
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTiming matters: pre-assembly versus post-assembly functionalization of a polyoxovanadate–organic cuboid†
Ji
Guo
a,
Junrui
Liu
b,
Yingcui
Cui
a,
Chuanhong
Liu
a,
Yangming
Wang
a,
Mou
Wang
a,
Danmeng
Huang
a,
Guanying
Chen
 a,
Wei
Wang
*b,
Debin
Xia
a and
Xikui
Fang
*a
a,
Wei
Wang
*b,
Debin
Xia
a and
Xikui
Fang
*a
aMIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, China. E-mail: xkfang@hit.edu.cn
bCAS Key Laboratory of Design and Assembly of Functional Nanostructures, Fujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Xiamen, Fujian 361021, China. E-mail: wangwei@fjirsm.ac.cn
First published on 14th April 2022
Abstract
The pre-assembly and post-assembly approaches in the functionalization of a polyoxovanadate–organic cuboid, [{V6S}8(QPTC)8{V3}2]10−, are discussed. We have shown that the two pathways have led to distinctly different systems, with either an expanded or contracted interior void space, when phenylphosphonate is introduced at different stages of the self-assembly. One leaves the cuboid framework largely intact, whereas the other results in a compact, twisted cuboid. Kinetic factors will have to be considered in the equilibrium of these complex processes. Furthermore, the exceptional stability of these polyoxometalate–organic systems facilitates mass spectrometric characterization, which confirms the composition of the complexes and also indicates that the methoxide groups on the vanadium cluster nodes are labile. The results will help deepen the mechanistic understanding of the formation mechanisms of polyoxovanadate-based metal–organic cages and other functionalized polyoxovanadate clusters in general.
Introduction
Introducing chemical functionalities has become a widespread tool, in biology and chemistry alike, for regulating structures, properties, and functions. Phosphorylation, methylation, and acetylation of proteins,1 for instance, have played critical roles in the regulation of protein activities and almost all cellular processes. Inspired in part by these reactions in biology, the development of metal–organic frameworks,2 covalent organic frameworks,3 porous organic cages4 and metal–organic coordination cages5 has been increasingly focused on functionalization to impart new chemical and physical properties. Among these synthetic porous systems, metal–organic cage complexes have received much less attention than the others in this regard, largely due to the poor stability of the self-assembled coordination complexes.6The functionalization of metal–organic cages can follow two different paths: pre-assembly and post-assembly. In pre-assembly functionalization, functional groups are often covalently installed on the organic linkers before the coordination-driven self-assembly takes place. This is because the fragile and dynamic nature of metal–organic cages is usually not compatible with covalent-bond-forming reactions afterwards. This practice is best exemplified by the exo- and endohedral functionalization of Fujita's MnL2n-type spherical coordination polyhedra,7 upon reaction of pre-functionalized bis(pyridine) ligands and Pd2+ ions. The post-assembly approach, on the other hand, is often conducted on relatively stable metal–organic cages under mild reaction conditions. It enables the introduction of new functionalities, and often new properties, to a metal–organic cage without resorting to lengthy de novo ligand synthesis.5a For instance, Nitschke et al. demonstrated that post-synthetic functionalization may be utilized to modulate the stability,8 reactivity,9 and guest binding properties10 of coordination cages.
Little is known or discussed, however, about how pre- and post-assembly functionalization paths may differently affect the structures and properties of the final products. Most functionalization work was carried out either prior to or after self-assembly so that not much direct comparison is available. Even if both routes were attempted, in the very few reported cases,10a they had ended up with the same product. This is not surprising given the dynamic nature of coordination bonds; the thermodynamic product generally resulted upon equilibration through an error-checking pathway.
However, here we would like to present the first example where pre-assembly and post-assembly paths for the functionalization of a metal–organic cage have led to distinctly different systems. This follows a recent study of ours11 where endo-functionalization of polyoxovanadate–organic polyhedra (MOPs), a subclass of metal–organic cages in which the nodes are metal clusters, was demonstrated. Here we specifically compare the two different approaches of functionalization. These results suggest that the thermodynamic equilibrium may be influenced by kinetic stability of a pre-assembled MOP. This allows the system to stay in a kinetically trapped state if functional groups are installed post-synthetically. On the other hand, pre-assembly functionalization does not have to go through a significant activation barrier to break down the cage, leading to a thermodynamically stable state. In other words, timing matters when it comes to the introduction of functional groups into the MOP cages.
Results and discussion
The polyoxovanadate–organic cuboid
We have previously shown that coordinative functionalization can be a promising alterative to the covalent approach that has been more widely adopted. Thus, for a set of MOPs where the secondary building units (SBUs) are methoxide-decorated hexavanadate clusters, [VIV6O6(OCH3)9(μ6-SO4)]+ ({V6S}),12 replacing the central sulfate group with more strongly coordinating organophosphonates allows easy installation of functionalities within the cage cavities.11 For instance, in an ABTC-paneled, cubic MOP complex [{V6S}8(ABTC)6]16− (where ABTC is 3,3′,5,5′-azobenzene tetracarboxylate),12e phenylphosphonate or biphenylphosphonate functions can go into the internal cavity. However, larger terphenyl-substituted phosphonate would not fit in, and as a result, it inhibits the MOP formation if it were installed onto the hexavanadate cluster prior to the cage assembly.In this work, we introduce quaterphenyl-3,3′′′,5,5′′′-tetracarboxylate (QPTC4−, Fig. 1a) as the linker ligand, hoping to increase the size of the central cavity and to see how it impacts the endo-functionalization with phosphonates. The acid ligand H4QPTC was prepared following a reported procedure13 and confirmed by 1H NMR. Thus, when H4QPTC and VOSO4 were heated in a solvent mixture of DMF and MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v) under solvothermal conditions, prismatic, dark green crystals were obtained after cooling. X-ray structure determination revealed a MOP cage, 1, which is constructed from 18 building blocks: 8 QPTC linkers, 8 {V6S} and 2 {V3} SBUs (Fig. 1a). It crystallizes in the triclinic space group P
:2 v/v) under solvothermal conditions, prismatic, dark green crystals were obtained after cooling. X-ray structure determination revealed a MOP cage, 1, which is constructed from 18 building blocks: 8 QPTC linkers, 8 {V6S} and 2 {V3} SBUs (Fig. 1a). It crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with only one molecule in the unit cell (V = 17168 Å3) sitting on the crystallographic inversion center. The overall shape of 1 can be simplified as a rectangular cuboid; the ligand–node connectivities within it are illustrated by a cartoon model in Fig. 1b. Four QPTC panels are on the outer faces of the cuboid, interconnecting the eight {V6S} SBUs located at the vertices, while the other four QPTC ligands link the same {V6S} SBUs to two {V3} clusters inside the cuboid. Thus, the local symmetry of MOP 1 approximates to point group D2h.
, with only one molecule in the unit cell (V = 17168 Å3) sitting on the crystallographic inversion center. The overall shape of 1 can be simplified as a rectangular cuboid; the ligand–node connectivities within it are illustrated by a cartoon model in Fig. 1b. Four QPTC panels are on the outer faces of the cuboid, interconnecting the eight {V6S} SBUs located at the vertices, while the other four QPTC ligands link the same {V6S} SBUs to two {V3} clusters inside the cuboid. Thus, the local symmetry of MOP 1 approximates to point group D2h.
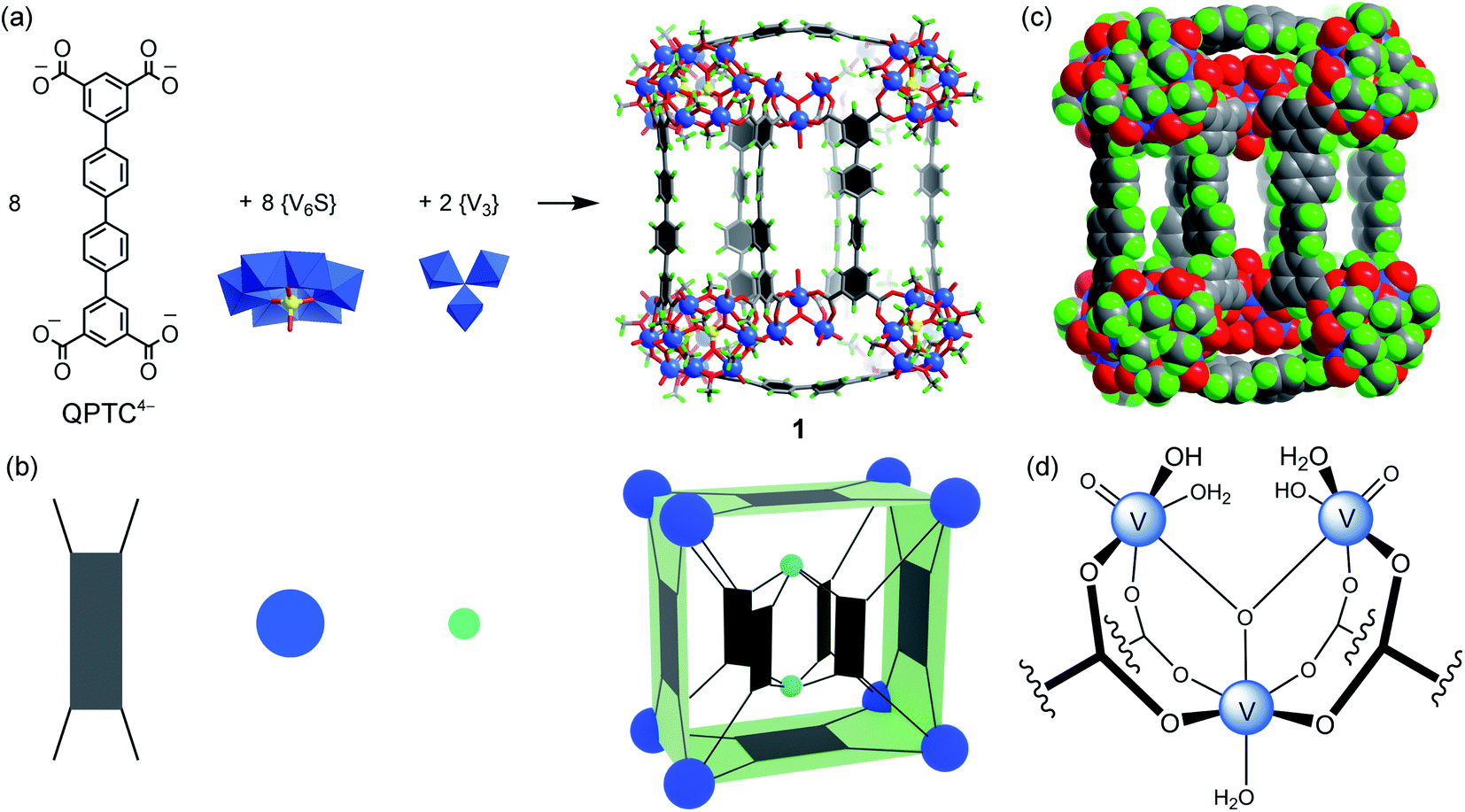 | ||
| Fig. 1 (a) The self-assembly of 1 from QPTC linkers and {V6S} and {V3} SBUs; (b) a simplified cartoon model of 1 showing the ligand–node connectivities; the dark blue spheres represent the {V6S} SBUs, the light blue spheres represent the {V3} SBUs and the four-pronged rectangles represent the QPTC linkers; (c) the space-filling model of 1 illustrating the three compartments of its interior cavity (V blue; S yellow; O red; C dark gray; H green). Counter ions and solvent molecules are omitted for clarity; (d) chemical structure of the {V3} SBU. | ||
The hexanuclear {V6S} clusters on the cuboid vertices are tritopic, each linked to three QPTC ligands. All six vanadium ions comprising the {V6S} nodes are in the +4 oxidation state as seen before.12 The trinuclear, oxo-centered {V3} SBUs (Fig. 1d), on the other hand, have never been observed in other polyoxovanadate-based MOPs. Each {V3} SBU is connected with four QPTC ligands via bridging carboxylates. Bond valence sum (BVS) calculations indicated that its three vanadium sites are all V(V), giving the formula [VV3O3(OH)2(H2O)3]7+ for this tetratopic {V3} SBU. Mixed-valence vanadium sites in 1 were also verified by X-ray photoelectron spectroscopy (XPS) analysis (Fig. S15†), which yielded V 2p3/2 and V 2p1/2 peaks that can be deconvoluted into those for V(IV) (at 516.14 and 523.47 eV) and V(V) (at 517.3 and 524.62 eV). Accordingly, the formula of polyanion 1 can be expressed as [{V6S}8(QPTC)8{V3}2]10−.
The interior cavity of MOP 1 is separated by the six parallel-aligned QPTC ligands into three compartments (Fig. 1c), with a total solvent-accessible volume of 1099 Å3 (Fig. S24†).14 The 10− charge of complex anion 1 is balanced by tetramethylammonium (TMA+) counter ions, which are believed to come from thermal decomposition of DMF solvent molecules in the presence of CH3OH.15 Four of the TMA+ ions are located within the interior cavities, with each filling the void space between two adjacent {V6S} clusters (Fig. S22a†), likely drawn in by electrostatic attraction to the sulfate anions on the hexavanadate SBUs.
1H NMR is of limited use in characterizing the solution speciation of 1 due to the paramagnetic effect of the V(IV) centers. However, the stability of MOP 1 in solution was proven by high-resolution electrospray ionization mass spectrometry (ESI-MS). Recorded in a mixture of DMF/CH3CN, the mass spectrum of 1 in the negative-ion mode is shown in Fig. 2a. A collection of multiply charged ions were detected at m/z = 1837.4278, 1564.3512, 1359.5480 and 1200.2509 for [(TMA+)10−n[1–(˙CH3)22]·(DMF)6]n− (n = 6–9), corresponding to the dissociation of counter ions (TMA+). The rather broad envelope of peaks for each charge state is a result of sequential loss of ˙CH3 (15 Da, Fig. S14†) radicals from the methoxide-bridged {V6S} clusters, indicating that the methoxide ligands on the {V6S} SBUs are fairly labile. The loss of neutral methyl radicals, however, does not change the charge state or affect the framework integrity of the MOP structure. The base peak at m/z = 1359.5480 could be assigned to {[TMA+]2[1–(˙CH3)22]·(DMF)6}8−, and the isotopic resolution is in good agreement with the theoretical distribution (Fig. 2a, inset). The most intense peaks with 9– and 8– charge states having only one or two associated TMA+ counter cations indicate that the four encapsulated TMA+ ions are easily removed in solution.
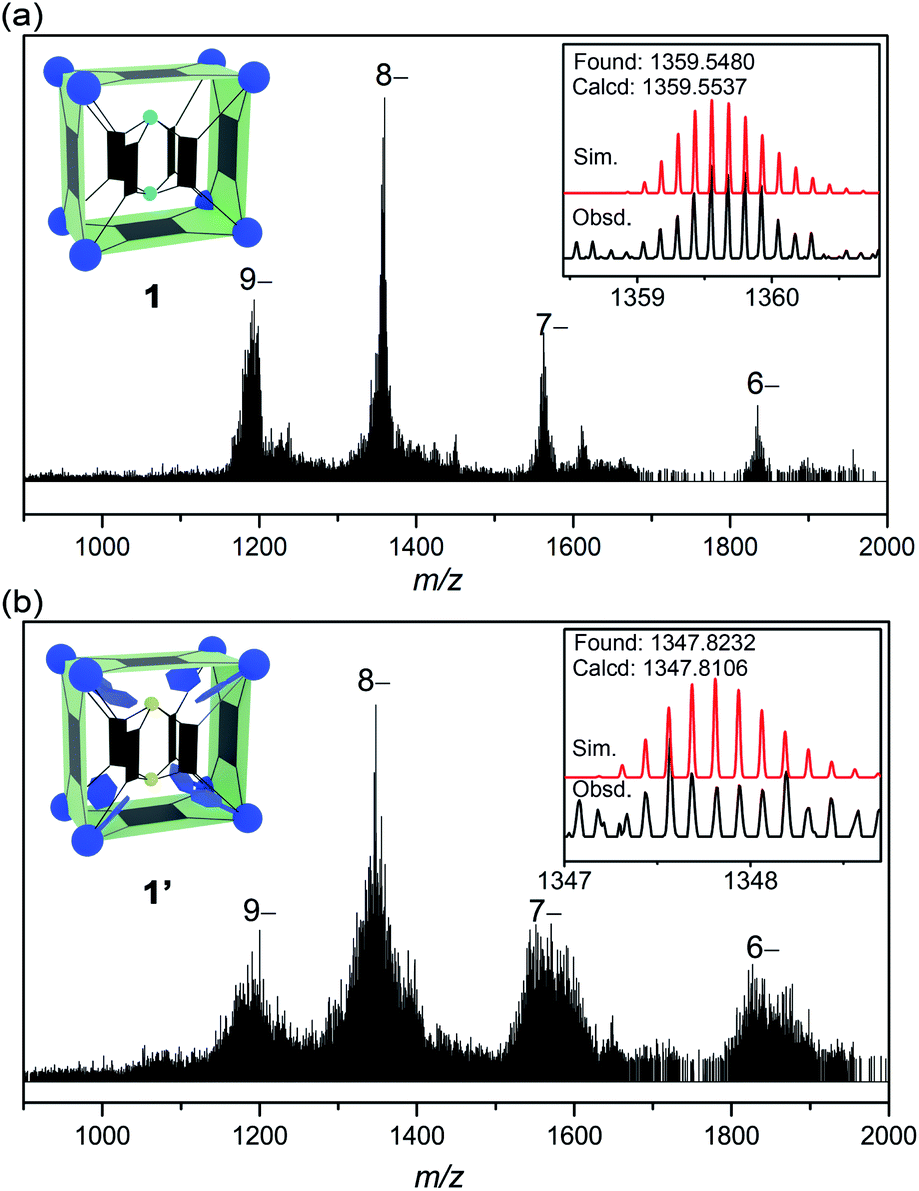 | ||
| Fig. 2 The ESI-MS spectra of 1 (a) and 1′ (b), with the insets showing the observed and simulated isotope patterns of the peak envelopes containing the base peaks (8– ions). | ||
The post-assembly functionalization
To achieve post-assembly functionalization, stoichiometric amounts (8:1) of phenylphosphonic acid and a crystal sample of the cuboid MOP 1 were suspended in DMF/MeOH, and the solvothermal reaction was carried out again under the same conditions as before (see the ESI†). An endo-functionalized MOP derivative was thus obtained, with a yield of 51%. This post-functionalized complex, 1′, again crystallizes in the triclinic space group P (Z = 1), but it has a larger unit cell volume of 19856 Å3, reflecting the incorporation of bulky phenylphosphonate groups.
The overall structure of the new cuboid 1′ (Fig. 3) is broadly similar to that of 1, but its interior is different in a few ways. As expected, phenylphosphonate replaces the sulfate ion in each of the eight {V6S} clusters to give the functionalized SBU, [VIV6O6(OCH3)9(μ6-PhPO3)]+ ({V6P}, Fig. 3c). The steric effect due to the inclusion of bulky phenylphosphonates increases the distances between adjacent hexavanadate clusters. This distance is 8.53 Å in 1 (as measured by S⋯S separation, Fig. S22a†), but increases to either 8.80 Å or 9.15 Å in 1′ (P⋯P separation, Fig. S18b†), depending on the π–π stacking conformation16 of the two phenyl rings from adjacent phosphonates (Fig. S22d†). Furthermore, the two inner {V3} SBUs in 1 are now changed into {V2} clusters, [VIV2O2(OCH3)2]2+, in complex 1′. The two V centers in this dinuclear cluster (Fig. 3d) are dually bridged by two methoxide groups. Four QPTC ligands are bound to it in either a chelating or monodentate mode, instead of the bridging mode seen in the {V3} clusters of 1. Again, here the oxidation state of the V sites was established by both XPS measurements (Fig. S16†) and BVS calculations. Although the structural roles of {V3} and {V2} are similar, the {V2} SBUs noticeably increase the distances between the six parallel QPTC ligands and consequently expand the interior cavity of MOP 1′. As a result, even with the space between the adjacent {V6P} SBUs completely filled by phenylphosphonate groups, the interior void volume for 1′ increases to 1213 Å3 (Fig. S24†), from the 1099 Å3 for 1.14 In the solid state structure, we have located two TMA+ counter ions and two ordered DMF molecules (Fig. 3a) in the expanded cavity of 1′.
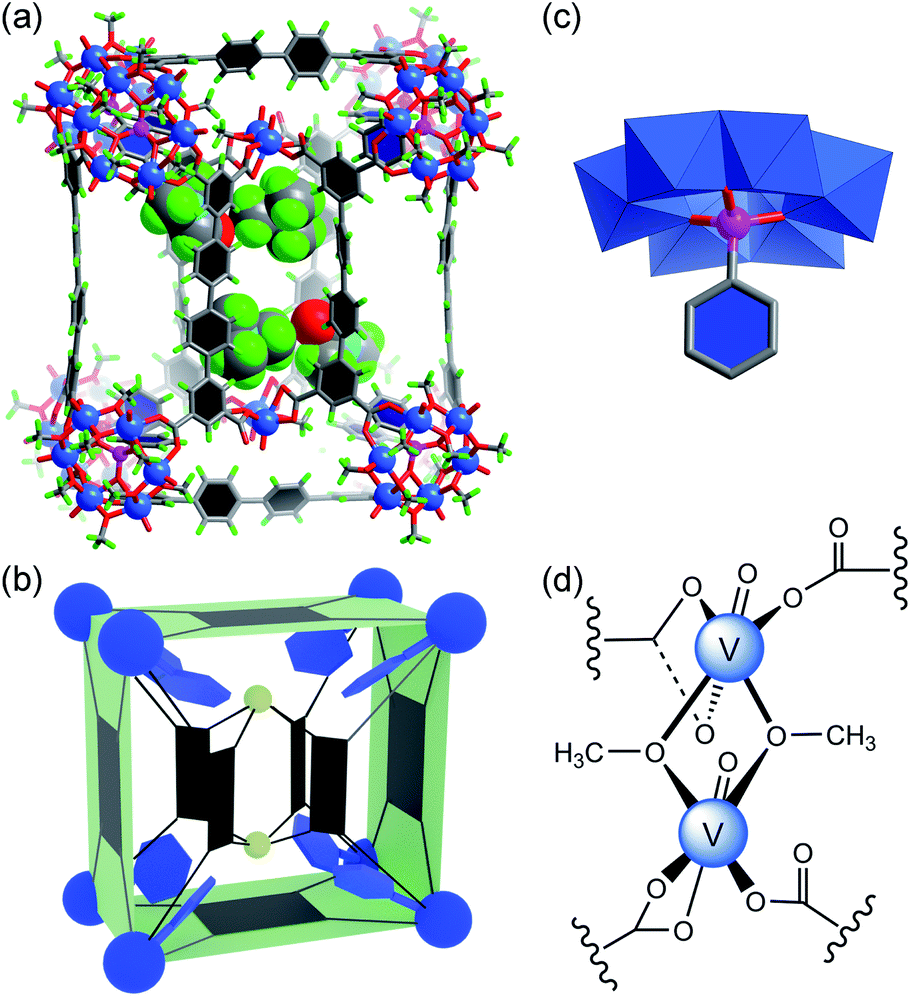 | ||
| Fig. 3 (a) The crystal structure of 1′ (ball-and-stick model), showing the encapsulated TMA+ counter ions and DMF solvent molecules (space-filling model) in the expanded cavity; the color code is the same as in Fig. 1, with P atoms as pink spheres and N atoms as light blue spheres; (b) the cartoon model of 1′, where the phenylphosphonate functional groups are shown as blue hexagons and the two {V2} clusters as small greenish-yellow spheres; (c) the functionalized {V6P} SBU, where the central sulfate ion is replaced by phenylphosphonate; hydrogen atoms are not shown for clarity; (d) chemical structure of the {V2} SBU. | ||
Although the 1H NMR spectra of complexes 1 and 1′ themselves were not informative due to paramagnetism, treating them with a base broke down the MOP structures and allowed oxidation of the V(IV) centers in air. Recording the NMR signals afterwards did provide confirmation of the identity of organic ligands and their relative ratios, if multiple ligands are present. The 1H NMR spectra of NaOD-digested samples of 1 and 1′ are displayed in Fig. S9 and 10.† The 1H NMR spectrum of 1′ clearly shows signals from both QPTC and phenylphosphonate, the ratio of which was determined to be ∼1:1 by 1H NMR integrals, agreeing with the solid state structure. The 31P NMR spectrum of base-digested 1′ also supported the presence of phenylphosphonate functions, with a single peak at 11.30 ppm (Fig. S12†).
The ESI-MS spectrum of 1′ (Fig. 2b) is similar to that of 1, but with a broader peak profile. The series of intense peaks at m/z = 1821.7842, 1550.9461, 1347.8232 and 1189.8036 can be correctly assigned to {[TMA+]20−n[1′–(˙CH3)68]}n− (n = 6–9). However, the overall isotope distribution pattern does not match well with theoretical simulation (Fig. 2b, inset). This indicates that multiple components contribute to the observed pattern, leading to overlapping isotope distributions. The molecular weights of these components likely differ only by a few Da, so that for a given isotopic envelop, multiple intense peaks are seen, instead of a single Gaussian distribution. Our attempts to model the data with more than one component have been unsatisfactory so far. When a mixture of components all undergo ˙CH3 fragmentation, that would make it very difficult or impossible to identify each of the steps in the loss of ˙CH3.
The pre-assembly functionalization
For pre-assembly functionalization, the functional groups will need to be installed prior to the self-assembly process. Thanks to the strong coordination ability of phosphonates, this can be simply achieved with one-pot reactions.11 Thus, solvothermal reaction of VOSO4 with H4QPTC was carried out in the presence of phenylphosphonic acid in DMF/MeOH, whereby green plate crystals were obtained (see the ESI†).Single-crystal X-ray diffraction analysis reveals a new MOP complex, 2, which is distinctly different from 1′. MOP 2 is constructed from 8 {V6P} SBUs and 8 QPTC ligands (Fig. 4); no {V3} or {V2} clusters are present. It features a twisted cuboid shape and exhibits ligand–node connectivities that are very different from those of 1 and 1′. The distortion from a rectangular cuboid lowers the symmetry of the MOP cage to C2, and thus, unlike centrosymmetric 1 and 1′, complex 2 is chiral. As it crystallizes in the space group C2/c, both enantiomers are present in the unit cell as a racemic mixture.
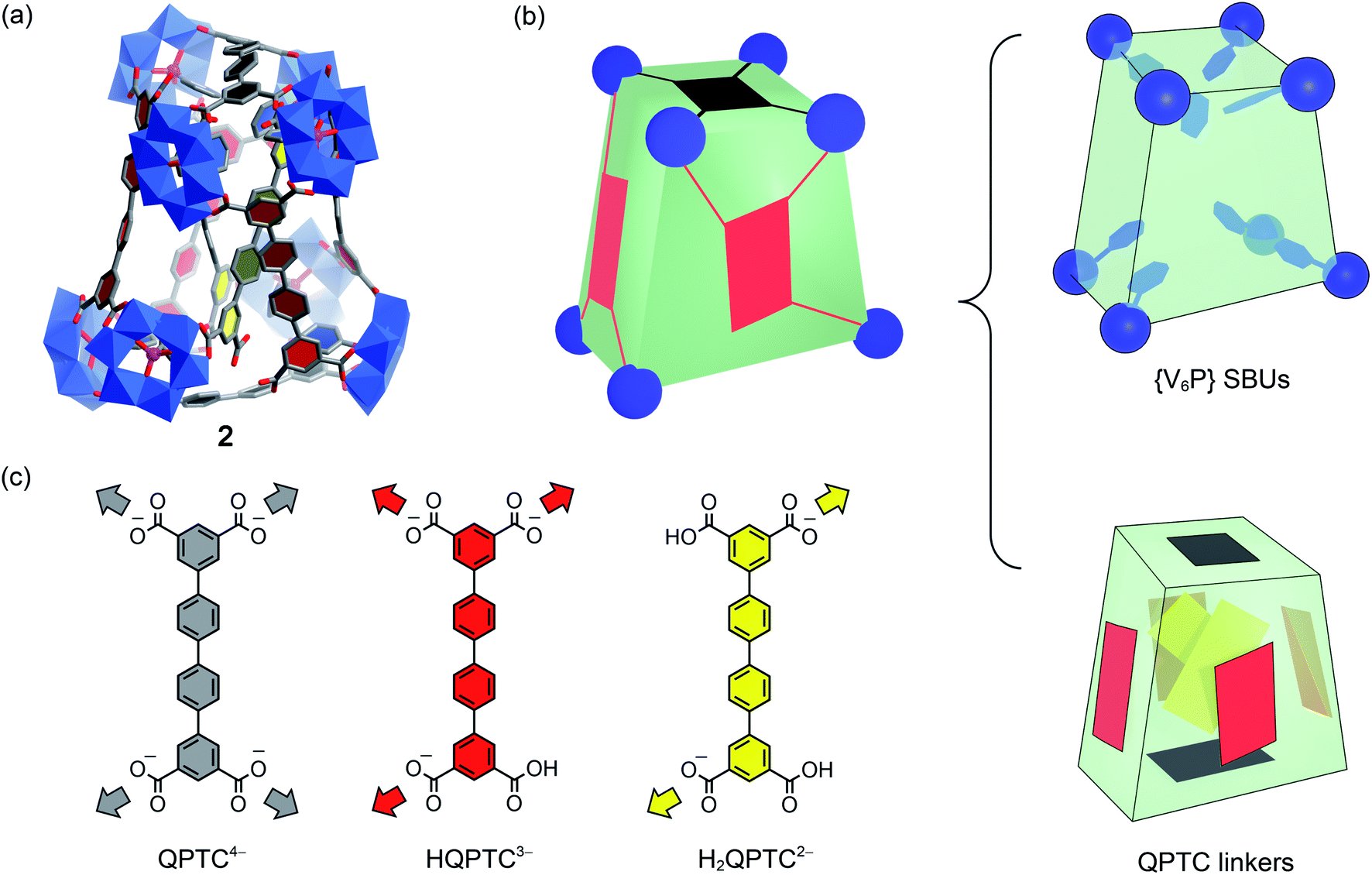 | ||
| Fig. 4 (a) The crystal structure of 2; the color code is the same as before, with P atoms as violet spheres. Counter cations, hydrogen atoms, and CH3 groups on the {V6P} clusters are omitted for clarity; (b) the cartoon model of 2 with its eight {V6P} SBUs, and the QPTC linkers are separately shown on the right to elucidate the interior structure; (c) the three different modes of coordination displayed by QPTC ligands in 2 are colored differently. | ||
What also makes 2 unique among polyoxovanadate-based MOPs is that it features multiple free carboxylic acid groups. In 2, the 8 QPTC linkers display three different coordination modes (Fig. 4c). The top and bottom QPTC panels (in black) both coordinate with four {V6P} SBUs, as tetratopic QPTC4−. The four linkers on the side faces of the twisted cuboid are monoprotonated, tritopic HQPTC3− (in red). The last two are diprotonated H2QPTC2− (in yellow) crisscrossing inside the MOP cage; each of them connects to two diagonally opposite SBUs on the twisted cuboid, thus leaving two free carboxylic acid groups. In all, there are eight free carboxylic acid units in the MOP complex 2, [{V6P}8H8(QPTC)8]16−.
The IR spectrum of 2 (Fig. S1†) also supports the existence of uncoordinated carboxylic acids. In addition to the same νa(COO) bands at 1670–1575 cm−1 for coordinated carboxylates, as in 1 and 1′, MOP 2 exhibits a new, higher frequency νa(COO) vibration at 1727 cm−1, which is attributed to uncoordinated carboxylic acids.17 Through 1H NMR of NaOD-digested 2, it is again determined that the ratio of QPTC to phenylphosphonate is approximately 1:1 (Fig. 5a). The signal at 8.26 ppm is attributed to free formate, a byproduct coming from MeOH oxidation under the solvothermal conditions18 or from the decomposition of the solvent DMF,15 and have been observed by us before.11 The 31P NMR spectrum (Fig. S13†) also confirms the presence of phosphonate functions.
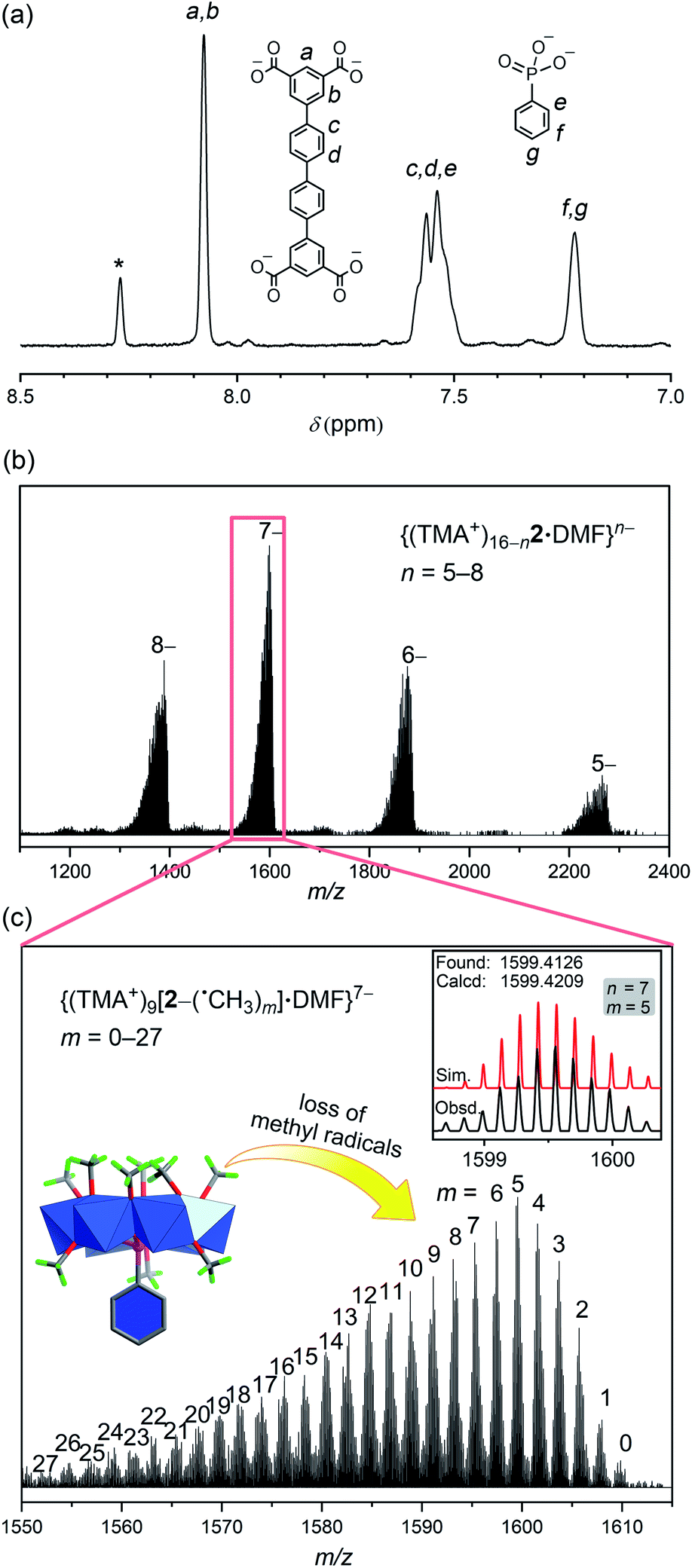 | ||
| Fig. 5 (a) The 1H NMR spectrum of a base-digested sample of 2 in NaOD/D2O (*: formate) confirmed the ratio between QPTC and phosphonate functional groups; (b) the negative-ion ESI-MS spectrum of complex 2 in DMF/CH3CN; (c) zoomed view of the 7– peak envelope showing the sequential loss of m number of ˙CH3 radicals from the {V6P} SBUs of 2 (inset: the observed and simulated isotope patterns for the peak envelope where n = 7 and m = 5). | ||
In the ESI-MS spectrum of 2 (Fig. 5b), a set of well resolved peaks with continuous charge states from 5– to 8– are detected along with fragments corresponding to losing different numbers of methyl radicals. For instance, for the 7– charge state, the broad signal in the 1550–1610 m/z region (Fig. 5c) can be deconvoluted into dozens of smaller, even-spaced isotopic peak envelopes; each corresponds to a step in the sequential loss of the m number of neutral ˙CH3 radicals from the {V6P} clusters. A total of 72 methoxide groups are attached to the 8 {V6P} SBUs of 2. Up to 27 of them are involved in the fragmentation processes, while the parent ion (m = 0) is also detected as low-intensity signals. These clean and intense MS peaks are also a testament to the exceptional stability of the MOP framework of 2.
The structure of 2 is very compact; it has a much smaller interior void space of 523 Å3 (see Fig. S24†), less than half of those in 1 or 1′. For 2, the phenyl rings from QPTC ligands and phenylphosphonates are engaged in extensive π–π stacking interactions, leaving little space in between. In contrast, the QPTC ligands in 1 and 1′ are all well separated from each other. Despite the considerable difference in the size and shape of their interior cavities, all three MOPs exhibited similar behavior in the removal of organic dyes: methylene blue (MB+), Sudan III (SD), acid orange 7 (AO−), as monitored by UV-vis spectroscopy. When fresh crystals of the MOPs were soaked in ethanol solutions of the above dyes, all MOP samples selectively took up MB+ but absorbed little of the neutral SD and anionic AO− (Fig. S6–8†). The results suggest that the absorption processes are electrostatic instead of supramolecular host–guest complexation. The positively charged MB dye ions are believed to be attracted by the anionic MOPs and substitute some of the TMA+ counter ions in the channels (Fig. S25†) between them. The channel space accounts for up to 53.0% of the unit cell volumes in the crystals, meaning the dye molecules can move rather freely within the channel void. As a result, the MOPs all displayed similar absorption behavior.
The reaction pathways: some considerations
It is now clear that pre-assembly and post-assembly functionalization of 1 has led to markedly different products (Fig. 6). In the post-assembly route, replacing sulfates with phenylphosphonate functions leaves the cuboid structure of MOP 1 largely intact. On the other hand, pre-assembly incorporation of phenylphosphonates has resulted in a twisted cuboid with vastly different ligand connectivities. To put it in another way, one is like remodelling an old house, whereas the other is like constructing a new house from scratch with a completely different design, all from the same materials.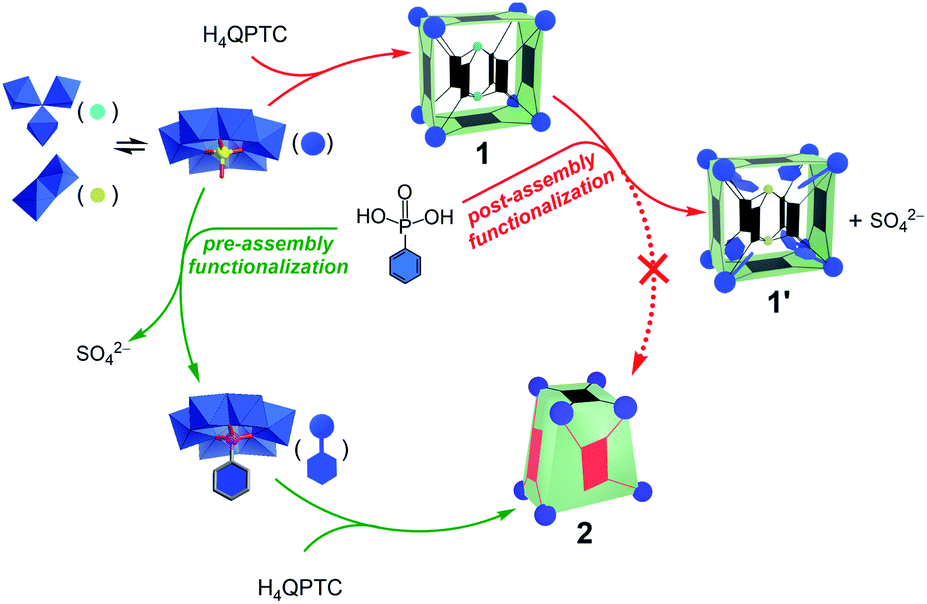 | ||
| Fig. 6 Reaction pathways leading to different products for pre-assembly versus post-assembly functionalization. | ||
As the precursors and reaction conditions are the same for both pathways, we suspect that kinetic factors may have played a crucial role in determining the final self-assembled structures. Due to the dynamic character of metal–ligand coordination bonds, the formation of metal–organic cages is generally believed to operate under thermodynamic control;5a that is, all constituents or building blocks are in equilibrium through dynamic combinatorial chemistry.19 This is likely the case for the pre-assembly route of functionalization, which has led directly to the low-energy product, 2. In comparison to 1′, the lower number of building blocks (entropic gain) and extensive π–π stacking interactions between the QPTC ligands (enthalpic gain) in 2 indicate that it is thermodynamically favored. This being said, kinetic products built from multitopic precursors and exhibiting multiple bonds can be stabilized. In post-assembly functionalization of 1, equilibrium involving ligand–SBU coordinative bond breakage could fall into kinetic control due to tetratopicity of the QPTC ligands. This is exacerbated by the large number of the linkers in the complex and the rigidity of the linkers, which makes new bond formation difficult and helps old bonds re-form. This kinetic trap would thus prevent a complete reshuffle of the ligands and SBUs into the thermodynamically stable complex 2 (Fig. 6).
A final note concerns the size of the function groups. When larger phosphonate functions, e.g., biphenyl-4-phosphonate and p-terphenyl-4-phosphonate, were introduced, the end results of functionalization also hinged on the reaction pathways chosen. These bulkier functional groups, if installed prior to the self-assembly process, inhibited the formation of the cuboid cages. This is consistent with our previous observation11 that the steric effect of organophosphonates on the {V6P} SBUs would prevent the MOPs from being assembled. On the other hand, it is a different story if the post-assembly pathway was selected. It was found that, after solvothermal reaction of pre-formed MOP 1 with the same biphenyl or terphenyl phosphonates, these bulkier functions failed to either replace the sulfates or to destroy the cuboid cage of 1. We established, by X-ray diffraction, that the crystals isolated after post-functionalization reactions were still MOP 1, i.e., no sulfate ions in its {V6S} SBUs were replaced. The 1H and 31P NMR spectra of base-digested samples (Fig. S18 and 19†) also confirmed the absence of any phosphonate functions. These results again support the kinetic stability of 1 and highlight the importance of kinetic factors that might influence the reaction outcome.
Conclusions
In summary, we have compared the pre-assembly and post-assembly approaches in the functionalization of a polyoxovanadate–organic cuboid. This is, to the best of our knowledge, the first example where the two pathways in metal–organic cage functionalization have led to distinctly different products. Thermodynamic and kinetic factors, therefore, will likely both have to be considered when it comes to the equilibrium of these complex systems. Rational design in the future may require computational modeling to gain a better understanding of the underlying driving forces that shape the energy landscape in the self-assembly processes and to predict the reaction outcome.All three compounds, including the native cuboid cage 1, post-functionalized 1′, and pre-functionalized 2, have been fully characterized. It is worth noting that ESI-MS of many polyoxovanadates and functionalized derivatives produced vast and often unidentifiable fragments.20 However, these polyoxovanadate–organic polyhedra display exceptional stability and facilitate mass spectrometric characterization. We believe that these results will help deepen the mechanistic understanding of the formation mechanisms of polyoxovanadate-based MOPs and other functionalized polyoxovanadate clusters.21 Mass spectrometry may also give valuable insights into polyoxometalate systems22 where NMR can sometimes offer very little information due to paramagnetism.
Data availability
All experimental details and supporting analytical data are available in the ESI.†Author contributions
J. G. and X. F. conceived the project, designed and supervised the study. J. G., J. L., C. L., Y. W., M. W. and D. H. synthesized the samples, carried out the single-crystal X-ray diffraction analysis and the supporting synthetic/characterization work, including thermogravimetric analysis, IR and UV. J. G., W. W., D. X. and G. C. led efforts associated with the NMR and XPS measurements. J. G., W. W. and X. F. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21671048, 51972084, and 21871025), the Fundamental Research Funds for the Central Universities, China (AUGA5710094420), the Youth Innovation Promotion Association CAS (2021302), and Natural Science Foundation of Heilongjiang Youth Fund (YQ2021B002). X. F. and G. C. also acknowledge the support of the Heilongjiang Touyan Innovation Team Program.Notes and references
- (a) Y.-C. Wang, S. E. Peterson and J. F. Loring, Cell Res., 2014, 24, 143–160 CrossRef CAS PubMed; (b) B. J. Grant, A. A. Gorfe and J. A. McCammon, Curr. Opin. Struct. Biol., 2010, 20, 142–147 CrossRef CAS PubMed.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- J. L. Segura, S. Royuela and M. M. Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- H. Wang, Y. Jin, N. Sun, W. Zhang and J. Jiang, Chem. Soc. Rev., 2021, 50, 8874–8886 RSC.
- (a) D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626–644 RSC; (b) P. M. Bogie, T. F. Miller and R. J. Hooley, Isr. J. Chem., 2019, 59, 130–139 CrossRef CAS.
- S. Mollick, S. Fajal, S. Mukherjee and S. K. Ghosh, Chem. - Asian J., 2019, 14, 3096–3108 CrossRef CAS PubMed.
- (a) K. Suzuki, J. Iida, S. Sato, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2008, 47, 5780–5782 CrossRef CAS PubMed; (b) S. Sato, J. Iida, K. Suzuki, M. Kawano, T. Ozeki and M. Fujita, Science, 2006, 313, 1273–1276 CrossRef CAS PubMed; (c) K. Suzuki, K. Takao, S. Sato and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2544–2545 CrossRef CAS PubMed; (d) Q.-F. Sun, S. Sato and M. Fujita, Chem. Lett., 2011, 40, 726–727 CrossRef CAS; (e) D. Fujita, A. Takahashi, S. Sato and M. Fujita, J. Am. Chem. Soc., 2011, 133, 13317–13319 CrossRef CAS PubMed; (f) D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T. Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato and M. Fujita, Nat. Commun., 2012, 3, 1093–1099 CrossRef PubMed; (g) K. Harris, Q.-F. Sun, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 12497–12499 CrossRef CAS PubMed; (h) K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- D. A. Roberts, A. M. Castilla, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 8201–8204 CrossRef CAS PubMed.
- D. A. Roberts, B. S. Pilgrim, J. D. Cooper, T. K. Ronson, S. Zarra and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 10068–10071 CrossRef CAS PubMed.
- (a) C. T. McTernan, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 6837–6842 CrossRef CAS PubMed; (b) T. K. Ronson, B. S. Pilgrim and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 10417–10420 CrossRef CAS PubMed.
- J. Guo, Q. Chang, Z. Liu, Y. Wang, C. Liu, M. Wang, D. Huang, G. Chen, H. Zhao, W. Wang and X. Fang, Chem. Sci., 2021, 12, 7361–7368 RSC.
- (a) Y.-T. Zhang, X.-L. Wang, S.-B. Li, Y.-R. Gong, B.-Q. Song, K.-Z. Shao and Z.-M. Su, Chem. Commun., 2016, 52, 9632–9635 RSC; (b) Y.-T. Zhang, S.-B. Li, X.-L. Wang, Y.-R. Gong, K.-Z. Shao and Z.-M. Su, Dalton Trans., 2016, 45, 14898–14901 RSC; (c) Y.-R. Gong, W.-C. Chen, L. Zhao, K.-Z. Shao, X.-L. Wang and Z.-M. Su, Dalton Trans., 2018, 47, 12979–12983 RSC; (d) Y. Gong, Y. Zhang, C. Qin, C. Sun, X. Wang and Z. Su, Angew. Chem., Int. Ed., 2019, 58, 780–784 CrossRef CAS PubMed; (e) Y. Gong, C. Qin, Y. Zhang, C. Sun, Q. Pan, X. Wang and Z. Su, Angew. Chem., Int. Ed., 2020, 59, 22034–22038 CrossRef CAS PubMed.
- X. Lin, I. Telepeni, A. J. Blake, A. Daily, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159–2171 CrossRef CAS PubMed.
- The size of void space was calculated by using the 3V web-based cavity calculator. See: N. R. Voss and M. Gerstein, Nucleic Acids Res., 2010, 38, W555–W562 CrossRef CAS PubMed.
- (a) X. Hang, B. Liu, X. Zhu, S. Wang, H. Han, W. Liao, Y. Liu and C. Hu, J. Am. Chem. Soc., 2016, 138, 2969–2972 CrossRef CAS PubMed; (b) J.-Q. Liu, J. Wu, J. Wang, L. Lu, C. Daiguebonne, G. Calvez, O. Guillou, H. Sakiyama, N. S. Weng and M. Zeller, RSC Adv., 2014, 4, 20605–20611 RSC.
- (a) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed; (b) L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, John Wiley & Sons, 2009, Complexes of Carboxylic Acids, pp. 64–67 Search PubMed.
- V. V. Kaichev, G. Y. Popova, Y. A. Chesalov, A. A. Saraev, D. Y. Zemlyanov, S. A. Beloshapkin, A. Knop-Gericke, R. Schlögl, T. V. Andrushkevich and V. I. Bukhtiyarov, J. Catal., 2014, 311, 59–70 CrossRef CAS.
- (a) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (b) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- N. M. Al Hasan, G. E. Johnson and J. Laskin, J. Am. Soc. Mass Spectrom., 2013, 24, 1385–1395 CrossRef CAS PubMed.
- S. Chakraborty, B. E. Petel, E. Schreiber and E. M. Matson, Nanoscale Adv., 2021, 3, 1293–1318 RSC.
- (a) H. N. Miras and L. Cronin, in New Strategies in Chemical Synthesis and Catalysis, ed. B. Pignataro, Wiley-VCH, 2012, pp. 1–32 Search PubMed; (b) A. J. Surman, P. J. Robbins, J. Ujma, Q. Zheng, P. E. Barran and L. Cronin, J. Am. Chem. Soc., 2016, 138, 3824–3830 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, physical characterization and X-ray analysis. CCDC 2144006–2144008. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00533f |
| This journal is © The Royal Society of Chemistry 2022 |