
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of a promising oxychalcogenide infrared nonlinear optical crystal†
Yansong
Cheng
,
Hongping
Wu
 ,
Hongwei
Yu
*,
Zhanggui
Hu
,
Jiyang
Wang
and
Yicheng
Wu
,
Hongwei
Yu
*,
Zhanggui
Hu
,
Jiyang
Wang
and
Yicheng
Wu
Tianjin Key Laboratory of Functional Crystal Materials, Institute of Functional Crystal, Tianjin University of Technology, Tianjin 300384, China. E-mail: hwyu15@gmail.com
First published on 12th April 2022
Abstract
Oxychalcogenides with the performance-advantages of both chalcogenides and oxides are emerging materials class for infrared (IR) nonlinear optical (NLO) crystals that can expand the wavelength of solid-state lasers to IR regions and are of importance in industrial and civil applications. But rationally designing a high-performance oxychalcogenide NLO crystal remains a great challenge. Herein, we chose the melilite-type Sr2ZnSi2O7 as the structure template. Through part isovalent substitution of S2− for O2− anions, the first hetero-anionic thiostannate Sr2ZnSn2OS6 with wide IR transmission has been synthesized. More importantly, compared to the maternal oxide, Sr2ZnSi2O7, the second harmonic generation (SHG) response of Sr2ZnSn2OS6 is enhanced by two orders of magnitude. In addition, Sr2ZnSn2OS6 can exhibit a large band-gap and high laser damage threshold. These advantages make Sr2ZnSn2OS6 a promising IR NLO crystal. Our research will provide insights into the rational design of new IR NLO crystals.
Introduction
Mid-infrared (MIR) sources covering 3 to 5 μm and 8 to 12 μm are of great interest in industrial and civil applications such as molecular spectroscopy, remote sensing, free-space communication, and environmental monitoring.1–5 However, it is difficult for solid-state lasers to directly radiate MIR coherent light. Nevertheless, relying on frequency conversion techniques of nonlinear optical (NLO) crystals, such as optical parametric oscillators (OPOs) and optical parametric amplifiers (OPAs), is an effective way for solid-state lasers to generate MIR radiation. In these processes, the performances of NLO crystals determine the conversion efficiency of IR coherent light. So, they play a strategically important role in the development of IR lasers. In the past three decades, although a series of high-performance NLO crystals have been discovered and widely commercialized in the ultraviolet and visible regions, including β-BaB2O4 (BBO),6 LiB3O5 (LBO),7 CsLiB6O10 (CLBO),8 KH2PO4 (KDP)9 and KTiOPO4 (KTP),10 NLO crystals available in the infrared (IR) region are still seldom, and they are mainly some chalcopyrite-type chalcogenides, such as AgGaQ2 (Q = S, Se)11,12 and ZnGeP2.13 Although these crystals feature very large second harmonic generation (SHG) coefficients, the low laser damage thresholds (LDTs) of AgGaQ2 (Q = S, Se) and harmful two-photon absorption (TPA) of ZnGeP2 limit their wide applications,14 particularly under high-power conditions. So, exploring new IR NLO crystals with high LDTs and large band gaps is still a current research interest.15–18For the design of new IR NLO crystals, metal chalcogenides, oxides and halides are the three most important materials. Regarding chalcogenides, they generally exhibit large SHG responses and wide IR transparency,19,20 while low LDTs and undesired TPA caused by their small band-gaps as well as the difficulty in crystal growth are the main problems restricting their wide applications, just as observed in typical AgGaQ2 (Q = S, Se) and ZnGeP2.21 Compared to chalcogenides, metal oxides can exhibit larger band-gaps and thus higher LDTs as well as easier crystal growth.22 But the poor IR transparency and relatively weak SHG responses make their application limited within 5 μm.23 Therefore, it is clear that chalcogenides can exhibit larger SHG response and better IR transmission, while metal oxides have advantages in band-gaps and LDTs. Therefore, combining two types of anions may be an expectant strategy for designing high-performance IR NLO crystals with balanced properties. Up to now, although some oxychalcogenide IR NLO crystals, e.g. SrZn2OS2 (Eg = 3.86 eV, 0.06× AgGaS2),24 Sr5Ga8O3S14 (Eg = 3.90 eV, 0.8× AgGaS2),25 SrGeOSe2 (Eg = 3.16 eV, 1.3× AgGaS2),26 BaGeOSe2 (Eg = 3.20 eV, 1.1× AgGaS2),27 and Sr6Cd2Sb6O7S10 (Eg = 1.89 eV, 4.0× AgGaS2),28 have been reported, these compounds are still less investigated than their maternal oxides or chalcogenides. In particular, how to optimize the structure of oxychalcogenides to use the advantages of oxides and chalcogenides to the maximum through a rational structure design is still indistinct.
In this research, we choose the melilite-type Sr2ZnSi2O7 as a structural template to design a new oxychalcogenide through rational chemical substitution. Sr2ZnSi2O729 crystallizes in the noncentrosymmetric (NCS) space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21m. Its structure features a pentagon-like layer, which endows its structure with better flexibility and designability. But remarkably, the SHG response of Sr2ZnSi2O7 is only 35.0× α-SiO2,30 almost two orders of magnitude smaller than that of AgGaS2. And owing to the absorption of the metal–oxygen bonds, it also has poor IR transmission. In our study, we used Sn atoms to substitute Si in melilite-type Sr2ZnSi2O7, first, because Sn atoms are heavier and more polarizable than Si atoms, which is helpful for the material to exhibit a wider IR transmission and larger SHG response.31 Furthermore, with the help of the unique pentagonal layer configurations, we expect to use heavier and more polarizable S atoms to selectively substitute the terminal O atoms in the pentagonal layers as well as the bridging O atoms in relatively light Zn–O–Sn bonds, whereas retaining the O atoms in Sn–O–Sn bonds, which have the heaviest equivalent mass for metal–oxygen bonds in the structure and have the smallest effect on IR transmission of the materials.32,33 Meanwhile, the hetero-anionic [SnOS3] tetrahedra may exhibit large polarization, which will favor the material to generate a large SHG response.25 By doing these, we have successfully synthesized the first hetero-anionic thiostannate, Sr2ZnSn2OS6. It exhibits not only a wide IR transmission, but also a large SHG response, ∼0.7× AgGaS2, which is two orders of magnitude larger than that of maternal Sr2ZnSi2O7. These results indicate that Sr2ZnSn2OS6 is a promising IR NLO crystal. Herein, we will report its synthesis, structure, and NLO properties.
21m. Its structure features a pentagon-like layer, which endows its structure with better flexibility and designability. But remarkably, the SHG response of Sr2ZnSi2O7 is only 35.0× α-SiO2,30 almost two orders of magnitude smaller than that of AgGaS2. And owing to the absorption of the metal–oxygen bonds, it also has poor IR transmission. In our study, we used Sn atoms to substitute Si in melilite-type Sr2ZnSi2O7, first, because Sn atoms are heavier and more polarizable than Si atoms, which is helpful for the material to exhibit a wider IR transmission and larger SHG response.31 Furthermore, with the help of the unique pentagonal layer configurations, we expect to use heavier and more polarizable S atoms to selectively substitute the terminal O atoms in the pentagonal layers as well as the bridging O atoms in relatively light Zn–O–Sn bonds, whereas retaining the O atoms in Sn–O–Sn bonds, which have the heaviest equivalent mass for metal–oxygen bonds in the structure and have the smallest effect on IR transmission of the materials.32,33 Meanwhile, the hetero-anionic [SnOS3] tetrahedra may exhibit large polarization, which will favor the material to generate a large SHG response.25 By doing these, we have successfully synthesized the first hetero-anionic thiostannate, Sr2ZnSn2OS6. It exhibits not only a wide IR transmission, but also a large SHG response, ∼0.7× AgGaS2, which is two orders of magnitude larger than that of maternal Sr2ZnSi2O7. These results indicate that Sr2ZnSn2OS6 is a promising IR NLO crystal. Herein, we will report its synthesis, structure, and NLO properties.
Results and discussion
The polycrystalline sample of Sr2ZnSn2OS6 was obtained by a solid-state reaction in a sealed silica tube (for more details see the ESI†). The purity of the sample was confirmed by powder X-ray diffraction (PXRD). In order to investigate the thermal behavior of Sr2ZnSn2OS6, we carried out a series solid state reactions with different calcination temperatures and performed their powder XRD from 500 to 800 °C (Fig. S1†). It is clear that Sr2ZnSn2OS6 is thermally stable until 750 °C. After 750 °C, Sr2ZnSn2OS6 decomposes into SrS, SnS and ZnS (Fig. S1†). The millimeter-size crystal of Sr2ZnSn2OS6 was grown by the solution method with SrS as the flux (Fig. S2†), and the specific crystal photograph is shown in Fig. S3.† The structure was determined by single crystal XRD and the crystallographic data are given in Table S1.†Sr2ZnSn2OS6 crystallizes in the NCS tetragonal space group P21m. In the asymmetric unit of Sr2ZnSn2OS6, there are one unique Sr, one Zn, one Sn, one O and two S atom(s). Among them, the Sn atom is coordinated by one O atom and three S atoms to form the SnOS3 tetrahedron with the distance of d(Sn–O) = 2.029(4) Å and Sn–S distances in the range of 2.321(2)–2.366(16) Å, respectively. The Zn atom is coordinated with four S atoms forming the typical ZnS4 tetrahedron with identical Zn–S distance, d(Zn–S) = 2.333(16) Å (Fig. 1a). In the structures, three SnOS3 tetrahedra and two ZnS4 tetrahedra are connected to form an interesting pentagon ring (Fig. 1c). In the pentagon ring, the SnOS3 tetrahedron connects with a ZnS4 tetrahedron by sharing the S atoms, and the two SnOS3 tetrahedra are connected by sharing the O atoms. These pentagons further form two-dimensional (2D) [ZnSn2OS6]∞ layers (Fig. 1b), which are separated by Sr2+ cations to form the structure of Sr2ZnSn2OS6 (Fig. 1d). Regarding the Sr2+ cations, they are located in an eight-coordinated SrOS7 bicapped trigonal prism with the d(Sr–O) = 2.788(5) Å and d(Sr–S) = 3.113(2)–3.150(17) Å, respectively (Table S2†). These bond distances are comparable to those of other reported compounds.21 Bond valence calculations show that the bond valence sums (BVSs) for each atom are 2.12, 2.00, 4.04, and 1.73 for Sr2+, Zn2+, Sn4+, and O2−, respectively. The BVSs of S2− anions are in the range of 2.08–2.14 (Table S3†). All the BVSs are consistent with the expected valences.
 | ||
| Fig. 1 (a) [SnOS3] and [ZnS4] tetrahedra. (b) The 2-dimensional [ZnSn2OS6]4− layers composed of pentagons. (c) The pentagonal ring composed of [SnOS3] and [ZnS4] tetrahedra. (d) The crystal structure of Sr2ZnSn2OS6. (e and f) Structural evolution from Sr2ZnSi2O7 to Sr2ZnSn2OS6. (g) The pentagon consisting of Zn–O–Si and Si–O–Si bonds. (h) The pentagon consisting of Zn–S–Sn and Sn–O–Sn bonds. | ||
Structurally, Sr2ZnSn2OS6 can be seen as a homologue of Sr2ZnSi2O7 (Fig. 1e–h). Compared with Sr2ZnSi2O7, Sr2ZnSn2OS6 exhibits the following structural features: (i) the Si4+ cations in Sr2ZnSi2O7 were substituted by the heavier Sn4+ cations, which are the heaviest cations but have similar coordination to Si4+ in group IV; (ii) all the terminal O atoms in the Zn–O or Sn–O bonds, and all the bridging O atoms in the Zn–O–Sn bonds are substituted by the heavier S atoms, while the O atoms in the Sn–O–Sn bonds are retained (Fig. 1e and f). The above substitution will be favorable for Sr2ZnSn2OS6 to exhibit wide IR transmission. Generally, the IR absorption of materials mainly originates from the vibration of the groups. Thus, in Sr2ZnSn2OS6, the large equivalent mass of SnOS3 and ZnS4 groups will be favorable for widening the IR transmission of materials. In Sr2ZnSi2O7, the Si–O bonds have the lightest equivalent mass, so they have the greatest effect on IR transmission. Using the heavier Sn4+ cations to substitute Si4+ can reduce the effect. As shown in Fig. S4,† the peaks at 1123, 963, 894, and 832 cm−1 correspond to the asymmetric and symmetric stretching vibrations of Si–O bonds. The peak at 587 cm−1 can be assigned to the bending vibrations of Si–O bonds in Sr2ZnSi2O7. Furthermore, a micron sized crystal was used to measure the IR transmission spectrum of Sr2ZnSn2OS6. Similar measurements have also been found in the previous research.34–37 It is clear that there is no obvious absorption in the range from 1800 to 800 cm−1, corresponding to 5.5 to 12.5 μm (Fig. 2a). The absorption peak at 800 cm−1 can be assigned to the stretching vibration of the Sn–O–Sn bond.38 In addition, except for the Si4+ cations, the terminal O atoms and bridging O atoms in those bonds with low equivalent mass (e.g. the O in the Zn–O–Sn bonds) are also more prone to vibrate than the bridging O atoms in the heavy Sn–O–Sn bonds. Therefore, substituting them with the relatively heavy S atoms would also be helpful to further widen the IR transmission region of the material. The rational substitution from Sr2ZnSi2O7 to Sr2ZnSn2OS6 can be attributed to the minimum distortion of pentagonal rings. In Sr2ZnSi2O7, the pentagonal ring is composed of four long Si–O–Zn bonds and one short Si–O–Si bond (Fig. 1g). From Sr2ZnSi2O7 to Sr2ZnSn2OS6, substituting O atoms with S atoms in Sn–O–Zn bonds and retaining O atoms in the Sn–O–Sn bonds can best retain a similar pentagonal-ring-configuration, i.e. also including four long Sn–S–Zn bonds and one short Sn–O–Sn bond (Fig. 1h).
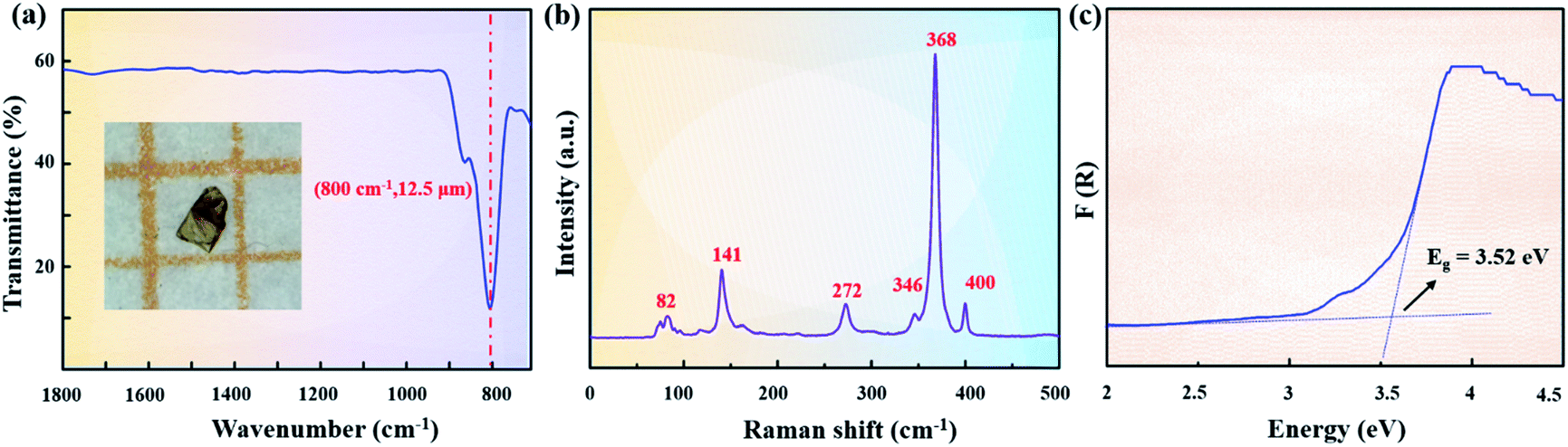 | ||
| Fig. 2 (a) IR transmission spectrum from single crystals, (b) Raman spectrum, and (c) UV-vis-NIR diffuse reflectance spectrum of Sr2ZnSn2OS6. | ||
Furthermore, Raman spectroscopy of Sr2ZnSn2OS6 is shown in Fig. 2b. In the Raman spectra, the absorption peaks at 400, 368, and 346 cm−1 can be attributed to the characteristic absorption of Sn–S modes, the peaks around 272 cm−1 should be from the Zn–S bonds and the absorption peaks below 200 cm−1 (including 141 and 82 cm−1) are mainly attributed to Sr–S vibrations.39–41
The UV-vis-NIR diffuse reflectance spectrum in the range of 200–2500 nm was also measured by using a Shimadzu SolidSpec-3700DUV spectrophotometer (Fig. 2c). It was transformed into an F(R) versus (hν) curve based on the Kubelka–Munk function.42 Extrapolating the linear part of the rising curve to zero, an experimental band gap of 3.52 eV can be obtained. The band gap of Sr2ZnSn2OS6 is much larger than those in commercial AgGaS2 (2.56 eV)11 and AgGaSe2 (1.83 eV).12 Since the large bandgap will reduce the proportion of two photon or multiphoton absorption, the large band-gap is generally helpful for producing a large LDT.43 So, the LDT of Sr2ZnSn2OS6 was also measured by using a Q-switched pulse laser (1064 nm, 10 ns, 10 Hz) with a powder AgGaS2 sample as the reference. It indicates that Sr2ZnSn2OS6 has a good laser damage resistance, and the measured LDT (131 MW cm−2) is about 10 times that of AgGaS2 (12 MW cm−2).44
Sr2ZnSn2OS6 crystallizes in the NCS space group P21m. Its SHG response was measured based on the Kurtz–Perry method by using a laser with a wavelength of 2.09 μm and with powder AgGaS2 as the reference.45 The sample of AgGaS2 was purchased from Sichuan Ninth Research Institute (Fig. S5†) and the powder AgGaS2 sample was obtained by grinding the AgGaS2 crystal. As shown in Fig. 3a, the SHG intensities of Sr2ZnSn2OS6 increase with its particle size. So, Sr2ZnSn2OS6 can achieve type-I phase-matching. The SHG response with the particle size in the range of 150–180 μm is approximately 0.7× AgGaS2 (Fig. 3b), indicating that Sr2ZnSn2OS6 exhibits moderate SHG response. Apart from the large SHG response, a suitable birefringence is also necessary for the application of NLO crystals. Hence, we measured the birefringence of Sr2ZnSn2OS6 by using a cross-polarizing microscope. The orientation of such a utilized crystal was determined as [100] by using the “Index Crystal Faces” program in Bruker SMART APEX III. The observed color of Sr2ZnSn2OS6 in cross-polarized light is second-order yellow. The thickness of the crystal used for the measurement is 8 μm (Fig. S6†). According to the Michel-Levy chart, the retardation (R value) is 920 nm. Thus, the birefringence of Sr2ZnSn2OS6 in the visible region is 0.12, which is large enough for ensuring the phase matching of Sr2ZnSn2OS6.
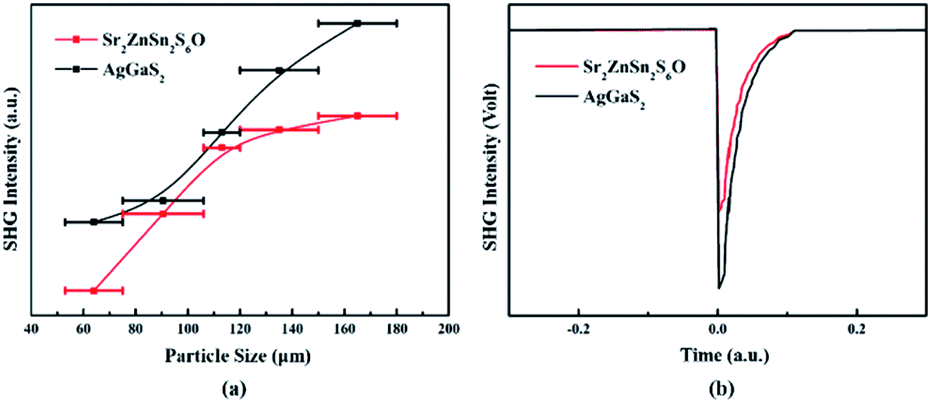 | ||
| Fig. 3 (a) SHG intensity versus particle size for Sr2ZnSn2OS6 and AgGaS2. (b) Comparison of SHG intensities for Sr2ZnSn2OS6 and AgGaS2 at the particle sizes of 150–180 μm. | ||
The above measurements indicate that Sr2ZnSn2OS6 not only exhibits well-balanced NLO properties, including a wide transmission region, large band-gap, and high LDT as well as moderate birefringence and SHG response (Fig. 4a), but also presents the first Sn-containing oxychalcogenide IR NLO with [SnOS3] hetero-anionic tetrahedra (Fig. 4b).46–52 In order to better understand the origin of these excellent NLO properties, especially the moderate SHG response and birefringence, we further calculated the electronic structure of Sr2ZnSn2OS6 through the first-principles calculation.
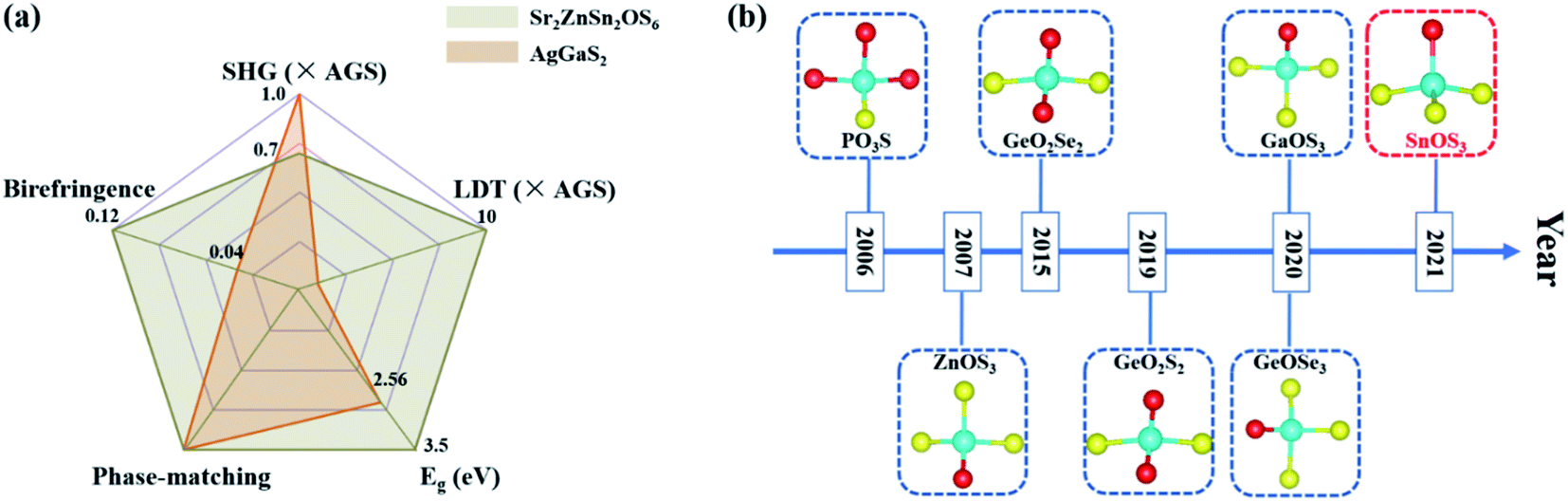 | ||
| Fig. 4 (a) The excellent overall NLO performances of Sr2ZnSn2OS6. (b) Timeline of the hetero-anionic NLO-active unit discovered in oxychalcogenides. | ||
As shown in Fig. 5a, Sr2ZnSn2OS6 is a direct-gap compound and the calculated band gap is 2.71 eV, which is smaller than the experimental value (3.52 eV). The smaller calculated band gap can be attributed to the limitation of the density functional theory (DFT) method.53 The density of states (DOS) and partial densities of states (PDOS) of Sr2ZnSn2OS6 are shown in Fig. 5b. The top of the valence band (VB) is predominately composed of O 2p and S 3p states, mixed with small amounts of Sn 5p, while the bottom of the conductive band (CB) mainly originates from O 2p, S 3p, and Sn 5s,5p states. As we know, the optical properties of a material are mainly affected by the electronic transitions between the energy levels close to the forbidden band. So, the SHG response and birefringence will be mainly determined by [SnOS3] and [ZnS4] tetrahedra (Fig. S7†). Particularly for [SnOS3] tetrahedra, owing to the part substitution of O and S, the dipole moment of the SnOS3 tetrahedron is obviously larger than that of ZnS4 (Table S5†). It indicates that SnOS3 tetrahedra have the main contribution to the SHG response and birefringence of Sr2ZnSn2OS6. Meanwhile, the dipole moments of ZnO4 and SiO4 in Sr2ZnSi2O7 are also calculated (Fig. S8†). By comparing the dipole moments of SiO4 (6.29 D) and SnOS3 (6.77 D), it can be found that the mixed-anion SnOS3 units can exhibit larger polarity in the structure. These results indicate that the rational substitutions of S for O and Sn for Si have contributed the enhancement of the NLO response in Sr2ZnSn2OS6.
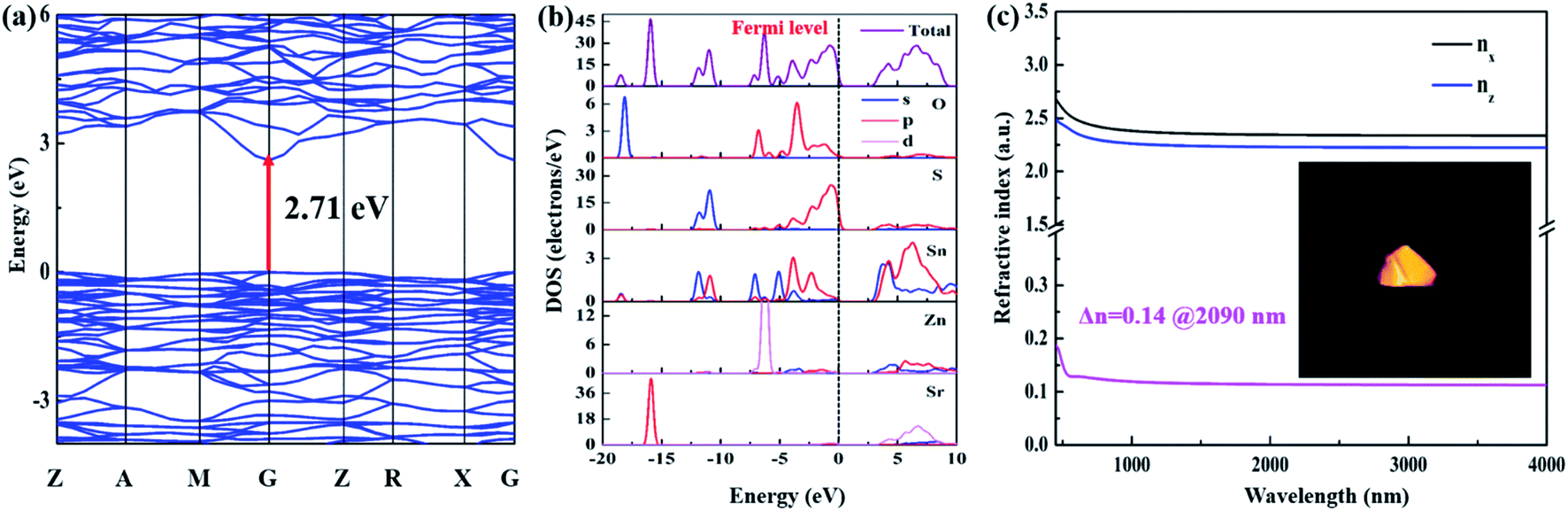 | ||
| Fig. 5 (a) Calculated band structures of Sr2ZnSn2OS6. (b) PDOS and TDOS plot of Sr2ZnSn2OS6. (c) The calculated refractive dispersion curve of Sr2ZnSn2OS6 (the inset image shows the crystal for the birefringence determination). | ||
Based on the calculated electronic structure, the NLO coefficient and birefringence of Sr2ZnSn2OS6 can also be calculated. Sr2ZnSn2OS6 belongs to the point group of 2m. It only has one non-zero independent SHG tensor, d14 under the restriction of Kleinman symmetry. The calculation shows d14 = 4.48 pm V−1, which is comparable with the experimental results. Moreover, the refractive indices and birefringence (Δn) were also calculated. The calculated birefringence is shown in Fig. 5c, and the birefringence Δn = 0.14 at 2090 nm. These calculated results are also essentially consistent with the experimental ones.
Conclusions
In summary, based on the rational partial substitution of S2− for O2− anions in Sr2ZnSi2O7, the first Sn-containing oxychalcogenide IR NLO crystal Sr2ZnSn2OS6 has been successfully designed and synthesized. It can exhibit excellent NLO properties, including a wide IR transmission region, large band gap (3.52 eV), and high LDT (10× AGS) as well as a suitable SHG response (0.7× AGS) and birefringence (0.14 @ 2090 nm), which are comparable with other excellent oxychalcogenides (Table 1). These results show that Sr2ZnSn2OS6 would be a promising IR NLO crystal. The excellent NLO properties of Sr2ZnSn2OS6 can be attributed to the rational substitution of S2− for O2−, i.e. the terminal O atoms and the bridging O atoms in the light Zn–O–Sn bonds, which would lead to an insufficient IR transparency window, have been substituted by the heavier S, while the bridging O atoms in the heavy Sn–O–Sn bonds, which have a weak effect on the IR transparent region, are retained for constructing the distorted SnOS3 groups to produce a moderate SHG response. These findings will be helpful to provide some new insights for the design of oxychalcogenide IR NLO crystals.| Crystals | Space group | IR absorption edge (μm) | E g (eV) | SHG (×AGS) | PM/NPM |
|---|---|---|---|---|---|
| a PM = phase matchability, NPM = non-phase matchability. | |||||
| Sr2ZnSn2OS6 |
P21m |
12.5 | 3.52 | 0.7 | PM |
| Sr6Cd2Sb6O7S10 | Cm | 15.5 | 1.89 | 4 | PM |
| SrZn2OS2 | Pmn21 | — | 3.86 | 0.06 | PM |
| Sr3Ge2O4Se3 | R3m | — | 2.96 | 0.8 | PM |
| BaGeOSe2 | P212121 | 13.3 | 3.2 | 1.1 | PM |
| SrGeOSe2 | P212121 | 12.6 | 3.16 | 1.3 | PM |
| BaGeOS2 | P212121 | — | 4.1 | 0.5 | NPM |
| SrGeOS2 | P212121 | — | 3.9 | 0.4 | NPM |
| Sr5Ga8O3S14 | P21212 | 13.4 | 3.9 | 0.8 | NPM |
Data availability
Supporting data for this article is presented in the ESI.†Author contributions
Conceptualization and supervision: H. W. Yu and H. P. Wu; synthesis and characterization (single-crystal XRD, powder XRD, IR, UV-vis-NIR and SHG measurements): Y. S. Cheng; all authors proof-read, provided comments, and approved the final version of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 22071179, 52172006, 51972230, 51890864, 61835014, 51890865) and Natural Science Foundation of Tianjin (Grant No. 21JCJQJC00090 and 20JCJQJC00060).Notes and references
- P. Becker, Adv. Mater., 1998, 10, 979–992 CrossRef CAS
.
- R. E. Sykora, K. M. Ok, P. S. Halasyamani and T. E. Albrecht-Schmitt, J. Am. Chem. Soc., 2002, 124, 1951–1957 CrossRef CAS PubMed
- H. Wu, S. Pan, K. R. Poeppelmeier, H. Li, D. Jia, Z. Chen, X. Fan, Y. Yang, J. M. Rondinelli and H. Luo, J. Am. Chem. Soc., 2011, 133, 7786–7790 CrossRef CAS PubMed
- X. Wang, Y. Wang, B. Zhang, F. Zhang, Z. Yang and S. Pan, Angew. Chem., Int. Ed., 2017, 56, 14119–14123 CrossRef CAS PubMed
- B. Zhang, G. Shi, Z. Yang, F. Zhang and S. Pan, Angew. Chem., Int. Ed., 2017, 56, 3916–3919 CrossRef CAS PubMed
- C. T. Chen, B. C. Wu, A. D. Jiang and G. M. You, Sci. China, Ser. B: Chem., 1985, 28, 235–243 Search PubMed
- C. T. Chen, Y. C. Wu, A. D. Jiang, B. C. Wu, G. M. You, R. K. Li and S. J. Lin, J. Opt. Soc. Am. B, 1989, 6, 616–621 CrossRef CAS
- G. Ryu, C. S. Yoon, T. P. J. Han and H. G. Gallagher, J. Cryst. Growth, 1998, 191, 492–500 CrossRef CAS
- H. Siegfried, Z. Kristallogr.–Cryst. Mater., 1964, 120, 401–414 CrossRef
- J. D. Bierlein and H. Vanherzeele, J. Opt. Soc. Am. B, 1989, 6, 622–633 CrossRef CAS
- A. O. Okorogu, S. B. Mirov, W. Lee, D. I. Crouthamel, N. Jenkins, A. Y. Dergachev, K. L. Vodopyanov and V. V. Badikov, Opt. Commun., 1998, 155, 307–312 CrossRef CAS
- G. Boyd, H. Kasper, J. McFee and F. Storz, IEEE J. Quantum Electron., 1972, 8, 900–908 CrossRef CAS
- G. D. Boyd, E. Buehler and F. G. Storz, Appl. Phys. Lett., 1971, 18, 301–304 CrossRef CAS
- K. Wu, B. Zhang, Z. Yang and S. Pan, J. Am. Chem. Soc., 2017, 139, 14885–14888 CrossRef CAS PubMed
- H. Chen, Y. Y. Li, B. X. Li, P. F. Liu, H. Lin, Q. L. Zhu and X. T. Wu, Chem. Mater., 2020, 32, 8012–8019 CrossRef CAS
- B. B. Zhang, G. Q. Shi, Z. H. Yang, F. F. Zhang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 3916–3919 CrossRef CAS PubMed
- J. Lu, J. N. Yue, L. Xiong, W. K. Zhang, L. Chen and L. M. Wu, J. Am. Chem. Soc., 2019, 141, 8093–8097 CrossRef CAS PubMed
- G. H. Zou, N. Ye, L. Huang and X. S. Lin, J. Am. Chem. Soc., 2011, 133, 20001–20007 CrossRef CAS PubMed
- Y. Wang, B. Zhang, Z. Yang and S. Pan, Angew. Chem., Int. Ed., 2018, 57, 2150–2154 CrossRef CAS PubMed
- H. Yu, H. Wu, S. Pan, Z. Yang, X. Su and F. Zhang, J. Mater. Chem., 2012, 22, 9665–9670 RSC
- L. Kang, M. Zhou, J. Yao, Z. Lin, Y. Wu and C. Chen, J. Am. Chem. Soc., 2015, 137, 13049–13059 CrossRef CAS PubMed
- H. Yu, W. Zhang and P. S. Halasyamani, Cryst. Growth Des., 2016, 16, 1081–1087 CrossRef CAS
- G. Han, Y. Wang, X. Su, Z. Yang and S. Pan, Sci. Rep., 2017, 7, 1901 CrossRef PubMed
- Y. Tsujimoto, C. A. Juillerat, W. Zhang, K. Fujii, M. Yashima, P. S. Halasyamani and H.-C. zur Loye, Chem. Mater., 2018, 30, 6486–6493 CrossRef CAS
- R. Wang, Y. Guo, X. Zhang, Y. Xiao, J. Yao and F. Huang, Inorg. Chem., 2020, 59, 9944–9950 CrossRef CAS PubMed
- M. Y. Ran, Z. Ma, H. Chen, B. Li, X. T. Wu, H. Lin and Q. L. Zhu, Chem. Mater., 2020, 32, 5890–5896 CrossRef CAS
- B. W. Liu, X. M. Jiang, G. E. Wang, H. Y. Zeng, M. J. Zhang, S. F. Li, W. H. Guo and G. C. Guo, Chem. Mater., 2015, 27, 8189–8192 CrossRef CAS
- R. Wang, F. Liang, F. Wang, Y. Guo, X. Zhang, Y. Xiao, K. Bu, Z. Lin, J. Yao, T. Zhai and F. Huang, Angew. Chem., Int. Ed., 2019, 58, 8078–8081 CrossRef CAS PubMed
- M. Mutailipu, Z. Li, M. Zhang, D. Hou, Z. Yang, B. Zhang, H. Wu and S. Pan, Phys. Chem. Chem. Phys., 2016, 18, 32931–32936 RSC
- H. Yu, W. Zhang, J. Young, J. M. Rondinelli and P. S. Halasyamani, J. Am. Chem. Soc., 2015, 138, 88–91 CrossRef PubMed
- K. Chen, C. Lin, S. Zhang, G. Peng, Y. Chen, D. Zhao, H. Fan, S. Yang, X. Wen, M. Luo, Z. Lin and N. Ye, Chem. Mater., 2021, 33, 6012–6017 CrossRef CAS
- X. Chen, Q. Jing and K. M. Ok, Angew. Chem., Int. Ed., 2020, 59, 20323–20327 CrossRef CAS PubMed
- X. Luo, Z. Li, F. Liang, Y. Guo, Y. Wu, Z. Lin and J. Yao, Inorg. Chem., 2019, 58, 7118–7125 CrossRef CAS PubMed
- W. Xing, F. Liang, C. Tang, E. Uykur, Z. Lin, J. Yao, W. Yin and B. Kang, ACS Appl. Mater. Interfaces, 2021, 13, 37331–37338 CrossRef CAS PubMed
- J. Chen, H. Chen, F. Xu, L. Cao, X. Jiang, S. Yang, Y. Sun, X. Zhao, C. Lin and N. Ye, J. Am. Chem. Soc., 2021, 143, 10309–10316 CrossRef CAS PubMed
- Y. Hu, C. Wu, X. Jiang, Z. Wang, Z. Huang, Z. Lin, X. Long, M. G. Humphrey and C. Zhang, J. Am. Chem. Soc., 2021, 143, 12455–12459 CrossRef CAS PubMed
- W. Xing, C. Tang, N. Wang, C. Li, E. Uykur, J. Wu, Z. Lin, J. Yao, W. Yin and B. Kang, Adv. Opt. Mater., 2021, 9, 2100563 CrossRef CAS
- P. Pascuta, L. Pop, S. Rada, M. Bosca and E. Culea, Vib. Spectrosc., 2008, 48, 281–284 CrossRef CAS
- J. He, Y. Guo, W. Huang, X. Zhang, J. Yao, T. Zhai and F. Huang, Inorg. Chem., 2018, 57, 9918–9924 CrossRef CAS PubMed
- K. Wu, Z. Yang and S. Pan, Angew. Chem., Int. Ed., 2016, 55, 6713–6715 CrossRef CAS PubMed
- X. Luo, Z. Li, F. Liang, Y. Guo, Y. Wu, Z. Lin and J. Yao, Inorg. Chem., 2019, 58, 7118–7125 CrossRef CAS PubMed
- E. L. Simmons, Appl. Opt., 1975, 14, 1380–1386 CrossRef CAS PubMed
- T. W. Walker, A. H. Guenther and P. Nielsen, IEEE J. Quantum Electron., 1981, 17, 2053–2065 Search PubMed
- K. Wu, Z. Yang and S. Pan, Chem. Mater., 2016, 28, 2795–2801 CrossRef CAS
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798–3813 CrossRef CAS
- X. Luo, F. Liang, M. Zhou, Y. Guo, Z. Li, Z. Lin, J. Yao and Y. Wu, Inorg. Chem., 2018, 57, 9446–9452 CrossRef CAS PubMed
- P. A. Maggard, T. S. Nault, C. L. Stern and K. R. Poeppelmeier, Solid State Chem., 2003, 175, 27–33 CrossRef CAS
- J. Zhang, Z. Zhang, W. Zhang, Q. Zheng, Y. Sun, C. Zhang and X. Tao, Chem. Mater., 2011, 23, 3752–3761 CrossRef CAS
- W. Xing, P. Fang, N. Wang, Z. Li, Z. Lin, J. Yao, W. Yin and B. Kang, Inorg. Chem., 2020, 59, 16716–16724 CrossRef CAS PubMed
- H. Yan, Y. Matsushita, K. Yamaura and Y. Tsujimoto, Angew. Chem., Int. Ed., 2021, 60, 26561–26565 CrossRef CAS PubMed
- Y. F. Shi, W. B. Wei, X. T. Wu, H. Lin and Q. L. Zhu, Dalton Trans., 2021, 50, 4112–4118 RSC
- X. Zhang, Y. Xiao, R. Wang, P. Fu, C. Zheng and F. Huang, Dalton Trans., 2019, 48, 14662–14668 RSC
- R. W. Godby, M. Schlter and L. J. Sham, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 6497–6650 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2132019. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00099g |
| This journal is © The Royal Society of Chemistry 2022 |