
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcceptorless dehydrogenative synthesis of primary amides from alcohols and ammonia†
Jie
Luo‡
,
Quan-Quan
Zhou‡
,
Michael
Montag
 ,
Yehoshoa
Ben-David
and
David
Milstein
*
,
Yehoshoa
Ben-David
and
David
Milstein
*
Department of Molecular Chemistry and Materials Science, Weizmann Institute of Science, Rehovot 76100, Israel. E-mail: david.milstein@weizmann.ac.il
First published on 2nd March 2022
Abstract
The highly desirable synthesis of the widely-used primary amides directly from alcohols and ammonia via acceptorless dehydrogenative coupling represents a clean, atom-economical, sustainable process. Nevertheless, such a reaction has not been previously reported, and the existing catalytic systems instead generate other N-containing products, e.g., amines, imines and nitriles. Herein, we demonstrate an efficient and selective ruthenium-catalyzed synthesis of primary amides from alcohols and ammonia gas, accompanied by H2 liberation. Various aliphatic and aromatic primary amides were synthesized in high yields, with no observable N-containing byproducts. The selectivity of this system toward primary amide formation is rationalized through density functional theory (DFT) calculations, which show that dehydrogenation of the hemiaminal intermediate into primary amide is energetically favored over its dehydration into imine.
Introduction
Ammonia is the simplest, most abundant precursor for the industrial preparation of nitrogen-containing compounds, but its application in selective homogeneously catalyzed organic synthesis by metal complexes has been challenging, partly due to the formation of stable Werner-type ammine complexes.1 In 2008, our group reported an atom- and step-economical synthesis of primary amines from alcohols and ammonia, catalyzed homogeneously by an acridine-based PNP-type ruthenium pincer complex.2a Since then, environmentally benign processes, which involve coupling of alcohols and ammonia, and generate no hazardous waste, have been developed to access amines, imines (including N-heterocycles) and nitriles.2 The first mechanistic steps common to these transformations are alcohol dehydrogenation into aldehyde or ketone,3 followed by nucleophilic attack of ammonia on the newly-formed carbonyl, leading to a hemiaminal intermediate. The latter subsequently undergoes facile dehydration into imine, which typically reacts further to yield various N-containing products (Scheme 1a). In the case of primary alcohols, an alternative route can be envisioned, wherein the hemiaminal undergoes dehydrogenation rather than dehydration,4 thereby affording a primary amide.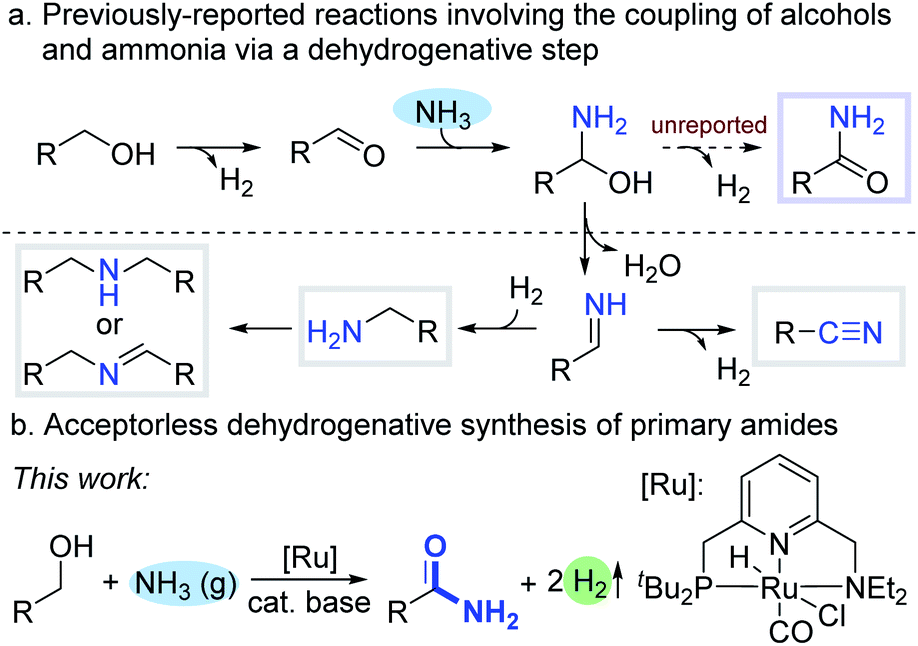 | ||
| Scheme 1 Direct dehydrogenative coupling of alcohols and ammonia for the synthesis of primary amides. | ||
Primary amides feature extensively in organic synthesis, and their ubiquity in pharmaceuticals, natural products, agrochemicals and biologically-active molecules has made their preparation the focus of widespread attention.5 Classical synthetic methodologies involve the amidation of carboxylic acid derivatives or hydration of nitriles – processes that either generate considerable waste or require the use of pre-prepared starting materials.6 Consequently, there is high demand for more sustainable and efficient means of accessing primary amides from readily-available sources.7 From this perspective, the synthesis of primary amides directly from alcohols and ammonia is highly desirable, since these precursors are abundant and inexpensive. Nevertheless, the existing methods for this transformation require stoichiometric amounts of additives, such as expensive or harmful oxidants or organic hydrogen acceptors, making them neither sustainable nor atom-economical.8 For instance, Grützmacher and co-workers reported an efficient homogeneous rhodium-catalyzed coupling of alcohols and ammonia into primary amides,8b but this required a 5-fold excess of methyl methacrylate as a hydrogen scavenger. Some catalytic systems employ O2 as the hydrogen acceptor, with concomitant generation of water, thereby making them more environmentally friendly.8j–l A notable example was reported by Mizuno and co-workers, who implemented a heterogeneous catalysis approach, using manganese-oxide-based molecular sieves under 3 bar of O2.8j However, a fundamental drawback of all acceptor-based systems, apart from the inherent need for additives, is that hydrogen atoms from the substrate are transferred to the acceptor, rather than being released as hydrogen gas – a valuable commodity chemical in its own right. An acceptorless process, whereby alcohols and ammonia are coupled into primary amides with liberation of H2 as a byproduct, would therefore be highly advantageous, but this has never been reported. One of the challenges in developing such a process is that the hemiaminal intermediate can easily dehydrate into an imine upon heating,8j thereby inducing side reactions. Furthermore, reactions involving H2 evolution usually benefit from an open system, whereas utilizing gaseous NH3 as a reactant typically requires a closed system.
Herein, we report the unprecedented synthesis of primary amides directly from alcohols and gaseous ammonia, with concomitant evolution of H2, using a pyridine-based PNN–ruthenium pincer complex (Ru-1) as the catalyst, combined with catalytic amounts of base (Scheme 1b). This system exhibits excellent chemoselectivity toward the generation of primary amides, rather than other N-containing compounds, and enables the synthesis of various aliphatic and aromatic primary amides in generally high yields.
Results and discussion
Establishment and optimization of the catalytic reaction conditions
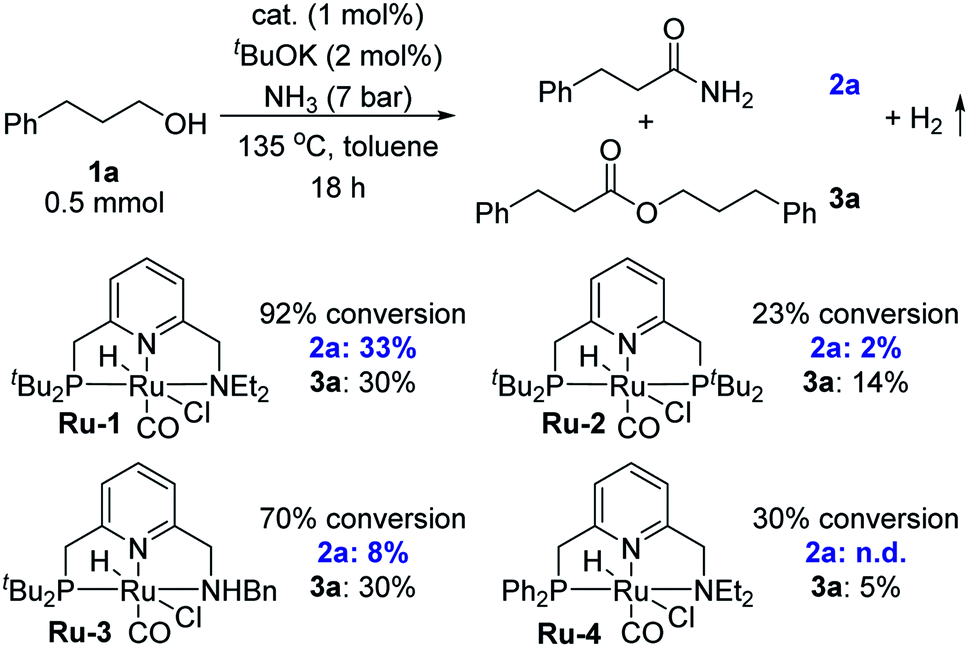
To realize the synthesis of primary amides by dehydrogenative coupling of alcohols and ammonia, we reasoned that a pincer complex could constitute an appropriate catalyst, since such complexes have usually been stable in the presence of ammonia.2 Several pyridine-based ruthenium-containing pincer complexes, especially those already known to generate secondary or tertiary amides from alcohols,3b,4 were examined as catalysts in order to preferentially achieve the dehydrogenation of the ammonia-derived hemiaminal intermediate (Scheme 1a). 3-Phenyl-1-propanol (1a) was chosen as the model substrate for the preliminary experiments. In each of these trials, a toluene solution of 1a containing 1 mol% catalyst and 2 mol% tBuOK, was placed under 7 bar of ammonia inside a 90 mL Fischer–Porter tube at room temperature, and was then heated at 135 °C by immersion in an oil heating bath (Table 1). Interestingly, using our PNN–Ru complex Ru-1 (ref. 3a) as the catalyst, 92% conversion was achieved after heating for 18 h, with the target product 2a being observed in 33% yield, along with 30% of ester 3a (accounting for 60% of the consumed substrate). No other N-containing side products, such as amines, imines or nitriles, were observed by GC-MS analysis of the crude reaction mixture. This attests to the excellent activity of this catalytic system toward dehydrogenation rather than dehydration. By contrast, applying the PNP-Ru complex Ru-2 resulted in low conversion of 1a and a marginal yield of amide 2a. Complex Ru-3,4e,f which is an analog of Ru-1 with an NHBn group (Bn = benzyl), exhibited the same alcohol esterification reactivity as Ru-1, but was inferior with respect to primary amide synthesis.9 Finally, using the new complex Ru-4, which is a PPh2-substituted variant of Ru-1, afforded no observable amide. It should be noted that a bipyridine-based PNN-Ru pincer complex, previously reported to catalyze the formation of tertiary amides from alcohols and amines,4c was also employed as a catalyst for the coupling of alcohols and ammonia, but only secondary amines and imines were obtained.2d| a Conditions: 1a (0.5 mmol), cat. (1 mol%), tBuOK (2 mol%), toluene (2 mL), NH3 (7 bar), 135 °C, 18 h. Conversions and yields were determined by NMR spectroscopy using benzyl benzoate as internal standard. |
|---|
|
In contrast to previously reported catalytic reactions wherein organoamines were used as coupling partners, the current system showed no improvement in ester-to-amide conversion upon prolonging the reaction time, possibly because ammonia is not nucleophilic enough to amidate esters under the applied conditions.10 We therefore undertook systematic optimization of the catalytic conditions in order to improve the yields of primary amides, using 1a as substrate and Ru-1 as catalyst. Firstly, the effect of base was evaluated. No reaction was observed in the absence of base, but when KOH was used, alcohol conversion and product yield were slightly lower than with tBuOK (Table 2, entries 1–3). These findings clearly indicate that the base is necessary for catalysis to occur. Moreover, when the amount of tBuOK was reduced from 2 to 1 equiv relative to the catalyst, conversion of alcohol decreased from 92 to 80% (entry 4). We also examined alternative solvents, namely, THF and hexamethyldisiloxane (HMDSO), but no improvement was observed in amide yield (entries 5 and 6). The yield of 2a was found to be inversely related to temperature. Thus, increasing the temperature from 135 to 150 °C slightly decreased the yield from 33 to 30% (entry 7), whereas lowering the temperature to 120 °C raised the yield of 2a to 40%, while decreasing the yield of ester 3a to 20% (entry 8). The improvement in amide yield at 120 °C might be due to enhanced ammonia solubility upon cooling.11a We reason that the concentration of ammonia in solution directly affects the formation of hemiaminal from the in situ-generated aldehyde, which is further influenced by the competition between ammonia and alcohol, the latter being responsible for the ester byproducts. Based on these assumptions, and the fact that ammonia solubility varies with solvent,11b the effect of solvent composition was investigated at 120 °C. Since ammonia miscibility is usually higher in ethers or alcohols than in benzene-derived solvents, we studied equivolume mixtures of toluene with THF, tAmOH (Am = amyl) or tBuOH. As shown in Table 2, of the three toluene/cosolvent mixtures examined (entries 9–11), the one involving tAmOH provided the best results, with an enhanced amide yield of 56%, and only 11% of the ester (entry 10). It should be noted that using pure tAmOH resulted in low conversion (40%), and therefore incorporating toluene is crucial for ensuring significant conversion. Interestingly, doubling the volume of the toluene/tAmOH mixture, while halving the concentration of substrate 1a, improved the yield of primary amide to 71%, whereas the yield of ester dropped to only 3% (entry 13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solventb (mL) | T °C | Conversion (%) | Yield (%) | |
| 2a | 3a | ||||
| a Conditions: 1a (0.5 mmol), Ru-1 (1 mol%), tBuOK (2 mol%), solvent as indicated, NH3 (7 bar), temperature as indicated, 18 h. For reactions at 120 °C: Ru-1 (2 mol%), tBuOK (4 mol%), 36 h. Conversions and yields were determined by NMR spectroscopy using benzyl benzoate as internal standard. b Volumetric ratios (mL/mL). c No base was added. d KOH (2 mol%) was added instead of tBuOK. e 1 mol% tBuOK was used. f Pressure was released after 24 h of heating, and then NH3 was refilled to 7 bar, and heating was resumed for 12 h. g Temperature was increased to 150 °C for the final 12 h. h Isolated yield. | |||||
| 1 | Toluene (2) | 135 | 92 | 33 | 30 |
| 2c | Toluene (2) | 135 | n.d. | n.d. | n.d. |
| 3d | Toluene (2) | 135 | 83 | 29 | 27 |
| 4e | Toluene (2) | 135 | 80 | 23 | 28 |
| 5 | THF (2) | 135 | 71 | 12 | 28 |
| 6 | HMDSO (2) | 135 | 87 | 13 | 36 |
| 7 | Toluene (2) | 150 | 92 | 30 | 31 |
| 8 | Toluene (2) | 120 | 84 | 40 | 20 |
| 9 | Toluene/THF (1/1) | 120 | 72 | 15 | 28 |
| 10 | Toluene/tAmOH (1/1) | 120 | 80 | 56 | 11 |
| 11 | Toluene/tBuOH (1/1) | 120 | 66 | 33 | 14 |
| 12 | Toluene/tAmOH (2/1) | 120 | 78 | 66 | 5 |
| 13 | Toluene/tAmOH (4/2) | 120 | 78 | 71 | 3 |
| 14f | Toluene/tAmOH (4/2) | 120 | 88 | 83 | 3 |
| 15f,g | Toluene/tAmOH (4/2) | 120 | 95 | 87h | 4 |
Considering that combining Ru-1 with a base also affords an efficient hydrogenation catalyst,12 we reasoned that under the current conditions, which involve the use of a sealed reaction vessel, the generated hydrogen gas that accumulates inside it might counteract the forward dehydrogenation reaction. Thus, a technically-modified procedure was employed, whereby the gaseous contents of the Fischer–Porter tube were vented after 24 h of heating at 120 °C, followed by refilling the flask with 7 bar of ammonia, and heating for another 12 h. As expected, this improved the yield of 2a to 83%, while the yield of ester remained at 3% (entry 14). Repeating the same procedure, but raising the temperature to 150 °C during the final 12 h, increased the conversion to 95%, with 2a isolated in 87% yield (entry 15).
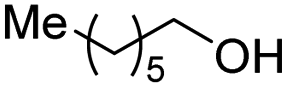
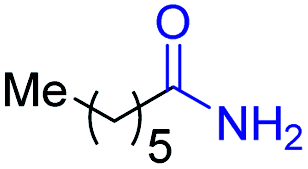
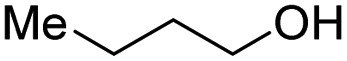
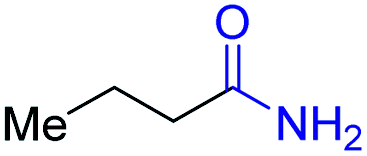
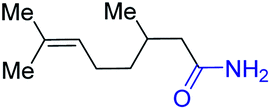
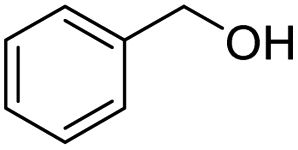
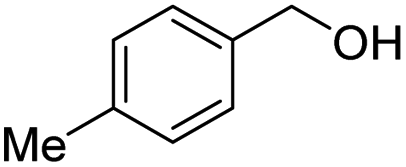
With the optimal catalytic conditions in hand, we explored the substrate scope of the amidation reaction catalyzed by Ru-1 (Table 3). Linear primary alcohols of different chain lengh, namely, 1-heptanol, 1-butanol, and 1-propanol, gave the respective products, 2b–d, in 81–87% isolated yield. The compatibility of the catalytic system with various functional groups was also examined. Increased steric hindrance near the –CH2OH moiety, i.e., replacing H by CH3 at the β-position, decreased the yield of the target product (2e, 51%). By contrast, tertiary amine and methoxy groups were well-tolerated (2f, 81%; 2g, 85%). Remarkably, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds of olefinic substrates were also preserved despite the presence of H2 in the reaction vessel. Thus, using 4-hexen-1-ol as starting material, a mixture of primary amides was obtained due to alkene isomerization, with the internal alkenes being the major products (2h, 61% in total). In the case of citronellol, the trisubstituted double bond was unaffected, and the corresponding primary amide (2i) was isolated in 61% yield. Benzyl alcohol and its para-methyl, -methoxy, -chloro and -fluoro derivatives were also studied as substrates under the same reaction conditions, and the corresponding benzamides were produced in high yields without a significant substituent effect (2j–n, 82–91%). Finally, in addition to alcohols, 3-phenylpropionaldehyde was also examined as a starting material, but its self-condensation was the primary outcome.
C double bonds of olefinic substrates were also preserved despite the presence of H2 in the reaction vessel. Thus, using 4-hexen-1-ol as starting material, a mixture of primary amides was obtained due to alkene isomerization, with the internal alkenes being the major products (2h, 61% in total). In the case of citronellol, the trisubstituted double bond was unaffected, and the corresponding primary amide (2i) was isolated in 61% yield. Benzyl alcohol and its para-methyl, -methoxy, -chloro and -fluoro derivatives were also studied as substrates under the same reaction conditions, and the corresponding benzamides were produced in high yields without a significant substituent effect (2j–n, 82–91%). Finally, in addition to alcohols, 3-phenylpropionaldehyde was also examined as a starting material, but its self-condensation was the primary outcome.
| Entry | Alcohol | Product | Isolated yield |
|---|---|---|---|
| a Conditions: alcohol 1 (0.5 mmol), Ru-1 (2 mol%), tBuOK (4 mol%), toluene (4 mL), tAmOH (2 mL), NH3 (7 bar), 120 °C. Pressure was released after 24 h at 120 °C, and then NH3 was refilled to 7 bar, and the reaction mixture was heated at 150 °C for 12 h. Residual alcohol substrates and homocoupled ester byproducts were detected in all cases. | |||
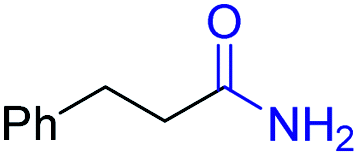
| 1 |
|
|
2a, 87% |
| 2 |
|
|
2b, 82% |
| 3 |
|
|
2c, 87% |
| 4 |
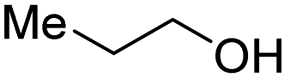
|
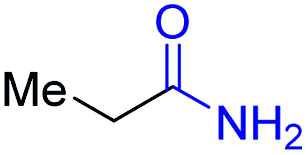
|
2d, 81% |
| 5 |
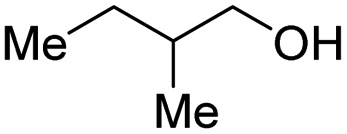
|
|
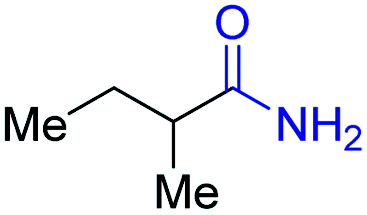
2e, 51% |
| 6 |
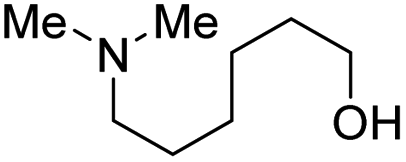
|
|
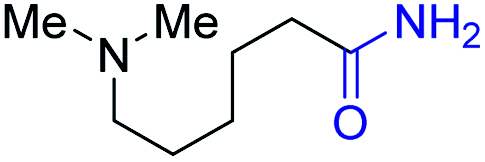
2f, 81% |
| 7 |
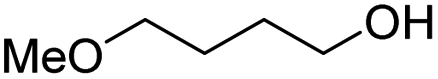
|
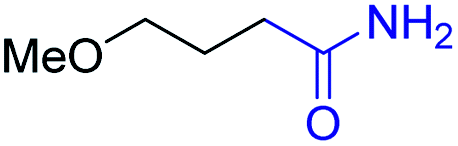
|
2g, 85% |
| 8 |
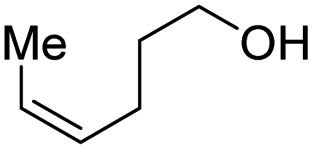
|
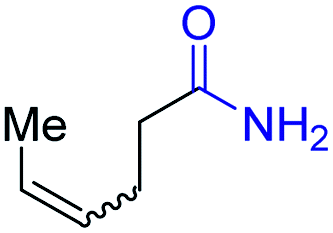
|
2h, 61% |
| 9 |
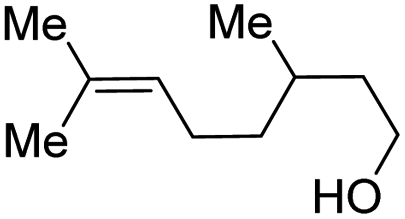
|
|
2i, 61% |
| 10 |
|
|
2j, 82% |
| 11 |
|
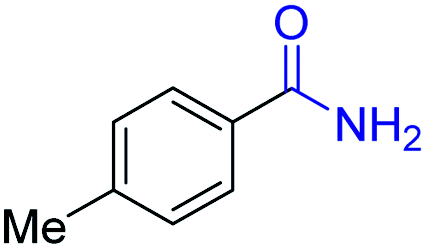
|
2k, 88% |
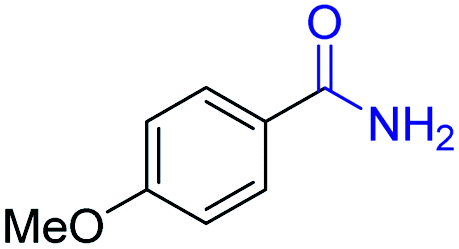
| 12 |
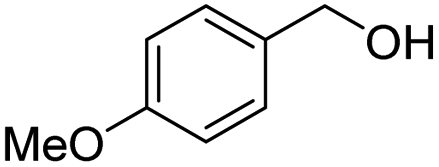
|
|
2l, 91% |
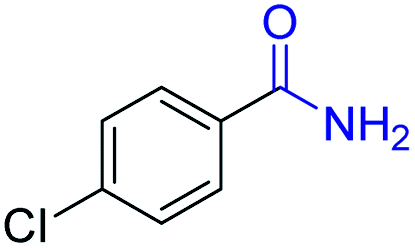
| 13 |
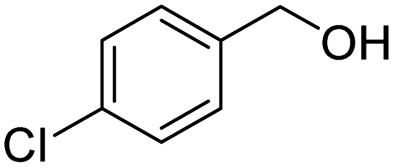
|
|
2m, 85% |
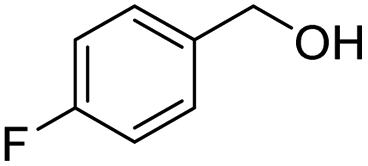
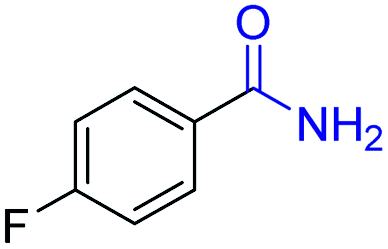
| 14 |
|
|
2n, 92% |
Mechanistic investigation
In an attempt to elucidate the underlying mechanism of the amidation process, we studied the roles played by the various components of the catalytic system, employing both experimental and computational tools. As part of these efforts, the organic species present in the catalytic reaction mixture were monitored by a set of parallel catalytic experiments, carried out under identical reaction conditions (as per entry 13, Table 2), but sampled at different time intervals. The time-variation of the residual alcohol substrate 1a, as well as the yields of primary amide 2a and ester 3a, are presented in Fig. 1. These data clearly indicate that alcohol consumption is correlated with amide formation, and that the reaction rate gradually decreases with time.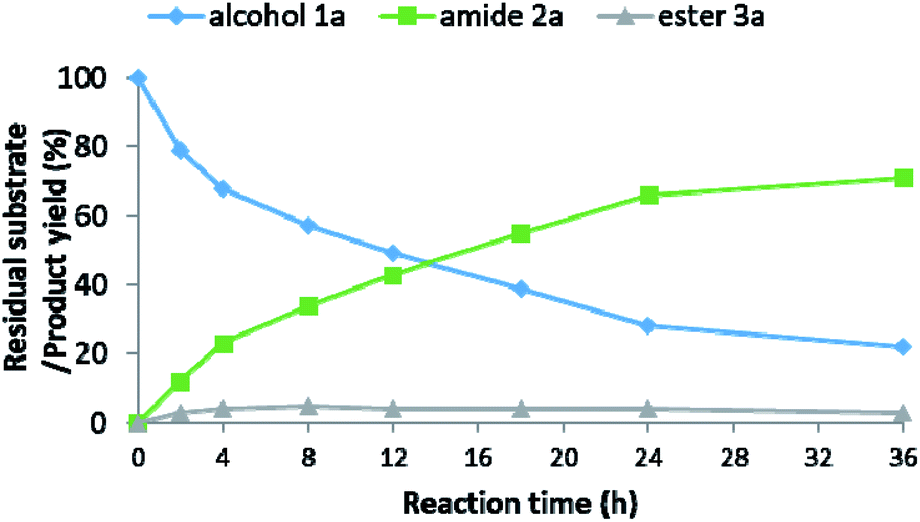 | ||
| Fig. 1 Amidation reaction progress. | ||
The ester byproduct reached a yield of 3% within the first 2 h, and this remained roughly constant throughout the catalytic run. This cannot simply be ascribed to the gradual decrease in alcohol concentration, since an appreciable amount of alcohol remained available in the mixture well after the initial 2 h, thereby allowing further esterification to take place. Instead, considering the known chemistry of Ru-1 and related complexes, it is likely that the continuous generation of ester was offset by its hydrogenation back to the alcohol,12 induced by the accumulation of H2 gas in the closed reaction vessel, or by direct amidation under the pressurized NH3. The ability of our catalyst to promote facile ester hydrogenation was demonstrated by the fact that hexanol was obtained in 27% yield after heating hexyl hexanoate at 120 °C under only ∼0.3 bar of H2 (∼20 mL H2, representing ∼80% H2 yield in the actual catalytic reaction; Scheme 2a). By contrast, when the ester was exposed to 7 bar of NH3 under similar conditions, but in the absence of H2, only trace amounts of the corresponding amide were detected by GC-MS after completion of the catalytic procedure (Scheme 2b). In a third experiment, the ester was treated simultaneously with ∼0.3 bar of H2 and 7 bar of NH3 under the optimal catalytic conditions (as per entry 15, Table 2), and this afforded the amide in 48% yield (Scheme 2c). Furthermore, we exposed primary amide 2a to ∼0.3 bar of H2 and 7 bar of NH3 under similar catalytic conditions, but observed only trace amounts of alcohol (∼1%) after 18 h of heating (Scheme 2d).13 Taken together, these results imply that the ester byproduct formed during the catalytic process is funneled back into the catalytic cycle by its hydrogenation into alcohol, rather than direct amidation. Moreover, while ester formation is reversible under the applied catalytic conditions, amide formation is virtually irreversible, eventually leading the primary amide to become the dominant product.
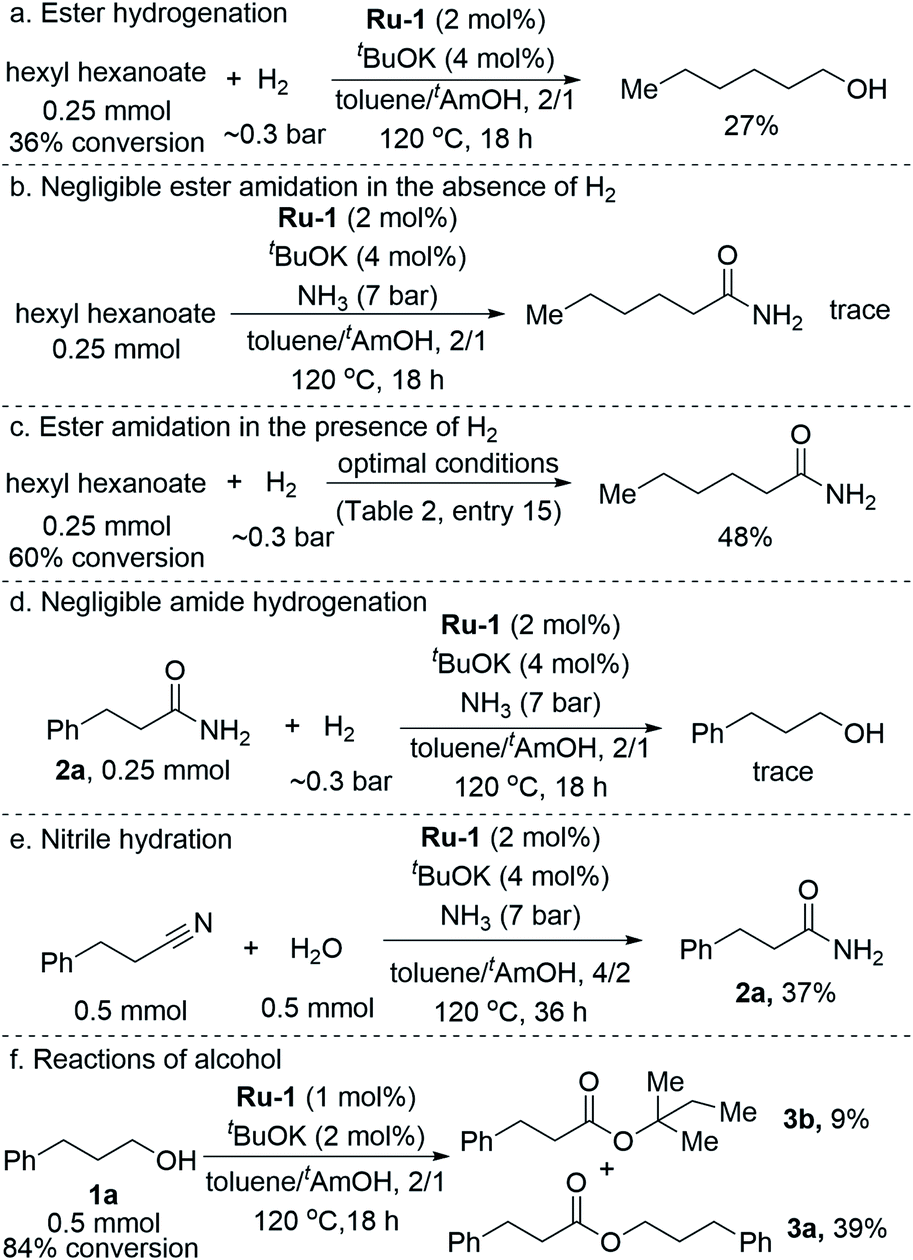 | ||
| Scheme 2 Control experiments. | ||
To supplement the above experiments, which addressed the species observed within the catalytic reaction mixture, we also explored the involvement of nitriles. These have not been observed in the current system, but they are nonetheless plausible reaction intermediates, because they can be generated by hemiaminal dehydration into imines, followed by dehydrogenation, and may subsequently undergo rehydration to produce the primary amides. Moreover, the current catalyst has recently been shown to promote nitrile hydration into amides.6d To probe this alternative pathway, 3-phenylpropionitrile was subjected to the catalytic conditions (as per entry 13, Table 2) in the presence of 1 equiv. of added water (Scheme 2e). However, primary amide 2a was generated in only 37% yield, compared to 71% when alcohol 1a was used as substrate, indicating that the rate of nitrile hydration is significantly lower than that of amide generation from alcohol and ammonia under the same conditions. This result, combined with the fact that no nitrile was observed in the current catalytic process, clearly demonstrate that nitrile hydration is not the dominant reaction pathway (see ESI, Section 3.2,† for further details).
As described above, utilizing tAmOH as a cosolvent to toluene was found to substantially improve the yield of amide. In order to probe the role of tAmOH, a catalytic experiment was carried out, employing the usual toluene/tAmOH solvent mixture and 1a as the substrate, but in the absence of ammonia (Scheme 2f). Despite the high concentration of tAmOH, only a small portion of it coupled with 1a to afford the tertiary ester 3b in 9% yield, whereas the major product, obtained in 39% yield, was ester 3a. In fact, under the optimal catalytic conditions, in the presence of ammonia, and regardless of the identity of the primary alcohol substrate, esters of tAmOH were not observed, showing that any involvement of this cosolvent as a substrate is not competitive with the amidation reaction. Furthermore, when the amidation of 1a was carried out after replacing tAmOH by 1,4-dioxane, which is both less polar and aprotic, only a small decrease in the yield of 2a was observed (80%, with 6% of 3a). This demonstrates that polarity and hydrogen-bonding are not decisive factors in the ability of tAmOH to promote alcohol amidation.
The above results led us to surmise that tAmOH enhances the solubility of ammonia in the reaction medium, thereby allowing amidation to outcompete the reversible esterification reaction. In the absence of sufficient literature data for ammonia solubility in the relevant solvents, we assessed it semi-quantitatively at room temperature by exposing a given solvent to 10 bar of NH3, and calculating the concentration of ammonia from the pressure drop. The obtained data, compiled in Fig. 2, indicate correlation between the ability of a given solvent to dissolve ammonia and the amide yield obtained in that solvent. Thus, NH3 is less soluble in toluene than in the 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 toluene/tAmOH mixture used in our catalytic experiments, and this is consistent with the observed improvement in amide yield upon going from toluene to toluene/tAmOH. On the other hand, HMDSO, in which the amide yield was poor, exhibits the lowest ammonia solubility of all examined solvents. Furthermore, the solubility of NH3 in dioxane is only a little lower than in tAmOH, and the same is true of their respective mixtures with toluene, and this is in line with the relatively small drop in amide yield when tAmOH was replaced by dioxane. Thus, the above findings support our assertion that using tAmOH as a cosolvent improves the yield of amide by increasing ammonia solubility.
:2 toluene/tAmOH mixture used in our catalytic experiments, and this is consistent with the observed improvement in amide yield upon going from toluene to toluene/tAmOH. On the other hand, HMDSO, in which the amide yield was poor, exhibits the lowest ammonia solubility of all examined solvents. Furthermore, the solubility of NH3 in dioxane is only a little lower than in tAmOH, and the same is true of their respective mixtures with toluene, and this is in line with the relatively small drop in amide yield when tAmOH was replaced by dioxane. Thus, the above findings support our assertion that using tAmOH as a cosolvent improves the yield of amide by increasing ammonia solubility.
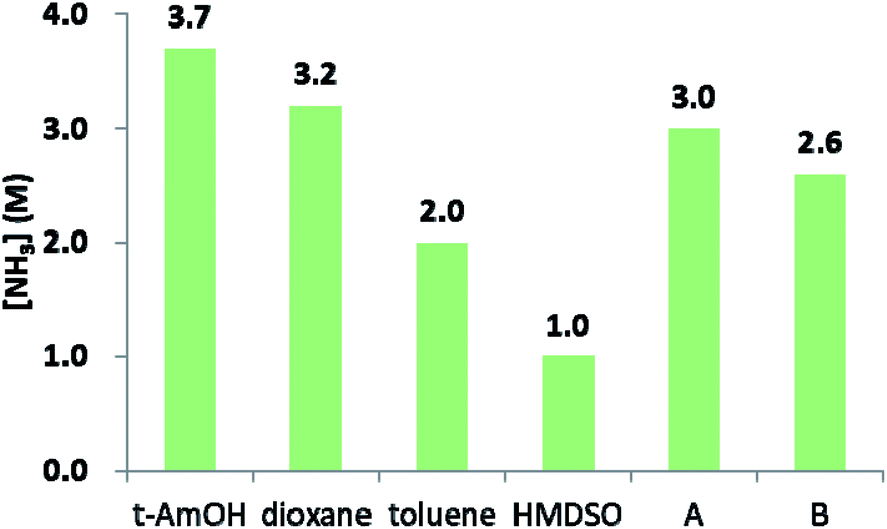 | ||
| Fig. 2 Ammonia concentrations in the catalytically-relevant solvents at room temperature, after initial introduction of 10 bar of NH3. A, toluene/tAmOH, 4:2; B, toluene/1,4-dioxane, 4:2 (volumetric ratios, mL/mL). | ||
In order to clarify those mechanistic aspects that directly involve the coordination sphere of Ru-1, we also explored possible catalytically-related species (Scheme 3). As previously reported, the catalytic cycle commences when Ru-1 undergoes base-induced dearomatization to afford the well-documented catalytically-active complex Ru-5 (Scheme 3a).3a When a C6D6 solution of this complex was exposed to 3 bar of NH3, the ammonia-adduct Ru-6 immediately formed, as evidenced by a clear color change from purple to brown. Removal of ammonia under reduced pressure regenerated Ru-5, showing that formation of Ru-6 is reversible (see Fig. S1–S7† for details). Interestingly, Ru-5 was found to be inert toward excess tAmOH (180 equiv.) at room temperature, indicating that this cosolvent does not directly interfere with the catalytic cycle. By contrast, Ru-5 is highly reactive toward primary alcohols, affording the ruthenium-alkoxide species Ru-7 (Scheme 3b) by adding their O–H bond across the metal–ligand framework,12e as presently demonstrated by the reaction of Ru-5 with 3 equiv of 1-butanol. The resulting n-butoxide complex is unstable at room temperature in C6D6, gradually converting into dihydride Ru-8 (ref. 12c) (major species), as well as aldehyde adduct Ru-9 (ref. 14) (minor species), which can also be obtained directly from Ru-5 and the aldehyde, and is proposed to be an off-cycle species. The very generation of Ru-8 and Ru-9 strongly indicates that Ru-7 undergoes facile β-hydride elimination to produce the aldehyde, which under the catalytic conditions would subsequently react with ammonia. Based on the above results, it is proposed that conversion of alcohol into aldehyde is not the rate-determining step in this system, since it readily occurs at room temperature.
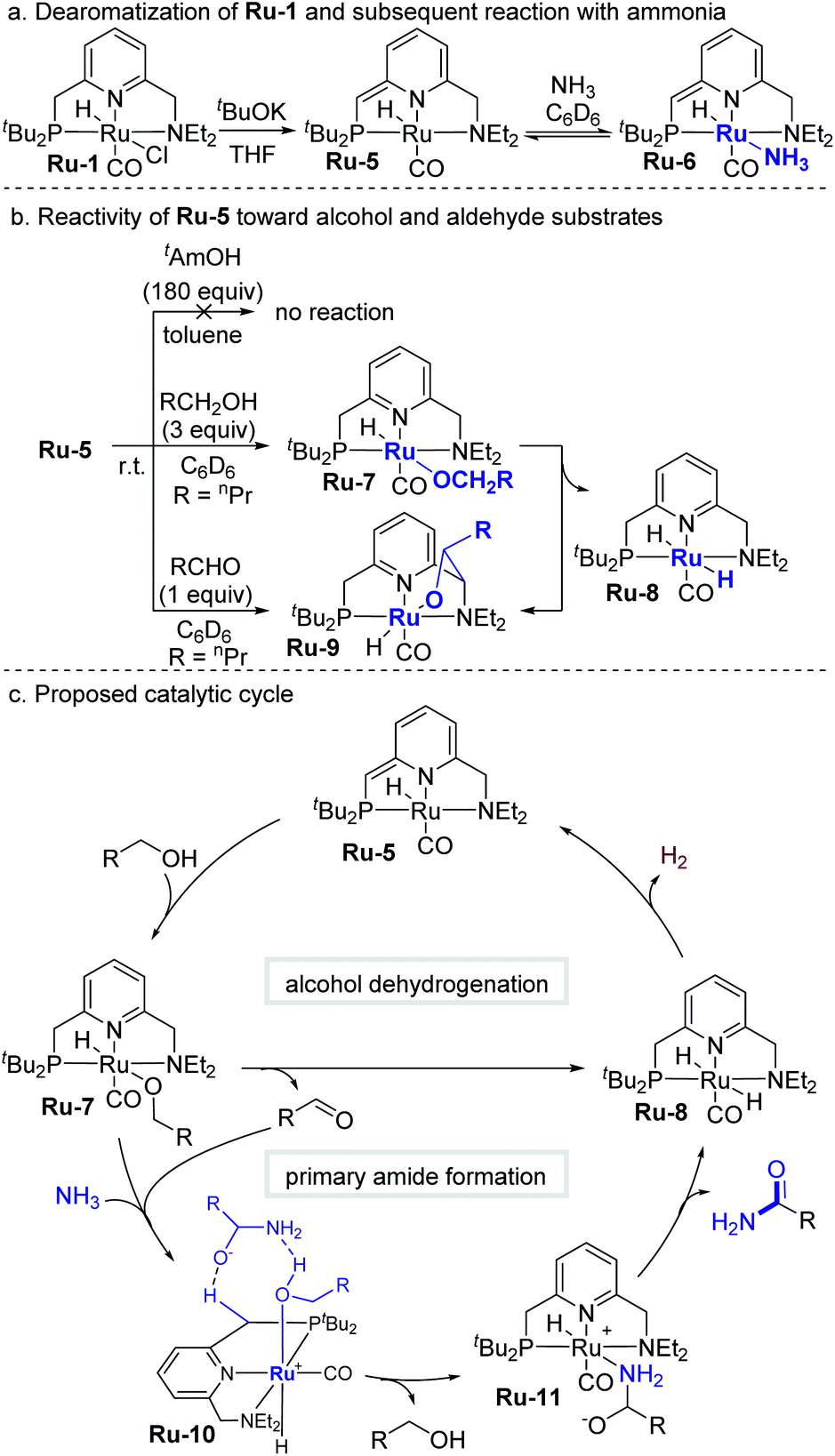 | ||
| Scheme 3 Mechanistic studies and proposed catalytic cycle. | ||
A plausible catalytic cycle for the transformation mediated by Ru-5 is outlined in Scheme 3c. This reaction pathway was investigated by density functional theory (DFT) calculations, based on previously-reported works,15 with ethanol as a minimal alcohol model, and toluene as an implicit solvent (Fig. 3). Given that the alkoxide complex Ru-7 was observed to form under the applied catalytic conditions (Fig. S17†),16 and based on a report involving a comparable osmium-alkoxide pincer complex,15c we propose that Ru-7 promotes the coupling of ammonia and aldehyde through a concerted outer–sphere transition state (TS7,10, Fig. 3). This leads to a hydrogen-bonded intermediate Ru-10, which releases one molecule of alcohol to generate the hemiaminalate complex Ru-11. This, in turn, undergoes β-hydride elimination to afford Ru-8 and liberate the primary amide product. According to the computed energy profile, the conversion of aldehyde to primary amide is thermodynamically favored by 6.4 kcal mol−1, with an overall kinetic barrier of 26.8 kcal mol−1, corresponding to the alcohol-assisted extrusion of H2 from Ru-8.17 This indicates that the generation of primary amides from aldehydes and ammonia is energetically highly-feasible in the current system, in agreement with our experimental findings (see Fig. S57–S63† for other computed pathways, such as those involving pincer ligand arm-opening18 and ammonia activation19).
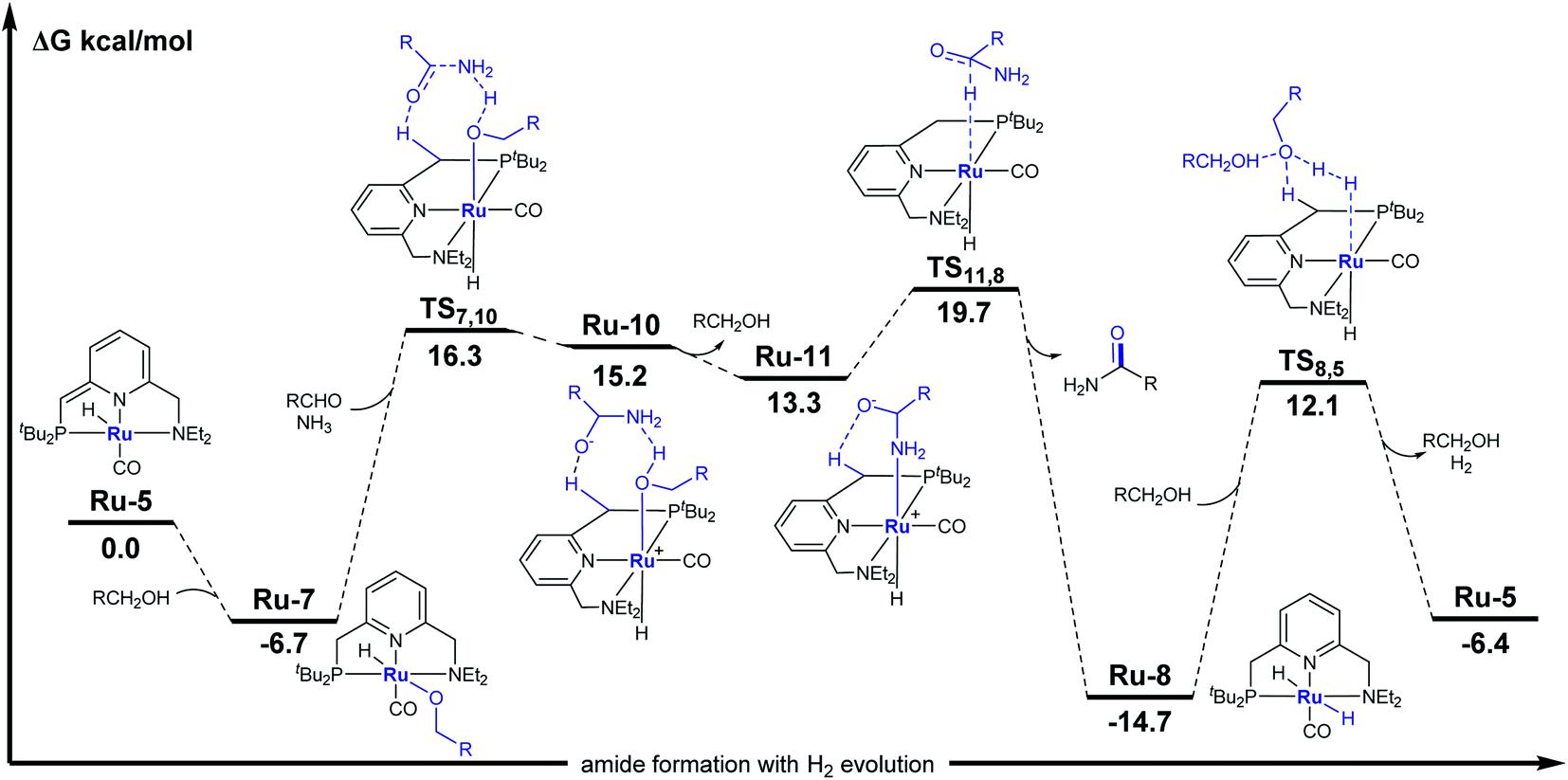 | ||
| Fig. 3 Computed energy profile for primary amide formation by the current catalytic system. Ethanol (R = CH3) was used as a minimal alcohol model, and toluene as an implicit solvent. Mass balance is ensured throughout. | ||
In an attempt to further rationalize the selectivity of the current catalytic system toward primary amide formation, we carried out a computational study of the alternative imine-producing pathway, involving aldehyde and ammonia, which could lead to other N-containing compounds.20 The resulting energy profiles, which are presented in Fig. S64 and S65,† show that this pathway is thermodynamically uphill by 3.9 kcal mol−1, with an overall kinetic barrier of 37.0 kcal mol−1, which is substantially higher than the direct amidation pathway shown in Fig. 3.21 Therefore, primary amide generation by the present PNN–ruthenium system is both kinetically and thermodynamically more favorable than imine formation, and this explains the high selectivity of this system toward the synthesis of primary amides rather than other N-containing products.
Conclusion
We have presented an environmentally-benign and highly selective synthesis of primary amides through an unprecedented ruthenium-catalyzed acceptorless dehydrogenative coupling of alcohols and ammonia. The yield of primary amides was found to depend strongly on ammonia solubility in the reaction medium, and utilizing a toluene/tAmOH mixture allowed high amide yields to be achieved, with only marginal yields of ester byproducts. The PNN–ruthenium catalyst used in our system efficiently promotes the dehydrogenation of the ammonia-derived hemiaminal intermediate, rather than its dehydration, thereby selectively forming primary amides instead of other N-containing compounds, such as amines, imines or nitriles, which are typically generated by other catalytic systems. Based on DFT calculations, we attribute this selectivity to both kinetic and thermodynamic preference for the generation of primary amides over that of imines, which are also the source of other N-containing products.Data availability
Experimental details, GC trace, compound characterization data, NMR spectra, computational details.Author contributions
D. M. and J. L. conceived the project and designed the experiments. J. L. and Q.-Q. Z. performed the experiments, analyzed the data. J. L. performed the computational studies. M. M. provided insightful discussions on the project. Y. B.-D. synthesized the ligands used in this study. J. L., M. M. and D. M. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the European Research Council (ERC AdG 692775). D. M. holds the Israel Matz Professorial Chair of Organic Chemistry. J. L. is thankful to the Feinberg Graduate School, Weizmann Institute of Science, for a Senior Postdoctoral Fellowship.Notes and references
- (a) D. M. Roundhill, Chem. Rev., 1992, 92, 1–27 CrossRef CAS; (b) K. J. McCullough, in Encyclopedia of Reagents for Organic Synthesis, ed. L. A. Paquette, Wiley: Chichester, U.K., 2001, ch. Ammonia Search PubMed; (c) J. L. Klinkenberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 86–95 CrossRef CAS PubMed; (d) i. Kim, H. J. Kim and S. Chang, Eur. J. Org. Chem., 2013, 3201–3213 CrossRef.
- (a) C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661–8664 CrossRef CAS PubMed; (b) D. Pingen, C. Müller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130–8133 CrossRef CAS PubMed; (c) X. Ye, P. N. Plessow, M. K. Brinks, M. Schelwies, T. Schaub, F. Rominger, R. Paciello, M. Limbach and P. Hofmann, J. Am. Chem. Soc., 2014, 136, 5923–5929 CrossRef CAS PubMed; (d) E. Balaraman, D. Srimani, Y. Diskin-Posner and D. Milstein, Catal. Lett., 2015, 145, 139–144 CrossRef CAS; (e) K.-i. Fujita, S. Furukawa, N. Morishima, M. Shimizu and R. Yamaguchi, ChemCatChem, 2018, 10, 1993–1997 CrossRef CAS; (f) P. Daw, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2018, 140, 11931–11934 CrossRef CAS PubMed; (g) P. Daw, A. Kumar, N. A. Espinosa-Jalapa, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2019, 141, 12202–12206 CrossRef CAS PubMed; (h) Y. Wang, S. Furukawa, Z. Zhang, L. Torrente-Murciano, S. A. Khan and N. Yan, Catal. Sci. Technol., 2019, 9, 86–96 RSC; (i) Y. Wang, S. Furukawa and N. Yan, ACS Catal., 2019, 9, 6681–6691 CAS.
- (a) J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed; (b) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed; (c) B. Gnanaprakasam, J. Zhang and D. Milstein, Angew. Chem., Int. Ed., 2010, 49, 1468–1471 CrossRef CAS PubMed; (d) E. Balaraman, E. Khaskin, G. Leitus and D. Milstein, Nat. Chem., 2013, 5, 122–125 CrossRef CAS PubMed; (e) R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed; (f) N. A. Espinosa-Jalapa, A. Kumar, G. Leitus, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11722–11725 CrossRef CAS PubMed; (g) J. Luo, M. Rauch, L. Avram, Y. Diskin-Posner, G. Shmul, Y. Ben-David and D. Milstein, Nat. Catal., 2020, 3, 887–892 CrossRef CAS.
- (a) H. Zeng and Z. Guan, J. Am. Chem. Soc., 2011, 133, 1159–1161 CrossRef CAS PubMed; (b) M. H. G. Prechtl, K. Wobser, N. Theyssen, Y. Ben-David, D. Milstein and W. Leitner, Catal. Sci. Technol., 2012, 2, 2039–2042 RSC; (c) D. Srimani, E. Balaraman, P. Hu, Y. Ben-David and D. Milstein, Adv. Synth. Catal., 2013, 355, 2525–2530 CrossRef CAS; (d) D. Spasyuk, C. Vicent and D. G. Gusev, J. Am. Chem. Soc., 2015, 137, 3743–3746 CrossRef CAS PubMed; (e) Y.-Q. Zou, Q.-Q. Zhou, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. Sci., 2020, 11, 7188–7193 RSC; (f) S. Kar, Y. Xie, Q.-Q. Zhou, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 7383–7393 CrossRef CAS PubMed.
- (a) The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Material Science, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, Wiley, New York, 2000 Search PubMed; (b) J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS PubMed; (c) B. L. Bray, Nat. Rev. Drug Discovery, 2003, 2, 587–593 CrossRef CAS PubMed; (d) T. A. Dineen, M. A. Zajac and A. G. Myers, J. Am. Chem. Soc., 2006, 128, 16406–16409 CrossRef CAS PubMed; (e) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed; (f) M. Szostak, M. Spain, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2014, 136, 2268–2271 CrossRef CAS PubMed; (g) M. Ganesan and P. Nagaraaj, Org. Chem. Front., 2020, 7, 3792–3814 RSC; (h) R. Thakur, Y. Jaiswal and A. Kumar, Tetrahedron, 2021, 93, 132313 CrossRef CAS.
- (a) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (b) W. K. Fung, X. Huang, M. L. Man, S. M. Ng, M. Y. Hung, Z. Lin and C. P. Lau, J. Am. Chem. Soc., 2003, 125, 11539–11544 CrossRef CAS PubMed; (c) T. Hirano, K. Uehara, K. Kamata and N. Mizuno, J. Am. Chem. Soc., 2012, 134, 6425–6433 CrossRef CAS PubMed; (d) B. Guo, J. G. de Vries and E. Otten, Chem. Sci., 2019, 10, 10647–10652 RSC; (e) Q.-Q. Zhou, Y.-Q. Zou, S. Kar, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 10239–10245 CrossRef CAS PubMed.
- (a) H. Fujiwara, Y. Ogasawara, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 5202–5205 CrossRef CAS PubMed; (b) L. Cao, J. Ding, M. Gao, Z. Wang, J. Li and A. Wu, Org. Lett., 2009, 11, 3810–3813 CrossRef CAS PubMed; (c) S. C. Ghosh, J. S. Y. Ngiam, A. M. Seayad, D. T. Tuan, C. L. L. Chai and A. Chen, J. Org. Chem., 2012, 77, 8007–8015 CrossRef CAS PubMed; (d) Q. Song, Q. Feng and K. Yang, Org. Lett., 2014, 16, 624–627 CrossRef CAS PubMed; (e) M. Sharif, J.-L. Gong, P. Langer, M. Beller and X.-F. Wu, Chem. Commun., 2014, 50, 4747–4750 RSC; (f) V. G. Jadhav, J. M. Bhojane and J. M. Nagarkar, RSC Adv., 2015, 5, 6636–6641 RSC; (g) K. Murugesan, T. Senthamarai, M. Sohail, M. Sharif, N. V. Kalevarua and R. V. Jagadeesh, Green Chem., 2018, 20, 266–273 RSC; (h) R. Ray, A. S. Hazari, S. Chandra, D. Maiti and G. K. Lahiri, Chem.–Eur. J., 2018, 24, 1067–1071 CrossRef CAS PubMed; (i) S. Shang, P.-P. Chen, L. Wang, Y. Lv, W.-X. Li and S. Gao, ACS Catal., 2018, 8, 9936–9944 CrossRef CAS.
- (a) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 73–75 CrossRef CAS PubMed; (b) T. Zweifel, J.-V. re Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559–563 CrossRef CAS PubMed; (c) R. Ohmura, M. Takahata and H. Togo, Tetrahedron Lett., 2010, 51, 4378–4381 CrossRef CAS; (d) J.-F. Soulé, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 18550–18553 CrossRef PubMed; (e) R. Das and D. Chakraborty, Catal. Commun., 2012, 26, 48–53 CrossRef CAS; (f) X.-F. Wu, M. Sharif, J.-B. Feng, H. Neumann, A. Pews-Davtyan, P. Langerb and M. Beller, Green Chem., 2013, 15, 1956–1961 RSC; (g) R. A. Molla, K. Ghosh, K. Tuhina and S. M. Islam, New J. Chem., 2015, 39, 921–930 RSC; (h) Z. Xie, R. Chen, Z. Du, L. Kong, Z. Li, Z. Li, N. Wang and J. Liu, Asian J. Org. Chem., 2017, 6, 157–160 CrossRef CAS; (i) R. Ray, A. S. Hazari, S. Chandra, D. Maiti and G. K. Lahiri, Chem.–Eur. J., 2018, 24, 1067–1071 CrossRef CAS PubMed; (j) K. Yamaguchi, H. Kobayashi, T. Oishi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 544–547 CrossRef CAS PubMed; (k) K. Yamaguchi, H. Kobayashi, Y. Wang, T. Oishi, Y. Ogasawara and N. Mizuno, Catal. Sci. Technol., 2013, 3, 318–327 RSC; (l) R. Nie, J. Shi, S. Xia, L. Shen, P. Chen, Z. Hou and F.-S. Xiao, J. Mater. Chem., 2012, 22, 18115–18118 RSC.
- Ru-3 is known to catalyze both the formation of esters from alcohols, and their amidation with organoamines to afford secondary amides (see ref. 4f). It appears that ammonia is not nucleophilic enough to amidate esters under the current catalytic conditions.
- (a) B. Gnanaprakasam and D. Milstein, J. Am. Chem. Soc., 2011, 133, 1682–1685 CrossRef CAS PubMed; (b) H. Sun, M. I. Page, J. H. Atherton and A. Hall, Catal. Sci. Technol., 2014, 4, 3870–3878 RSC.
- (a) C. L. Young and P. G. T. Fogg, International Union of Pure and Applied Chemistry (IUPAC), in Solubility Data Series: Ammonia, Amines, Phosphine, Arsine, Stibine, Silane, Germane and Stannane in Organic Solvents, ed. A. S. Kertes, Pergamon Press, Oxford, U.K., 1985, vol. 21, pp. 1–79 Search PubMed; (b) I. Short, A. Sahgal and W. Hayduk, J. Chem. Eng. Data, 1983, 28, 63–66 CrossRef CAS.
- (a) J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113–1115 CrossRef CAS PubMed; (b) E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS PubMed; (c) E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609–614 CrossRef CAS PubMed; (d) J. R. Khusnutdinova, J. A. Garg and D. Milstein, ACS Catal., 2015, 5, 2416–2422 CrossRef CAS; (e) P. Hu, E. Fogler, Y. Diskin-Posner, M. A. Iron and D. Milstein, Nat. Commun., 2015, 6, 6859–6865 CrossRef CAS PubMed.
- (a) V. Papa, J. R. Cabrero-Antonino, E. Alberico, A. Spanneberg, K. Junge, H. Jungea and M. Beller, Chem. Sci., 2017, 8, 3576–3585 RSC; (b) M. Subaramanian, G. Sivakumar, J. K. Babu and E. Balaraman, Chem. Commun., 2020, 56, 12411–12414 RSC.
- (a) C. A. Huff, J. W. Kampf and M. S. Sanford, Chem. Commun., 2013, 49, 7147–7149 RSC; (b) M. Montag, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2012, 134, 10325–10328 CrossRef CAS PubMed.
- (a) H. Li, X. Wang, F. Huang, G. Lu, J. Jiang and Z.-X. Wang, Organometallics, 2011, 30, 5233–5247 CrossRef CAS; (b) H. Li and M. B. Hall, ACS Catal., 2015, 5, 1895–1913 CrossRef CAS; (c) D. G. Gusev, ACS Catal., 2017, 7, 6656–6662 CrossRef CAS; (d) D. G. Gusev, Organometallics, 2020, 39, 258–270 CrossRef CAS.
- Ru-6 was not observed to form under the catalytic conditions, and is proposed to be an off-cycle species.
- Although not considered here, the concentrations and pressures of NH3 and H2 can strongly affect ΔG, as has been discussed elsewhere: (a) K. Krogh-Jespersen, M. Czerw, N. Summa, K. B. Renkema, P. D. Achord and A. S. Goldman, J. Am. Chem. Soc., 2002, 124, 11404–11416 CrossRef CAS PubMed; (b) M. Rauch, J. Luo, L. Avram, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 2795–2807 CrossRef CAS PubMed.
- (a) L. Li, M. Lei, L. Liu, Y. Xie and H. F. Schaefer III, Inorg. Chem., 2018, 57, 8778–8787 CrossRef CAS PubMed; (b) P. M. Fanara, S. N. MacMillan and D. C. Lacy, Organometallics, 2020, 39, 3628–3644 CrossRef CAS.
- E. Khaskin, M. A. Iron, L. J. W. Shimon, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2010, 132, 8542–8543 CrossRef CAS PubMed.
- The formation of esters from alcohols, which is the main process competing with alcohol amidation, has been thoroughly studied in previous works involving the same catalyst. See ref. 15.
- (a) D. Jiang, S. Wang, H. Li, L. Xu, X. Hu, B. Barati and A. Zheng, ACS Sustainable Chem. Eng., 2021, 9, 3095–3103 CrossRef CAS; (b) H. Li, X. Wang, M. Wen and Z.-X. Wang, Eur. J. Inorg. Chem., 2012, 5011–5020 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc07102e |
| ‡ Jie Luo and Quan-Quan Zhou contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |