
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe underappreciated influence of ancillary halide on metal–ligand proton tautomerism†
Anant Kumar
Jain
 ,
Michael R.
Gau
,
Patrick J.
Carroll
and
Karen I.
Goldberg
*
,
Michael R.
Gau
,
Patrick J.
Carroll
and
Karen I.
Goldberg
*
Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. E-mail: kig@sas.upenn.edu
First published on 20th June 2022
Abstract
Syntheses of Vaska-type complexes [IrP2X(CO)] (P = phosphine, X = halide) with all four common halides (fluoride, chloride, bromide, and iodide) was attempted using a protic and hemilabile imidazolyl di-tert-butyl phosphine ligand. In the solid-state, all four complexes were found to be ionic with the halides in the outer-sphere, and the fourth coordination site of the square plane occupied by the imidazole arm of the ligand. In solution, however, the chloride complex was found to be in equilibrium with an octahedral IrIII–H species at room temperature. For the bromide and iodide analogs, the corresponding IrIII–H species were also observed but only after heating the solutions. The neutral IrI Vaska's analogs for X = Cl, Br, and I were obtained upon addition of excess halide salt, albeit heating was required for X = Br and I. The IrIII–H species are proposed to originate from tautomerization of minor amounts of the electron rich neutral Vaska analog (halide inner-sphere and phosphines monodentate) that are in equilibrium with the ionic species. Heating is required for the larger anions of bromide and iodide to overcome a kinetic barrier associated with their movement to an inner-sphere position prior to tautormerization. For the fluoride analog, the IrIII–H was not observed, attributable to strong hydrogen bonding interactions of the imidazolyl proton with the fluoride anion.
Introduction
Metal–Ligand Proton Tautomerism (MLPT) is the interconversion between a metal hydride and its corresponding ligand protonated congener (eqn (1)). MLPT is increasingly being identified as a key feature in enzymatic as well as synthetic transformations such as hydrogen evolution and cooperative bond activation.1–12 Involvement of an MLPT equilibrium has been shown to be important in accessing lower energy pathways for these reactions. | (1) |
MLPT equilibria can be intentionally incorporated to improve existing catalysis involving metal hydride intermediates by installing a basic site on the ligand backbone.3,6,13–15 Alternatively, catalysis involving low-valent metal center intermediates can be improved by adding a proton-responsive site on the ligand. Thus, strategies to control the position of an MLPT equilibrium (i.e. to favor either the ligand protonated side or the metal protonated side) can be of great use to catalyst design. Recently several such strategies have been reported. For example, changing the identity of the metal in a catalyst was used to increase the rate of dehydrogenation of dimethylamine-borane.10 The explanation for the rate increase was that switching from Ir to the less basic Rh favored the ligand protonated side of the MLPT equilibrium which provided an alternative lower energy pathway for the reaction. Recently, we found that an MLPT equilibrium was involved in a rarely observed dinuclear reductive elimination of dihydrogen from a third-row metal-hydride. Here, the concentration of an exogenous electron donating ligand (4-tert-butyl pyridine) was used to control the position of the MLPT equilibrium; a higher concentration of 4-tert-butyl pyridine favored the metal protonated (metal-hydride) side of an Ir-complex possessing a protic ligand.9 In a related rhenium system, use of electron withdrawing ligands on a ReIII–H species was shown to influence the MLPT equilibrium.16 Computations showed that stronger π-acid ligands resulted in a smaller energy difference between the ReIII–H and the ligand protonated tautomer. Another strategy to control the position of the MLPT equilibrium involved addition of an exogenous hydrogen bonding acceptor (such as BF4− or CF3SO3−) that engaged in favorable interactions with the acidic proton on the ligand backbone, thus favoring the ligand protonated side.9,17,18
Some common themes emerge from the strategies described above to control the position of an MLPT equilibrium. One can either change the basicity of the metal by changing the identity of the metal, or its ancillary ligands, or add a hydrogen bond acceptor to stabilize ligand based protons. An even more powerful strategy is to combine two of these modifiers. Halides in this regard are particularly promising in that they can act as a ligand (and so change the sterics and electronics at the metal center), and also engage, to different extents based on the identity of the halide, in hydrogen bonding interactions.19–27 Thus, halides, among the most common ligands/counter ions found in transition metal catalysis, can play an important role in controlling the position of the MLPT equilibrium.
In this report, the attempted syntheses of Vaska-type IrI complexes bearing protic imidazolyl phosphine ligands with all four common halides (fluoride, chloride, bromide, and iodide) are described. While in many reactions halides are merely “spectators”, here we show that the diversity of electronics and sterics offered by the halides, along with varying degrees of hydrogen bonding propensities, can be rationally utilized to control the position of the MLPT equilibrium. The bifunctional (2-(di-tert-butylphosphaneyl)-1H-imidazole) ligand (LH) was chosen because of its ability to not only engage in hydrogen bonding interactions, but also undergo proton-transfer to electron-rich metal centers28 – a criterion we sought for this study on metal–ligand proton tautomerism. Furthermore, work from Grotjahn and co-workers has also demonstrated the ability of LH to act in a bidentate fashion in solution.29
Herein, all four halide complexes, which contain the Vaska-type complexes' ligand set of two phosphines, a carbonyl, and a halide, were found to be ionic in the solid-state with the halide ion in the outer-sphere and the fourth coordination site of the square plane occupied by the imidazole groups of one of the phosphine ligands (e.g.[LHIr][Cl] in Fig. 1). In solution, however, varying amounts of IrIII–H species, a consequence of MLPT equilibrium described below, were observed at room temperature or after heating. The concentration of the IrIII–H was dependent upon the identity of the halide.
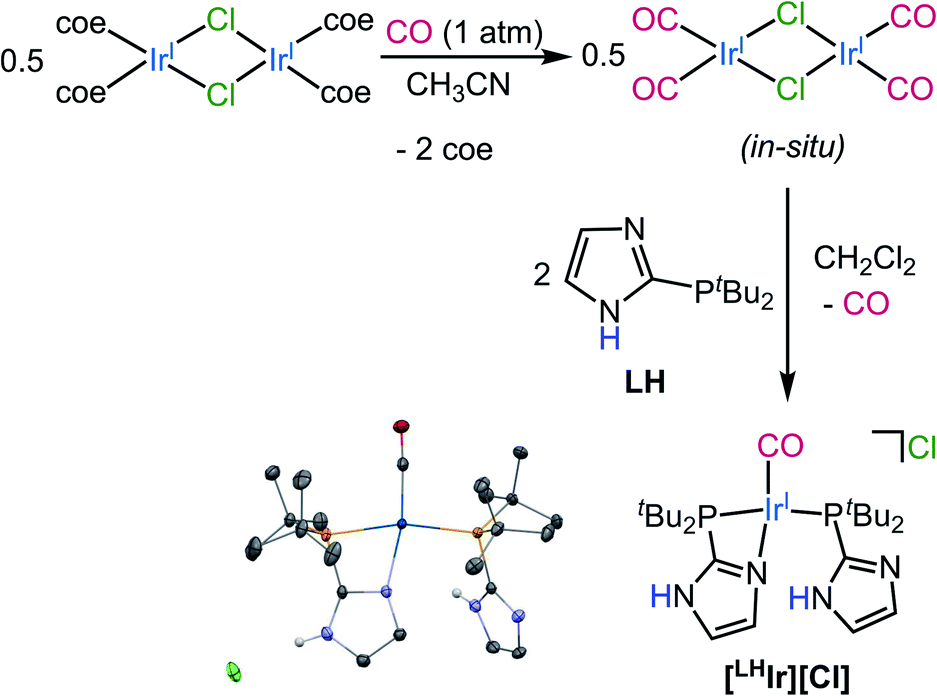 | ||
| Fig. 1 Synthesis of [LHIr][Cl] from in situ generated [Ir(CO)2Cl]2 dimer. Thermal ellipsoid plot of [LHIr][Cl] is also shown with 50% probability. H-atoms except for N–H's are omitted for clarity. | ||
Results and discussion
Synthesis and solid/solution-state structure of [LHIr][Cl]
Synthesis of the Vaska-type complex [IrP2X(CO)] where X = Cl was attempted following an adapted literature procedure;30 four molar equivalents of the protic phosphine ligand LH,28 were added to the in situ generated dimeric precursor [Ir(CO)2Cl]2 in dichloromethane (Fig. 1). Solid-state analysis of the product revealed that the Cl anion is present as a counterion with Ir preferentially binding the imidazole of one of the phosphines, thereby generating the ionic complex [LHIr][Cl] (Fig. 1). In a DCM-d2 solution of the isolated product, however, signals corresponding to two species (one major and one minor) were observed in the 1H NMR spectrum (Fig. S1†). The major set of signals included four singlets of equal intensity in the aromatic region, consistent with the identity of the ionic [LHIr][Cl] complex (as observed in the solid-state). The minor set of signals include a triplet at δ −17.22 (JH–P = 12 Hz) along with other aromatic and aliphatic resonances, indicating that these set of signals belong to an Ir–H species.31To confirm that the major set of 1H NMR spectral signals are correctly attributed to the ionic species [LHIr][Cl] that was observed in the solid state, the related ionic compound [LHIr][PF6] was synthesized. As hexafluorophosphate is a weakly coordinating anion,32 the 1H NMR spectral signals for the [LHIr] cation in both complexes should be virtually identical if the chloride in [LHIr][Cl] exists as a non-coordinating anion in solution. [LHIr][PF6] was prepared by the following route. First, two molar equivalents of LH were added to an acetone solution of [Ir(coe)2(acetone)2][PF6] (coe = cyclooctene), resulting in a quantitative conversion to [Ir(LH)2][PF6] (Ir-1). Then, an acetone solution of Ir-1 was exposed to an atmosphere of carbon monoxide (CO) gas resulting in the formation of [LHIr][PF6] complex (Fig. 2) which was fully characterized. The 1H NMR spectrum of [LHIr][PF6] in DCM-d2 (Fig. S8†) consists of four separate singlets in the aromatic region which fully overlap with the four aromatic signals assigned to the ionic [LHIr][Cl] species as described above,33 thus confirming the validity of the peak assignments for the ionic [LHIr][Cl] species in solution.
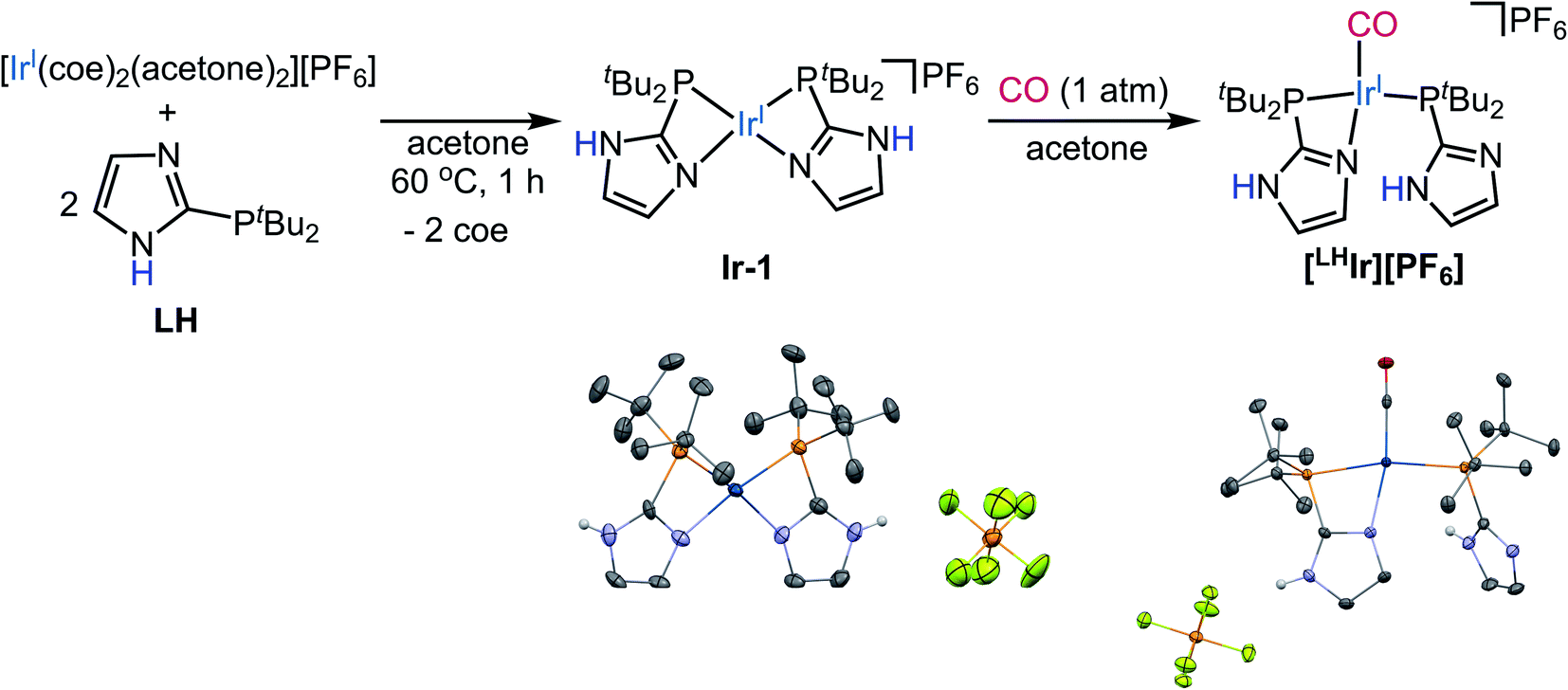 | ||
| Fig. 2 Synthesis of [LHIr][PF6]viaIr-1 in acetone. Thermal ellipsoid plots of Ir-1 and [LHIr][PF6] are shown with 50% probability. H-atoms except for N–H's and co-crystallized solvent molecules are omitted for clarity. | ||
It is proposed that the Ir–H observed upon dissolution of [LHIr][Cl] originates via proton-transfer from the imidazolyl phosphine ligand to the Ir metal center. The chloride is also likely to be coordinated to generate a stable 18-electron octahedral species ([H–LIr–Cl] in Scheme 1). The two bulky phosphine ligands in the proposed hydride species [H–LIr–Cl] are expected to occupy positions trans to each other, and with the deprotonated imidazole also coordinated to the Ir center, a PNP-type meridional plane is generated. With this arrangement of the phosphines, the possibilities for the other ligands within the [H–LIr–Cl] complex include: (1) H and Cl trans to each other; (2) H and CO trans to each other; (3) Cl and CO trans to each other. Analysis of all the mononuclear and neutral complexes of the type [Ir(PEP)(CO)(H)(X)] (E = monoanionic C or N; X = F, Cl, Br, I) in the Cambridge Structural Database (CSD) reveals that there are 15 examples of the first configuration, 6 examples of the second configuration, and none for the third configuration.34 In the 15 examples where H and X are trans to each other,35–45 the δIr–H (t) is observed between −15 to −20 with 2JH–P < 15 Hz. For the 6 complexes with H and CO trans to each other, δIr–H (t) is reported between −5 and −10, and 2JH–P between 14–18 Hz.45–51 With δIr–H (t) −17.22 and 2JH–P = 12 Hz for the neutral [H–LIr–Cl], the data are most consistent with H and Cl trans to each other (as depicted in Scheme 1).
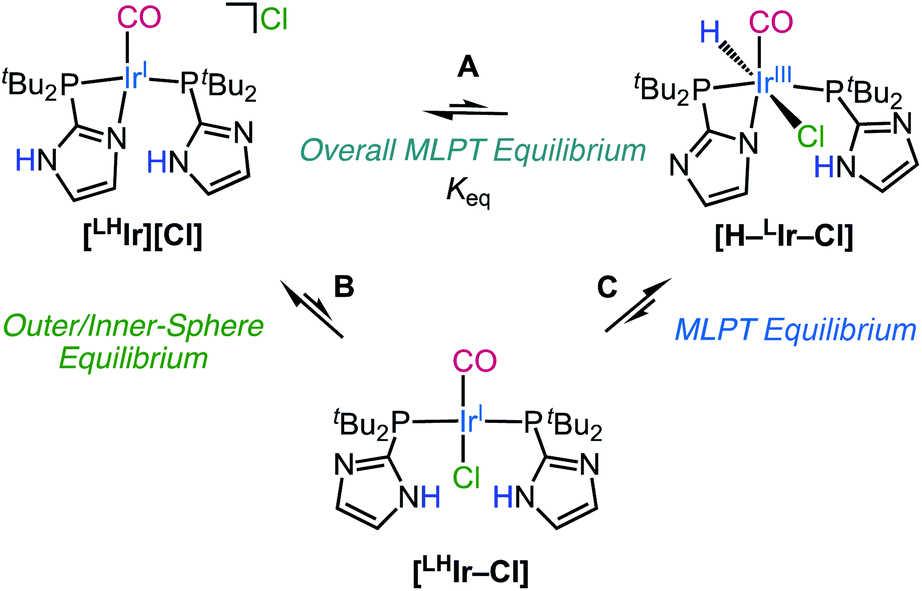 | ||
| Scheme 1 Proposed MLPT equilibria of [LHIr][Cl] in DCM-d2. | ||
To consider how [H–LIr–Cl] might originate from [LHIr][Cl] in solution, one can either propose that proton-transfer from the imidazole to the Ir occurs first and is followed by Cl− coordination, or that Cl− coordination is followed by ligand to metal proton-transfer. Based on literature precedent wherein addition of electron density to the metal center (thereby increasing its basicity) has been shown to trigger proton-transfer from the ligand to the metal,9 it is expected that proton-transfer from the imidazole to the more electron rich neutral Ir center in [LHIr–Cl] (Scheme 1, C) should be more favorable than proton-transfer to the cationic Ir center in [LHIr][Cl]. In light of this proposal, it was of interest to explore the effect of added Cl anion. Upon addition of 10 molar equivalents of tetraethylammonium chloride (NEt4Cl) salt to a DCM-d2 solution of [LHIr][Cl], full conversion to a new species with signals consistent with that expected for the neutral Vaska-type [LHIr–Cl] complex was observed. The new species is symmetric showing only two resonances in the aromatic region of the 1H NMR spectrum (Fig. S26†) and a singlet in the 31P{1H} NMR spectrum (Fig. S27†). There was no upfield signal in the 1H NMR spectrum for Ir–H. A decrease in the carbonyl stretching frequency (in DCM-d2) from 1967 cm−1 in [LHIr][Cl] to 1956 cm−1 in the presence of excess Cl anions is also consistent with generation of the more electron rich neutral Vaska-type [LHIr–Cl] complex.
Thus, it appears that addition of Cl anions to the solution of [LHIr][Cl] formed the Vaska-type [LHIr–Cl] complex (Scheme 1, B). This observation supports the proposal that chloride can enter the coordination sphere of the square planar [LHIr][Cl] species. Interesting however is to consider why proton-transfer from this presumably more electron rich [LHIr–Cl] species to form [H–LIr–Cl] (Scheme 1, C) was not observed. The chemistry of the analogous halide complexes with bromide and iodide shed light on this issue.
Evaluation of the role of other halides on MLPT equilibrium
The fluoro, bromo, and iodo-analogs of [LHIr][Cl] were prepared by salt-metathesis reactions of [LHIr][PF6] with a suitable tetraalkylammonium halide (NR4X) salt in acetonitrile (Fig. 3). Solid-state analysis of the resulting three complexes revealed that the halide remains outer-sphere in every case with Ir preferentially binding an imidazole on one of the phosphine ligands. | ||
| Fig. 3 (Top) Synthesis of [LHIr][Br], [LHIr][I], and [LHIr][F]via salt-metathesis in acetonitrile. (Bottom) Thermal ellipsoid plots of [LHIr][Br] (left), [LHIr][I] (middle), and [LHIr][F] (right) are shown with 50% probability. H-atoms except for N–H's and co-crystallized solvent molecules are omitted for clarity. | ||
As both bromide and iodide are stronger σ-donor ligands than chloride,19 it was expected that when the solid samples of [LHIr][X] (X = Br, I) were dissolved in solution, the corresponding hydride [H–LIr–X] species should again be observed. However, when the solids [LHIr][Br] and [LHIr][I] were dissolved in DCM-d2, no upfield signal corresponding to an Ir–H was observed in the 1H NMR spectrum (Fig. S11 and S15†). The 1H, 13C{1H} and 31P{1H} NMR spectra of both closely matched with those of the [LHIr][PF6] complex suggesting that [LHIr][Br] and [LHIr][I] retain their ionic form in solution, consistent with the solid-state structure. However, after DCM-d2 solutions of both [LHIr][Br] and [LHIr][I] were heated at 60 °C in a J. Young NMR tube for 7 days and 2 days respectively, signals corresponding to the minor hydride species [H–LIr–X] were observed (Fig. S14 and S18†) at room temperature. The major constituent in solution was still the ionic complex ([LHIr][Br] or [LHIr][I]) as indicated by its characteristic four seprate signals in the aromatic region. Longer heating times resulting in no further changes to the ratios of the Ir species indicated that the solutions had reached equilibrium. Thus, both [LHIr][Br] and [LHIr][I] complexes equilibrate with their metal protonated [H–LIr–X] congener in solution, but in contrast to the establishment of equilibrium at room temperature for the chloride analog, heating is required. This result implies that there is a greater kinetic barrier to generate the IrIII–H species for the bromide and iodide [LHIr][X] complexes than for the chloride complex.
The effect of added bromide and iodide anions to the respective solutions of [LHIr][Br] and [LHIr][I] complexes was also evaluated. When 10 molar equivalents of tetrabutylammonium bromide (NBu4Br) salt was added to the DCM-d2 solution of [LHIr][Br], only a very slow reaction occurred at room temperature. This result contrasts with the similar experiment with the chloride analog where rapid conversion to the neutral Vaska-type IrI complex was observed. Heating the bromide solution at 60 °C for one hour resulted in a complete conversion to a symmetric species. The two new resonances in the aromatic region of the 1H NMR spectrum (Fig. S29†) and a singlet in the 31P{1H} NMR spectrum (Fig. S30†) are consistent with formation of the neutral [LHIr–Br] complex.
When 10 molar equivalents of tetrabutylammonium iodide (NBu4I) salt were added (in the dark) to a DCM-d2 solution of [LHIr][I], there was again a very slow reaction at room temperature. Heating the solution at 60 °C for one hour resulted in formation of the [LHIr–I] complex; however, minor signals corresponding to the hydride complex [H–LIr–I] were also observed (Fig. S31†). Signals for the ionic [LHIr][I] had completely disappeared, and the solution consisted of approximately 90% [LHIr–I] + 10% [H–LIr–I].
Discussion of the MLPT equilibria for the chloride, bromide, and iodide analogs
It is interesting that the addition of excess halide to the chloride and bromide complexes led to exclusive production of the Vaska type neutral complexes [LHIr–X], while the iodide complex formed predominantly [LHIr–I] with a small amount of the hydride congener [H–LIr–I]. To explain this discrepancy, we must consider the electronics of σ-donation as well as the steric influences among the halides which both increase as F < Cl < Br < I.19 Notably, this ordering is opposite to their propensity for hydrogen bonding acceptance which decreases as F ≫ Cl ≥ Br > I.52 It is reasonable to argue then that the excess Cl anions when added to a solution of [LHIr][Cl] likely engage in strong hydrogen bonding interactions with the acidic imidazolyl protons of the neutral [LHIr–Cl] species. This hydrogen-bonding should inhibit proton-transfer to generate [H–LIr–Cl]. Evidence for this hydrogen bonding proposal can be provided by the significantly downfield shifted resonance of the N–H protons (δ 13.46) in the in situ generated [LHIr–Cl] complex (Fig. S26†) compared to those of [LHIr][Cl] (δ 9.56, 12.24) (Fig. S1†).53 Furthermore, in related studies of MLPT, Rauchfuss, Bullock, and our group have previously demonstrated that the MLPT equilibrium can be perturbed towards the ligand-protonated side by addition of an exogenous hydrogen bonding acceptor such as BF4− or CF3SO3− that can engage in favorable hydrogen bonding interactions with the acidic proton on the ligand backbone.9,17,18 A similar hydrogen bonding stabilization of the imidazolyl protons is proposed here that disrupts the MLPT equilibrium of [LHIr–Cl] with [H–LIr–Cl] (Scheme 1, C) upon addition of excess Cl anions, precluding the formation of latter.The result of the addition of excess Br anions to the solution of [LHIr][Br] was very similar to that of the chloride case as only the neutral Vaska-type [LHIr–Br] complex was again observed. This result can be similarly attributed to hydrogen bonding assisted stabilization of the acidic imidazolyl protons with the excess halide anions in solution, especially since chloride and bromide have similar hydrogen bonding abilities.52 Indeed the 1H NMR shift for the N–H proton of the generated [LHIr–Br] in the presence of a large excess of bromide was quite far downfield (ca. 13.01 ppm, Fig. S29†). One notable difference between the chloride and bromide systems however is that the reaction of the ionic [LHIr][Cl] with excess chloride took place at room temperature while the same reaction for the bromide analog required heating to 60 °C. This difference suggests that a higher kinetic barrier is associated with the bromide ion to move inner-sphere, likely due to its larger size. Also worth noting here is that heating to 60 °C was also required to generate the IrIII–H tautomer for the bromide complex. These data are consistent with the proposal that coordination of the halide occurs prior to proton migration from the ligand to the metal to form the IrIII–H (equilibrium B followed by C in Scheme 1).
In contrast to the chloride and bromide systems, addition of excess I anions to the solution of [LHIr][I] and heating to 60 °C resulted in a mix of both [LHIr–I] and [H–LIr–I]. Similar to the observations in bromide system, heating was required to promote the reaction of [LHIr][I] with the large iodide anion. Here however, the IrIII–H congener, [H–LIr–I], is observed alongside [LHIr–I]. Iodide has a lower affinity for hydrogen bonding than bromide and chloride, and iodide is also a stronger σ-donor ligand than bromide and chloride.19 The result of the weaker hydrogen bonding and stronger σ-donation is that the proton is free to migrate from the imidazole to the electron rich Ir center forming the IrIII–H tautomer [H–LIr–I].
Finally, consider the results of dissolution of the original [LHIr][X] complexes, without heating for the chloride and after heating for the bromide and iodide. In all cases, a mixture of mixture of [LHIr][X] and [H–LIr–X] were observed, with the IrIII–H as the minor species. The respective Keq values for the overall MLPT equilibrium (Scheme 1, A) are 8.4 ± 0.5 M−1, 10.6 ± 0.7 M−1 and 21.0 ± 0.3 M−1 for the chloride, bromide and iodide respectively (Tables S1–S3†).
It is notable that in these Ir-complexes, in contrast to the classic Vaska system, there is a strong preference for the ionic [LHIr][X] species over the neutral [LHIr–X]. Sterics are likely playing the key role in this preference. The tBu groups on the phosphines provide substantial steric hindrance to the coordination of the halides.42,54 Remarkably, there are no crystallographically characterized complexes of the type [Ir(PtBu2R)2(CO)(X)] (X = Br, I; R = any group) to date.55 Spectroscopically, only 4 such complexes have been characterized and in all 4 of those complexes, the R-group was very small in size (hydrogen or methyl).56 Additionally, work from both Grotjahn and Love has shown that larger phosphine substituents favor the formation of “strained” 4-membered PN-chelates.57,58 The bulky R-groups on phosphine are proposed to lower the ring strain through the Thorpe–Ingold effect compensating for the steric repulsion offered by the same R-groups. Thus, overall, the sterics of the tBu groups and the Thorpe–Ingold effect both favor the ionic [LHIr][X] species over the neutral [LHIr–X] species in these Ir-complexes.
The fluoride analog
Lastly, when the smallest halide, fluoride, was employed in this system, somewhat different behavior was observed. While four singlets in the aromatic region of the 1H NMR spectrum were still present in the DCM-d2 solution of [LHIr][F] (Fig. S19†), these signals were significantly offset from those of the [LHIr][PF6] complex. No upfield signals corresponding to an Ir–H were present and heating the solution for two weeks at 60 °C resulted in no change to the spectrum.The solid-state structure of [LHIr][F] may provide clues to its different solution behavior relative to the other halide complexes. The N⋯F distance is considerably shorter than the N⋯X distances in the other [LHIr][X] complexes (Table 1), indicative of an extremely strong hydrogen bonding interaction between the fluoride anion and the imidazolyl proton. Given the high propensity of fluoride to form hydrogen bonds, it is reasonable to propose that this strong hydrogen bonding is maintained in solution and the [LHIr][F] complex exists as a hydrogen bonded adduct ([LHIr·F]) in solution (shown in Scheme 2). Indeed, the acidic proton of the coordinated imidazole involved in the hydrogen bond exhibits a broad feature that appears very far downfield in 1H NMR spectrum (ca. 16.15 ppm, Fig. S19†). Also consistent is that the other spectral signals for the complex lie in between those of the protonated [LHIr] cation ([LHIr][PF6]) and its deprotonated derivative ([LIr]). The deprotonated version of the [LHIr] cation was prepared by addition of triethylamine (NEt3) base to the [LHIr][PF6] complex in acetonitrile as shown in Fig. 4. The deprotonated [LIr] complex was characterized by X-ray crystallography as well as by multi-nuclear NMR spectroscopy in DCM-d2 solution. Overlaying the 1H NMR spectra of [LHIr][F] complex with the [LHIr][PF6] (protonated) complex and [LIr] (deprotonated) complex reveals that the four aromatic resonances in the 1H NMR spectrum of the [LHIr][F] complex lie in between the protonated and the deprotonated species (Fig. 5). Similar observations were made with respect to the 31P{1H} and 13C{1H} NMR spectral data as well. Finally, the IR data (Table 1) in DCM-d2 also supports strong hydrogen bonding for the fluoride complex – the CO stretching frequency of the [LHIr][F] complex in solution (νCO = 1952 cm−1) lies in between the CO stretching frequency of the protonated [LHIr][PF6] (νCO = 1973 cm−1) and deprotonated [LIr] complex (νCO = 1947 cm−1).59
| Complex | N⋯X distance (Å) | ν CO (cm−1, in CD2Cl2) |
|---|---|---|
| [LHIr][PF6] | N/A | 1973 |
| [LHIr][I] | 3.51 | 1970 |
| [LHIr][Br] | 3.26 | 1969 |
| [LHIr][Cl] | 3.07 | 1967 |
| [LHIr][F] | 2.52 | 1952 |
| [LIr] | N/A | 1947 |
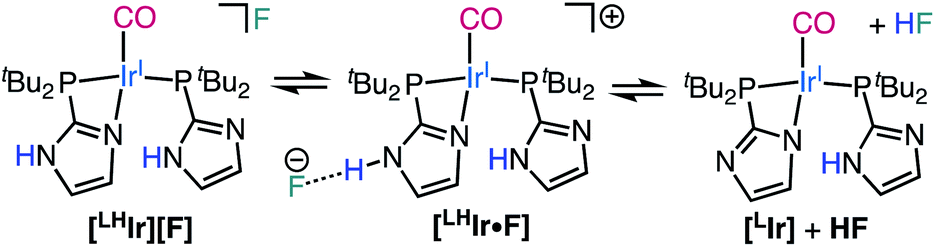 | ||
| Scheme 2 Proposed hydrogen bonding/acid–base equilibria operative for [LHIr][F] in DCM-d2. | ||
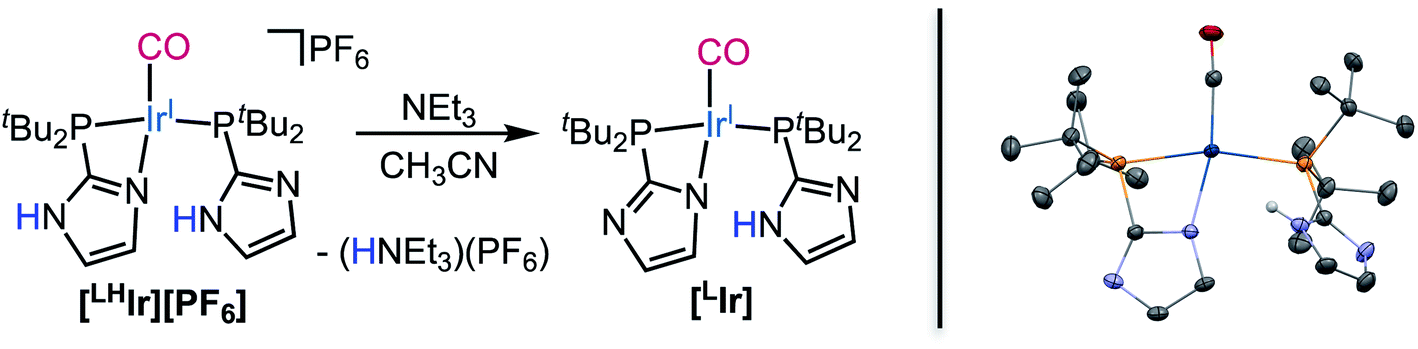 | ||
| Fig. 4 (Left) Synthesis of [LIr] complex by deprotonation of [LHIr][PF6] using NEt3. (Right) Thermal ellipsoid plot of [LIr] shown with 50% probability. H-atoms except for N–H and co-crystallized solvent molecules are omitted for clarity. | ||
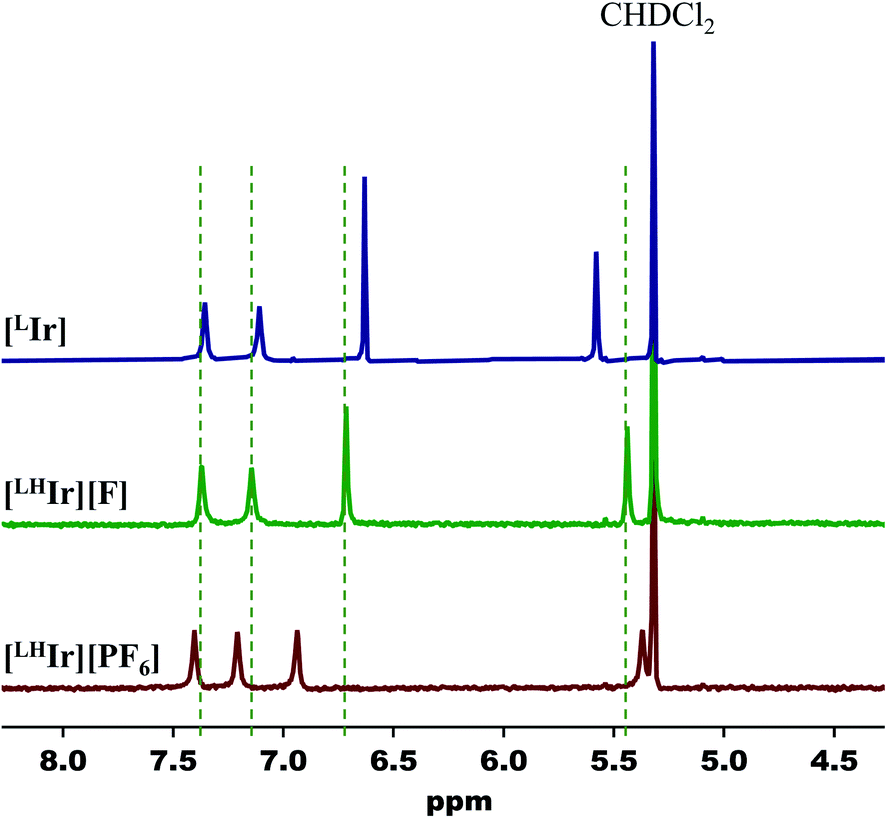 | ||
| Fig. 5 Partial 1H NMR spectra (400 MHz, DCM-d2) of [LHIr][PF6], [LHIr][F], and [LIr]. Dashed lines are centered at the signals of [LHIr][F]. | ||
When 10 molar equivalents of tetramethylammonium fluoride (NMe4F) salt were added to the DCM-d2 solution of [LHIr][F], complete conversion to a new species was observed after one day at room temperature. This new species exhibited four aromatic singlets in the 1H NMR spectrum (Fig. S33†) with weak coupling to fluorine nuclei (JH–F values between 1.3 Hz to 1.7 Hz). In the 31P{1H} NMR spectrum, two sets of doublets with a large JP–P (260 Hz) were observed (Fig. S34†), consistent with two trans phosphines in different environments. Signals for HF2− were detected in both 1H and 19F NMR spectrum (JH–F = 124 Hz) of the reaction mixture (Fig. S35†).60 Thus, addition of excess fluoride to [LHIr][F] has led to deprotonation; the resulting spectral signals are of the deprotonated [LIr] complex (Scheme 2) involved in hydrogen bonding interactions with the excess fluoride in solution. Indeed, the same NMR spectra were obtained after a day when excess NMe4F salt was added separately to a solution of isolated [LIr] complex at room temperature, consistent with the hypothesis that addition of excess fluoride to [LHIr][F] resulted in its deprotonation.61
Conclusions
Halides are among the most common spectators or ancillary ligands present in transition metal-based catalysis. The range of sterics and electronics, as well as the hydrogen bonding potential, that the halides offer is often underappreciated. In this report, the study of Vaska-type complexes with protic phosphine ligands and all four common halides (F, Cl, Br, I) allows for insight into the role of sterics and electronics of the ancillary halide on metal–ligand proton tautomerism.The four Vaska-analogs were isolated in their ionic form [LHIr][X] as solids, with an imidazole on one of the phosphines occupying the fourth coordination site and the halide as the counterion. In solution, the ligand protonated [LHIr][X] (X = Cl, Br, I) complexes were found to be in an equilibrium with their neutral metal protonated [H–LIr–X] congeners. The evidence is consistent with generation of the hydride [H–LIr–X] complex from [LHIr][X] first involving coordination of the anion to the metal center to generate an electron rich neutral Vaska-type species [LHIr–X]. For the larger anions, bromide and iodide, there is a sizable kinetic barrier to this associative reaction and heating of their solutions is required to reach the neutral [LHIr–X] species. Once the halide coordinates to the metal and [LHIr–X] is formed, proton-transfer (or tautomerization) to form [H–LIr–X] occurs rapidly. The IrI neutral species [LHIr–X] are apparently less thermodynamically favored than their isomers, [LHIr][X] and [H–LIr–X].
When excess halide was added to the solutions of [LHIr][X] (X = Cl, Br, I), the IrIII–H species [H–LIr–X] was observed only for X = I. This result is readily explained on the basis of extensive hydrogen bonding of the imidazolyl protons with the chloride and bromide anions stabilizing their [LHIr–X] isomers such that the proton-transfer to the Ir center is disfavored. This hypothesis is supported by the downfield shift of the N–H proton of the chloride and bromide. Additional support for more extensive hydrogen bonding for the chloride and bromide relative to the iodide is found by comparison of the N⋯X distances in the [LHIr][X] complexes (Table 1). Also working to favor the tautomerization to the hydride species for the iodide congener is the greater σ-donor strength of the iodide relative to bromide and chloride.
The fluoride analog [LHIr][F] exists as a hydrogen bonded adduct [LHIr·F] in solution; the acidic proton of the coordinated imidazole and fluoride anion engage in a very strong hydrogen bond as indicated by an extremely downfield N–H resonance. Rather than adding to the metal to form the Vaska-type neutral [LHIr–F] analog, addition of excess fluoride to a solution of [LHIr][F] instead results in deprotonation with production of HF2− and the deprotonated species [LIr].
Beyond the outer/inner-sphere and the consequent metal–ligand proton tautomeric equilibrium (Scheme 3, horizontal equilibria) which is shown to be operative for the [LHIr][X] (X = Cl, Br, I) complexes, additional hydrogen bonding/acid–base equilibria are also operative in solution (Scheme 3, vertical equilibria). The extent of these latter equilibria is clearly more prevalent for the fluoride analog as fluoride is the strongest hydrogen bond acceptor and the strongest base among the halides. However, the lower CO stretching frequencies for the iodo (νCO = 1970 cm−1), bromo (νCO = 1969 cm−1), and chloro (νCO = 1967 cm−1) [LHIr][X] complexes (Table 1) as compared to that of the protonated [LHIr][PF6] species (νCO = 1973 cm−1) suggests that some small amount of a hydrogen bonding equilibria is present for all halide complexes.62
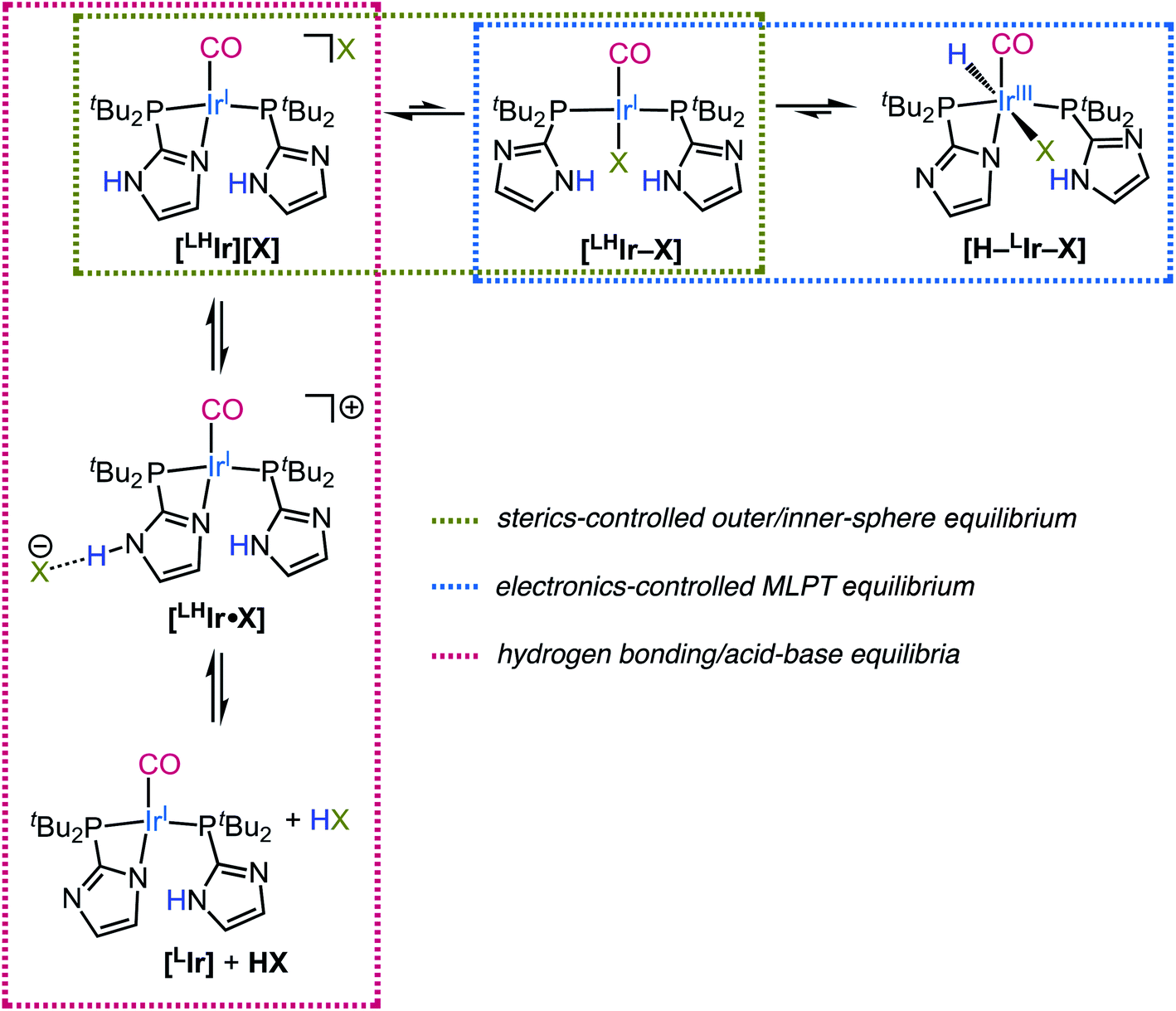 | ||
| Scheme 3 The outer/inner-sphere, MLPT, and hydrogen bonding/acid–base equilibria operative in solution for [LHIr][X] complexes. | ||
Thus overall, it can be noted that each of the [LHIr][X] complex equilibrates with multiple species in solution depending on the identity of X (Scheme 3). The different sterics, electronics, and hydrogen bonding propensities of the different halides can thus provide access to reactions involving an MLPT equilibrium to different extents. Using such insight, the community can better attempt to mimic the remarkable transformations of Nature which employ proton responsive moieties in the active site of enzymes to provide low energy pathways for selective and efficient reactivity.
Data availability
The experimental procedures and characterization data are available in the ESI.†Author contributions
A. K. J. carried out the syntheses and characterization of the reported complexes. The crystal structure data collection and analyses were performed by M. R. G. and P. J. C. The project was supervised by K. I. G., and the manuscript was written and edited by A. K. J. and K. I. G. All authors approved the final manuscript version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the US Department of Energy (DE-SC0018057), and the Vagelos Institute of Energy Science and Technology (fellowship to A. K. J.). Accurate Mass Measurements were carried out by Dr Charles W. Ross III (University of Pennsylvania). Elemental Analysis was performed by Dr William Brennessel at the CENTC Elemental Analysis Facility, University of Rochester (funded by NSF CHE-0650456). Prof. Eric J. Schelter's group (University of Pennsylvania) is thanked for sharing their IR instrument. Dr Jun Gu and Dr Chad W. Lawrence (University of Pennsylvania) are acknowledged for collecting the 13C{1H} NMR spectra. The NSF Major Research Instrumentation Program (NSF CHE-1827457), the NIH Supplemental Awards 3R01GM118510-03S1 and 3R01GM087605-06S1, and the Vagelos Institute of Energy Science and Technology supported the purchase of the NMR instruments used in this study. Judith Currano is acknowledged for assisting with online database and literature searches. Dr Jonathan L. Kuo and Dr Eric S. Cueny are thanked for helpful discussions and guidance.References
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- B. E. Barton, M. T. Olsen and T. B. Rauchfuss, J. Am. Chem. Soc., 2008, 130, 16834–16835 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- K. Weber, T. Krämer, H. S. Shafaat, T. Weyhermüller, E. Bill, M. van Gastel, F. Neese and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 20745–20755 CrossRef CAS PubMed.
- J. W. Slater, S. C. Marguet, S. L. Cirino, P. T. Maugeri and H. S. Shafaat, Inorg. Chem., 2017, 56, 3926–3938 CrossRef CAS PubMed.
- A. Aster, S. Wang, M. Mirmohades, C. Esmieu, G. Berggren, L. Hammarström and R. Lomoth, Chem. Sci., 2019, 10, 5582–5588 RSC.
- L. M. A. Quintana, S. I. Johnson, S. L. Corona, W. Villatoro, W. A. Goddard, M. K. Takase, D. G. VanderVelde, J. R. Winkler, H. B. Gray and J. D. Blakemore, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6409–6414 CrossRef PubMed.
- S. I. Johnson, H. B. Gray, J. D. Blakemore and W. A. Goddard, Inorg. Chem., 2017, 56, 11375–11386 CrossRef CAS PubMed.
- J. L. Kuo and K. I. Goldberg, J. Am. Chem. Soc., 2020, 142, 21439–21449 CrossRef CAS PubMed.
- S. Pal, S. Kusumoto and K. Nozaki, Organometallics, 2018, 37, 906–914 CrossRef CAS.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- T. P. Gonçalves, I. Dutta and K.-W. Huang, Chem. Commun., 2021, 57, 3070–3082 RSC.
- A. D. Wilson, R. K. Shoemaker, A. Miedaner, J. T. Muckerman, D. L. DuBois and M. R. DuBois, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6951–6956 CrossRef CAS PubMed.
- J. Y. Yang, R. M. Bullock, W. J. Shaw, B. Twamley, K. Fraze, M. R. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2009, 131, 5935–5945 CrossRef CAS PubMed.
- E. S. Wiedner, J. Y. Yang, S. Chen, S. Raugei, W. G. Dougherty, W. S. Kassel, M. L. Helm, R. M. Bullock, M. R. DuBois and D. L. DuBois, Organometallics, 2012, 31, 144–156 CrossRef CAS.
- T. D. Lohrey, J. I. Fostvedt, R. G. Bergman and J. Arnold, Chem. Commun., 2020, 56, 3761–3764 RSC.
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, J. Am. Chem. Soc., 2012, 134, 18843–18852 CrossRef CAS PubMed.
- G. M. Chambers, S. I. Johnson, S. Raugei and R. M. Bullock, Chem. Sci., 2019, 10, 1410–1418 RSC.
- K. Fagnou and M. Lautens, Angew. Chem., Int. Ed., 2002, 41, 26–47 CrossRef CAS.
- P. M. Maitlis, A. Haynes, B. R. James, M. Catellani and G. P. Chiusoli, Dalton Trans., 2004, 3409–3419 RSC.
- J. P. Flemming, M. C. Pilon, O. Y. Borbulevitch, M. Y. Antipin and V. V. Grushin, Inorg. Chim. Acta, 1998, 280, 87–98 CrossRef CAS.
- K. R. Flower, L. G. Leal and R. G. Pritchard, J. Organomet. Chem., 2005, 690, 3390–3396 CrossRef CAS.
- Y. Deng, G. Zhang, X. Qi, C. Liu, J. T. Miller, A. J. Kropf, E. E. Bunel, Y. Lan and A. Lei, Chem. Commun., 2015, 51, 318–321 RSC.
- E. W. Dahl and N. K. Szymczak, Angew. Chem., Int. Ed., 2016, 55, 3101–3105 CrossRef CAS PubMed.
- D. Hill, C. Delaney, M. Clark, M. Eaton, B. Hassan, O. Hendricks, D. K. Dang and R. U. Kirss, RSC Adv., 2017, 7, 34425–34434 RSC.
- J. R. Wilson, M. Zeller and N. K. Szymczak, Chem. Commun., 2021, 57, 753–756 RSC.
- D. C. Newman, M. R. Gau, P. J. Carroll and K. I. Goldberg, Organometallics, 2021, 40, 1806–1810 CrossRef CAS.
- D. B. Grotjahn, Y. Gong, A. G. DiPasquale, L. N. Zakharov and A. L. Rheingold, Organometallics, 2006, 25, 5693–5695 CrossRef CAS.
- D. B. Grotjahn, J. E. Kraus, H. Amouri, M.-N. Rager, A. L. Cooksy, A. J. Arita, S. A. Cortes-Llamas, A. A. Mallari, A. G. DiPasquale, C. E. Moore, L. M. Liable-Sands, J. D. Golen, L. N. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2010, 132, 7919–7934 CrossRef CAS PubMed.
- D. K. Dutta, B. Deb, B. J. Sarmah, J. D. Woollins, A. M. Z. Slawin, A. L. Fuller and R. A. M. Randall, Eur. J. Inorg. Chem., 2011, 2011, 835–841 CrossRef.
- Two sets of signals (one major and one minor) were also observed in the 13C{1H} NMR spectra. In the 31P{1H} NMR spectrum, however, signals (two sets of doublets) only for the major species (ionic [LHIr][Cl]) were observed. Signals corresponding to the minor Ir–H species could not be resolved due to its low concentraion as well as potential overlap with the signals for the major species (ionic [LHIr][Cl]). See ESI† for more details.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- Slight chemical shift differences (∼0.1 ppm for aromatic/aliphatic and ∼1.0 ppm for N–H resonances) are observed likely due to hydrogen bonding associated with Cl anion in solution.
- CSD accessed on July 27, 2021. Complexes for which their NMR data have been reported were shortlisted.
- U. Behrens and L. Dahlenburg, J. Organomet. Chem., 1976, 116, 103–111 CrossRef CAS.
- H. A. Mayer, R. Fawzi and M. Steimann, Chem. Ber., 1993, 126, 1341–1346 CrossRef CAS.
- S. Nemeh, R. J. Flesher, K. Gierling, C. Maichle-Mössmer, H. A. Mayer and W. C. Kaska, Organometallics, 1998, 17, 2003–2008 CrossRef CAS.
- S.-M. Kuang, D. A. Edwards, P. E. Fanwick and R. A. Walton, Inorg. Chim. Acta, 2003, 343, 275–280 CrossRef CAS.
- K. Ruhland and E. Herdtweck, Adv. Synth. Catal., 2005, 347, 398–404 CrossRef CAS.
- J. J. Adams, A. Lau, N. Arulsamy and D. M. Roddick, Organometallics, 2011, 30, 689–696 CrossRef CAS.
- A. F. Hill and C. M. A. McQueen, Organometallics, 2012, 31, 8051–8054 CrossRef CAS.
- J. M. Goldberg, G. W. Wong, K. E. Brastow, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, Organometallics, 2015, 34, 753–762 CrossRef CAS.
- L. Maser, C. Schneider, L. Vondung, L. Alig and R. Langer, J. Am. Chem. Soc., 2019, 141, 7596–7604 CrossRef CAS PubMed.
- C. Azerraf and D. Gelman, Organometallics, 2009, 28, 6578–6584 CrossRef CAS.
- K. J. Jonasson, A. V. Polukeev and O. F. Wendt, RSC Adv., 2015, 5, 15534–15538 RSC.
- W. J. Youngs, B. L. Simms and J. A. Ibers, J. Organomet. Chem., 1984, 272, 295–307 CrossRef CAS.
- M. Schulz and H. Werner, Organometallics, 1992, 11, 2790–2795 CrossRef CAS.
- Y. Segawa, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 9201–9203 CrossRef CAS PubMed.
- A.-K. Jungton, P. Kläring, T. Braun and A. Eißler, Z. Anorg. Allg. Chem., 2012, 638, 505–511 CrossRef CAS.
- P. Kläring, A.-K. Jungton, T. Braun and C. Müller, Eur. J. Inorg. Chem., 2012, 2012, 1430–1436 CrossRef.
- S. De-Botton, S. Cohen and D. Gelman, Organometallics, 2018, 37, 1324–1330 CrossRef CAS.
- L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1, 277–290 CrossRef CAS.
- To prove that such a shift of the N–H resonance in the presence of excess Cl anions is not due to the increased ionic strength of the solution, the in situ generated [LHIr–Cl] complex was treated with more ions in the form of (NBu4)(PF6). Upon addition of 10 molar equivalents of (NBu4)(PF6) salt to a solution of [LHIr–Cl] complex, the N–H resonance shifts by only 0.03 ppm (Fig. S28†). Thus, the observed shift in N–H resonance (>1 ppm) is not attributable to an increase in the ionic strength of the solution.
- J. M. Goldberg, L. M. Guard, G. W. Wong, D. F. Brayton, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, Organometallics, 2020, 39, 3323–3334 CrossRef CAS.
- CSD accessed on July 27, 2021.
- C. H. Bushweller, C. D. Rithner and D. J. Butcher, Inorg. Chem., 1986, 25, 1610–1616 CrossRef CAS.
- D. B. Grotjahn, Y. Gong, L. Zakharov, J. A. Golen and A. L. Rheingold, J. Am. Chem. Soc., 2006, 128, 438–453 CrossRef CAS PubMed.
- E. G. Bowes, D. D. Beattie and J. A. Love, Inorg. Chem., 2019, 58, 2925–2929 CrossRef CAS PubMed.
- The CO strecthing frequency for the fluoride complex is significantly closer to that of the deprotonated [LIr] species than the protonated [LHIr][PF6] species, likely a result of the extremely strong hydrogen bonding interaction with the fluoride anion in solution..
- K. O. Christe and W. W. Wilson, J. Fluorine Chem., 1990, 47, 117–120 CrossRef CAS.
- Several attempts to grow single crystals of the hydrogen bonded adduct of [LIr] suitable for X-ray diffraction were made, but to no avail. In one particular instance, however, vapor diffusion of the DCM solution with pentane produced very minor amounts of single crystals which were studied via X-ray diffraction. The resulting species was found to be a DCM adduct of the deprotonated species, Ir-2 (Fig. S42†). While Ir-2 is unlikely to be the proposed hydrogen bonded adduct of [LIr], it does suggest the generation of the neutral [LIr] species in solution (which in turn crystallizes as a DCM adduct).
- Similar systematic reductions in the N–X bond lengths from iodide to fluoride are evident in the solid-state structures of [LHIr][X]complexes (Table 1).
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and spectroscopic characterization of new complexes, NMR spectra for probing the equilibria of all halide complexes, Keq values for the MLPT equilibrium, and crystallographic data. CCDC 2123867–2123874. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00279e |
| This journal is © The Royal Society of Chemistry 2022 |