
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid access to t-butylalkylated olefins enabled by Ni-catalyzed intermolecular regio- and trans-selective cross-electrophile t-butylalkylation of alkynes†
Yi-Zhou
Zhan
,
Huan
Meng
and
Wei
Shu
 *
*
Shenzhen Grubbs Institute, Department of Chemistry, Guangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen 518055, Guangdong, P. R. China. E-mail: shuw@sustech.edu.cn
First published on 30th March 2022
Abstract
Among the carbo-difunctionalization of alkynes, the stereoselective dialkylation of alkynes is the most challenging transformation due to associated competitive side reactions and thus remains underdeveloped. Herein, we report the first Ni-catalyzed regio- and trans-selective cross-dialkylation of alkynes with two distinct alkyl bromides to afford olefins with two aliphatic substituents. The reductive conditions circumvent the use of organometallic reagents, enabling the cross-dialkylation process to occur at room temperature from two different alkyl bromides. This operationally simple protocol provides a straightforward and practical access to a wide range of stereodefined dialkylated olefins with broad functional group tolerance from easily available starting materials.
Introduction
Multi-alkyl substituted olefins with defined stereochemistry are ubiquitous subunits in natural products, bioactive molecules and conjugated organic materials,1 and also serve as important precursors for other functional groups due to their orthogonal reactivity with respect to carbonyls and other polar functional groups.2 Although numerous methods to construct olefins have been well-established, the stereoselective synthesis of multi-substituted olefins, particularly multialkyl-substituted ones, remains an unmet challenge.3 Over the past decades, the carbo-difunctionalization of alkynes represents one of the most step-economic and efficient ways to construct multi-substituted olefins as they selectively forge two carbon–carbon bonds and build up molecular complexity from simple starting materials in one single step.4 Among these, the Ni-catalyzed cis-selective carbo-difunctionalization of alkynes has been extensively explored using different strategies, such as the carbonickelation of alkynes,5 migratory insertion of alkynes into the nickelacycle intermediates,6 cyclonickelation of alkynes with unsaturated systems,7 and olefin isomerization by photo-redox/nickel dual catalysis.8 On the contrary, the Ni-catalyzed trans-selective carbo-difunctionalization of alkynes is still nontrivial.4 One strategy for the trans-selective carbo-difunctionalization of alkynes relies on the Z/E isomerization of the key alkenylnickel intermediates from the carbo-metallation of alkynes to form thermodynamically more stable alkenylnickel species (Scheme 1a). Arai,9 Martin and Montgomery10 developed the Ni-catalyzed trans-selective difunctionalization of alkynes, in which the E/Z isomerization of alkenylnickel species was dominated by the steric hindrance of substrates. Moreover, Liu,11 Lam,12 Kong13 and others14 disclosed a Z/E isomerization driven by the formation of chelation in the E-alkenyl-nickel species facilitated by a tethered chelation in the substrate to deliver the trans-alkylarylation of alkynes. Unfortunately, this strategy could not be applied to intermolecular three-component reactions. The other strategy is the Ni-initiated trans-radical addition onto alkynes, followed by a Ni-catalyzed coupling with organometallic reagents (Scheme 1b). Kambe developed a Ni-catalyzed regioselective alkylarylation of alkynes using alkyl iodides and aryl metallic reagents (M = Mg, Zn).15 Using a similar strategy, Nevado et al. disclosed an Ni-catalyzed intermolecular three-component coupling reaction of terminal alkynes with alkyl halides and arylboronic acids (M = B) for the trans-selective synthesis of 1,1-bisarylolefins.16 This approach provides a valuable platform to install C(sp3)-components onto alkynes with defined regio- and stereoselectivity. To date, the above-mentioned two strategies are still not able to access the trans-dialkylation of alkynes due to associated challenges, such as β-hydrogen elimination of alkylnickel intermediates, homocoupling of alkyl precursors, and hydroalkylation of alkynes (Scheme 1c).17 Moreover, the regio- and stereoselectivity issues of intermolecular reactions impose an additional challenge for the dialkylation of alkynes. To this end, the use of two distinct alkyl electrophiles to sequentially couple across alkynes under reductive conditions renders an appealing alternative to access multialkylated olefins with defined stereochemistry.18 As our continuous interest in earth-abundant metal-catalyzed reductive selective transformations,19 we herein report the Ni-catalyzed radical addition of one alkyl bromide onto alkynes, followed by a Ni-catalyzed coupling with another alkyl halide to afford the dialkylation of alkynes with exquisite trans-selectivity under reductive conditions (Scheme 1d). This three-component reaction enables the sequential installation of two distinct alkyl bromides onto alkynes, allowing for the regio- and diastereoselective dialkylation of alkynes without the use of organometallic reagents at room temperature.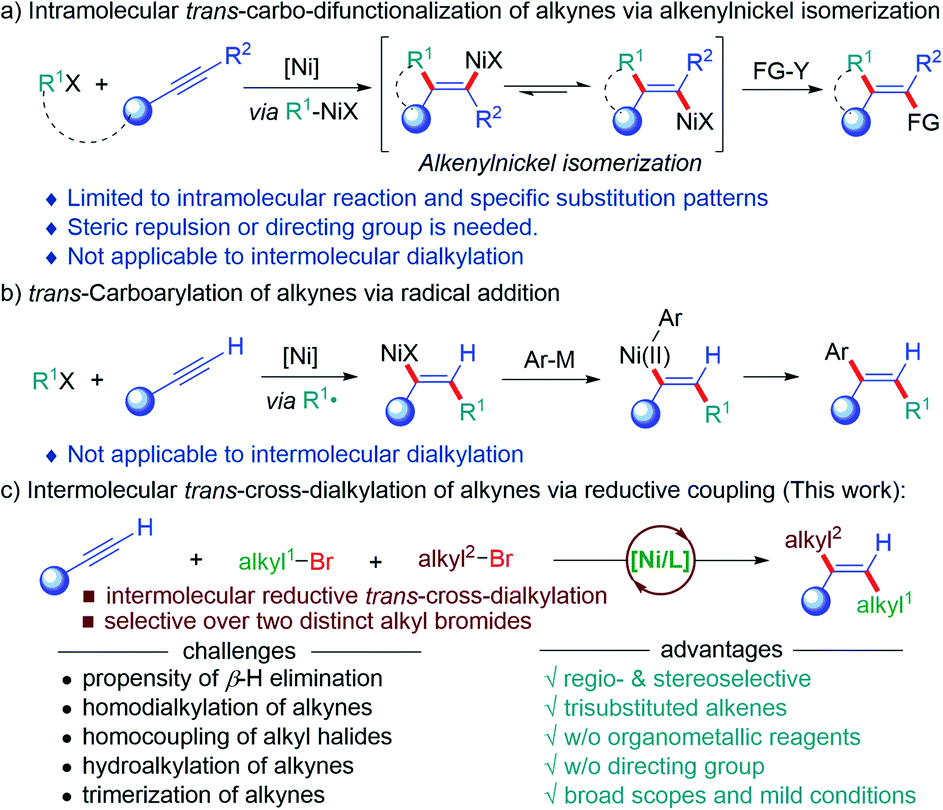 | ||
| Scheme 1 Ni-catalyzed trans-selective carbo-difunctionalization of alkynes. | ||
Results and discussion
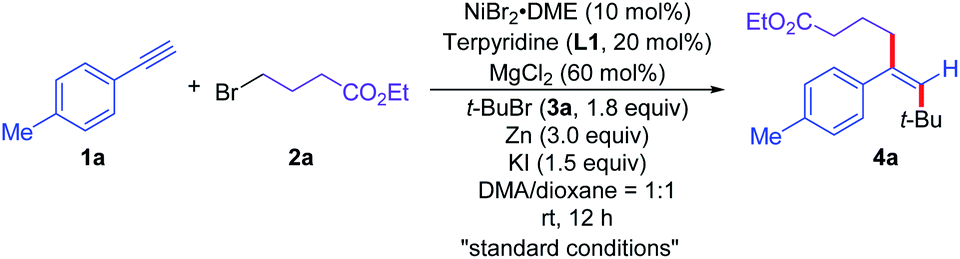
To test the feasibility of this proposal, we commenced the dialkylation reaction of alkynes using alkyne (1a), ethyl 4-bromobutanate (2a), and tert-butyl bromide (3a) as the prototype substrates under reductive conditions. After the evaluation of a wide range of parameters, the use of NiBr2·DME (10 mol%), terpyridine (L1, 20 mol%), MgCl2 (60 mol%), zinc (3.0 equiv.), potassium iodide (1.5 equiv.) in the mixture of N,N-dimethylacetamide (DMA) and 1,4-dioxane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature was defined as standard conditions for the reaction, affording the cross-electrophile trans-cross-dialkylation product 4a in 75% isolated yield (Table 1, entry 1). No desired product 4a was detected in the absence of L1, while 4a was formed in 53% yield with 10 mol% of L1 (Table 1, entries 2 and 3). Other nickel-based precatalysts could also mediate the reaction to afford 4a, albeit in lower efficiency (Table 1, entries 4–6). Replacing the reductant with manganese furnished comparable outcome, delivering 4a in 74% yield (Table 1, entry 7). Both nickel catalyst and reductant are required for this reductive cross-dialkylation reaction. No desired reaction occurred in the absence of nickel catalyst or reductant (Table 1, entry 8). The use of potassium iodide significantly enhanced the reactivity of the cross-dialkylation reaction probably due to the slow halide exchange with the alkyl bromides (Table 1, entries 9 and 10). The reaction furnished 4a in 71% yield without MgCl2 (Table 1, entry 11).20 The reaction delivered diminished yields in either DMA or 1,4-dioxane, affording the desired product 4a in 68% and 25% yields, respectively (Table 1, entries 12 and 13).21
:1) at room temperature was defined as standard conditions for the reaction, affording the cross-electrophile trans-cross-dialkylation product 4a in 75% isolated yield (Table 1, entry 1). No desired product 4a was detected in the absence of L1, while 4a was formed in 53% yield with 10 mol% of L1 (Table 1, entries 2 and 3). Other nickel-based precatalysts could also mediate the reaction to afford 4a, albeit in lower efficiency (Table 1, entries 4–6). Replacing the reductant with manganese furnished comparable outcome, delivering 4a in 74% yield (Table 1, entry 7). Both nickel catalyst and reductant are required for this reductive cross-dialkylation reaction. No desired reaction occurred in the absence of nickel catalyst or reductant (Table 1, entry 8). The use of potassium iodide significantly enhanced the reactivity of the cross-dialkylation reaction probably due to the slow halide exchange with the alkyl bromides (Table 1, entries 9 and 10). The reaction furnished 4a in 71% yield without MgCl2 (Table 1, entry 11).20 The reaction delivered diminished yields in either DMA or 1,4-dioxane, affording the desired product 4a in 68% and 25% yields, respectively (Table 1, entries 12 and 13).21
|
|
||
|---|---|---|
| Entries | Variations from “standard conditions” | Yield of 4aa |
|
a The reaction was conducted using 0.2 mmol of 1a, 0.6 mmol of 2a, 0.36 mmol of 3a in a mixture of DMA and 1,4-dioxane (1:1, 0.033 M) under indicated conditions for 24 h. N.R. = No reaction. L1 = 2,2':6′,2′′-terpyridine. Yield was determined by GC analysis using n-dodecane as internal standard. Isolated yield after flash chromatography is shown in the parentheses.
|
||
| 1 | None | 77% (75%) |
| 2 | w/o L1 | N.R. |
| 3 | L1 (10 mol%) | 53% |
| 4 | NiCl2·DME instead of NiBr2·DME | 73% |
| 5 | NiBr2 instead of NiBr2·DME | 57% |
| 6 | NiI2 instead of NiBr2·DME | 56% |
| 7 | Mn instead of Zn | 74% |
| 8 | w/o “Ni” or Zn | N.R. |
| 9 | NaI instead of KI | 49% |
| 10 | w/o KI | 42% |
| 11 | w/o MgCl2 | 71% |
| 12 | DMA as solvent | 68% |
| 13 | 1,4-Dioxane as solvent | 25% |
With the optimized conditions in hand, we turned to evaluate the scope of the reaction. First, the scope of alkynes (1) was examined (Scheme 2). Various aromatic alkynes with para-substituents were all good substrates for this transformation, affording the desired dialkylated olefins in moderate to good yields (4b–4l). Electron-donating groups, such as alkyl, dimethylamino, silyl groups (4c–4g) and electron-withdrawing (4h–4l) groups, such as halogens, trifluoromethyl, esters on aromatic rings were all well-tolerated. Aromatic alkynes with meta-substituents could be tolerated in the reaction, delivering the desired products (4m–4p) in 60–68% yields. In addition, more steric congested ortho-substituted aromatic alkynes could be transformed to corresponding olefins (4q–4s) in 60–68% yields. Multi-substituted aromatic alkynes were converted to trisubstituted olefins (4t and 4u) in 67% yield. Moreover, this reaction conditions tolerated more sensitive functional groups. Acidic proton containing ketone was compatible in the reaction, affording corresponding ketone containing olefin (4v) in 48% yield. Free aniline and N–H containing amide were well-tolerated under the reaction conditions, furnishing corresponding olefins (4w and 4x) in 54% yield. Heteroaromatic alkynes, such as quinoline, pyridine, indole, and thiophene, were all converted smoothly to corresponding heteroaryl dialkylated olefins (4y–4ab) in synthetic useful yields. Notably, various 1,3-enynes could be converted to deliver multi-substituted conjugated dienes (4ac–4ae) in 48–58% yields. Internal alkynes showed no reactivity to the desired reaction due to the increased steric hindrance for radical addition onto alkynes. Moreover, aliphatic alkynes proved unsuccessful for the reaction, partially due to the low stability and reactivity of resulting vinyl radical intermediates for Ni-catalyzed Csp2–Csp3 cross-coupling reaction. Furthermore, the reaction could be scaled up to 2.0 mmol scale to deliver 4a in 72% yield, without erasing the efficiency of this transformation.
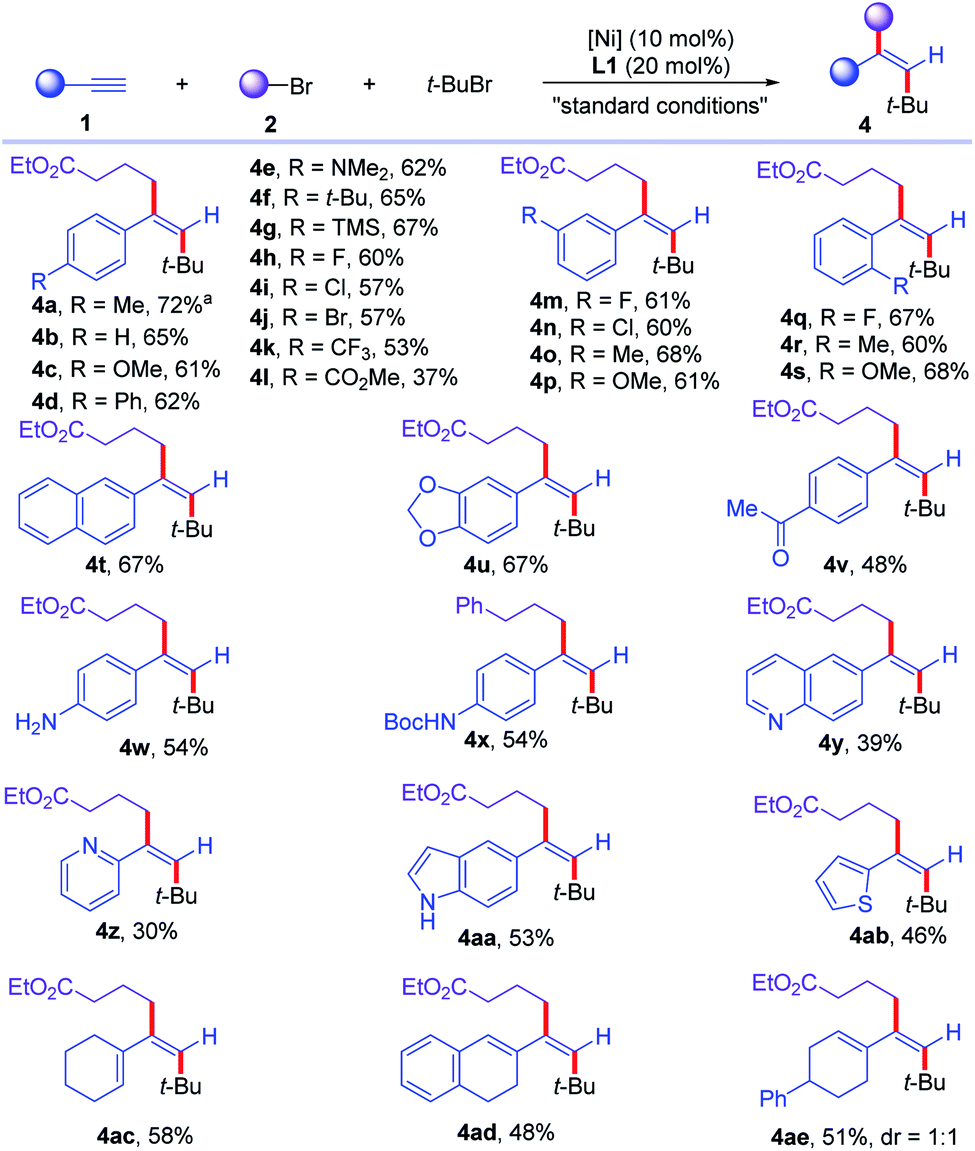 | ||
| Scheme 2 Scope for the cross-electrophile dialkylation of alkynes with respect to alkynes. For standard conditions, see Table 1. a The reaction was conducted on 2.0 mmol scale. | ||
Next, the scope of the first component of alkyl bromides (2) was tested (Scheme 3). Unfunctionalized alkyl bromides with different alkyl chains were good substrates for the reaction, furnishing dialkylated olefins with different alkyl chains in 59–68% yields (5a–5d). Phenyl and diverse substituted aryl pendent alkyl bromides were transformed into corresponding olefins in 60–68% yields (5e–5h). Heteroaryl containing alkyl bromides were successfully converted to trans-dialkylated olefins (5i and 5j) in 54% and 51% yields. Acetals, ethers, ketones, chlorides, and nitrile in alkyl bromides were tolerated in the reaction, affording corresponding olefins (5k–5o) in 48–72% yields. Moreover, unsaturated carbon–carbon bonds, such as terminal olefins, terminal and internal alkynes were all compatible in this reaction, undergoing chemoselective dialkylation reaction to furnish desired products (5p–5r) in 50–62% yields. Bromo-aliphatic amides and alcohols with free N–H and O–H were compatible under the reaction conditions, affording the desired amides or alcohols pendant dialkylated olefins (5s–5u) in 61–67% yields. In addition, secondary bromide was successfully involved in the reaction, delivering the corresponding trans-dialkylated olefin 5v in 52% yield. Unfortunately, secondary and primary alkyl bromides failed to deliver the trisubstituted alkenes via a three-component cross-coupling reaction, partially due to the competitive cross coupling between two alkyl bromides.
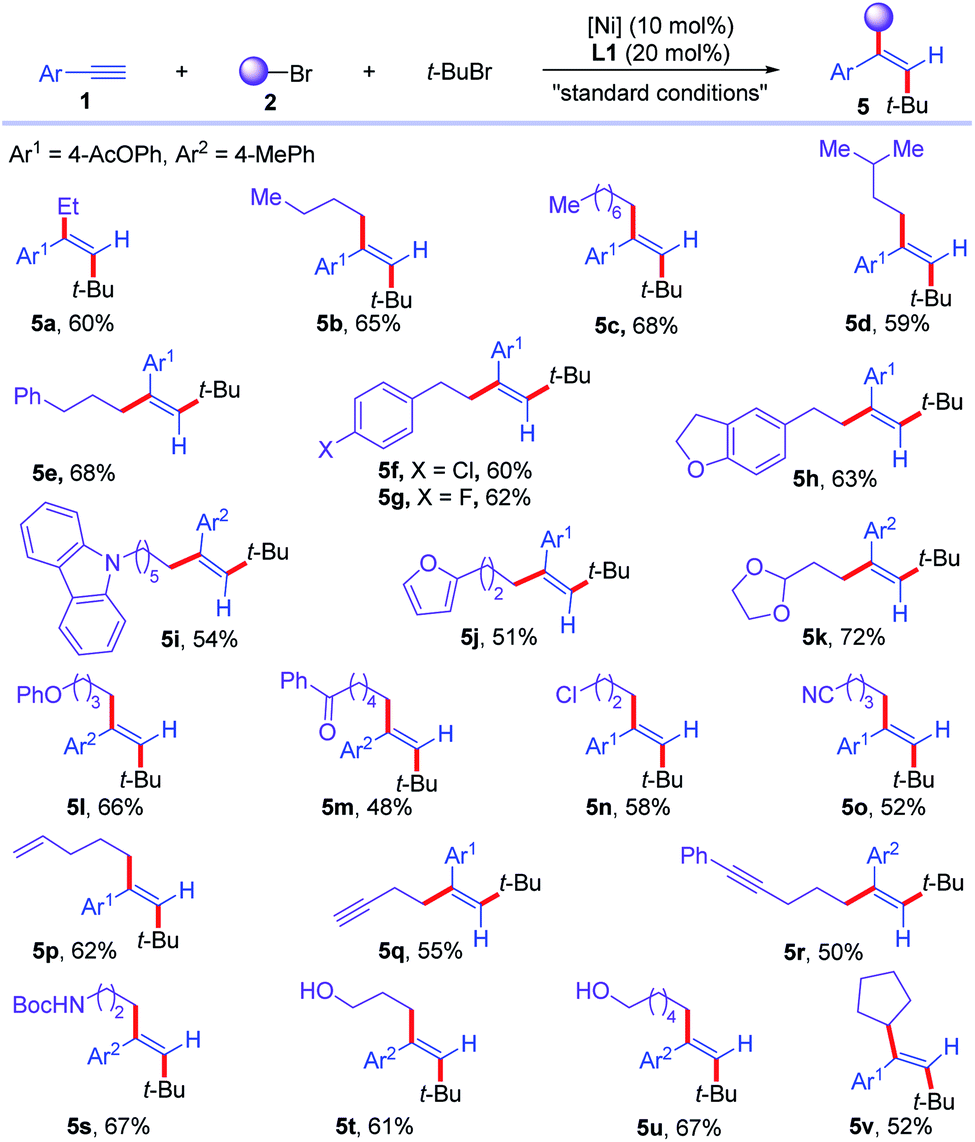 | ||
| Scheme 3 Scope of alkyl bromides for the cross-electrophile dialkylation of alkynes with respect to first alkyl bromides. For standard conditions, see Table 1. | ||
Then, the scope of the other component of alkyl bromide was examined (Scheme 4). Various tertiary alkyl bromides were good substrates for this reductive trans-dialkylation reaction of alkynes. Tertiary alkyl bromides with different alkyl chain lengths were all good substrates for this cross-dialkylation reaction (6a–6d). Chloro, phenyl, ester, and alcohol were well-tolerated in the reaction, delivering corresponding functionalized trans-dialkylated olefins in moderate yields (6e–6h). Cyclic tertiary alkyl bromide was converted to corresponding olefin 6i in 54% yield. Unfortunately, secondary alkyl bromides were unsuccessful to this reaction.
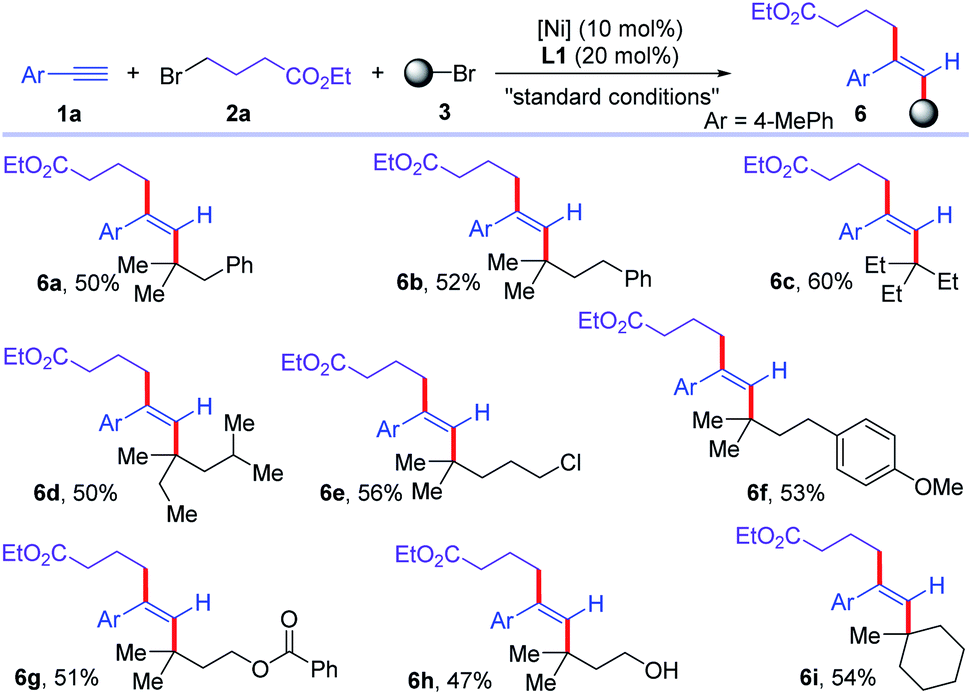 | ||
| Scheme 4 Scope of alkyl bromides for the cross-electrophile dialkylation of alkynes with respect to tertiary alkyl bromides. For standard conditions, see Table 1. | ||
Next, the application of this protocol to the late-stage functionalization of complex molecules, including natural products and drug molecules, was demonstrated (Scheme 5). Alkynes based on L-phenylalanine and sulbactam were successfully converted to corresponding trans-dialkylated olefins (7a and 7b) in 56% and 48% yields, respectively. Primary alkyl bromides from L-menthol, theobromine, isoxepac, estrone, and indometacin were all good substrates for this reaction, delivering corresponding trans-dialkylated olefins derived from various complex molecules (7c–7g) in 53–64% yields. Moreover, tetrahydrolinalool-derived tertiary alkyl bromide was successfully incorporated in the reaction to afford olefin 7h in 56% yield. The configuration of the products was further confirmed unambiguously by the X-ray diffraction analysis of 7d.
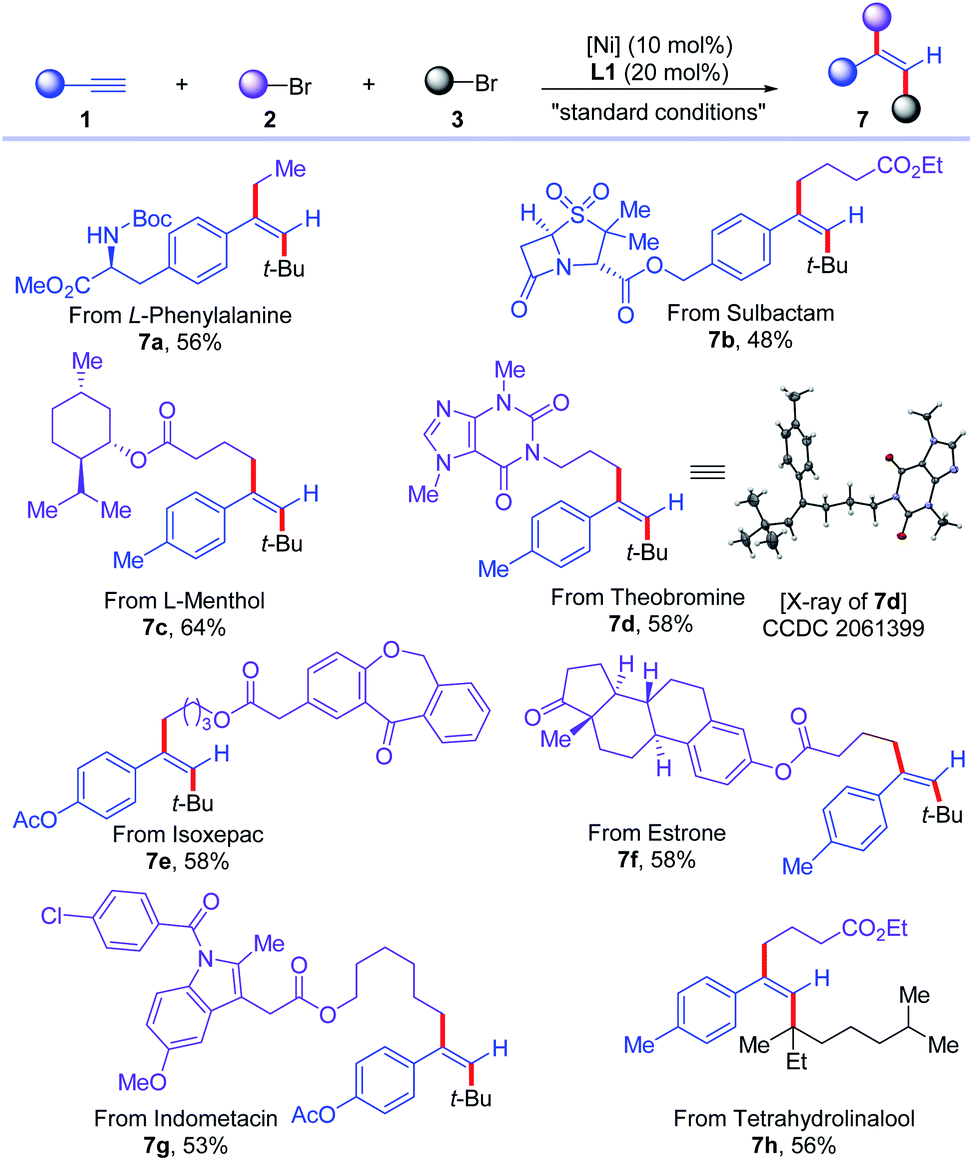 | ||
| Scheme 5 Application the cross-electrophile dialkylation of alkynes for complex molecules. For standard conditions, see Table 1. | ||
To gain some insights into the reaction mechanism, a series of control experiments were conducted (Scheme 6). First, the reaction of 1b with 3a and a radical clock substrate 8 under standard conditions did not deliver the regular coupling product 9′, affording the ring opening three-component cross-coupling product 9 in 32% yield (Scheme 6a). Moreover, the reaction of olefin pendant alkyl bromide (10) with alkyne (1b) and tert-butyl bromide (3a) delivered the ring-closing three-component dialkylation product 11 in 48% yield, without the formation of 11′ (Scheme 6a). When the reaction of 1c and 3a was carried out without 2a, the dimer (12) of vinyl radical from tert-butyl radical addition to alkyne along with the protonated product 13 were formed (Scheme 6b). In addition, the organozinc species was not detected under the standard conditions (Scheme 6c). These results indicated that the reaction proceeded via radical pathways and vinyl radical intermediates from the radical additional of tert-butyl radical to alkyne was involved.
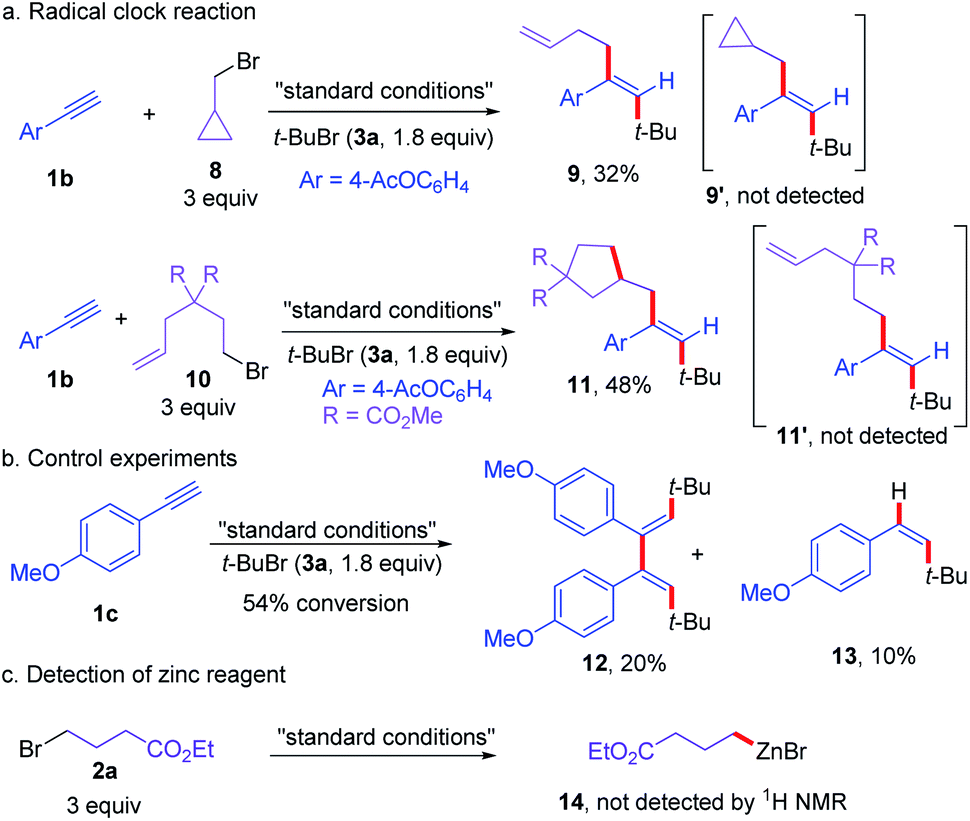 | ||
| Scheme 6 Mechanistic investigations and control experiments. | ||
Based on the mechanistic results and literature precedence,16a,22 a plausible mechanism for this reaction is proposed in Scheme 7. First, Ni(II) was reduced to give Ni(I) in the presence of zinc, which could react with the first alkyl bromide (3) to give alkyl radical intermediate M1 and Ni(II). M1 was added to an alkyne (1) to give a vinyl radical intermediate M2 by the formation of a Csp2–Csp3 bond, which could rebound with Ni(I) to generate the vinyl Ni(II) intermediate M3. The Ni(I)L approached M2 to give M3 in trans-selectivity due to the repulsion between alkyl1 and the ligand on nickel.16aM4 could be generated from M3 by single electron reduction, which could undergo further oxidative addition with the second alkyl bromide (2) to give Ni(III) intermediate M6viaM5. The final product was formed by the reductive elimination of M6 to form the second Csp2–Csp3 bond along with the regeneration of the Ni(I) intermediate.
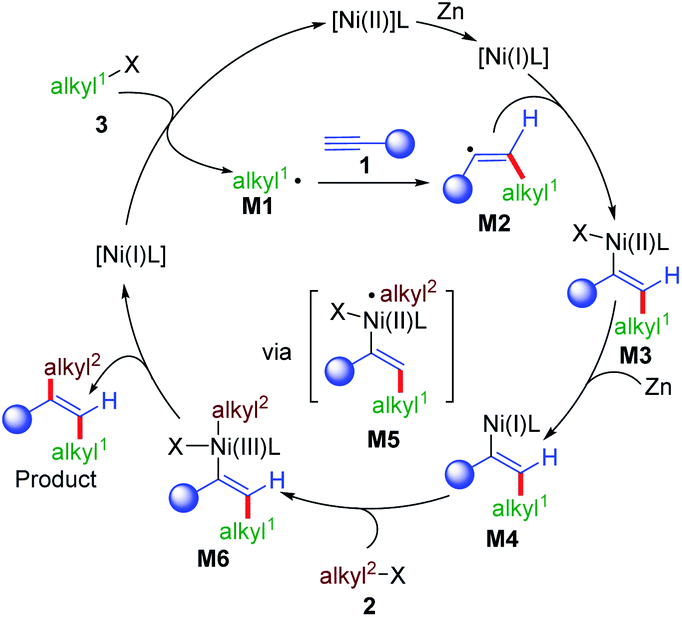 | ||
| Scheme 7 Proposed mechanism for the reaction. | ||
Conclusions
In summary, a Ni-catalyzed fully intermolecular trans-cross-dialkylation of alkynes with two different alkyl bromides has been developed for the first time. Two distinct alkyl bromides could be selectively employed as alkylating agents to sequentially install across alkynes to afford trans-dialkylated olefins. The reaction undergoes with exclusive regio- and diastereoselectivity under reductive conditions, circumventing the preformation of organometallic reagents. The key to the success of this protocol includes the judicious selection of ligand and reaction conditions to suppress the competitive β-hydride elimination of alkyl nickel intermediates as well as the homo-dialkylation reaction.Data availability
Experimental data has been provided as ESI.†Author contributions
Y. Z. Z. discovered the reaction. W. S. conceived and directed the project. Y. Z. Z. and H. M. performed the experiments and analysed the data. W. S. and Y. Z. Z. wrote the manuscript. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely acknowledge NSFC (21971101, 22171127, 21801126), Guangdong Basic and Applied Basic Research Foundation (2019A1515011976), Department of Education of Guangdong Province (2021KTSCX106), The Pearl River Talent Recruitment Program (2019QN01Y261), The Stable Support Plan Program of Shenzhen Natural Science Fund (No. 20200925152608001), Thousand Talents Program for Young Scholars, Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002). We acknowledge the assistance of SUSTech Core Research Facilities. We thank Dr Xiaoyong Chang (SUSTech) for X-ray crystallographic analysis of 7d (CCDC 2061399) and Dr Wen-Tao Zhao (SUSTech) for reproducing the results of 4v, 4y, 5i and 6e.Notes and references
-
(a) H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1304–1307 CrossRef CAS PubMed
; (b) V. C. Jordan, J. Med. Chem., 2003, 46, 1081–1111 CrossRef CAS PubMed
- Y. Yang, S.-L. Shi, D.-W. Niu, P. Liu and S. L. Buchwald, Science, 2015, 349, 62–66 CrossRef CAS PubMed
-
(a) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698–4745 CrossRef CAS PubMed
-
(a) S. E. Bottcher, L. E. Hutchinson and D. J. Wilger, Synthesis, 2020, 52, 2807–2820 CrossRef CAS
-
(a) Y. Nakao, S. Oda and T. Hiyama, J. Am. Chem. Soc., 2004, 126, 13904–13905 CrossRef CAS PubMed
-
(a) Y. Kajita, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2018, 130, 17226–17227 CrossRef PubMed
-
(a) J. Montgomery, Acc. Chem. Res., 2000, 33, 467–473 CrossRef CAS PubMed
-
(a) L. Guo, F. Song, S. Zhu, H. Li and L. Chu, Nat. Commun., 2018, 9, 4543 CrossRef PubMed
- T. Igarashi, S. Arai and A. Nishida, J. Org. Chem., 2013, 78, 4366–4372 CrossRef CAS PubMed
- X. Wang, Y. Liu and R. Martin, J. Am. Chem. Soc., 2015, 137, 6476–6479 CrossRef CAS PubMed
- X. Zhang, X. Xie and Y. Liu, Chem. Sci., 2016, 7, 5815–5820 RSC
-
(a) C. Clarke, C. A. Incerti-Pradillos and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 8068–8071 CrossRef CAS PubMed
-
(a) Z. Zhou, W. Liu and W. Kong, Org. Lett., 2020, 22, 6982–6987 CrossRef CAS PubMed
-
(a) G. R. Kumar, R. Kumar, M. Rajesh and M. S. Reddy, Chem. Commun., 2018, 54, 759–762 RSC
- J. Terao, F. Bando and N. Kambe, Chem. Commun., 2009, 45, 7336–7338 RSC
-
(a) Z. Li, A. Garcia-Dominguez and C. Nevado, Angew. Chem., Int. Ed., 2016, 55, 6938–6941 CrossRef CAS PubMed
-
(a) K. E. Poremba, S. E. Dibrell and S. E. Reisman, ACS Catal., 2020, 10, 8237–8246 CrossRef CAS PubMed
-
(a) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562–11565 CrossRef CAS PubMed
-
(a) Y.-Z. Zhan, N. Xiao and W. Shu, Nat. Commun., 2021, 12, 928 CrossRef CAS PubMed
- C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645–17651 CrossRef CAS PubMed
- For more detail on the condition optimization, see ESI†.
- B. Giese, Angew. Chem., Int. Ed. Engl., 1989, 28, 969–980 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2061399. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00487a |
| This journal is © The Royal Society of Chemistry 2022 |