
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled monolayer protection of chiral-imprinted mesoporous platinum electrodes for highly enantioselective synthesis†
Sopon
Butcha
 ab,
Véronique
Lapeyre
b,
Chularat
Wattanakit
*a and
Alexander
Kuhn
*ab
ab,
Véronique
Lapeyre
b,
Chularat
Wattanakit
*a and
Alexander
Kuhn
*ab
aSchool of Molecular Science and Engineering, School of Energy Science and Engineering, Vidyasirimedhi Institute of Science and Technology, 21210, Wangchan, Rayong, Thailand. E-mail: chularat.w@vistec.ac.th
bUniversity of Bordeaux, CNRS, Bordeaux INP, ISM UMR 5255, Site ENSCBP, 16 Avenue Pey Berland, 33607, Pessac, France. E-mail: kuhn@enscbp.fr
First published on 7th February 2022
Abstract
In modern chemistry, chiral (electro)catalysis is a powerful strategy to produce enantiomerically pure compounds (EPC). However, it still struggles with uncontrollable stereochemistry due to side reactions, eventually producing a racemic mixture. To overcome this important challenge, a well-controlled design of chiral catalyst materials is mandatory to produce enantiomers with acceptable purity. In this context, we propose the synergetic combination of two strategies, namely the elaboration of mesoporous Pt films, imprinted with chiral recognition sites, together with the spatially controlled formation of a self-assembled monolayer. Chiral imprinted metals have been previously suggested as electrode materials for enantioselective recognition, separation and synthesis. However, the outermost surface of such electrodes is lacking chiral information and thus leads to unspecific reactions. Functionalising selectively this part of the electrode with a monolayer of organosulfur ligands allows an almost total suppression of undesired side reactions and thus leads to a boost of enantiomeric excess to values of over 90% when using these surfaces in the frame of enantioselective electrosynthesis. In addition, this strategy also decreases the total reaction time by one order of magnitude. The study therefore opens up promising perspectives for the development of heterogeneous enantioselective electrocatalysis strategies.
Introduction
Chirality, especially at the molecular level, contributes to a large number of important functions, ranging from the biochemistry of life to the synthesis of pharmaceuticals, cosmetics, and other fine chemicals.1,2 There are various strategies that have been followed for the well-controlled production of chiral compounds, such as asymmetric synthesis, separation, resolution, and crystallization.3 Among them, enantioselective synthesis has been one of the most promising approaches to achieve product selectivity and minimize the formation of by-products.3a In this context, electrosynthesis of enantiomers is a very attractive and promising route, due to the fact that it is one of the green and cost-effective processes, producing various types of organic molecules without the need for additional chemical reagents.4 However, it may suffer from uncontrolled stereochemistry during the electrochemical conversion, eventually resulting in a low yield of the desired molecules.5In order to achieve a practically relevant enantiomeric excess (%ee > 90), a well-designed catalyst with high selectivity and long-term stability is a key challenge. Among numerous types of materials, metals or metal oxides with intrinsically chiral surface properties are considered as promising materials for asymmetric synthesis due to an easy fabrication, a rigid structure, and a simple regeneration process after the reaction.6,7 Very interesting examples of these types of materials are organically doped metals/metal oxides, such as bulk gold (Au) and silver (Ag),8 copper oxide (CuO) surfaces,9 and palladium (Pd) nanoparticles.10 Interestingly, chiral features are preserved in these matrices, even after the removal of the chiral template molecules. Such nanostructured surfaces display some promising features for various potential applications, ranging from chiral recognition to asymmetric synthesis.
Due to the above-mentioned general benefits of electrosynthesis, chiral materials have also been tested as enantioselective electrodes over the past decade.11,12 Recently, chiral-imprinted metal surfaces, combining in a synergetic way the benefits of chiral features with mesoporous structures have been successfully elaborated by electrodeposition of metals in the simultaneous presence of self-assembled non-ionic surfactants and chiral templates.13,14 Due to the interactions between a non-ionic surfactant, present as a columnar supramolecular structure and serving as meso-porogen, and chiral molecules, adsorbed at the external wall of the surfactant columns, mesopores with chiral recognition sites are generated in the internal part of the bulk metal during electrodeposition.15 Various chiral templates, such as pharmaceutical molecules i.e. 3,4-dihydroxyphenylalanine (DOPA),13,19 or active ingredients in cosmetic industry like mandelic acid (MA),14a,17 and phenylethanol (PE),14b,18,19 as well as amino acids e.g. tryptophan (Tryp),16 were employed to validate this strategy. It was possible to generate unique chiral properties, opening a wide variety of applications, ranging from enantioselective recognition,13 and chiral separation,16 to actuators,17 and asymmetric synthesis.14,18,19
In addition, several types of metal matrixes have been proposed for the imprinting, ranging from pure noble (platinum, Pt),14 and non-noble metals (nickel, Ni),18 to metal alloy structures.19 In some cases, excellent enantioselectivity of up to 90% could be obtained, due to the synergy between mesoporous channels, with improved mass transport and active surface area, and the chiral recognition sites. However, in order to achieve such high %ee values, pulsed electroreduction with very short pulse times had to be employed, which results in very long global synthesis times. Consequently, this can be a major roadblock for practical applications.
In this contribution, in order to achieve high enantioselectivity, combined with a reduced total synthesis time, an alternative strategy is proposed. As the above mentioned pulsed electroreduction is time-consuming, due to the required rather long relaxation period,14b it would be preferable to work under potentiostatic conditions. However, such a steady-state electrosynthesis process leads to a preferential formation of racemic mixtures at the non-imprinted outermost surface of the electrodes, thus lowering the global enantiomeric excess.14a To overcome this limitation, we propose here the selective modification of the outermost surface of chiral encoded metal surfaces with a self-assembled monolayer of organosulfur ligands, to specifically protect this part of the electrode from contributing to the global reaction. Such a rational design of the overall electrode structure drastically decreases non-stereospecific reactions and consequently gives access to high %ee values, combined with short reaction times.
Results and discussion
Synthesis and characterization of chiral-imprinted mesoporous Pt electrodes
Initially, bare chiral-imprinted mesoporous Pt electrodes have been synthesized by electrodeposition from a Pt precursor (hexachloroplatinic acid) in the simultaneous presence of a non-ionic surfactant, analogue to what has been reported earlier,15 and one of the chiral template molecules, (R)- or (S)-phenylethanol (PE). The latter is located at the external surface of the columnar supramolecular structure of the surfactant molecules, forming a lyotropic liquid crystal. This leads to the imprinting of the chiral information in the walls of the mesopores (Fig. S1†). As can be seen in the SEM images, the electrode presents not only an overall extremely smooth surface (Fig. 1A), but also a very regular thickness of approximately 1 μm (Fig. 1B). The TEM image also confirms a hexagonal mesoporous structure with a pore dimension of around 5 nm (Fig. 1C), which is very similar to what has been observed in previous studies.13a,14,15a–c In order to further investigate the beneficial effect of the porous structure, the active surface area is characterized by cyclic voltammetry in H2SO4 in a potential window ranging from −0.20 to 1.20 V (Fig. 1D). The typical anodic and cathodic peaks, related to adsorption and desorption of hydrogen are observed in the potential range below 0 V. The oxidation, starting at 0.70 V, and the reduction at 0.55 V can be associated with the Pt oxidation and PtO reduction, respectively.13a The specific surface area was calculated,20 demonstrating a greatly enhanced roughness factor of mesoporous Pt with respect to flat Pt by a factor of up to 170. | ||
| Fig. 1 Characterization of mesoporous Pt electrodes; (A) top-view and (B) cross-sectional SEM images of a mesoporous Pt electrode; (C) TEM image of an ultra-thin mesoporous Pt electrode; (D) cyclic voltammograms of a mesoporous Pt electrode (red) compared with flat Pt (black) in 0.5 M H2SO4 at a scan rate of 100 mV s−1. For the CV curve, the mesoporous Pt electrode was obtained via electrodeposition by injecting a charge density of 4 C cm−2. | ||
Surface functionalization with thiol molecules
Metal surfaces modified with organosulfur ligands have been widely utilized as selective catalysts.21 Among the various types of organosulfur ligands, linear alkanethiols with a medium chain length are often used for surface modification due to their advantages, including easy preparation and good stability of the strong covalent bond between metal and sulphur atoms.22 These advantages have been also confirmed by a study of alkane thiolate-protected Pt nanoparticles (NPs), showing that adsorbed sulphur species on Pt surfaces are indeed very stable against desorption even at high reduction potentials.23 Ligand coated surfaces are usually undesirable in electrocatalysis because the charge transfer between catalyst and reactant can be significantly slower, or completely shut down depending on the length of the ligand.21b In our case, we want to inhibit as much as possible the electron transfer at the monolayer modified sections of the electrode. Therefore, alkanethiols with a quite long chain should be in principle more efficient. Floridia et al. found out that medium (hexanethiol, C6-SH) and long (dodecanethiol, C12-SH) chain thiols adsorbed on Pt NPs exhibit similar stability against desorption at high reduction potentials (up to −1.8 V vs. SCE), but the quality of the self-assembled monolayers (SAMs) strongly depends on the chain length, with longer chains leading to higher coverage.23,24 Taking into account also solubility problems, encountered for long-chain thiols, one can conclude that, as a compromise, an intermediate chain length might be optimal to modify the mesoporous Pt surfaces. Accordingly, heptanethiol (C7-SH) was selected for functionalizing the external surfaces of chiral-imprinted mesoporous Pt electrodes. With this strategy, enantioselectivity should increase even for steady-state electrochemical synthesis, since undesired reactions at the external surface are prevented due to the selective blocking of non-imprinted sites by the thiol layer.In order to passivate the outer surface by organosulfur ligands, two strategies have been tested. The first one relies on the self-assembly of thiol ligands from solution, whereas the second one is based on microcontact printing (μCP).25a,b For the first method, an electrode, which still contains surfactant and chiral template molecules inside the mesopores, is exposed to a solution of heptanethiol for a given time to allow the self-assembly to occur at the outer surface, without taking the risk that the thiol layer also adsorbs inside the mesopores. Subsequently, the templates and left-over thiol molecules are eliminated by rinsing with ethanol and MilliQ water (Fig. 2A).26 For the μCP approach, heptanethiol is loaded on a commercial rubber stamp, which is then pressed on the external surface of an already cleaned Pt electrode with empty mesopores. During a predefined contact time, the thiol molecules are transferred to the outermost surface of the mesoporous Pt electrode, forming a SAM as illustrated in Fig. 2B.25c,26
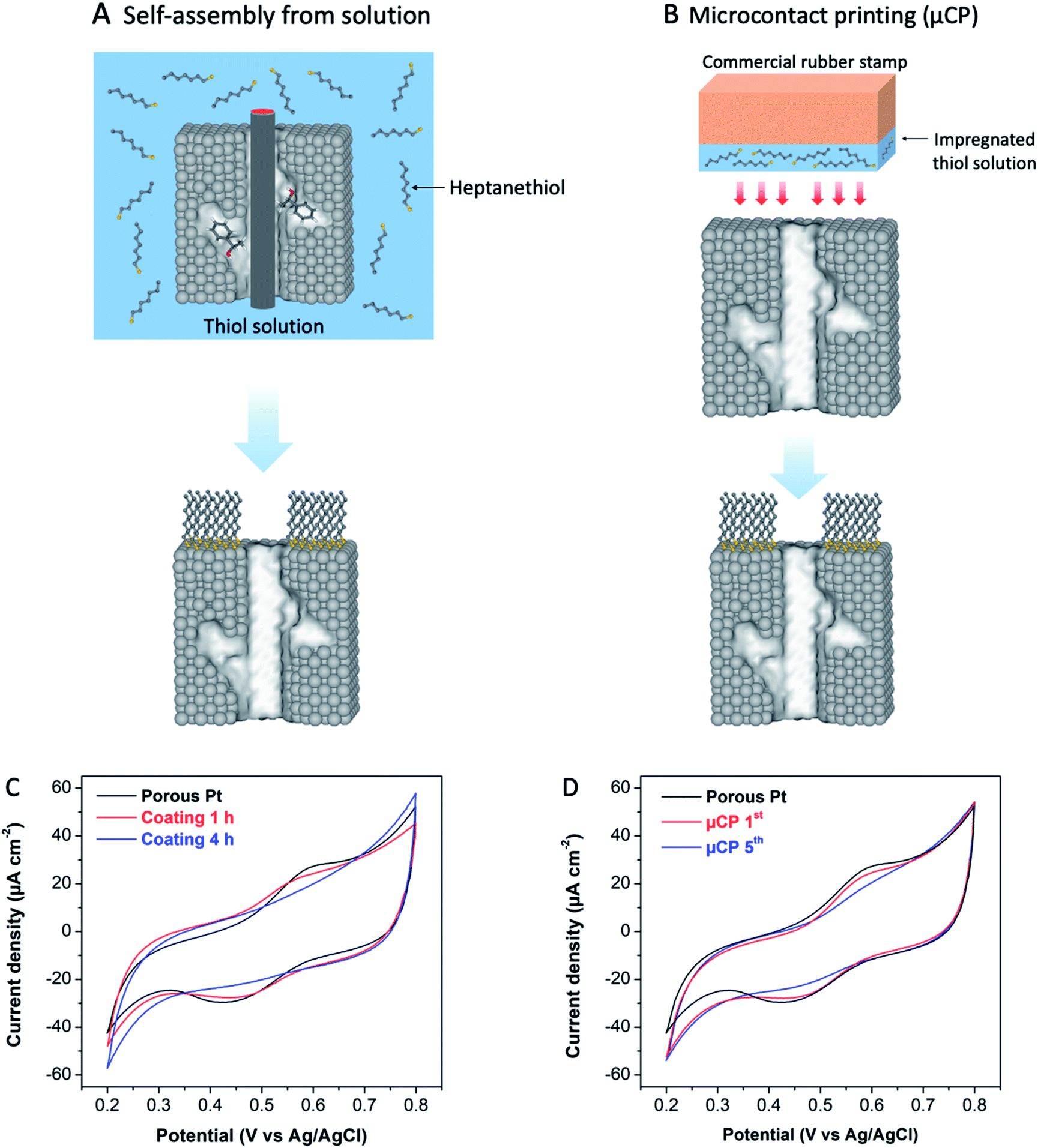 | ||
| Fig. 2 Graphical illustration of the two thiol-coating strategies and the resulting surface blocking properties on mesoporous Pt electrodes; (A) and (B) thiol coating via self-assembly from solution and microcontact printing (μCP), respectively; (C) and (D) cyclic voltammograms of the solution-coated and μCP-coated mesoporous Pt electrodes in 10 mM K4Fe(CN)6/1 M HNO3 at a scan rate of 100 mV s−1 (black: uncoated electrode; blue: electrode with the final coating). | ||
To investigate the accessibility of the thiol-protected Pt surfaces for electroactive molecules, the behaviour of ferro/ferricyanide in HNO3 solution was studied by cyclic voltammetry. As can be seen in Fig. 2C, the characteristic peaks corresponding to the oxidation/reduction of the ferro/ferricyanide redox couple ([Fe(CN)6]4−/[Fe(CN)6]3−) appear at 0.60 and 0.45 V, respectively, for a bare porous Pt electrode (black). For the surface, modified with a thiol coating by following the solution approach depicted in Fig. 2B, the redox peaks completely disappear after 4 h of thiol adsorption, indicating a total blocking, due to heptanethiol adsorption on mesoporous Pt. In the case of μCP, when the number of printing cycles is increased from one (μCP 1st) to five (μCP 5th), the outermost surface of porous Pt is again completely blocked (Fig. 2D). It is noteworthy that the faradaic peak currents of a solution based redox couple appear to be in general rather small for porous electrodes due to the high active internal surface area, leading to a dominating very large capacitive current (Fig. 2C and D). The observed total blocking is therefore a direct consequence of the adsorbed thiol molecules, which hinder the electron transfer between the redox couple and the outermost part of the Pt layer.24,27
Enantioselective synthesis
The beneficial effects of the thiol coated surface can be illustrated when using the chiral-encoded mesoporous Pt electrodes as working electrodes for the enantioselective synthesis of 1-phenylethanol enantiomers via the electroreduction of a prochiral molecule, acetophenone, by an overall two-electron-two-proton transfer (Fig. S2†). Conventional steady state electrosynthesis with a global synthesis time of 13 h is carried out under conditions that were previously optimized to ensure good product selectivity (pH 4.0).19To confirm the formation of 1-phenylethanol enantiomers, the resulting mixture was extracted and subsequently analyzed by HPLC, equipped with a chiral column, allowing to separate the starting compound acetophenone, (S)-PE and (R)-PE, with retention times of 10.50, 13.00 and 13.75 min, respectively (Fig. S3†). As expected, in the case of bare (S)-PE imprinted mesoporous Pt (0.150 weight ratio of PE/PtCl62− in the plating mixture), the resulting % enantiomeric excess (%ee) of (S)-PE is very modest (29% ± 3.2) (Fig. 3A and C (black)). In strong contrast to this, after functionalization of the Pt surface with thiol molecules by self-assembly from solution, an extremely high %ee of up to 93% is obtained (Fig. S4†). The %ee decreases from 93 to 89, 84 and 79% when increasing the time of exposure to the thiol solution from 30 min to 1, 2 and 4 h, respectively. This relates to the fact that the non-ionic surfactant, which is initially still present inside the Pt mesopores, can gradually dissolve during the thiol coating process. As a consequence, thiol molecules not only adsorb at the outermost electrode surface, but also inside the mesopores, thus blocking also the enantioselective part of the electrode.
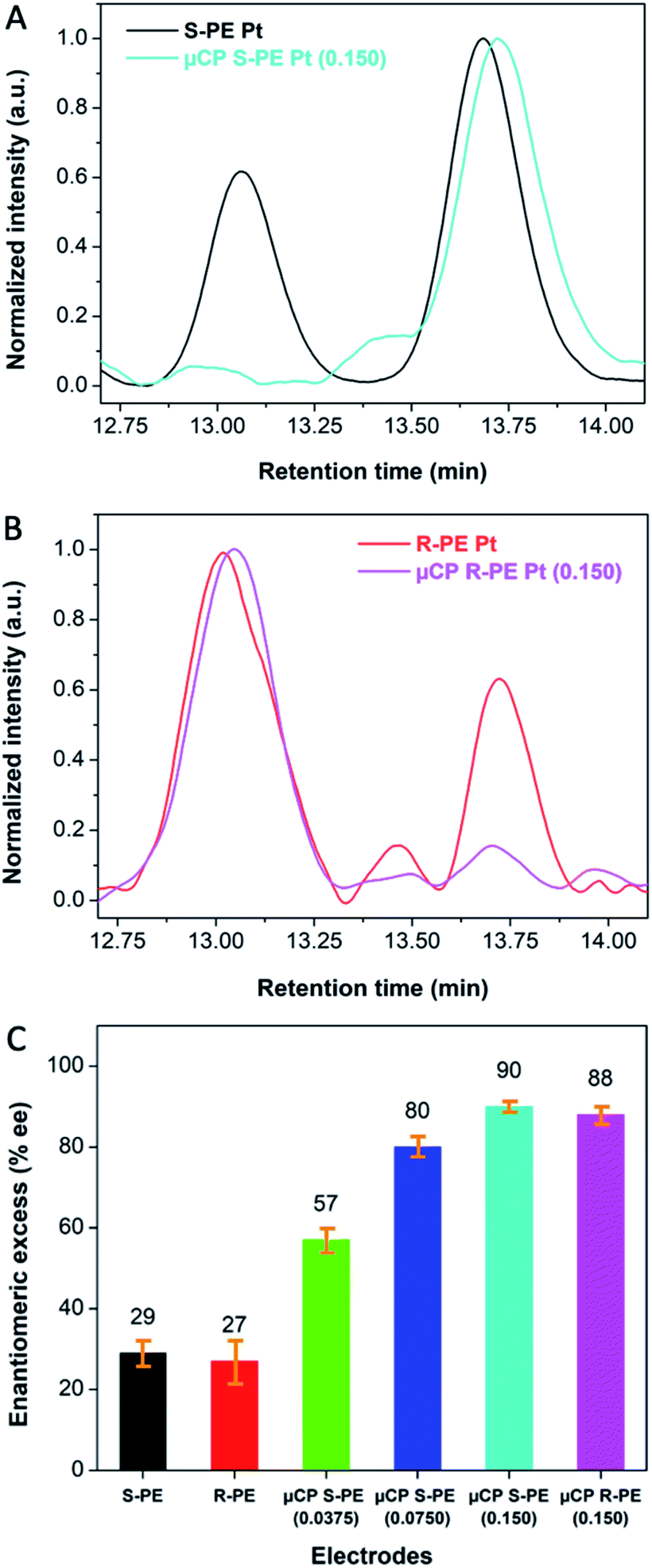 | ||
| Fig. 3 Characterisation of enantioselectivity; (A) and (B) HPLC chromatograms of the product solutions obtained by electrosynthesis performed with (S)-PE and (R)-PE imprinted mesoporous Pt electrodes, respectively, that are either non-coated or coated with thiols by using μCP; (C) summary of enantiomeric excess (%ee), obtained after asymmetric electrosynthesis carried out with bare (S)-PE (black) and (R)-PE (red) imprinted mesoporous Pt electrodes (0.150 weight ratio PE/PtCl62−); with (S)-PE imprinted mesoporous Pt electrodes functionalized with thiol ligands by μCP, using different (S)-PE/PtCl62− weight ratios in the plating mixture, 0.0375 (green), 0.0750 (dark blue) and 0.150 (light blue); with a (R)-PE imprinted mesoporous Pt electrode obtained from a plating solution with 0.150 weight ratio (R)-PE/PtCl62− and modified with thiol molecules by μCP (pink). | ||
In order to avoid this drawback, we additionally examined the surface modification based on the μCP strategy. In this case, the thiol molecules should not be able to passivate the inside of the mesopores, because they can only adsorb at the outermost surface during contact with the stamp. Consequently, the very high average % ee value of 90% ± 1.3 for (S)-PE does not change, even if the electrode is modified several times with the stamp (Fig. 3A and C (light blue) and Fig. S5†). This can be ascribed to a thiol coverage limited to a confined space even when repeating the μCP several times.25
In order to further confirm that thiol molecules can also adsorb inside the mesopores when using the classic solution coating, whereas adsorption only occurs at the outermost surface with the μCP approach, we recorded CV curves in 0.5 M H2SO4 for three different cases (Fig. S6†): (i) non-coated porous Pt; (ii) thiol-coating via the solution coating approach for 4 h; (iii) thiol-coating via microcontact printing for five-cycles. A decrease in the faradaic current associated with the H-adsorption/desorption region (−0.2 to 0.0 V) of the thiol-coated electrodes was observed with respect to the bare mesoporous Pt. However, the active surface area of electrodes coated by the solution approach is much lower than the one obtained via microcontact printing. It is therefore reasonable to assume that the μCP approach is more controllable and very efficient for generating a thiol layer exclusively at the outermost surface of the mesoporous electrode. Therefore, this strategy was selected to perform the further experiments.
After having obtained the very high %ee values for (S)-PE imprinted electrodes, non-imprinted mesoporous Pt was used in a control experiment. As expected, no selectivity for neither (S)- nor (R)-PE could be observed (Fig. S7† (orange)). Moreover, the opposite chiral configuration, (R)-PE imprinted mesoporous Pt with the same weight ratio of PE/PtCl62− (0.150), was used as a working electrode. Fig. 3B shows the chromatograms of product solutions obtained with bare (red) and μCP functionalized electrodes (pink). Respective %ee values of 27 ± 5.5% and 88 ± 2.1% were obtained (Fig. 3C), reflecting a symmetric situation with respect to (S)-PE encoded Pt. The drastic improvement of %ee after thiol functionalization is also confirmed for different (S)-PE/PtCl62− weight ratios as illustrated in Fig. S7† and Fig. 3C. It was found that %ee values of (S)-PE are significantly enhanced from 57 ± 2.8% to 80 ± 2.5% and 90 ± 1.3% when increasing the weight ratio from 0.0375 (green) to 0.0750 (dark blue) and 0.150 (light blue), respectively, due to the generation of a higher fraction of chiral recognition sites in the mesopores.
In order to demonstrate the general validity of the approach, not only acetophenone, but also an acetophenone derivative, 4-bromoacetophenone (4-Br-AP), has been tested as a prochiral precursor. By fine-tuning the conditions of the asymmetric electrosynthesis via the electroreduction of 4-Br-AP to 1-(4-bromophenyl) ethanol (4-Br-PE) (Fig. S8†), using thiol-coated mesoporous Pt electrodes obtained with the μCP approach, again a high enantiomeric excess of around 90% (±5.0%) could be successfully achieved. In addition, the perfectly symmetric situation is observed when employing the opposite enantiomer for the imprinting (Fig. S9†).
Apart from the enantiomeric excess, the overall yield of an enantioselective electrosynthesis is also an important aspect. In electrochemical synthesis the global yield obviously depends on how long the potential is applied, which current is flowing and which initial concentration of prochiral precursor is used. It also is a direct function of the size of the electrodes with respect to the global volume of electrolyte in the cell. All these parameters have not been optimized in the present proof-of-concept study, as the main focus was a maximization of the enantioselectivity. Therefore, the total conversion when using the present experimental set-up is rather low, however high selectivity can be obtained with the thiol functionalization as shown in Fig. S10† and summarized in Table S1.† One reason might be that in the present set-up there is no separation between the anodic and the cathodic compartment, meaning that the reduced product can be, at least partially, oxidized back at the anode, leading to a kind of electrochemical short-circuit, consuming current without a net formation of product. The consequence is a decrease in global conversion, but further developments, for example in terms of cell design, such as the introduction of a membrane separating the anodic and cathodic compartment, might allow to considerably increase these values.
As mentioned above, the %ee of non-coated electrodes is rather low, even with pulsed electrosynthesis when using a longer pulse duration (60 s). However, the %ee can be greatly improved when employing the thiol-coated mesoporous Pt (>90%), even in a potentiostatic experiment. Even though the thiol coating decreases the global active surface area of the electrode, causing a lower overall conversion of acetophenone, the production rate of a specific chiral compound is not decreasing, and can be even further improved by almost 30% under certain conditions (Table S1,† entry 1 and 2.1). When comparing solution coated electrodes with those modified by μCP for similar %ee and conversion values (around 90% and 0.7%, respectively), the μCP approach leads to a significantly higher efficiency in terms of production rate (Table S1,† entry 2.2, 3.1 and 3.2). Furthermore, when in both cases the external surface of the mesoporous Pt is completely blocked by the thiol layer, as confirmed by the absence of faradaic current of the Fe2+/Fe3+ redox couple (Fig. 2C and D), the rate of selective enantiomer production is around 20% higher when using μCP (Table S1,† entry 2.3 and 3.3). This again confirms that the controlled formation of a thiol layer, mainly on the outermost part of the mesoporous Pt electrode, can enhance the production efficiency of chiral compounds (Fig. S6†). It should be noted that the production rate of chiral molecules using pulse electrosynthesis is much lower due to the very long synthesis time (Table S1,† entry 4). Thus, compared with our previous studies using pulse synthesis,14b,19,28 the thiol protection strategy simultaneously leads to a high %ee and an improved production rate.
In order to further investigate the stability and reusability of the thiol-modified catalyst surface, an electrode was rinsed in water overnight after the first catalytic run, and then used again in a new electrocatalytic cycle. A decrease in enantioselectivity was observed, due to a partial desorption of the protecting SAM layer. However, the thiol layer can be completely regenerated by repeating the μCP modification. Consequently, a high %ee of over 90% can be maintained in subsequent runs (Fig. S11†).
Another point, that merits further consideration, is the mechanism of such an enantioselective reduction.29 The hydrogenation of prochiral compounds, like asymmetric ketones, usually produces racemic mixtures, because traditional metal catalysts are achiral. However, introducing chiral information on the metal surface can eventually generate an enantiomeric excess.29a As shown in Fig. S1C,† the presence of a chiral template can generate a chiral environment along the mesopore wall when using a lyotropic liquid crystal to control the mesoporous network inside the metal. The hydrophilic part of the chiral template molecule interacts directly with the non-ionic surfactant column, and therefore the Pt matrix grows around the aromatic unit, resulting in the formation of a specific chiral cavity corresponding to the shape and size of the chiral template, as described in some earlier work.14a When the chiral metal matrix is then employed as an electrode, the aromatic moiety of the prochiral reactant preferentially adsorbs in the deeper part of the cavity, exposing the carbonyl moiety to an asymmetric environment at the entrance of the chiral cavity,29d decorating the wall of the mesopore channel. In the case of the electroreduction of acetophenone, the hydrogenation of the carbonyl group to alcohol requires the addition of two electrons and two protons, forming the favourable enantiomer of the chiral products, either (R)- or (S)-PE, according to the preferential configuration of the chiral active site (Fig. S2†). As illustrated by the mechanism proposed in Fig. S12,† after the specific adsorption of the prochiral molecule at the asymmetric sites of the electrode, the polarized carbonyl can accept an electron and a proton producing a phenolic radical intermediate.29c At this stage, dimerization of the phenolic radical species might occur, eventually producing by-products. Thus, the pH of the electrolyte plays a crucial role after this electron transfer step.29b If the process is carried out in the presence of a sufficient concentration of protons, such as in the acidic solution of our study (pH 4), the reaction is shifted to the preferential formation of chiral products during the acceptance of a supplementary electron and proton. However, for higher pH values, where protons are lacking, a significant amount of side product is formed, as we found out during optimization experiments.19
Under such optimized conditions, the functionalization of the Pt electrodes with a thiol layer seems to almost completely suppress undesired reactions, also at the outermost surface of the Pt electrode. These findings demonstrate the benefits of the SAM strategy, allowing to perform experiments under steady-state conditions, instead of using the time-consuming pulsed electrosynthesis method,14b and nevertheless reaching extremely high %ee values. With this novel approach, both, the global reaction time and stereoselectivity are for the first time in the same range as the values reported for heterogeneous asymmetric hydrogenation of ketones on Pt catalysts,30 and it allows benefiting from the well-known advantages of electrochemistry in terms of tuneability of the driving force and a perfectly controllable design of the catalyst layer.
Conclusions
In this contribution, functionalization of chiral imprinted mesoporous Pt electrodes with an organosulfur ligand, either by self-assembly from solution or via a microcontact printing approach, leads to a greatly enhanced enantioselectivity when using such designer electrodes for the synthesis of chiral compounds. The results reveal that controlled tailoring of these materials allows reaching very high product selectivity, combined with reduced reaction times. Modification of the Pt electrodes with an outer heptanethiol layer boosts the excess of the desired enantiomer to up to 90%, which is a three-fold increase compared to a bare mesoporous electrode. The global reaction time is efficiently decreased from one week down to several hours, as it is no longer necessary to use the pulsed electrosynthesis concept reported previously, but constant potential electrosynthesis can be employed. These encouraging results open up interesting perspectives in the field of enantioselective synthesis, as the developed approach has a potential to be used for practical applications after careful optimization of all the experimental parameters, in order to ensure also a high global yield.Experimental
Chemicals
Hexachloroplatinic acid hydrate (H2PtCl6·xH2O), polyethylene glycol hexadecyl ether (Brij®C10), (R)- and (S)-phenylethanol, acetophenone, (S)- and (R)-1-(4-bromophenyl) ethanol, isopropyl alcohol, ethyl alcohol, heptane, ammonium chloride (NH4Cl), potassium hexacyanoferrate (II) trihydrate (K4Fe(CN)6·3H2O), nitric acid (HNO3) and heptanethiol were obtained from the Merck Sigma-Aldrich. 4-Bromoacetophenone, and sulfuric acid (H2SO4) were purchased from the TCI and Alfa, respectively. MilliQ water (18.2 MΩ cm) was used for all experiments. All chemicals were directly employed without further purification.Synthesis of chiral-imprinted mesoporous Pt electrodes
The experiments were carried out with a potentiostat (Metrohm μAutolab Type III). Typically, Ag/AgCl (sat. KCl), Pt mesh and a cleaned gold-coated glass slide were used as the reference, counter and working electrodes, respectively, in a three-electrode system. Firstly, a gold-coated glass slide was cleaned by sonication in isopropyl alcohol, rinsed with MilliQ water several times and dried under N2 gas flow to remove the impurities. Then, a lyotropic liquid crystal (LLC) gel for electrodeposition was prepared by mixing 39 wt% of nonionic surfactant (Brij®C10), 28 wt% of chloroplatinic acid (H2PtCl6·xH2O), 28 wt% of MilliQ water and the chiral templates, either 1-phenylethanol (PE) with a weight ratio of PE/PtCl62− varying in the range from 0.0375 to 0.150 or 1-(4-bromophenyl) ethanol (4-Br-PE) with a weight ratio of 0.150 4-Br-PE/PtCl62−. In order to obtain a homogeneous precursor gel, the components were continuously mixed for 10 min, then heated at 40 °C in the oven for 20 min, and this cycle was repeated three times. The electrodeposition was performed on the cleaned electrodes (working area 0.25 cm−2) by chronoamperometry with a charge density of 4 C cm−2 at −0.05 V (Ag/AgCl). Subsequently, the electrodes were rinsed and soaked in MilliQ water overnight to remove the templates. For the non-imprinted mesoporous Pt electrodes, the same process was followed, but without adding any chiral templates to the LLC precursor gel.Surface functionalization via self-assembly from solution
After electrodeposition, the excess template present at the external surface of the Pt electrodes was removed by rinsing with MilliQ water for approximately 15 min. Subsequently, the electrodes, which still contain the template molecules inside the mesopores, were immediately immersed into a solution of 10 mM heptanethiol in ethyl alcohol for various times (30 min, 1, 2 and 4 h) at room temperature. Finally, the as-prepared electrodes were rinsed with ethanol and stored in MilliQ water overnight for removal of the templates located inside mesopores. The electrodes were kept in the dark before using them for electrosynthesis.Surface functionalization via microcontact printing (μCP)
A commercial rubber stamp was used for μCP. The stamp was impregnated with a 50 mM heptanethiol/ethanol solution with a loading density of 1 μl cm−2. Ethanol present on the stamp was evaporated in ambient conditions for 60 s. Then, the dried stamp was immediately pressed on the bare chiral-encoded mesoporous Pt for 10 s. Total coating times were varied by repeating the above-mentioned steps for up to five cycles. The thiol-coated Pt electrodes were dried in the dark before using them in electrosynthesis.Characterization
The topology and morphology of the Pt electrodes were characterized by scanning electron microscopy (SEM) with a Hitachi TM-1000 tabletop microscope and transmission electron microscopy (TEM) with a JEOL JEM-2100 microscope at 200 kV. For TEM measurements, ultra-thin films of mesoporous Pt were prepared by electrodeposition with a charge density of 0.4 C cm−2. The films were removed from the Au-coated glass slide by gently scraping. Then, they were floated in ethyl alcohol and transferred onto TEM grids. Cyclic voltammetry was employed to measure the active surface area of the Pt electrodes in 0.5 M H2SO4 at 100 mV s−1, and to study the properties of the adsorbed heptanethiol layers in 10 mM K4Fe(CN)6 dissolved in 1 M HNO3 at a scan rate of 100 mV s−1.Enantioselective synthesis of 1-phenylethanol (PE)
Selective chiral electrosynthesis of 1-phenylethanol (PE) was investigated by continuous potentiostatic reduction of acetophenone at −0.40 V for 13 h in a stirred solution (250 rpm) of 5 mM acetophenone in 1 M NH4Cl (7 mL) as supporting electrolyte with a pH adjusted to 4.0 (Fig. S13†). The synthesized products were extracted with heptane and analyzed by a high-performance liquid chromatography (HPLC) equipment composed of a JASCO LC-Net II/ADC, a chiral column (CHIRALPAK IB N-5, 250 × 4.6 mm inner diameter) and a photodiode array (PDA) detector at 210 nm, using a mixture of 8/92 (v/v) isopropyl alcohol/heptane as mobile phase at a flow rate of 0.5 mL min−1.Data availability
All experimental supporting data associated with this article are available in the main manuscript and in the ESI.†Author contributions
S. B.: performed all experiments, data collection, and writing of the manuscript. V. L.: HPLC investigation. C. W. and A. K.: supervision, funding acquisition and writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Somlak Ittisanronnachai, Dr Sunpet Assavapanumat, Dr Thittaya Yutthalekha, Dr Duangkamon Suttipat, Dr Jakkapan Kumsampao and Thassanant Atithep for advice with respect to characterization experiments. S. B. is grateful to Campus France, the French Embassy in Thailand and the Vidyasirimedhi Institute of Science and Technology (VISTEC) for a PhD cotutelle scholarship, and the National Research Council of Thailand (NRCT) for the graduate research fund. The project has been funded by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 741251, ERC Advanced Grant ELECTRA) and has also been supported by the bilateral CNRS IRP project ChiraChem. C. W. thanks the Mid-Career Research Grant 2020 from NRTC (NRCT5-RSA63025-03) and support from the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (B05F640207).References
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS PubMed.
- H. Blaser, F. Spindler and M. Studer, Appl. Catal., A, 2001, 221, 119–143 CrossRef CAS.
- (a) M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed; (b) R. Xie, L. Y. Chu and J. G. Deng, Chem. Soc. Rev., 2008, 37, 1243–1263 RSC; (c) Y. Wang and A. M. Chen, Org. Process Res. Dev., 2008, 12, 282–290 CrossRef.
- (a) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC; (b) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC; (c) F. Zhang and H. Li, Chem. Sci., 2014, 5, 3695–3707 RSC.
- M. Ghosh, V. S. Shinde and M. Rueping, Beilstein J. Org. Chem., 2019, 15, 2710–2746 CrossRef CAS PubMed.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- A. J. Gellman, ACS Nano, 2010, 4, 5–10 CrossRef CAS PubMed.
- H. Behar-Levy, O. Neumann, R. Naaman and D. Avnir, Adv. Mater., 2007, 19, 1207–1211 CrossRef CAS.
- (a) E. W. Bohannan, H. M. Kothari, I. M. Nicic and J. A. Switzer, J. Am. Chem. Soc., 2004, 126, 488–489 CrossRef CAS PubMed; (b) J. A. Switzer, H. M. Kothari, P. Poizot, S. Nakanishi and E. W. Bohannan, Nature, 2003, 425, 490–493 CrossRef CAS PubMed.
- L. D. Pachón, I. Yosef, T. Markus, R. Naaman, D. Avnir and G. Rothenberg, Nat. Chem., 2009, 1, 160–164 CrossRef PubMed.
- J. C. Moutet, E. Saintl-Aman, F. Tran-Van, P. Angibeaud and J. P. Utille, Adv. Mater., 1992, 4, 511–513 CrossRef CAS.
- (a) C. Wattanakit and A. Kuhn, Chem.–Eur. J., 2020, 26, 2993–3003 CrossRef CAS PubMed; (b) C. Wattanakit, Curr. Opin. Electrochem., 2018, 7, 54–60 CrossRef CAS.
- (a) C. Wattanakit, Y. B. Saint Côme, V. Lapeyre, P. A. Bopp, M. Heim, S. Yadnum, S. Nokbin, C. Warakulwit, J. Limtrakul and A. Kuhn, Nat. Commun., 2014, 5, 3325 CrossRef PubMed; (b) T. Yutthalekha, C. Warakulwit, J. Limtrakul and A. Kuhn, Electroanalysis, 2015, 27, 2209–2213 CrossRef CAS.
- (a) T. Yutthalekha, C. Wattanakit, V. Lapeyre, S. Nokbin, C. Warakulwit, J. Limtrakul and A. Kuhn, Nat. Commun., 2016, 7, 12678 CrossRef CAS PubMed; (b) C. Wattanakit, T. Yutthalekha, S. Asssavapanumat, V. Lapeyre and A. Kuhn, Nat. Commun., 2017, 8, 2087 CrossRef PubMed.
- (a) G. S. Attard, P. N. Bartlett, N. R. Coleman, J. M. Elliott, J. R. Owen and J. H. Wang, Science, 1997, 278, 838–840 CrossRef CAS; (b) P. A. Nelson, J. M. Elliott, G. S. Attard and J. R. Owen, Chem. Mater., 2002, 14, 524–529 CrossRef CAS; (c) S. A. G. Evans, J. M. Elliott, L. M. Andrews, P. N. Bartlett, P. J. Doyle and G. Denuault, Anal. Chem., 2002, 74, 1322–1326 CrossRef CAS PubMed; (d) T. Imokawa, K. J. Williams and G. Denuault, Anal. Chem., 2006, 78, 265–271 CrossRef CAS PubMed.
- S. Assavapanumat, T. Yutthalekha, P. Garrigue, B. Goudeau, V. Lapeyre, A. Perro, N. Sojic, C. Wattanakit and A. Kuhn, Angew. Chem., Int. Ed., 2019, 58, 3471–3475 CrossRef CAS PubMed.
- S. Assavapanumat, B. Gupta, G. Salinas, B. Goudeau, C. Wattanakit and A. Kuhn, Chem. Commun., 2019, 55, 10956–10959 RSC.
- S. Assavapanumat, M. Ketkaew, A. Kuhn and C. Wattanakit, J. Am. Chem. Soc., 2019, 141, 18870–18876 CrossRef CAS PubMed.
- S. Butcha, S. Assavapanumat, S. Ittisanronnachai, V. Lapeyre, C. Wattanakit and A. Kuhn, Nat. Commun., 2021, 12, 1314 CrossRef CAS PubMed.
- J. M. Doña Rodríguez, J. A. Herrera Melián and J. Pérez Peña, J. Chem. Educ., 2000, 77, 1195 CrossRef.
- (a) S. T. Marshall, M. O'Brien, B. Oetter, A. Corpuz, R. M. Richards, D. K. Schwartz and J. W. Medlin, Nat. Mater., 2010, 9, 853–858 CrossRef CAS PubMed; (b) L. Jin, B. Liu, S. S. Duay and J. He, Catalysts, 2017, 7, 44 CrossRef; (c) S. E. Eklund and D. E. Cliffel, Langmuir, 2004, 20, 6012–6018 CrossRef CAS PubMed.
- C. Vericat, M. Vela, G. Benitez, P. Carro and R. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC.
- M. A. Floridia Addato, A. Rubert, G. Benítez, E. Zelaya, G. Cabello, A. Cuesta, J. E. Thomas, A. Visintín, R. C. Salvarezza and M. H. Fonticelli, J. Phys. Chem. C, 2013, 117, 7589–7597 CrossRef CAS.
- M. A. Floridia Addato, A. A. Rubert, G. A. Benítez, M. H. Fonticelli, J. Carrasco, P. Carro and R. C. Salvarezza, J. Phys. Chem. C, 2011, 115, 17788–17798 CrossRef CAS.
- (a) J. L. Wilbur, A. Kumar, E. Kim and G. M. Whitesides, Adv. Mater., 1994, 6, 600–604 CrossRef CAS; (b) A. Perl, D. N. Reinhoudt and J. Huskens, Adv. Mater., 2009, 21, 2257–2268 CrossRef CAS; (c) A. Kuhn and D. Martel, ChemPhysChem, 2001, 2, 688–691 CrossRef CAS PubMed.
- D. Losic, J. Shapter and J. Gooding, Electrochem. Commun., 2001, 3, 722–726 CrossRef CAS.
- H. Finklea, K. Yoon, E. Chamberlain, J. Allen and R. Haddox, J. Phys. Chem. B, 2001, 105, 3088–3092 CrossRef CAS.
- S. Assavapanumat, S. Butcha, S. Ittisanronnachai, A. Kuhn and C. Wattanakit, Chem.–Asian J., 2021, 16, 3345–3353 CrossRef CAS PubMed.
- (a) F. Zaera, Acc. Chem. Res., 2009, 42, 1152–1160 CrossRef CAS PubMed; (b) Y. Kodama, M. Imoto, N. Ohta, A. Kitani and S. Ito, J. Electroanal. Chem., 2001, 507, 103–109 CrossRef CAS; (c) D. C. Cantu, A. B. Padmaperuma, M. T. Nguyen, S. A. Akhade, Y. Yoon, Y. G. Wang, M. S. Lee, V. A. Glezakou, R. Rousseau and M. A. Lilga, ACS Catal., 2018, 8, 7645–7658 CrossRef CAS; (d) C. J. Bondue and M. T. M. Koper, J. Am. Chem. Soc., 2019, 141, 12071–12078 CrossRef CAS PubMed; (e) I. Matanović, Nat. Catal., 2019, 2, 186–187 CrossRef.
- F. Meemken and A. Baiker, Chem. Rev., 2017, 117, 11522–11569 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional results of enantioselective electrosynthesis. See DOI: 10.1039/d2sc00056c |
| This journal is © The Royal Society of Chemistry 2022 |