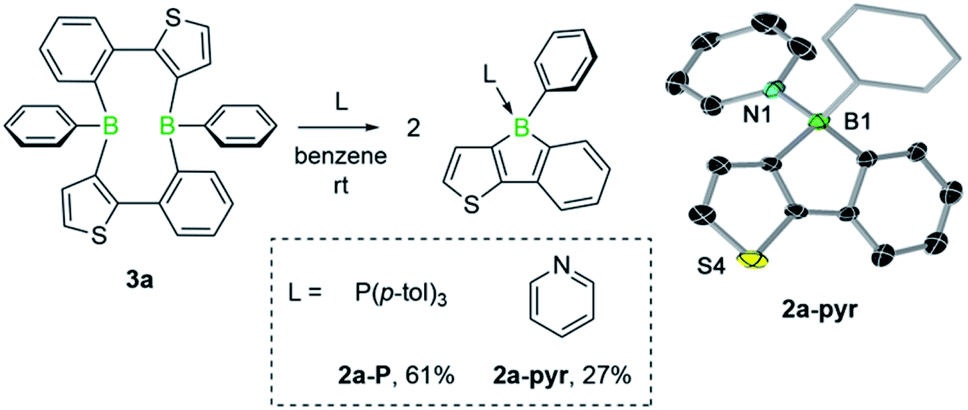 | ||
| Scheme 3 Lewis base adducts of 3a and molecular structure of 2a-pyr. Displacement ellipsoids are shown at the 50% probability level. The ellipsoids of the phenyl substituent, hydrogen atoms and solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (°): B1–N1 1.623(5), B1–C1 1.616(4); C1–B1–N1 109.46. | ||
Our observation that the boroles 2a, b exist in equilibrium with their cyclic dimers 3a, b led us to study the mechanism of the tin–boron exchange in more detail. We assumed that the tin–boron exchange would first yield the borole structures, later transforming into the ten-membered diboracyclic rings. To address this possibility, we carried out mechanistic computations on such a reaction pathway for the phenyl derivative (Fig. 2).
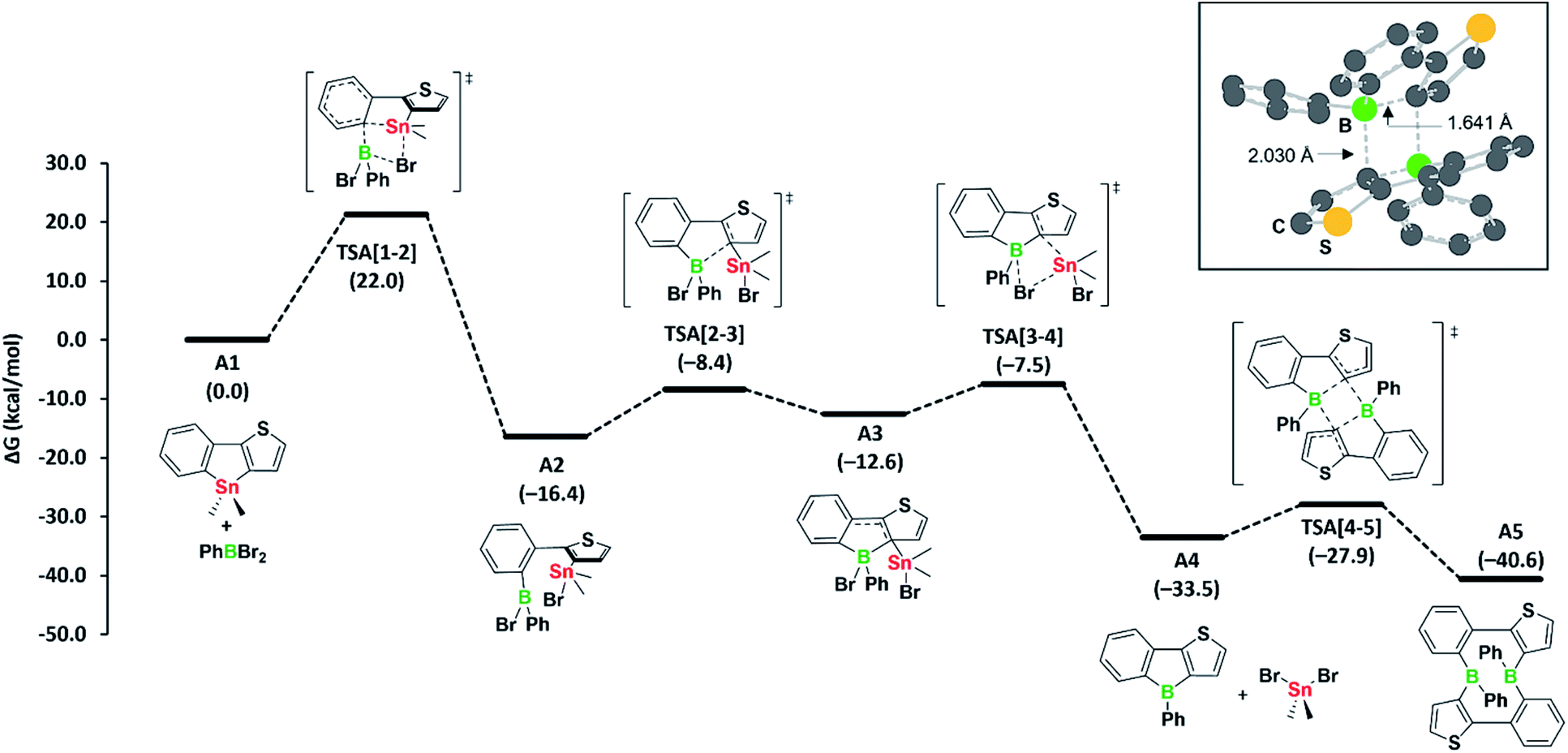 | ||
| Fig. 2 Computed mechanism for diboracycle formation via tin–boron exchange. Free energies in parentheses are in kcal mol−1. DFT-optimised structure with key distances of the transition state responsible for the borole-to-diboracycle interconversion (TSA[4-5]) is shown in the inset in the top right corner. | ||
The formation of the borole A4 (corresponding to 2a) and the by-product dibromo(dimethyl)stannane from the reaction of the stannole with dibromo(phenyl)borane is exergonic by −33.5 kcal mol−1. Kinetically, the first transmetalation step (A1 to A2) is rate-limiting, with a barrier of 22.0 kcal mol−1. This barrier is consistent with the reaction proceeding at room temperature. The remaining steps leading to borole A4 have only small barriers. Remarkably, the transformation of borole A4 to its dimer A5 (corresponding to 3a) via transition state TSA[4-5], involving a σ-bond metathesis between the B–C bonds of two boroles, shows that the barrier is small (ΔG‡ = 5.6 kcal mol−1). As the dimer A5 is only slightly more stable than borole A4 (ΔG = −7.1 kcal mol−1), its reversion to borole A4 is relatively facile due to the associated low barrier of only 12.7 kcal mol−1. Accordingly, the two species are expected to be in equilibrium at room temperature, consistent with the experimental observations. An alternative formation of dimer A5via intermolecular boron–tin exchange between two molecules of A2, which would circumvent a borole intermediate,6 might incur a large barrier, mainly due to the high entropic penalty associated with the intermolecular reaction. Thus, we have not further considered this possibility. Overall, we propose that the formation of the ten-membered diboracycles 3a and 3b from the tin–boron exchange reaction of stannole 1 proceeds via a borole intermediate.
The facile conversion of A4 to A5 can be mainly ascribed to the unfavourable antiaromatic character and low steric shielding in A4, both of which contribute to the ease of dimerisation. The nucleus-independent chemical shifts (NICS) computed for a small set of boroles show that the antiaromatic character within diarene-fused boroles increases with the number of thiophenes fused to the borole ring (see Table S1 in the ESI†), a result that is consistent with previous findings of Yamaguchi5 and He.6 These observed trends are also harmonious with the formation of diboracycles – and not boroles – from the tin–boron exchange of related doubly thiophene-fused stannoles, as the corresponding boroles would exhibit a strong antiaromatic character.6
Like non-fused boroles,2b,17 annulated boroles have been shown to undergo ring expansion with organic azides to generate BN heteroaromatic compounds.3,18 Deviations from the predominant denitrogenative pathway have been observed by Martin from the reaction between phenyl azide and 9-phenyl-9-borafluorene, the dibenzo analog of borole 2a.18b In this case, the reaction instead proceeded without extrusion of molecular nitrogen and afforded a diazene-functionalised BN-phenanthrene. We were thus eager to study the effect of the fused thiophene group on the reactivity of the “masked” borole 3a towards azides. Treatment of 3a with 1 equiv. of phenyl azide yielded a new product with one broad (δ = 52.2 ppm) and one sharp singlet (δ = 7.3 ppm) in the 11B NMR spectrum, indicative of the presence of both three- and four-coordinate boron centers, respectively. LIFDI-MS results were consistent with a 2:1 borole-azide adduct, specifying that the reaction proceeded without loss of dinitrogen. After work-up, product 4a was obtained as an orange solid in 67% yield. Due to the complex 1H NMR spectrum, the structure of 4a was finally revealed by a single-crystal X-ray diffraction study (Fig. 3). The solid-state structure confirmed the presence of an intact azide group, which was found to bridge the two boron atoms through the nitrogen atoms at either end. The polycyclic structure of 4a can be described as a heteropropellane as the C13–C16 bond is part of three different heterocyclic ring systems. As signaled by NMR spectroscopy, the molecular structure contains a boron atom in a trigonal-planar (sum of bond angles of 359.9(1)°) as well as in a tetrahedral geometry (sum of bond angles of 336.9(1)°). The latter is pyramidalised due to the coordination of the substituent-bearing nitrogen atom of the azide; the corresponding B–N bond length of 1.617(2) Å is typical of a dative boron–nitrogen interaction.19
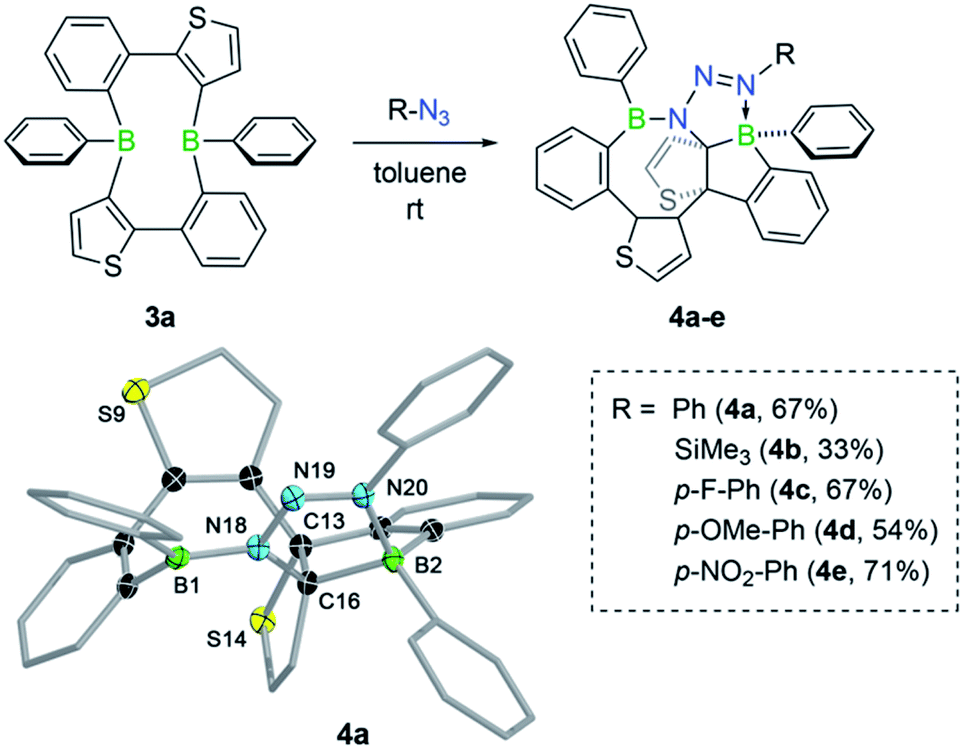 | ||
| Fig. 3 Reactivity of 3a towards a series of organic azides and molecular structure of 4a. Displacement ellipsoids are shown at the 50% probability level. Some ellipsoids and hydrogen atoms have been removed for clarity. Selected bond lengths (Å) and angles (°): B1–N18 1.456(2), B21–N20 1.617(2), N18–N19 1.339(2), N19–N20 1.277(2), N18–C16 1.491(2), C16–B21 1.661(2), C13–C16 1.592(2); N18–N19–N20 111.6(1). | ||
To determine if the reaction is more general in scope, we explored the reaction of 3a with a series of other organic azides, including aryl azides with different substitution patterns and trimethylsilyl azide (Fig. 3). In all cases, the reaction proceeded within minutes at room temperature and invariably yielded the appropriate heteropropellane compounds in moderate to good yields. The reaction outcome thus appears largely unaffected by the electronic and steric effects of the organic azides. Using an excess rather than one equivalent of the aryl azides led to the same outcome. The identities of products 4b–e were corroborated by single-crystal X-ray crystallography, heteronuclear NMR spectroscopy, and LIFDI mass spectrometry. The unusual propellane products 4a–e are distinct from the common ring expansion products derived from formal nitrene insertion into the antiaromatic borole and from other divergent outcomes of the reaction of boroles with organic azides.2b,3,17,18 However, the fact that only one borole unit underwent ring expansion with the organic azide could suggest that a reaction pathway involving an initial borole adduct of an eight-membered BN3C4 heterocycle, as previously observed by Martin,17b might be followed. To rationalise the mechanistic steps involved in forming the propellanes 4a–e, we performed DFT calculations on the reaction of 2a with phenyl azide. We considered borole 2a rather than the cyclic dimer 3a for the computations, as adduct formation with phenyl azide would preferentially occur with borole 2a, which is in equilibrium with 3a in the solution (vide supra). The computed mechanism, together with the free energy profile, is shown in Fig. 4. The first step entails a [2+3] addition between the endocyclic B–C bond of the borole and the outer nitrogen atoms of the azide, leading to the ring-expanded eight-membered BN3C4 heterocycle A6. Lewis acid complexation of A6 by a second borole unit through the more basic sp2 nitrogen atom yields adduct A7. Subsequent rotation of the borole unit around the newly formed B–N bond affords rotamer A8, which in turn triggers intramolecular B–C bond formation to give the bicyclic intermediate A9. This step corresponds to the re-formation of the previously broken B–C bond in the initial borole ring expansion with the azide. In the last step, the complexed borole in A9 expands its ring via C–C coupling between two thiophene rings to form the product A10 (corresponding to 4a). Thermodynamically, the overall reaction is highly favoured (ΔG = −37.9 kcal mol−1). Kinetically, the first and last steps of the reaction sequence are equally rate-determining with barriers of 12.6 kcal mol−1 and 12.7 kcal mol−1, respectively. These barriers can be easily attained under the reaction conditions and are in line with the rapidly occurring reaction at room temperature.
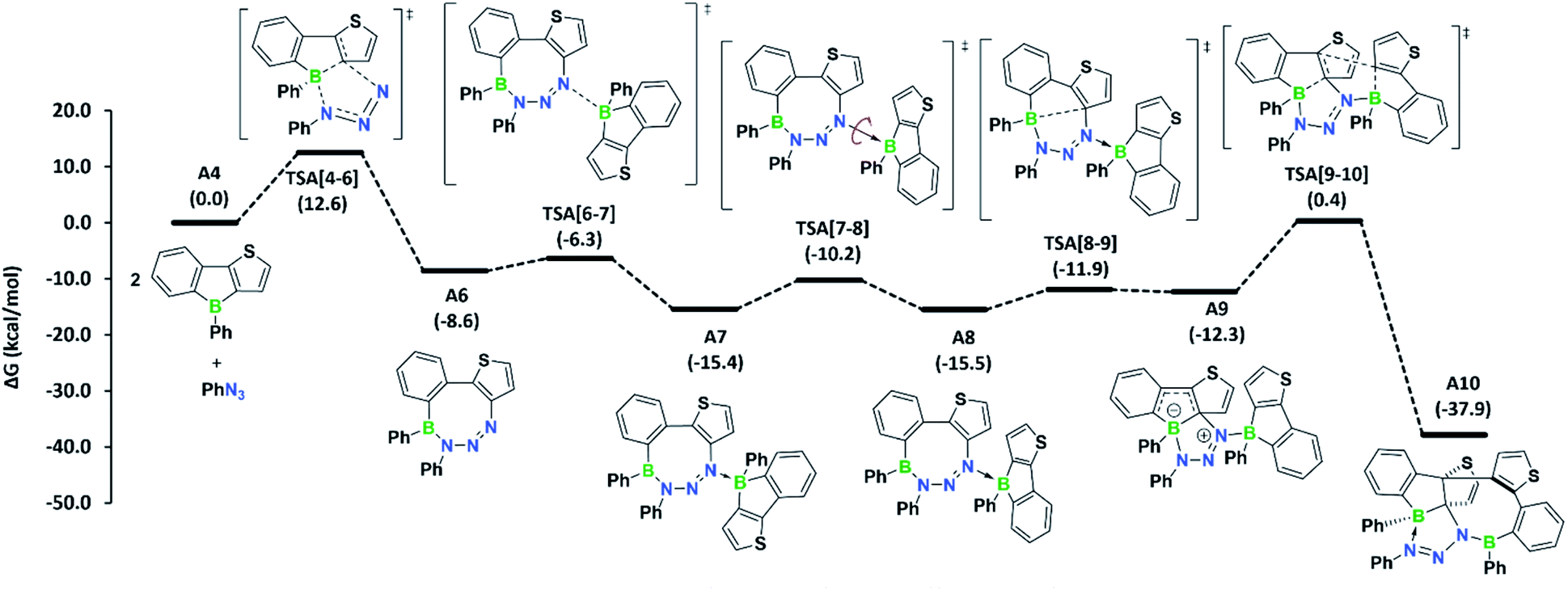 | ||
| Fig. 4 Mechanism computed for azide adduct formation at the B3LYP-D3BJ/6-311g++**/SMD(C6H6)//B3LYP-D3BJ/6-31g** level of theory. Free energies in parentheses are in kcal mol−1. | ||
Conceptually, much of the reaction path resembles that derived from the reaction of boroles with azides, resulting in the formation of 1,2-azaborinines.17b,e In fact, an adduct similar to A8 has been isolated by the group of Martin as a kinetic product in the reaction of pentaphenylborole with trimethylsilyl azide, which yields an 1,2-azaborinine as the thermodynamic product.17b
Thermodynamically, in our case, the formation of the corresponding 1,2-azaborinine derivative from borole 2a and phenyl azide is remarkably exergonic (ΔG = −95.7 kcal mol−1), and thus its formation is highly favoured over heteropropellane A10. Yet, the lack of 1,2-azaborinine formation, even after heating the reaction solution to 110 °C for 12 h, signifies that the formation of A9, and eventually A10, is highly favoured due to the low energy barriers associated with their formation. Moreover, the reactivity of 3a towards organic azides further establishes the potential of biaryl-bridged bis(boranes) to act as sources of monomeric boroles.
Conclusions
This study on the tin–boron exchange of a mixed thiophene-benzo-fused stannole with a series of sterically varied aryldibromoboranes has shown that mainly sterics of the B-aryl substituent dictate the product formation between boroles and diboracycles, their macrocyclic dimers. With decreasing steric demand of the boron substituent, the reaction diverts away from the anticipated tin–boron exchange product – the boroles – and produces diboracycles instead. NMR spectroscopic data and computational insights show that the two products are in equilibrium at room temperature, demonstrating that borole dimerisation and dissociation can occur reversibly. Reactivity studies towards Lewis bases and organic azides uncovered that these cyclic dimers behave as “masked” boroles. With a series of organic azides, an unprecedented mode of ring expansion afforded heteropropellanes containing all three nitrogen atoms of the azide. Mechanistic investigations by quantum chemical calculations revealed that the reaction proceeds through a borole-stabilised eight-membered BN3C4 heterocycle, followed by B–C bond formation and C–C coupling of the two thiophene units. Remarkably, the formation of heteropropellanes from the reaction of thiophene-benzo fused boroles with organic azides seems quite general for a variety of azide substituents.Data availability
Full experimental and computational details are provided as part of the ESI.†Author contributions
H. B. supervised the project. S. F., L. H., and M. B. carried out the synthetic work. S. F. and K. R. carried out the X-ray crystallographic analyses. M. F. and H. H. contributed to the photophysical studies. I. K. carried out the CV measurements. A. J. carried out the computational studies. I. K. and A. J. prepared the manuscript. S. F., A. J., M. F. and I. K. prepared the ESI. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Julius-Maximilians-Universität Würzburg for the financial support of this work. A. J. thanks the Alexander von Humboldt Foundation (Germany) and NSERC (Canada) for postdoctoral fellowships. H. H. thanks the German Research Foundation (DFG) for funding through the Heisenberg Programme HE 6171/9-1 and M. F. thanks the Deutsche Bundesstiftung Umwelt (DBU) for a doctoral fellowship.Notes and references
- For the first report, see: J. J. Eisch, N. K. Hota and S. Kozima, J. Am. Chem. Soc., 1969, 91, 4575–4577 CrossRef CAS.
- For the most recent borole reviews, see: (a) H. Braunschweig, I. Krummenacher and J. Wahler, Adv. Organomet. Chem., 2013, 61, 1–53 CrossRef CAS; (b) J. H. Barnard, S. Yruegas, K. Huang and C. D. Martin, Chem. Commun., 2016, 52, 9985–9991 RSC; (c) B. Su and R. Kinjo, Synthesis, 2017, 49, 2985–3034 CrossRef CAS; (d) W. Zhang, B. Zhang, D. Yu and G. He, Sci. Bull., 2017, 62, 899–900 CrossRef CAS; (e) A. Wakamiya, in Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jäkle, John Wiley & Sons Ltd., Hoboken (New Jersey), 2018, vol. 1, pp. 1–26 Search PubMed; (f) Y. Su and R. Kinjo, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC.
- For recent reviews on fused borole derivatives, see: (a) J. He, F. Rauch, M. Finze and T. B. Marder, Chem. Sci., 2021, 12, 128–147 RSC; (b) X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed.
- Z. Zhang, R. M. Edkins, M. Haehnel, M. Wehner, A. Eichhorn, L. Mailänder, M. Meier, J. Brand, F. Brede, K. Müller-Buschbaum, H. Braunschweig and T. B. Marder, Chem. Sci., 2015, 6, 5922–5927 RSC.
- (a) A. Iida and S. Yamaguchi, J. Am. Chem. Soc., 2011, 133, 6952–6955 CrossRef CAS PubMed; (b) A. Iida, A. Sekioka and S. Yamaguchi, Chem. Sci., 2012, 3, 1461–1466 RSC.
- W. Zhang, D. Yu, Z. Wang, B. Zhang, L. Xu, G. Li, N. Yan, E. Rivard and G. He, Org. Lett., 2019, 21, 109–113 CrossRef CAS PubMed.
- D. J. Morrison, S. D. Riegel, W. E. Piers, M. Parvez and R. McDonald, Chem. Commun., 2006, 2875–2877 RSC.
- A. Hübner, Z.-W. Qu, U. Englert, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2011, 133, 4596–4609 CrossRef PubMed.
- S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Commun., 2018, 54, 6808–6811 RSC.
- W. Haubold, J. Herdtle, W. Gollinger and W. Einholz, J. Organomet. Chem., 1986, 315, 1–8 CrossRef CAS.
- L. Kaufmann, H. Vitze, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2008, 27, 6215–6221 CrossRef CAS.
- (a) J. D. Hoefelmeyer and F. P. Gabbäi, J. Am. Chem. Soc., 2000, 122, 9054–9055 CrossRef CAS; (b) S. Biswas, I. M. Oppel and H. F. Bettinger, Inorg. Chem., 2010, 49, 4499–4506 CrossRef CAS PubMed.
- (a) M. F. Smith, S. J. Cassidy, I. A. Adams, M. Vasiliu, D. L. Gerlach, D. A. Dixon and P. A. Rupar, Organometallics, 2016, 35, 3182–3191 CrossRef CAS; (b) S. J. Cassidy, I. Brettell-Adams, L. E. McNamara, M. F. Smith, M. Bautista, H. Cao, M. Vasiliu, D. L. Gerlach, F. Qu, N. I. Hammer, D. A. Dixon and P. A. Rupar, Organometallics, 2018, 37, 3732–3741 CrossRef CAS; (c) F. Rauch, S. Fuchs, A. Friedrich, D. Sieh, I. Krummenacher, H. Braunschweig, M. Finze and T. B. Marder, Chem.–Eur. J., 2020, 26, 12794–12808 CrossRef CAS PubMed; (d) J. He, F. Rauch, A. Friedrich, J. Krebs, I. Krummenacher, R. Bertermann, J. Nitsch, H. Braunschweig, M. Finze and T. B. Marder, Angew. Chem., Int. Ed., 2021, 60, 4833–4840 CrossRef CAS PubMed; (e) J. He, F. Rauch, I. Krummenacher, H. Braunschweig, M. Finze and T. B. Marder, Dalton Trans., 2021, 50, 355–361 RSC.
- For pyridine-borole adducts, see: (a) H. Braunschweig and T. Kupfer, Chem. Commun., 2008, 4487–4489 RSC; (b) H. Braunschweig, C.-W. Chiu, A. Damme, K. Ferkinghoff, K. Kraft, K. Radacki and J. Wahler, Organometallics, 2011, 30, 3210–3216 CrossRef CAS; (c) H. Braunschweig, A. Damme, D. Gamon, T. Kupfer and K. Radacki, Inorg. Chem., 2011, 50, 4250–4252 CrossRef CAS PubMed; (d) K. Ansorg, H. Braunschweig, C.-W. Chiu, B. Engels, D. Gamon, M. Hugel, T. Kupfer and K. Radacki, Angew. Chem., Int. Ed., 2011, 50, 2833–2836 CrossRef CAS PubMed; (e) H. Braunschweig, A. Damme, D. Gamon, H. Kelch, I. Krummenacher, T. Kupfer and K. Radacki, Chem.–Eur. J., 2012, 18, 8430–8436 CrossRef CAS PubMed; (f) H. Braunschweig, C.-W. Chiu, D. Gamon, M. Kaupp, I. Krummenacher, T. Kupfer, R. Müller and K. Radacki, Chem.–Eur. J., 2012, 18, 11732–11746 CrossRef CAS PubMed; (g) H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, C. Hörl, I. Krummenacher, T. Kupfer, L. Mailänder and K. Radacki, J. Am. Chem. Soc., 2012, 134, 20169–20177 CrossRef CAS PubMed; (h) H. Braunschweig, C.-W. Chiu, A. Damme, B. Engels, D. Gamon, C. Hörl, T. Kupfer, I. Krummenacher, K. Radacki and C. Walter, Chem.–Eur. J., 2012, 18, 14292–14304 CrossRef CAS PubMed; (i) A. Fukazawa, J. L. Dutton, C. Fan, L. G. Mercier, A. Y. Houghton, Q. Wu, W. E. Piers and M. Parvez, Chem. Sci., 2012, 3, 1814–1818 RSC; (j) H. Braunschweig, C.-W. Chiu, D. Gamon, K. Gruss, C. Hörl, T. Kupfer, K. Radacki and J. Wahler, Eur. J. Inorg. Chem., 2013, 1525–1530 CrossRef CAS; (k) V. A. K. Adiraju and C. D. Martin, Dalton Trans., 2017, 46, 10324–10331 RSC.
- For phosphine-borole adducts, see: (a) J. M. Breunig, A. Hübner, M. Bolte, M. Wagner and H.-W. Lerner, Organometallics, 2013, 32, 6792–6799 CrossRef CAS; (b) S. Bontemps, M. Devillard, S. Mallet-Ladeira, G. Bouhadir, K. Miqueu and D. Bourissou, Inorg. Chem., 2013, 52, 4714–4720 CrossRef CAS PubMed; (c) L. E. Laperriere, S. Yruegas and C. D. Martin, Tetrahedron, 2019, 75, 937–943 CrossRef CAS; (d) W. Yang, K. E. Krantz, D. A. Dickie, A. Molino, D. J. D. Wilson and R. J. Gilliard Jr, Angew. Chem., Int. Ed., 2020, 59, 3971–3975 CrossRef CAS PubMed; (e) N. Ando, T. Yamada, H. Narita, N. N. Oehlmann, M. Wagner and S. Yamaguchi, J. Am. Chem. Soc., 2021, 143, 9944–9951 CrossRef CAS PubMed.
- (a) G. E. Herberich and H. Ohst, Chem. Ber., 1985, 118, 4303–4313 CrossRef CAS; (b) P. J. Fagan, E. G. Burns and J. C. Calabrese, J. Am. Chem. Soc., 1988, 110, 2979–2981 CrossRef CAS; (c) G. E. Herberich, M. Negele and H. Ohst, Chem. Ber., 1991, 124, 25–29 CrossRef CAS; (d) Z. Wang, Y. Zhou, K.-H. Lee, W. H. Lam, R. D. Dewhurst, H. Braunschweig, T. B. Marder and Z. Lin, Chem.–Eur. J., 2017, 23, 11587–11597 CrossRef CAS PubMed; (e) J. J. Baker, K. H. M. Al Furaiji, O. Tara Liyanage, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Chem.–Eur. J., 2019, 25, 1581–1587 CrossRef CAS PubMed; (f) X. Su, J. J. Baker and C. D. Martin, Chem. Sci., 2020, 11, 126–131 RSC.
- (a) H. Braunschweig, C. Hörl, L. Mailänder, K. Radacki and J. Wahler, Chem.–Eur. J., 2014, 20, 958–9861 Search PubMed; (b) S. A. Couchman, T. K. Thompson, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Chem. Commun., 2014, 50, 11724–11726 RSC; (c) H. Braunschweig, M. A. Celik, F. Hupp, I. Krummenacher and L. Mailänder, Angew. Chem., Int. Ed., 2015, 54, 6347–6351 CrossRef CAS PubMed; (d) H. Braunschweig, F. Hupp, I. Krummenacher, L. Mailänder and F. Rauch, Chem.–Eur. J., 2015, 21, 17844–17849 CrossRef CAS PubMed; (e) H. Braunschweig, M. A. Celik, T. Dellermann, G. Frenking, K. Hammond, F. Hupp, H. Kelch, I. Krummenacher, F. Lindl, L. Mailänder, J. H. Müssig and A. Ruppert, Chem.–Eur. J., 2017, 23, 8006–8013 CrossRef CAS PubMed.
- (a) M. Müller, C. Maichle-Mössmer and H. F. Bettinger, Angew. Chem., Int. Ed., 2014, 53, 9380–9383 CrossRef PubMed; (b) S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Commun., 2018, 54, 6808–6811 RSC; (c) W. Zhang, G. Li, L. Xu, Y. Zhuo, W. Wan, N. Yan and G. He, Chem. Sci., 2018, 9, 4444–4450 RSC.
- C. F. Pupim, A. J. L. Catão and A. López-Castillo, J. Mol. Model., 2018, 24, 283 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR and UV-vis spectra, X-ray crystallographic and computational details. CCDC 2126038–2126045. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06908j |
| This journal is © The Royal Society of Chemistry 2022 |