
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTorsional strain inversed chemoselectivity in a Pd-catalyzed atroposelective carbonylation reaction of dibenzothiophenium†
Qiuchi
Zhang
a,
Xiaoping
Xue
a,
Biqiong
Hong
b and
Zhenhua
Gu
 *ab
*ab
aDepartment of Chemistry, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 230026, P. R. China. E-mail: zhgu@ustc.edu.cn
bCollege of Materials and Chemical Engineering, Minjiang University, Fuzhou, Fujian 350108, P. R. China
First published on 1st March 2022
Abstract
A palladium-catalyzed enantioselective ring-opening/carbonylation of cyclic diarylsulfonium salts is reported. In comparison to thioethers, the sulfonium salts displayed high reactivity and enabled the reaction to be performed under mild conditions (room temperature). The steric repulsion of the two non-hydrogen substituents adjacent to the axis led cyclic diarylsulfonium salts to be distorted, which enabled the ring-opening reaction to proceed with significant preference for breaking the exocyclic C–S bond.
Introduction
Organosulfur compounds are important in synthetic organic chemistry, as they are both widely used as synthetic reagents and exist in many natural products and bioactive drugs. Importantly, dibenzo[b,d]thiophene and thianthrene derived sulfonium salts (I, II, III and IV) are widely used as electrophilic agents in organic synthesis, where the exo group usually works as the transferred functionality (Scheme 1a).1–4 The Umemoto reagent (I) is well-known as a powerful, thermally stable electrophilic trifluoromethylating agent. Sulfonium salts III and IV, prepared from the activation of sulfoxides, followed by the electrophilic thiolation of arenes, are efficient arylation transfer agents in transition-metal catalyzed cross-coupling, such as the Heck reaction, Sonogashira coupling, carbonylation etc. It was generally observed that the cleavage of the exocyclic S–aryl bond proceeded with significant preference for the cyclic S–aryl bond. In contrast, in 2015, Yorimitsu and co-workers reported a palladium-catalyzed ring-opening reaction of S–alkyl sulfonium salt (V), where the cyclic C–S bond was selectively cleaved to give acyclic biaryl compounds (Scheme 1b).5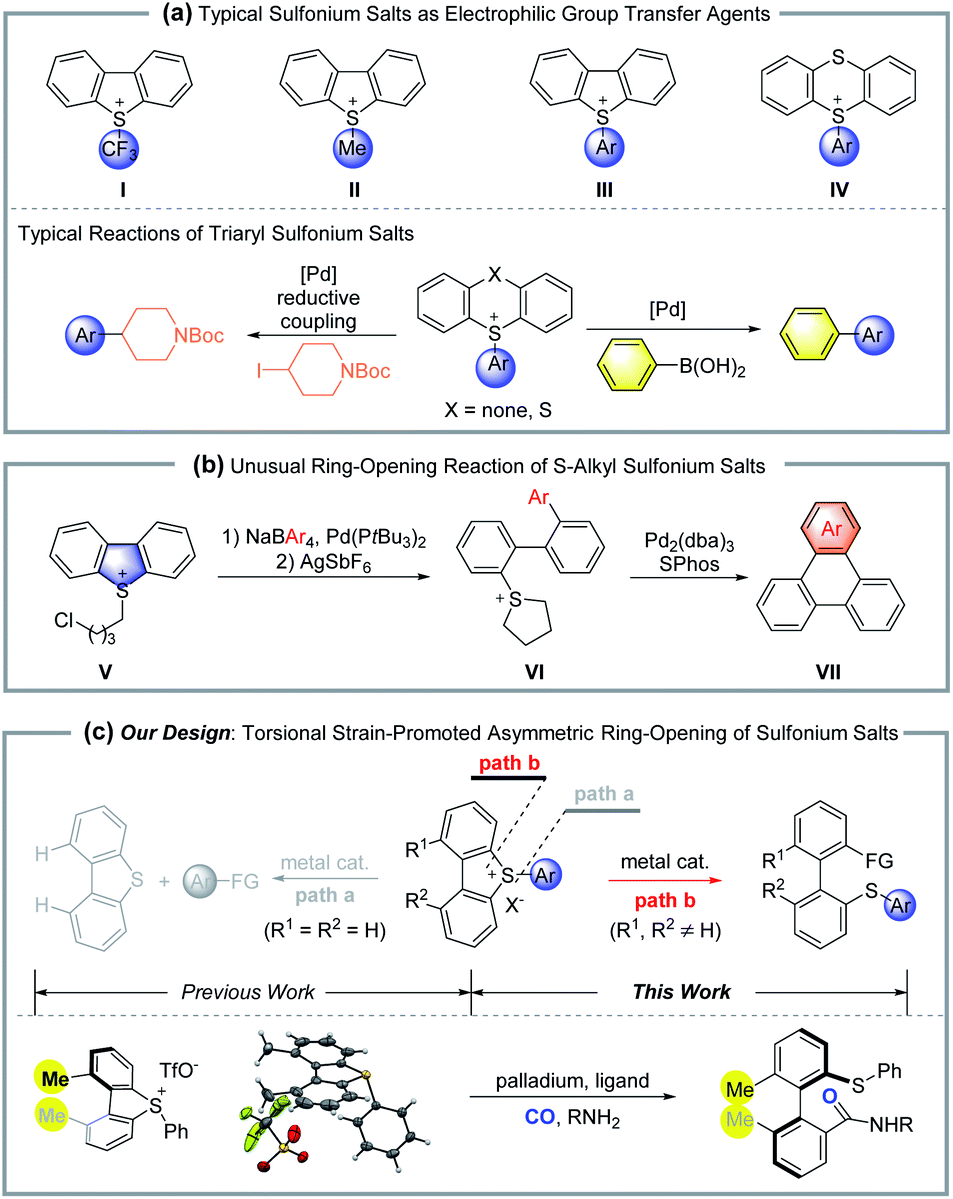 | ||
| Scheme 1 The selectivity of C–S bond cleavage for cyclic sulfonium salts. | ||
Atropisomers are an important class of compounds, which result from restricted rotation of a single C–C, C–N, C–O bond etc.6 They not only can be found in many bioactive natural products and drugs,7 but also are the leading skeleton for chiral ligands and catalysts. In view of the importance of axially chiral structures and their unique characters, several strategies have been developed for enantioselective synthesis of these kinds of compounds. Classic methods include asymmetric aryl–aryl cross-coupling,8de novo asymmetric aryl ring construction,9 desymmetrization or resolution,10 and point-to-axial chirality transfer.11 Bringmann and coworkers realized the asymmetric ring-opening reaction of lactones based on the fast dynamics of these biaryl fused lactones.12 Following that, the corresponding lactams, hemiacetals and N,O-acetals were successfully applied in atropisomer synthesis via ring-opening reactions.13 Recently, the ring-opening of diaryl fused iodonium salts,14 4,5-disubstituted 9H-fluoren-9-ols,15 silafluorenes16 and dinaphtho[2,1-b:1′,2′-d]furans17 showed excellent potential in functionalized axially chiral molecule synthesis.
In 2002, Hayashi reported a nickel-catalyzed atropisomer synthesis via the ring-opening reaction of dinaphtho[2,1-b:1′,2′-d]thiophene, which displayed low functional group tolerance by employing aryl Grignard reagents as the coupling partners.18 Alternatively, we reasoned that the corresponding sulfonium salts would show higher reactivity than dinaphthothiophene. Thus, the coupling partners can be extended to mild agents with excellent functional group tolerance. However, the concomitant challenge of this design is the chemoselectivity for cleavage of different C–S bonds (path a vs. path b) (Scheme 1c). Our previous studies indicated that the introduction of ortho, ortho′-disubstituents into dibenzo compounds would create torsional strain due to steric repulsion.14a Furthermore, the torsional strain would promote the subsequent ring-opening/bond breaking reactions. Herein, we report a palladium-catalyzed asymmetric ring-opening reaction of cyclic sulfonium salts for the synthesis of axially chiral biaryl thioethers, where the torsional strain of cyclic sulfonium not only promotes the C–S bond breaking, but also increases the chemoselectivity between different C–S bonds.
Results and discussion
Optimization
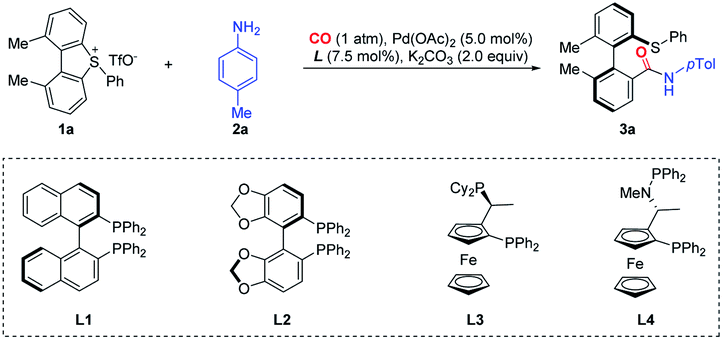
In the initial trials, we investigated a palladium-catalyzed ring-opening/carbonylation reaction by choosing 1,9-dimethyl-5-phenyl-5H-dibenzo[b,d]thiophen-5-ium triflate (1a) as the model substrate, whose distorted structure was confirmed by crystallographic analysis. In the atmosphere of CO (1 atm), the reaction catalyzed by Pd(OAc)2/(R)-BINAP (L1) gave moderate yield of ring-opening product 3a with 55% ee (Table 1, entry 1). In our survey of various chiral phosphine or related ligands, (Sa,R)-BoPhoz (L4) gave the best selectivity (entries 2–4). In terms of the solvent effect, THF displayed the same efficacy as 1,4-dioxane while using DMSO led to very low productivity (entries 5 and 6). Aromatic hydrocarbon solvents, i.e. toluene or p-cymene (pCM), led to a notable selectivity enhancement, which reached 73% ee and 84% ee, respectively (entries 7 and 8). Using a degassed and dried solvent does not provide a better ee value (entry 9). The reaction proceeded as efficiently at room temperature as at 60 °C (entry 10), however, further decreasing the temperature to 0 °C almost completely halted the reaction (entry 11). Further optimization of the solvent system indicated a beneficial effect of certain amounts of THF (p-cymene/THF), which improved both the yield and stereoselectivity and about 1% amount of THF showed the best result for stereoselectivity (entry 12–14). The base K3PO4 had a slightly better performance than K2CO3 (entry 15). Pd2dba3 gave a high yield and excellent enantioselectivity with a prolonged reaction time (entry 16).|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | T/°C | Yield/% | ee/% |
| a Conditions: 1a (43.9 mg, 0.10 mmol, 1.0 equiv.), 2a (21.4 mg, 0.20 mmol, 2.0 equiv.), CO (1 atm), Pd(OAc)2 (1.1 mg, 0.0050 mmol, 5.0 mol%), ligand (0.0075 mmol, 7.5 mol%), K2CO3 (27.6 mg, 0.20 mmol, 2.0 equiv.) in 1 mL of solvent for 24 h. b Reaction time is 3 days. c K3PO4 (1.0 equiv.) was used. d Pd2(dba)3 (2.5 mol%) was used. e The solvent was degassed and dried by 4 Å MS. | |||||
| 1 | L1 | Dioxane | 60 | 44 | 55 |
| 2 | L2 | Dioxane | 60 | 42 | 50 |
| 3 | L3 | Dioxane | 60 | 21 | 52 |
| 4 | L4 | Dioxane | 60 | 55 | 60 |
| 5 | L4 | THF | 60 | 48 | 60 |
| 6 | L4 | DMSO | 60 | NR | — |
| 7 | L4 | Toluene | 60 | 34 | 73 |
| 8 | L4 | pCM | 60 | 48 | 84 |
| 9 | L4 | pCMe | 60 | 61 | 78 |
| 10 | L4 | pCM | 25 | 53 | 82 |
| 11b | L4 | pCM | 0 | Trace | — |
| 12 | L4 |
pCM/THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
25 | 77 | 83 |
| 13 | L4 |
pCM/THF (4:1) |
25 | 76 | 85 |
| 14 | L4 |
pCM/THF (100:1) |
25 | 73 | 88 |
| 15c | L4 |
pCM/THF (100:1) |
25 | 80 | 89 |
| 16c,d | L4 |
pCM/THF (100:1) |
25 | 92 | 90 |
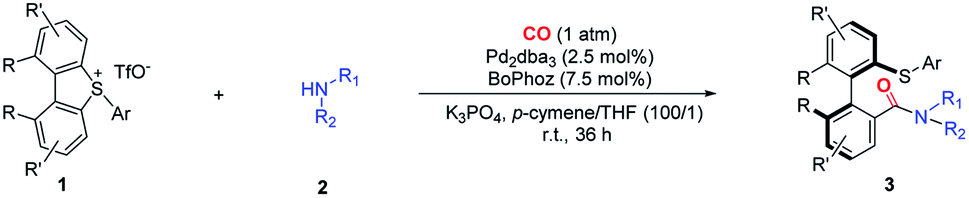
Substrate scope
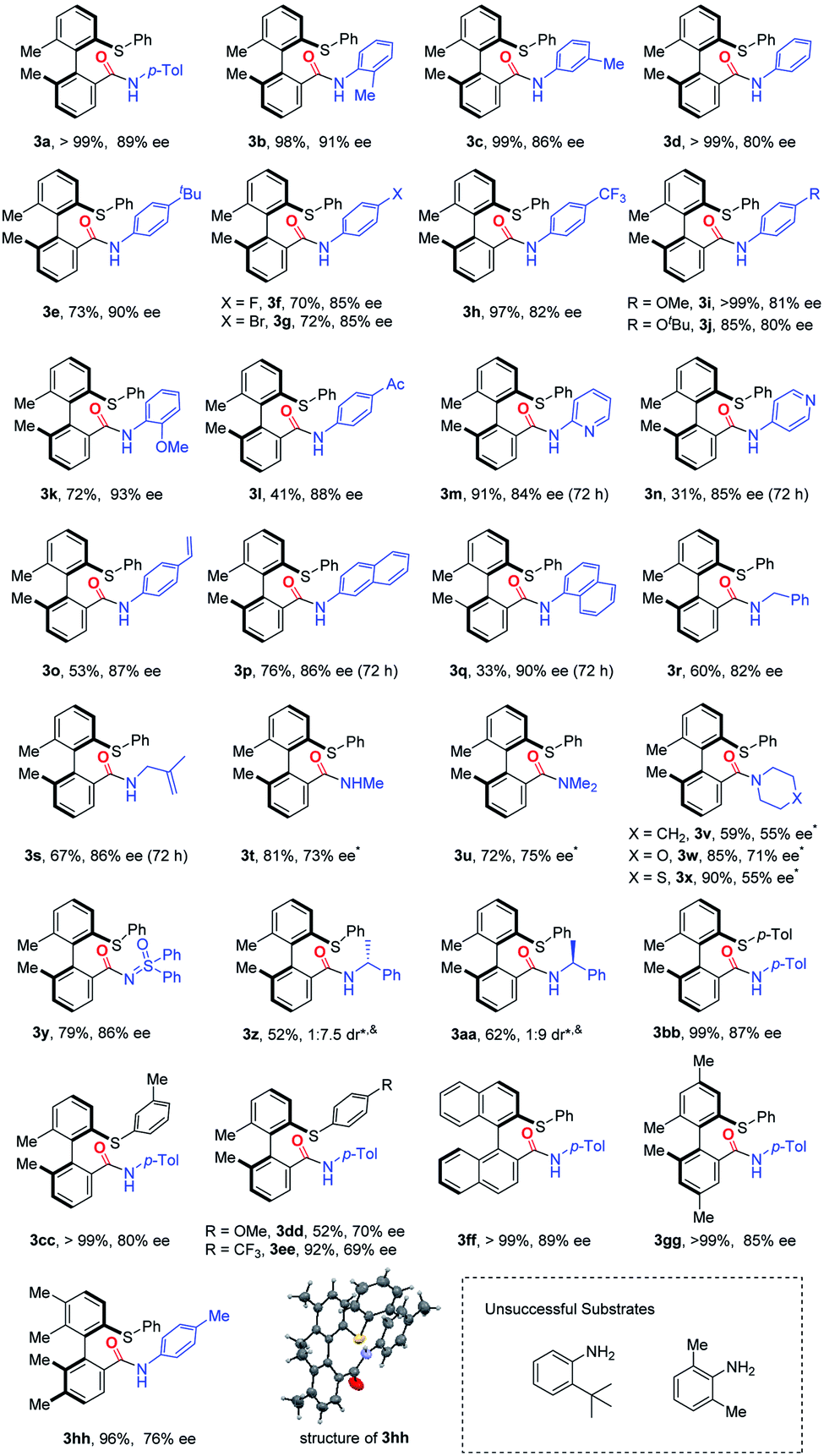
With the optimal conditions in hand, we started investigating the substrate scope of this asymmetric carbonylation reaction (Table 2). Substituted aromatic amines were tested, whose reactivities significantly depend on the nature of the substituents. Aniline derivatives bearing para, meta and ortho alkyl groups, as well as β-, or α-naphthalenamine, displayed good reactivity and selectivity, except the very bulky 2-tert-butyl aniline or 2,6-dimethyl aniline (3a–3e, 3p and 3q). Although it is a palladium(0)-catalyzed reaction, aryl–Br bonds and terminal alkene groups in the aniline substrates are tolerable, and the reaction provided comparable yields of the products (3g and 3o). Both electron-donating (3i, 3j) and electron-withdrawing (3h) groups at the para position of aniline slightly reduced stereoselectivity, while o-toluidine produced high enantioselectivity (3k). 2- and 4-Pyridinamines were equally effective substrates, although the latter gave the corresponding product in a dropped yield (31%) (3m and 3n).|
|
|---|
| a *60 °C for 24 h; &optically pure amine was used. |
|
This method is also amendable to apply to substrates with the amino group attached to sp3-hybridized carbon. Both benzylamine and 2-methylallylamine reacted smoothly with cyclic diarylsulfonium salts and the products were formed with good stereoselectivity (3r and 3s). Aliphatic amines showed relatively lower reactivity, which was possibly attributed to the potential saturated coordination of the metal center with aliphatic amines. Therefore, the corresponding reactions with aliphatic amines should be conducted at a higher temperature (60 °C) (3t–3x). Either primary or secondary (both acyclic and cyclic) amines gave decreased enantioselectivity. In addition, sulfoximine can take part in this reaction, and resulted in a similar yield and ee value in comparison with aromatic amines (3y). Pre-installed chiral elements at amines had a marginal effect on the stereoselectivity: both the reactions with (R)- and (S)-1-phenylethan-1-amine gave a pair of diastereomers with the dr values being 1:7.5 and 9:1, respectively (3z and 3aa).
We further studied the substituent effect of cyclic diarylsulfonium salts. Both electron-donating and electron-withdrawing substituents on the acyclic phenyl ring caused a decrease of the ee values (3bb–3ee). Replacement of the biphenyl skeleton with the binaphthyl structure produced a similar yield and enantioselectivity (3ff). Introducing p,p′-dimethyl groups to the biphenyl structure (3gg) did not have a significant effect on the results, while the corresponding m,m′-dimethyl substituted sulfonium salt gave a reduced ee value (76%) (3hh). Notably, a single crystal structure of 3hh was obtained by slow evaporation of a mixed solvent (DCM/Et3N = 1:3), which finally determined the absolute configuration of the axial chirality to be R.
In support of the role of torsional strain, we investigated the ring-opening reaction with sulfonium salt 1i, which bears two hydrogen atoms at the two adjacent positions of the biaryl axis (Scheme 2). Notably, the reaction became sluggish under standard conditions, and 50% of starting material 1i was recovered even with a prolonged reaction time (5 days). The loss of torsional strain results in no yield of the ring-opening product. The formation of N-(p-tolyl)benzamide 4a and dibenzothiophene 5a were confirmed by crude 1H NMR or GC-mass spectroscopy. These results indicated that 1,9-disubstituents on the sulfonium salt, which create the torsional strain, are crucial for both reactivity and selectivity for this ring-opening reaction.
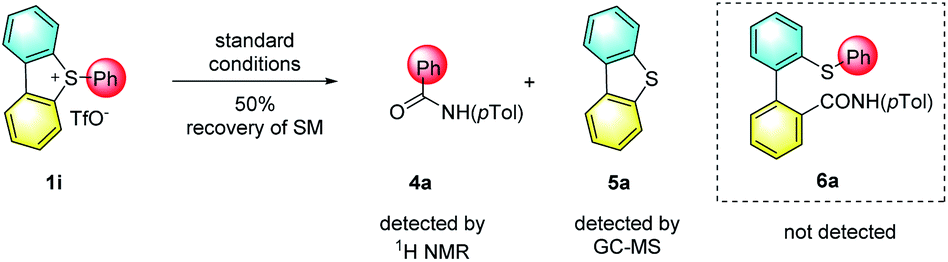 | ||
| Scheme 2 Reaction of sulfonium salt 1i. | ||
To further support our hypothesis that the torsional strain enhances the reactivity and selectivity, we calculated the torsional strain of 1a at the B3LYP level of theory.19 By following the calculation methods of ring strain for cyclic hydrocarbons,20 we determined the torsional strain of 1a by calculating the hydrogenation free energy difference between compound 1a and 1i by breaking one C–S bond. In order to avoid the discrepancy caused by the two methyl groups, we further calculated the hydrogenation free energy difference between compound 7a and 7i (Scheme 3). Thus, the torsional strain of 1a is estimated to be {ΔG(strain) = [ΔG(1i) − ΔG(1a)] − [ΔG(7i) − ΔG(7a)]} = 11.05 kcal mol−1.
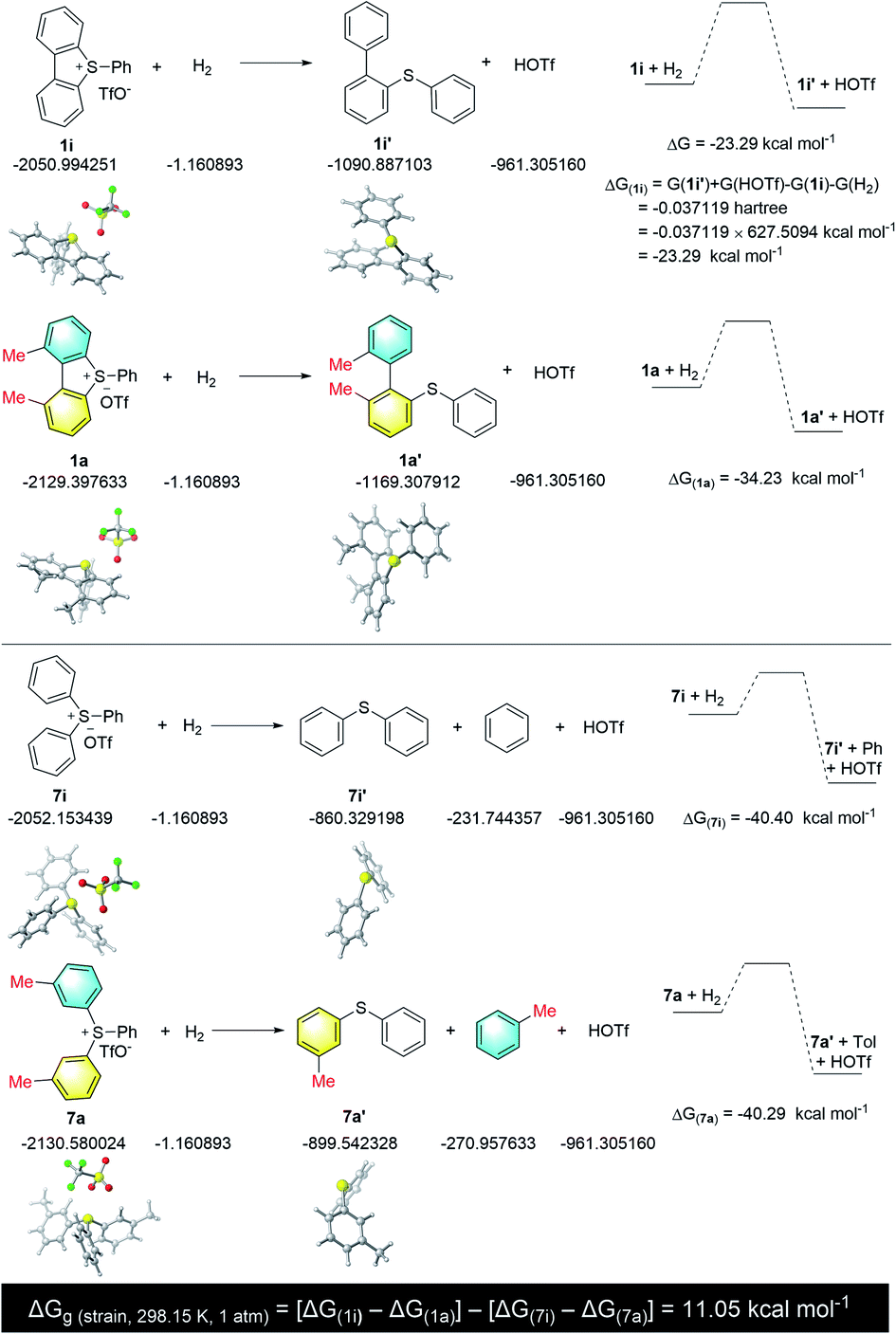 | ||
| Scheme 3 Calculation of torsional strain. | ||
Mechanistic studies
Following the above studies and previous reports,4,5 a tentative catalytic cycle with canonical organometallic steps is proposed (Scheme 4). The (R)-conformer and (S)-conformer of sulfonium salt 1 have a fast equilibrium at ambient temperature, and the oxidative addition of Pd(0)/(Sa,R)-BoPhoz and (R)-conformer is preferred over that of the (S)-conformer, which gave the palladium intermediate Int-1 with R axial chirality of the biaryl skeleton. Alternatively, the oxidative addition of Pd(0) with the exocyclic C–S bond would give Int-2 and is concomitant with the formation of dibenzothiophene. This undesired pathway became unfavourable due to torsional strain caused by the steric repulsion. Acylpalladium Int-3 would be formed upon the interaction of Int-1 and CO via a migration/insertion process. Subsequently, the amine coordination, followed by deprotonation and reductive elimination, produced amide 3 and therefore regenerated the Pd(0) catalyst.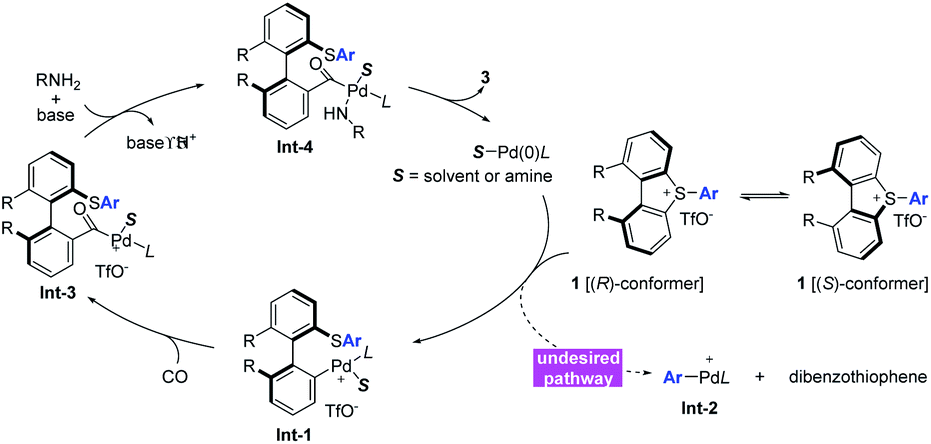 | ||
| Scheme 4 Plausible mechanism. | ||
Conclusions
We reported a palladium-catalyzed enantioselective ring-opening/carbonylation reaction of cyclic diarylsulfonium salts to form thioether-containing axially chiral biaryls. The success of this reaction benefited from the torsional strain created by steric repulsion, which enabled the high chemoselectivity of bond breaking between the exocyclic C–S bond and the cyclic one.Data availability
All experimental and characterization data, as well as DFT calculation data are available in the ESI.† Crystallographic data for compounds 1a and (R)-3hh have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under accession numbers CCDC 2129903 and 2129904.Author contributions
Q. Z. initiated this work. Q. Z. and B. H. performed the experiments. X. X. performed the DFT calculations. Q. Z. and Z. G. designed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Key R&D Program of China (No. 2021YFA1500100), the National Natural Science Foundation of China (No. 92056102, 21871241), USTC Research Funds of the Double First-Class Initiative (YD2060002014) and Minjiang University (32903602). We thank the Supercomputing Center of University of Science and Technology of China for providing the computational resources.Notes and references
- T. Umemoto, B. Zhang, T. Zhu, X. Zhou, P. Zhang, S. Hu and Y. Li, J. Org. Chem., 2017, 82, 7708–7719 CrossRef CAS.
- (a) D. C. Simkó, P. Elekes, V. Pázmándi and Z. Novák, Org. Lett., 2018, 20, 676–679 CrossRef PubMed; (b) K. K. Laali, J.-H. Chun and T. Okazaki, J. Org. Chem., 2007, 72, 8383–8393 CrossRef CAS PubMed.
- (a) T. Gendron, K. Sander, K. Cybulska, L. Benhamou, P. K. B. Sin, A. Khan, M. Wood, M. J. Porter and E. Årstad, J. Am. Chem. Soc., 2018, 140, 11125–11132 CrossRef CAS; (b) M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Nat. Catal., 2020, 3, 163–169 CrossRef CAS; (c) K. Kafuta, A. Korzun, M. Böhm, C. Golz and A. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 1950–1955 CrossRef CAS; (d) M. Hori, T. Kataoka, H. Shimizu, M. Miyagaki and T. Takagi, Chem. Pharm. Bull., 1974, 22, 2030–2041 CrossRef CAS; (e) E. M. Alvarez, T. Karl, F. Berger, L. Torkowski and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 13609–13613 CrossRef CAS; (f) P. Xu, D. Zhao, F. Berger, A. Hamad, J. Rickmeier, R. Petzold, M. Kondratiuk, K. Bohdan and T. Ritter, Angew. Chem., Int. Ed., 2020, 59, 1956–1960 CrossRef CAS.
- (a) F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223 CrossRef CAS; (b) X. Ming, Z. Tian and C. Zhang, Chem.–Asian J., 2019, 14, 3370–3379 CrossRef CAS; (c) Y. Ye, J. Zhu and Y. Huang, Org. Lett., 2021, 23, 2386–2391 CrossRef CAS; (d) J. Wu, Z. Wang, X. Chen, Y. Wu, D. Wang, Q. Peng and P. Wang, Sci. China: Chem., 2020, 63, 336–340 CrossRef CAS; (e) B. Lansbergen, P. Granatino and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7909–7914 CrossRef CAS; (f) A. Selmani and F. Schoenebeck, Org. Lett., 2021, 23, 4779–4784 CrossRef CAS.
- D. Vasu, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 7162–7166 CrossRef CAS PubMed.
- (a) G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615–624 CrossRef CAS PubMed; (b) J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, Angew. Chem., Int. Ed., 2009, 48, 6398–6401 CrossRef CAS; (c) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS; (d) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (e) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed; (f) A. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804–3815 RSC; (g) J. Feng and Z. Gu, SynOpen, 2021, 5, 68–85 CrossRef CAS; (h) J. K. Cheng, S.-H. Xiang, S. Li and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed.
- (a) J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, Angew. Chem., Int. Ed., 2009, 48, 6398–6401 CrossRef CAS; (b) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC.
- (a) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC; (b) D. Zhang and Q. Wang, Coord. Chem. Rev., 2015, 286, 1–16 CrossRef CAS; (c) T. Hayashi, K. Hayashizaki, T. Kiyoi and Y. Ito, J. Am. Chem. Soc., 1988, 110, 8153–8156 CrossRef CAS; (d) X. Shen, G. O. Jones, D. A. Watson, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2012, 132, 11278–11287 CrossRef PubMed; (e) G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570–573 CrossRef CAS PubMed; (f) Y.-H. Chen, D.-J. Cheng, J. Zhang, Y. Wang, X.-Y. Liu and B. Tan, J. Am. Chem. Soc., 2015, 137, 15062–15065 CrossRef CAS; (g) H. Yang, J. Sun, W. Gu and W. Tang, J. Am. Chem. Soc., 2020, 142, 8036–8043 CrossRef CAS; (h) S. Yan, W. Xia, S. Li, Q. Song, S.-H. Xiang and B. Tan, J. Am. Chem. Soc., 2020, 142, 7322–7327 CrossRef CAS PubMed.
- (a) K. Tanaka, Transition-Metal-Mediated Aromatic Ring Construction, John Wiley, 2013 CrossRef; (b) K. Tanaka, G. Nishida, A. Wada and K. Noguchi, Angew. Chem., Int. Ed., 2004, 43, 6510–6512 CrossRef CAS PubMed; (c) A. Link and C. Sparr, Angew. Chem., Int. Ed., 2014, 53, 5458–5461 CrossRef CAS; (d) T. Shibata, T. Fujimoto, K. Yokota and K. Takagi, J. Am. Chem. Soc., 2004, 126, 8382–8383 CrossRef CAS PubMed.
- (a) A. Ros, B. Estepa, P. Ramírez-López, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2013, 135, 15730–15733 CrossRef CAS; (b) V. Bhat, S. Wang, B. M. Stoltz and S. C. Virgil, J. Am. Chem. Soc., 2013, 135, 16829–16832 CrossRef CAS; (c) J. Zheng and S. L. You, Angew. Chem., Int. Ed., 2014, 53, 13244–13247 CrossRef CAS; (d) J. Wang, M. W. Chen, Y. Ji, S. B. Hu and Y. G. Zhou, J. Am. Chem. Soc., 2016, 138, 10413–10416 CrossRef CAS PubMed; (e) M. Hirai, S. Terada, H. Yoshida, K. Ebine, T. Hirata and O. Kitagawa, Org. Lett., 2016, 18, 5700–5703 CrossRef CAS PubMed; (f) G. Ma, J. Deng and M. P. Sibi, Angew. Chem., Int. Ed., 2014, 53, 11818–11821 CrossRef CAS PubMed; (g) C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871–13875 CrossRef CAS PubMed; (h) T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139–2144 CrossRef CAS; (i) Q. Dherbassy, G. Schwertz, M. Chessé, C. K. Hazra, J. Wencel-Delord and F. Colobert, Chem.–Eur. J., 2016, 22, 1735–1743 CrossRef CAS PubMed.
- (a) F. Guo, L. C. Konkol and R. J. Thomson, J. Am. Chem. Soc., 2011, 133, 18–20 CrossRef CAS PubMed; (b) V. S. Raut, M. Jean, N. Vanthuyne, C. Roussel, T. Constantieux, C. Bressy, X. Bugaut, D. Bonne and J. Rodriguez, J. Am. Chem. Soc., 2017, 139, 2140–2143 CrossRef CAS.
- (a) G. Bringmann, A. J. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed; (b) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (c) S. T. Toenjes and J. L. Gustafson, Future Med. Chem., 2018, 10, 409–422 CrossRef CAS PubMed.
- (a) K. Mori, T. Itakura and T. Akiyama, Angew. Chem., Int. Ed., 2016, 55, 11642–11646 CrossRef CAS; (b) J. Zhang and J. Wang, Angew. Chem., Int. Ed., 2018, 57, 465–469 CrossRef CAS PubMed; (c) D. Guo, Q. Peng, B. Zhang and J. Wang, Org. Lett., 2021, 23(20), 7765–7770 CrossRef CAS PubMed.
- (a) K. Zhao, L. Duan, S. Xu, J. Jiang, Y. Fu and Z. Gu, Chem, 2018, 4, 599–612 CrossRef CAS; (b) X. Xue and Z. Gu, Org. Lett., 2019, 21, 3942–3945 CrossRef CAS; (c) K. Zhu, K. Xu, Q. Fang, Y. Wang, B. Tang and F. Zhang, ACS Catal., 2019, 9, 4951–4957 CrossRef CAS; (d) L. Duan, K. Zhao, Z. Wang, F. Zhang and Z. Gu, ACS Catal., 2019, 9, 9852–9858 CrossRef CAS; (e) K. Zhu, Z. Song, Y. Wang and F. Zhang, Org. Lett., 2020, 22, 9356–9359 CrossRef CAS PubMed; (f) X. Zhang, K. Zhao, N. Li, J. Yu, L. Gong and Z. Gu, Angew. Chem., Int. Ed., 2020, 59, 19899–19904 CrossRef CAS PubMed; (g) K. Zhu, Z. Song, Y. Wang and F. Zhang, Org. Lett., 2020, 22, 9356–9359 CrossRef CAS PubMed; (h) J. Ke, B. Zu, Y. Guo, Y. Li and C. He, Org. Lett., 2021, 23, 329–333 CrossRef CAS PubMed; (i) K. Zhao, S. Yang, Q. Gong, L. Duan and Z. Gu, Angew. Chem., Int. Ed., 2021, 60, 5788–5793 CrossRef CAS PubMed.
- R. Deng, J. Xi, Q. Li and Z. Gu, Chem, 2019, 5, 1834 CAS.
- (a) J. Feng, X. Bi, X. Xue, N. Li, L. Shi and Z. Gu, Nat. Commun., 2020, 11, 4449 CrossRef CAS PubMed; (b) X. Bi, J. Feng, X. Xue and Z. Gu, Org. Lett., 2021, 23, 3201–3206 CrossRef CAS PubMed.
- J. Zhang, T. Sun, Z. Zhang, H. Cao, Z. Bai and Z.-C. Cao, J. Am. Chem. Soc., 2021, 143, 18380–18387 CrossRef CAS PubMed.
- T. Shimada, Y. H. Cho and T. Hayashi, J. Am. Chem. Soc., 2002, 124, 13396–13397 CrossRef CAS PubMed.
- (a) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. v. R. Schleyer, J. E. Williams and K. R. Blanchard, J. Am. Chem. Soc., 1970, 92, 2377–2386 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2129903 and 2129904. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00341d |
| This journal is © The Royal Society of Chemistry 2022 |