
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic discrimination of non-directing aryl and heteroaryl groups: enantioselective synthesis of bioactive indole-containing triarylmethanes†
Qiaolin
Yan‡
a,
Meng
Duan‡
bf,
Cien
Chen
cd,
Zhiqing
Deng
 de,
Mandi
Wu
cd,
Peiyuan
Yu
f,
Ming-Liang
He
*cd,
Guangyu
Zhu
*de,
K. N.
Houk
*b and
Jianwei
Sun
*ag
de,
Mandi
Wu
cd,
Peiyuan
Yu
f,
Ming-Liang
He
*cd,
Guangyu
Zhu
*de,
K. N.
Houk
*b and
Jianwei
Sun
*ag
aDepartment of Chemistry, the Hong Kong Branch of Chinese National Engineering Research Centre for Tissue Restoration & Reconstruction, The Hong Kong University of Science and Technology (HKUST), Clear Water Bay, Kowloon, Hong Kong SAR, China. E-mail: sunjw@ust.hk
bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, USA. E-mail: houk@ucla.edu
cDepartment of Biomedical Sciences, City University of Hong Kong, Kowloon Tong, Hong Kong SAR, China,. E-mail: mlhe7788@gmail.com
dCityU Shenzhen Research Institute, Shenzhen, China
eDepartment of Chemistry, City University of Hong Kong, Kowloon Tong, Hong Kong SAR, China. E-mail: guangzhu@cityu.edu.hk
fDepartment of Chemistry and Shenzhen Grubbs Institute, Guangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen, 518055, China
gShenzhen Research Institute, HKUST, No. 9 Yuexing 1st Rd, Shenzhen 518057, China
First published on 13th April 2022
Abstract
Despite the enormous developments in asymmetric catalysis, the basis for asymmetric induction is largely limited to the spatial interaction between the substrate and catalyst. Consequently, asymmetric discrimination between two sterically similar groups remains a challenge. This is particularly formidable for enantiodifferentiation between two aryl groups without a directing group or electronic manipulation. Here we address this challenge by using a robust organocatalytic system leading to excellent enantioselection between aryl and heteroaryl groups. With versatile 2-indole imine methide as the platform, an excellent combination of a superb chiral phosphoric acid and the optimal hydride source provided efficient access to a range of highly enantioenriched indole-containing triarylmethanes. Control experiments and kinetic studies provided important insights into the mechanism. DFT calculations also indicated that while hydrogen bonding is important for activation, the key interaction for discrimination of the two aryl groups is mainly π–π stacking. Preliminary biological studies also demonstrated the great potential of these triarylmethanes for anticancer and antiviral drug development.
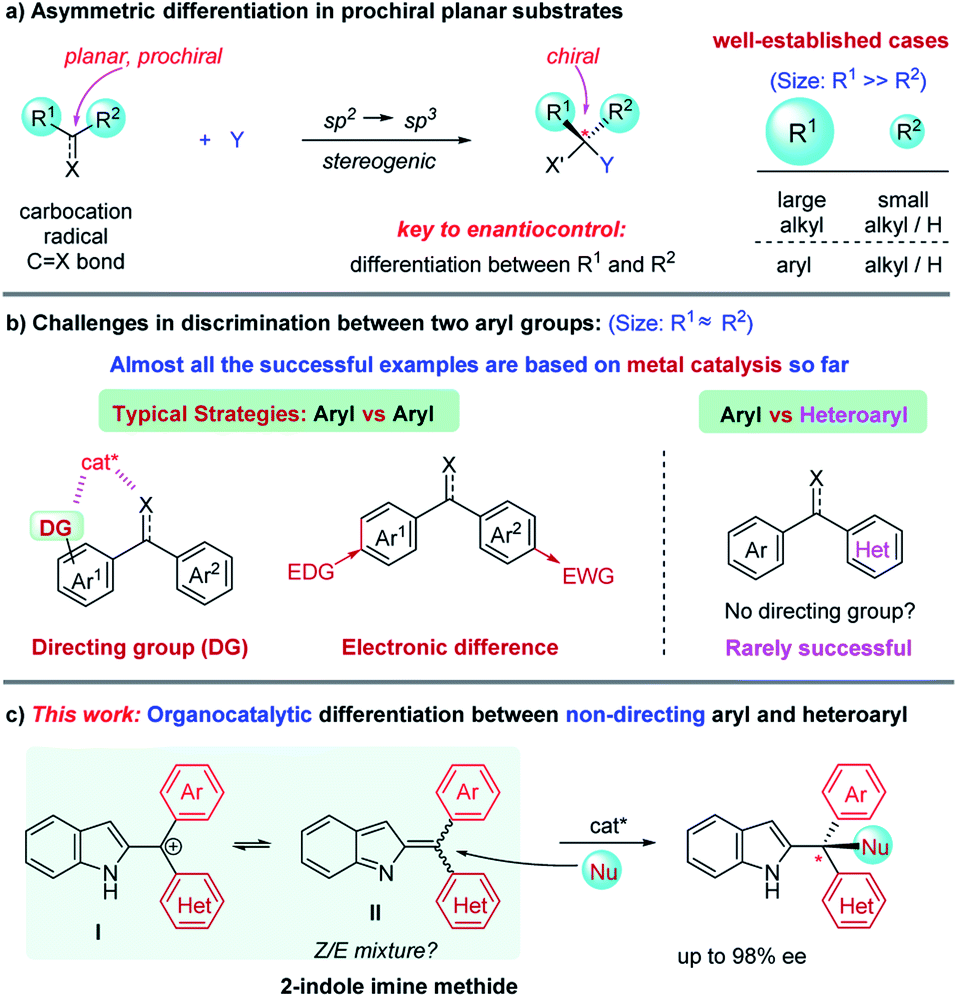
Asymmetric catalysis has evolved arguably into the most powerful method for the synthesis of enantioenriched molecules.1 It features high efficiency and atom-economy in principle as compared to other approaches such as chiral resolution and auxiliary-based asymmetric synthesis, thereby enabling increasing applications in industrial synthesis.2 In the past few decades, a wide range of chiral catalytic systems with diverse activation modes have been developed. However, the fundamental basis for enantiocontrol remains essentially unchanged, i.e., spatial interaction between the substrate and catalyst.1,2 For example, in the construction of a tetrahedral C(sp3)-chiral center from a prochiral C(sp2)-based planar substrate (e.g., carbocation, radical, carbonyl, and olefin), a chiral catalyst typically provides enantiodifferentiation by blocking one face of the plane and directing the reaction partner (Y) to approach towards the other face (Scheme 1a). To achieve this, the catalyst must be able to effectively discriminate between the two substituents (R1 and R2) on the prochiral carbon. Obviously, the larger the difference of these two substituents is, the better enantioselectivity will be expected. Consequently, it has been well-established to achieve high enantioselectivity for cases bearing two sterically different groups (e.g., alkyl/aryl vs. H and large alkyl vs. small alkyl). In contrast, for cases bearing two substituents of a similar size, it remains challenging.1
 | ||
| Scheme 1 Introduction to asymmetric differentiation in C(sp2)-prochiral centers. | ||
1,1-Diarylmethinyl stereocenters are a widely prevalent structural motif in various natural products and biologically important molecules.3 Asymmetric addition to the 1,1-diaryl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C = X (X = heteroatom) bonds represents one of the most direct approaches for the construction of this unit.4–8 However, this requires effective discrimination between two (often) sterically similar aryl groups, which represents a notable challenge in asymmetric catalysis (Scheme 1b).4 So far, success has mainly relied on the use of a directing group in one aryl group to allow catalyst recognition (e.g., by coordination) or electronic difference by incorporating electron-donating/withdrawing groups.6,7 Notably, the effective enantiodifferentiation between aryl and heteroaryl groups still remains challenging, particularly in the absence of a directing group.8 Moreover, despite the above-mentioned important progress, it is worth noting that almost all these examples relied on metal catalysis, and little success has been achieved by organocatalysis.4–8 In this context, here we describe organocatalytic discrimination of non-directing aryl and heteroaryl groups, providing access to highly enantioenriched triarylmethanes, and in view of the general diverse biological activities of triarylmethanes,9 we have also investigated the anticancer and antiviral activities of these products.
C and C = X (X = heteroatom) bonds represents one of the most direct approaches for the construction of this unit.4–8 However, this requires effective discrimination between two (often) sterically similar aryl groups, which represents a notable challenge in asymmetric catalysis (Scheme 1b).4 So far, success has mainly relied on the use of a directing group in one aryl group to allow catalyst recognition (e.g., by coordination) or electronic difference by incorporating electron-donating/withdrawing groups.6,7 Notably, the effective enantiodifferentiation between aryl and heteroaryl groups still remains challenging, particularly in the absence of a directing group.8 Moreover, despite the above-mentioned important progress, it is worth noting that almost all these examples relied on metal catalysis, and little success has been achieved by organocatalysis.4–8 In this context, here we describe organocatalytic discrimination of non-directing aryl and heteroaryl groups, providing access to highly enantioenriched triarylmethanes, and in view of the general diverse biological activities of triarylmethanes,9 we have also investigated the anticancer and antiviral activities of these products.
Indole imine methides (IIMs) have recently emerged as versatile intermediates for the asymmetric synthesis of enantioenriched indole derivatives, a family of useful units in medicinal chemistry.10–12 In particular, those with the methide motif adorned in the 2-position of indole are particularly useful to construct indole-fused polyheterocycles via asymmetric annulation processes, as pioneered by Shi and co-workers.10,11 In continuation of our interest in IIMs,12 we envisioned that these types of intermediates would be a good platform to study the power of organocatalysis for the challenging discrimination between aryl and heteroaryl groups lacking a directing group (Scheme 1c). However, additional challenges should be expected since this intermediate II is likely generated as a Z/E mixture, typically in equilibrium with carbocation I. Therefore, the equilibrium should be made in synergy with the nucleophilic addition step to allow dynamic asymmetric control in order to achieve high enantioselectivity.
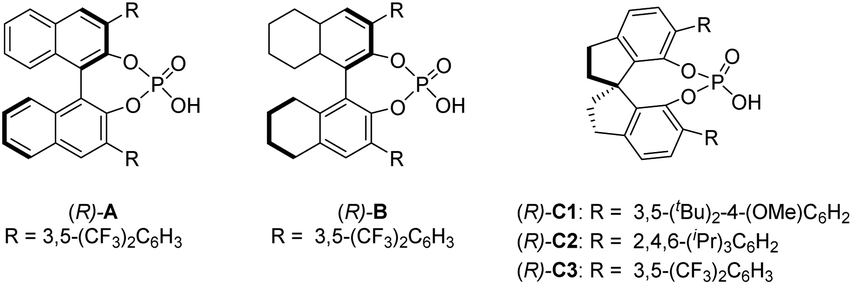
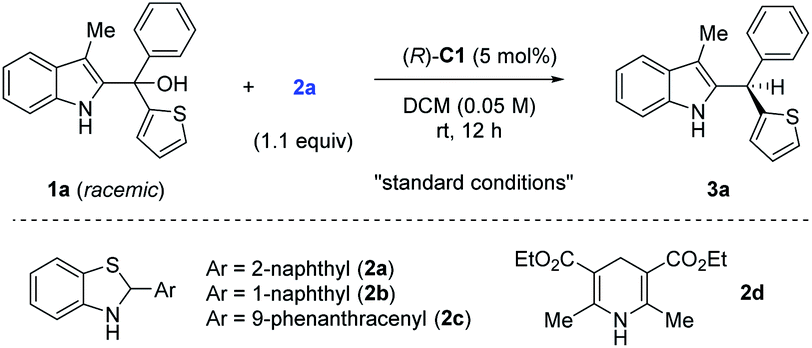
To test our hypothesis, we employed racemic tertiary alcohol 1a as the model precursor to the 2-indole imine methide intermediate. Notably, no directing group is incorporated in the two aryl groups (phenyl and thienyl) to be discriminated by the catalyst. Despite the above-mentioned substantial challenges in this asymmetric control, considerable efforts were devoted to condition optimization and ultimately led to excellent reaction efficiency and enantiocontrol (Table 1). Specifically, among the broad range of chiral acid catalysts evaluated, the SPINOL-derived chiral phosphoric acid C1 was identified as the optimal catalyst.13 With benzothiazoline 2a as the hydride source,14 the asymmetric reduction proceeded smoothly to form indole-containing triarylmethane 3a under mild conditions in essentially quantitative yield and 95% ee (entry 1, Table 1). For comparison, other conditions typically led to inferior results. For example, other SPINOL-based chiral phosphoric acids gave lower enantioselectivity (entries 2–5). In particular, the previously well-known STRIP catalyst C2 resulted in only 16% ee (entry 2). In addition, the catalyst chiral backbones were compared with the same 3,5-bis(trifluoromethyl)phenyl substituent in the 3,3′-positions (entries 3–5). While the SPINOL-based catalyst C3 gave 81% ee, the BINOL- and [H8]BINOL-derived analogues A and B did not show any asymmetric induction (<2% ee). This result not only highlighted the superiority of the spirocyclic skeleton, but also corroborated the elusive stereocontrol in this case. Other hydride sources were also examined (entries 6–8). Benzothiazolines 2b and 2c, bearing a different aryl substituent, led to lower chemoselectivity and enantioselectivity (entries 6 and 7). More surprisingly, Hantzsch ester 2d, the previously well-established hydride source,15 gave drastically low enantiocontrol (entry 8). Other solvents did not provide a better result either (entries 9–11). The reaction was very sensitive to coordinating solvents, such as ether and ethyl acetate, which completely shut down the reaction, presumably due to competing binding with the acid catalyst. Decreasing the reaction temperature to 0 °C maintained high enantioselectivity, but moderately affected the reaction rate (entry 12). Finally, at a higher concentration, slightly lower enantioselectivity was observed (entry 13).
|
|
|||
|---|---|---|---|
| Entry | Deviation from the “standard conditions” | Yieldb (%) | eeb (%) |
| a Reaction scale: 1a (25 μmol), hydride source (27.5 μmol), catalyst (2.5 μmol), solvent (0.5 mL). b Yield was determined by analysis of the 1H NMR spectrum of the crude reaction mixture with CH2Br2 as the internal standard. ee was determined by HPLC analysis on a chiral stationary phase. c A mixture of unidentifiable products was formed. d Clean conversion. The starting material accounts for the remainder of the mass balance. e Conversion <5%. | |||
| 1 | None | >95 | 95 |
| 2 | (R)-C2 instead of (R)-C1 | >95 | 16 |
| 3 | (R)-C3 instead of (R)-C1 | >95 | 81 |
| 4 | (R)-A instead of (R)-C1 | >95 | <2 |
| 5 | (R)-B instead of (R)-C1 | >95 | <2 |
| 6 | 2b instead of 2a | 11c | 80 |
| 7 | 2c instead of 2a | 15c | 55 |
| 8 | 2d instead of 2a | 78d | −9 |
| 9 | Et2O as solvent | <5e | — |
| 10 | Toluene as solvent | 87 | 89 |
| 11 | EtOAc as solvent | <5e | — |
| 12 | Run at 0 °C | 84d | 96 |
| 13 | c = 0.2 M | >95 | 93 |
|
|||
Under the optimized conditions, we examined the reaction scope with various substituted indole-derived tertiary alcohol substrates (Scheme 2). In general, this protocol provided efficient access to a wide range of highly enantioenriched indole-containing triarylmethanes. Substrates bearing electron-withdrawing and electron-donating groups at different positions were all suitable. The presence of a substituent at the 3-position of indole is not necessary (3f), although this position is nucleophilic and can potentially serve as a competitive intermolecular nucleophile. In addition to substitution at the 2-position of the thiophene ring in most cases, it is worth noting that substitution at the 3-position provided equally high enantioselectivity (3p). Finally, it is worth noting that other than these thiophene-containing examples, the discrimination between benzene and furan is also possible, leading to good enantiocontrol (3q). In all these cases, no directing group is needed to provide additional interaction (e.g. hydrogen bonding) with the catalyst in order to achieve high enantiocontrol. Finally, we also examined an example bearing an electron-rich aryl and electron-poor aryl group, which gave moderate enantioselectivity (3r), suggesting that the presence of a thienyl or furyl ring is important to achieve excellent enantiocontrol.
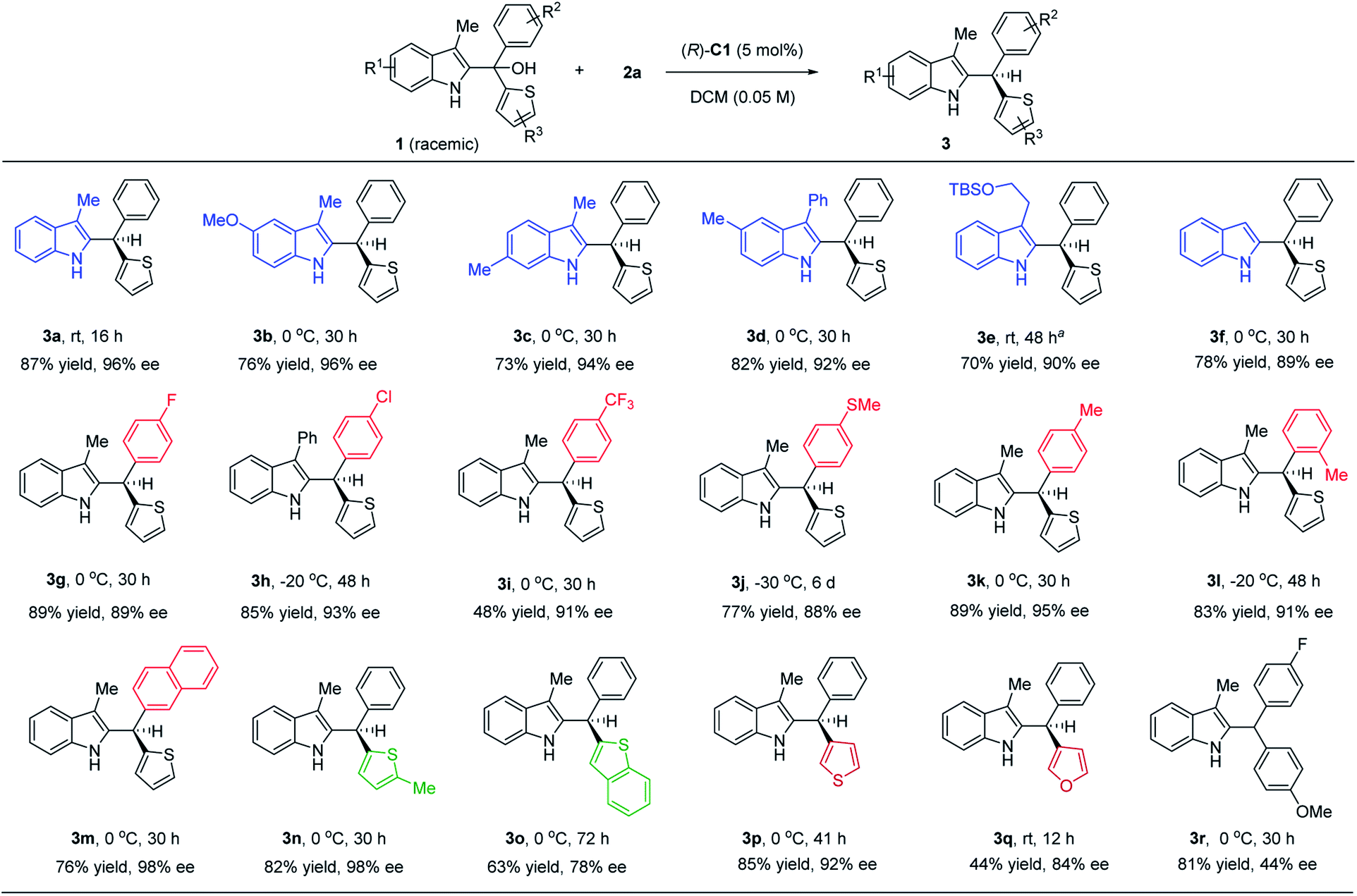 | ||
| Scheme 2 Reaction scope. Reaction scale: 1 (0.4 mmol), 2 (0.44 mmol), (R)-C1 (5 mol%), DCM (8.0 mL). aRun with 10 mol% of the catalyst. | ||
The robustness of this protocol was examined by stoichiometric adulteration of various additives bearing different functional groups (see the ESI† for details).16 In most cases, the excellent chemical efficiency and enantioselectivity were not obviously affected by the additives. Many of these additives contain highly polar and reactive functionalities that are typical strong hydrogen-bonding partners, such as primary amine, thiol, alcohol, carbonyl, sulfone, and boronic acid. This is particularly remarkable in view of the high possibility that hydrogen bonding is a key catalyst-substrate interaction in this process. Notably, from a different point of view, the little influence on enantiocontrol by polar additives might also imply that it is not hydrogen bonding, but other interactions such as π–π stacking, that provide the basis for asymmetric discrimination (vide infra). Nevertheless, these results clearly illustrated the excellent functional group tolerance and the robust enantiodifferentiation ability of this mild but powerful catalytic system.
A possible mechanism is proposed in Scheme 3a. We believe that this reaction begins with acid-catalyzed dehydration to from indolyl cation IM, paired with a phosphate counter anion. This ion pair might be in equilibrium (or pseudo resonance) with the activated indole imine methide form IM′. Subsequently, the hydride source approaches benzylic carbon to deliver the product 3.
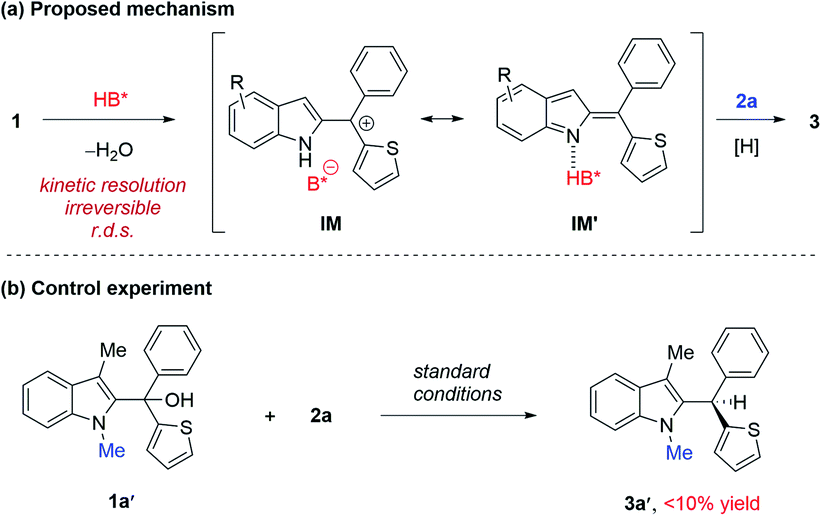 | ||
| Scheme 3 Proposed mechanism and a control experiment. | ||
We carried out a series of control experiments. First of all, under the standard conditions, the reaction with N-methylated substrate 1a′ did not proceed to form the desired product 3a′ (Scheme 3b). This result suggested that the free N–H motif in the indole moiety is essential for the observed reactivity, which is consistent with the intermediacy of 2-indole imine methide IM′, as this intermediate cannot be formed from 1a′. Next, the enantiomeric excess (ee) values of the substrate and product were both monitored during the reaction process (Fig. 1a). The product ee remained constant (95% ee) during the entire reaction, but substrate ee gradually increased over time. This enantioconvergent feature agrees with the initial formation of an achiral 2-indole imine methide intermediate followed by stereodefined asymmetric addition of a nucleophile. The observation of substrate enantioenrichment is indicative of kinetic resolution during the first step, which is likely irreversible. Taken together, a direct SN2 mechanism could be excluded. Furthermore, this reaction did not exhibit non-linear effects, suggesting that the enantiodetermining transition state likely involves only one catalyst molecule. Finally, kinetic studies indicated that this reaction exhibits zeroth order in the nucleophile and first order in the catalyst, which further confirmed that the first step is rate-determining and irreversible.
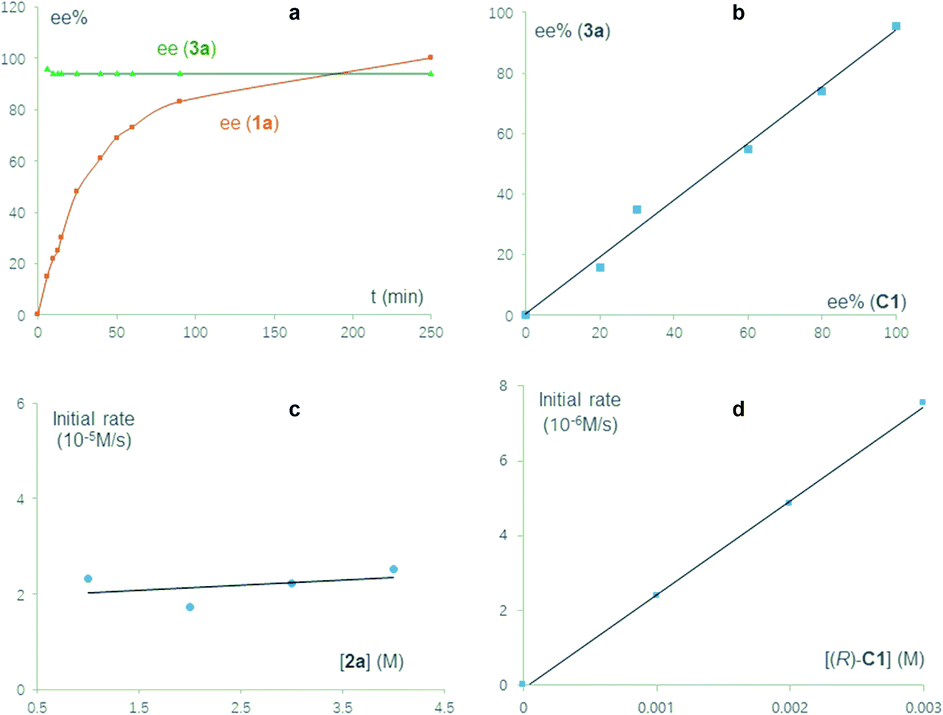 | ||
| Fig. 1 Mechanistic studies. (a) Time-dependence of substrate and product ee values. (b) Absence of non-linear effects. (c) Zeroth order in the nucleophile. (d) First order in the catalyst. | ||
To gain further insights into the factors that impact the enantioselectivity, the geometries of transition states TS-R and TS-S were compared (Fig. 2). No obvious steric clashes and hydrogen-bonding interaction difference between the catalyst and substrates are detected in these two competing transition states. Computational studies of the total Hirshfeld charges on the aryl groups show that the key interaction for discrimination of the two aryl groups is mainly π–π stacking. Thienyl is a better donor than phenyl so it donates more electrons to C+. In major TS-R, the electron-deficient thienyl (0.13 e) is in closer contact with the electron-rich benzo ring of benzothiazoline. By contrast, in minor TS-S, the phenyl group (0.04 e) forms a slip-stacked configuration with the benzene ring on hydride. As a result, the stronger π–π stacking stabilizes TS-R more than the weaker π–π stacking stabilizes TS-S. This conclusion rather than some interaction of the transition state with the catalyst was tested by calculations of the fixed transition state formed by removing the catalyst. Single-point ΔΔE‡ without optimization shows 2.5 kcal mol−1 advantage for the stronger attractive π–π stacking in TS-R. This is the significant contribution to the 3.6 kcal mol−1 preference for the formation of the R-product. Therefore, attractive π–π stacking plays a major role in the selectivity.
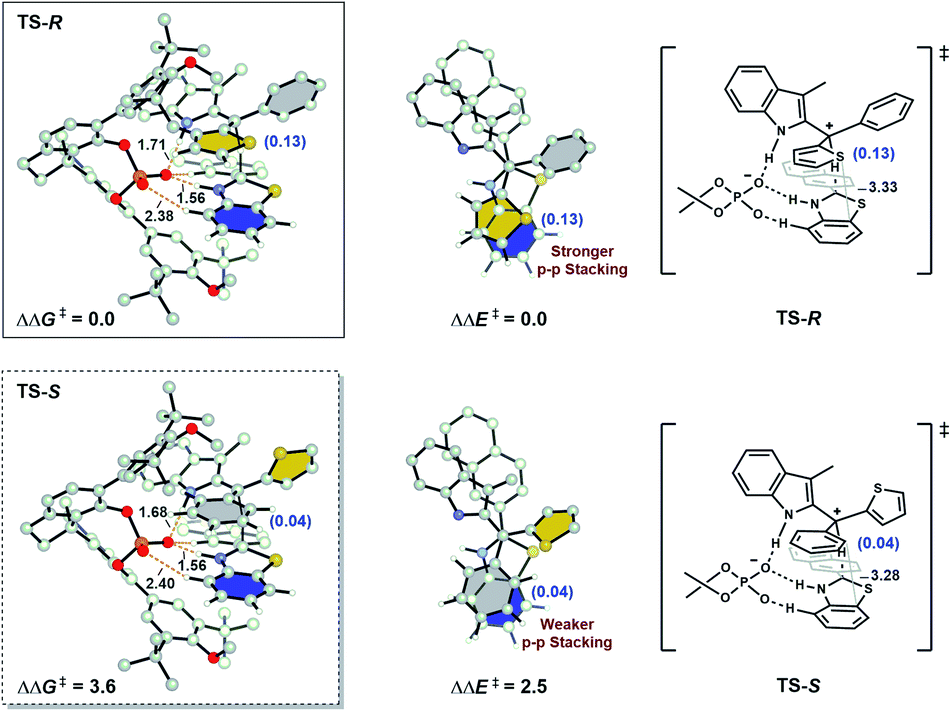 | ||
| Fig. 2 DFT-optimized stereo-determining transition structures. The distances are given in Ångstroms, and energies are given in kcal mol−1. Colored rings: grey, phenyl; yellow, thienyl; blue, benzo group on benzothiazoline. Numbers in parentheses are the total Hirshfeld charges on the aryl groups. | ||
Finally, to investigate the potential anticancer activity of the enantioenriched indole-containing triarylmethanes, we examined the cytotoxicity of the representative product 3d towards human cervical adenocarcinoma (HeLa), ovarian carcinoma (A2780), breast adenocarcinoma (MCF-7), colorectal carcinoma (HCT116), and lung carcinoma (A549) cells. A widely used anticancer drug, doxorubicin, was used as the control. As shown in Table 2, 3d exhibited significant cytotoxicity, with 50% cytotoxic concentration (CC50) values ranging from 5.6 to 18.2 μM. A549 lung carcinoma cells were found to be the most sensitive cell line towards 3d, and a normal cell line from the same origin (MRC-5 lung fibroblasts) was used to evaluate the cancer cell selectivity of 3d. The CC50 value in MRC-5 cells is 27.1 μM, corresponding to a selectivity index (SI; cytotoxicity in the normal cells/cytotoxicity in the cancer cells) of 4.8; whereas the SI value of doxorubicin is only 2.6. These preliminary results suggest that this class of compounds have promising potential for further development as anticancer drug candidates.
| Cell line | CC50 valuea (μM) | SIb | |||||
|---|---|---|---|---|---|---|---|
| HeLa | MCF-7 | A2780 | A549 | HCT116 | MRC-5 | ||
| a 50% cytotoxic concentration (CC50) values were determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in 72 h. The error bars were obtained as the standard deviation from the mean value based on three independent experiments. b Selectivity index, cytotoxicity in MRC-5 cells/cytotoxicity in A549 cells. | |||||||
| Doxorubicin | 1.4 ± 0.4 | 0.55 ± 0.11 | 0.36 ± 0.07 | 0.28 ± 0.06 | 1.4 ± 0.3 | 0.72 ± 0.14 | 2.6 |
| 3d | 18.2 ± 2.9 | 15.3 ± 2.3 | 8.9 ± 1.7 | 5.6 ± 0.9 | 9.7 ± 1.3 | 27.1 ± 3.4 | 4.8 |
We also tested the antiviral activity of another representative product 3a with enterovirus A71 (EV-A71) using the rhabdomyosarcoma (RD) cell line. The cytopathic effect (CPE) and intracellular viral RNA level were measured to reflect the antiviral effects. The CPE assay is commonly used to measure the virus-induced morphological change of host cells. Indeed, a strong CPE was observed after EV-A71 infection at a multiplicity of infection (MOI) of 0.01 for 36 hours. The morphology of RD cells changed from flat to round and even floated, indicating unhealthy and cell death. As shown in Fig. 3a, the CPE induced by EV-A71 infection was significantly reduced upon treatment with 3a. The antiviral effect was further measured by quantification of viral RNA genome reduction by RT-qPCR assays. We showed that the intracellular viral RNA level was decreased by 80–90% after treating with 3a at a concentration of 5–10 μM compared with untreated EV-A71 infected cells (Fig. 3b). The strong antiviral effect of 3a was also confirmed by viral titration. The virus titer was decreased by 35 fold upon treatment with 3a (Fig. 3c). Moreover, this compound showed low cytotoxicity according to the MTT assay (Table 3), thus indicating a high selectivity index and suggesting great potential of such molecules for antiviral drug development.
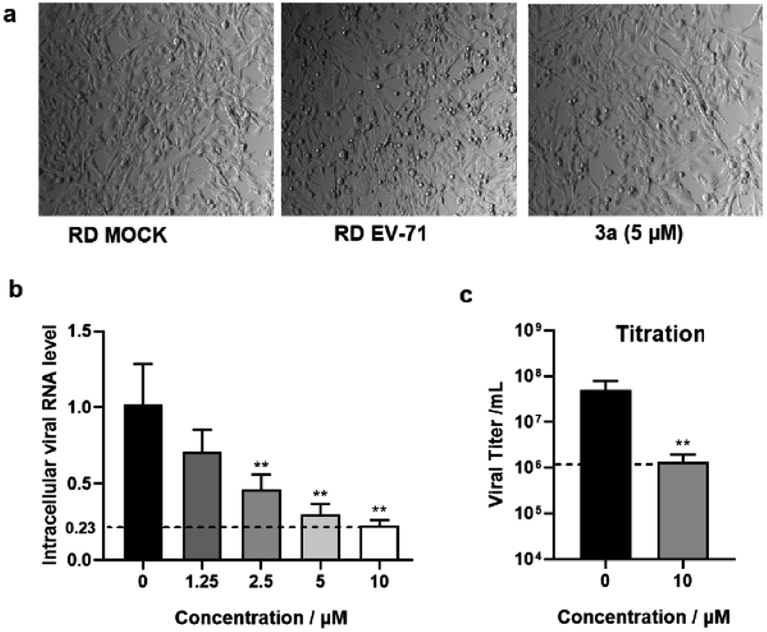 | ||
| Fig. 3 The antiviral effects of 3a shown by the CPE assay and intracellular viral RNA level. (a) RD cells were first treated with compounds at different concentrations and then infected with EV-A71 at a MOI of 0.01 after 2 hours. The cell morphology was observed 36 h post-infection. RD cells treated with DMSO only were set as Mock (or control). (b) Relative intracellular EV-A71 genome RNA level was determined by RT-qPCR. (c) The EV-A71 viral titer in the supernatant was measured by the 50% tissue culture infectious dose (TCID50) assay. Data are represented as mean ±SD (n = 3). **p < 0.01, compared with that of the not infected group. | ||
| Compound | CC50 (μM) | IC50 (μM) | Selectivity index |
|---|---|---|---|
| a CC50, 50% cytotoxic concentration tested by the viability assay with no viral infection. IC50, viral RNA copies deceased 50% compared with the control group (without compound treatment) in the secreted virions. A compound with a selectivity index (CC50/IC50) > 10 is assumed to be a potential candidate for further research analysis. | |||
| 3a | 55.46 | 2.27 | 24.43 |
In conclusion, despite the longstanding challenge in asymmetric discrimination between two sterically similar aryl groups and the dominant role of metal catalysis in limited previous studies, here we have demonstrated a new organocatalytic example with excellent efficiency and enantiocontrol. Versatile 2-indole imine methide bearing aryl and heteroaryl groups without a directing group was used as a platform for this study. The combined use of a superb chiral phosphoric acid catalyst and a benzothioazoline hydride source is critically important to the success. This protocol provided efficient access to a wide range of highly enantioenriched indole-containing triarylmethanes from the corresponding racemic tertiary alcohols. Mechanistic experiments, including control reactions and kinetic studies, provided important insights into the mechanism, which involves initial rate-determining dehydration (with concomitant substrate kinetic resolution) and subsequent enantioconvergent nucleophilic addition. Further DFT studies suggested that it is the π–π stacking, but not hydrogen bonding, that provides the key interaction for asymmetric discrimination between the phenyl and thienyl groups. This is also consistent with the robust enantiocontrol in the presence of various polar functional groups that are likely hydrogen-bond destroyers. Preliminary biological studies also demonstrated the great potential of these triarylmethanes for anticancer and antiviral drug development.
Data availability
Details of experimental procedures, characterizations, and copy of NMR spectra as well as HPLC traces are provided in the ESI.†Author contributions
Q. Y. performed the synthetic experiments. M.D. and P. Y. performed DFT calculations. C. C. and M. W. performed the antiviral experiments. Z. D. did the anticancer activity study. M. H. directed the antiviral study. G. Z. directed the anticancer activity study. K. N. H. directed the DFT calculations and mechanism analysis. J. S. conceived this work and directed the synthetic experiments. All the authors discussed the results and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (91956114 and 22077108), the Research Grants Council of Hong Kong (16303420, 11303320, 11104020, and 16309321), the Science and Technology Innovation Committee of Shenzhen (JCYJ20180507181627057 and JCYJ20200109141408054), and Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002), and the National Science Foundation (CHE-1764328 to K.N.H.). Computational work was supported by Center for Computational Science and Engineering at Southern University of Science and Technology, and the Extreme Science and Engineering Discovery Environment (XSEDE), which was supported by the National Science Foundation (OCI-1053575). We also thank Dr Herman H. Y. Sung and Dr Ian D. Williams for help with structure elucidation.References
- General reviews on asymmetric catalysis: (a) E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis I–III, Springer, Berlin, 1999 Search PubMed; (b) P. J. Walsh and M. C. Kozlowski, Fundamentals of Asymmetric Catalysis, University Science Books, Sausalito, CA, 2008 Search PubMed; (c) E. M. Carreira and H. Yamamoto, Comprehensive Chirality, 1st edn, Elsevier Science, 2012 Search PubMed.
- H. U. Blaser and H.-J. Federsel, Asymmetric Catalysis on Industrial Scale: Challenges, Approaches, and Solutions, Wiley-VCH, 2010 Search PubMed.
- (a) D. Ameen and T. J. Snape, Med. Chem. Commun., 2013, 4, 893–907 RSC; (b) S. Mondal, D. Roy and G. Panda, ChemCatChem, 2018, 10, 1941–1967 CrossRef CAS.
- T. Besset, R. Gramage-Doria and J. N. H. Reek, Angew. Chem., Int. Ed., 2013, 52, 8795–8797 CrossRef CAS PubMed.
- (a) Y.-Z. Sui, X.-C. Zhang, J.-W. Wu, S. Li, J.-N. Zhou, M. Li, W. Fang, A. S. C. Chan and J. Wu, Chem.–Eur. J., 2012, 18, 7486–7492 CrossRef CAS PubMed; (b) J. Mazuela, J. J. Verendel, M. Coll, B. Schäffner, A. Börner, P. G. Andersson, O. Pàmies and M. Diéguez, J. Am. Chem. Soc., 2009, 131, 12344–12353 CrossRef CAS PubMed; (c) J. Mazuela, P.-O. Norrby, P. G. Andersson, O. Pàmies and M. Diéguez, J. Am. Chem. Soc., 2011, 133, 13634–13645 CrossRef CAS PubMed; (d) T. Touge, H. Nara, M. Fujiwhara, Y. Kayaki and T. Ikariya, J. Am. Chem. Soc., 2016, 138, 10084–10087 CrossRef CAS PubMed; (e) T. Ohkuma, M. Koizumi, H. Ikehira, T. Yokozawa and R. Noyori, Org. Lett., 2000, 2, 659–662 CrossRef CAS PubMed; (f) H. Wang, Y. Zhang, T. Yang, X. Guo, Q. Gong, J. Wen and X. Zhang, Org. Lett., 2020, 22, 8796–8801 CrossRef CAS PubMed.
- For selected recent examples using directing groups: (a) S. Song, S.-F. Zhu, Y.-B. Yu and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 1556–1559 CrossRef CAS PubMed; (b) X. Wang, A. Guram, S. Caille, J. Hu, J. P. Preston, M. Ronk and S. Walker, Org. Lett., 2011, 13, 1881–1883 CrossRef CAS PubMed; (c) W. Liu, J. Guo, S. Xing and Z. Lu, Org. Lett., 2020, 22, 2532–2536 CrossRef CAS PubMed; (d) Y. Zheng, G. J. Clarkson and M. Wills, Org. Lett., 2020, 22, 3717–3721 CrossRef CAS PubMed.
- For selected examples using electronic effects: (a) A. Kokura, S. Tanaka, T. Ikeno and T. Yamada, Org. Lett., 2006, 8, 3025–3027 CrossRef CAS PubMed; (b) K. Yoo, H. Kim and J. Yun, Chem.–Eur. J., 2009, 15, 11134–11138 CrossRef CAS PubMed; (c) L.-L. Yang, D. Evans, B. Xu, W.-T. Li, M.-L. Li, S.-F. Zhu, K. N. Houk and Q.-L. Zhou, J. Am. Chem. Soc., 2020, 142, 12394–12399 CrossRef CAS PubMed; (d) M. Lee, Z. Ren, D. G. Musaev and H. M. L. Davies, ACS Catal., 2020, 10, 6240–6247 CrossRef CAS.
- Currently, successful differentiation between aryl and heteroaryl mostly involves a pyridine heterocycle due to its special coordinating ability. For selected examples, see: (a) C.-Y. Chen, R. A. Reamer, J. R. Chilenski and C. J. McWilliams, Org. Lett., 2003, 5039–5042 CrossRef CAS PubMed; (b) H. Yang, N. Huo, P. Yang, H. Pei, H. Lv and X. Zhang, Org. Lett., 2015, 17, 4144–4147 CrossRef CAS PubMed; (c) B. Wang, H. Zhou, G. Lu, Q. Liu and X. Jiang, Org. Lett., 2017, 19, 2094–2097 CrossRef CAS PubMed; (d) Y. Levedev, I. Polishchuk, B. Maity, M. D. V. Guerreiro, L. Cavallo and M. Rueping, J. Am. Chem. Soc., 2019, 141, 19415–19423 CrossRef PubMed; (e) F. Chen, D. He, L. Chen, X. Chang, D. Z. Wang, C. Xu and X. Xing, ACS Catal., 2019, 9, 5562–5566 CrossRef CAS; (f) S. Nian, F. Ling, J. Chen, Z. Wang, H. Shen, X. Yi, Y.-F. Yang, Y. She and W. Zhong, Org. Lett., 2019, 21, 5392–5396 CrossRef CAS PubMed; (g) D. He, X. Xu, Y. Lu, M.-J. Zhou and X. Xing, Org. Lett., 2020, 22, 8458–8463 CrossRef CAS PubMed.
- R. Kshatriya, V. P. Jejurkar and S. Saha, Eur. J. Org. Chem., 2019, 3818–3841 CrossRef CAS and references therein..
- Selected reviews on indole imine methides: (a) L. Wang, Y. Chen and J. Xiao, Asian J. Org. Chem., 2014, 3, 1036–1052 CrossRef CAS; (b) G.-J. Mei and F. Shi, J. Org. Chem., 2017, 82, 7695–7707 CrossRef CAS PubMed; (c) Y.-C. Zhang, F. Jiang and F. Shi, Acc. Chem. Res., 2020, 53, 425–446 CrossRef CAS PubMed; (d) J. Kikuchi and M. Terada, Chem.–Eur. J., 2021, 27, 10215–10225 CrossRef CAS PubMed.
- Selected examples on asymmetric reactions of 2-indole imine methides: (a) S. Qi, C.-Y. Liu, J.-Y. Ding and F.-S. Han, Chem. Commun., 2014, 50, 8605–8608 RSC; (b) X.-X. Sun, H.-H. Zhang, G.-H. Li, Y.-Y. He and F. Shi, Chem.–Eur. J., 2016, 22, 17526–17532 CrossRef CAS PubMed; (c) H.-H. Zhang, C.-S. Wang, C. Li, G.-J. Mei, Y. Li and F. Shi, Angew. Chem., Int. Ed., 2017, 56, 116–121 CrossRef CAS PubMed; (d) M. Sun, C. Ma, S.-J. Zhou, S.-F. Lou, J. Xiao, Y. Jiao and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 8703–8708 CrossRef CAS PubMed; (e) F. Goricke and C. Schneider, Org. Lett., 2020, 22, 6101–6106 CrossRef PubMed; (f) T.-Z. Li, S.-J. Liu, Y.-W. Sun, S. Deng, W. Tan, Y. Jiao, Y.-C. Zhang and F. Shi, Angew. Chem., Int. Ed., 2021, 60, 2355–2563 CrossRef CAS PubMed.
- For our efforts: (a) Z. Wang, F. Ai, Z. Wang, W. Zhao, G. Zhu, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 383–389 CrossRef CAS PubMed; (b) X. Li, M. Duan, Z. Deng, Q. Shao, M. Chen, G. Zhu, K. N. Houk and J. Sun, Nat. Catal., 2020, 3, 1010–1019 CrossRef CAS; (c) X. Li and J. Sun, Angew. Chem., Int. Ed., 2020, 59, 17049–17054 CrossRef CAS PubMed; (d) X. Li, M. Duan, P. Yu, K. N. Houk and J. Sun, Nat. Commun., 2021, 12, 4881 CrossRef CAS PubMed.
- For recent reviews and leading studies on chiral phosphoric acid catalysis, see: (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed; (c) D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626–9627 CrossRef CAS PubMed; (d) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed; (e) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS; (f) T. James, M. van Gemmeren and B. List, Chem. Rev., 2015, 115, 9388–9409 CrossRef CAS PubMed.
- (a) C. Zhu, K. Saito, M. Yamanaka and T. Akiyama, Acc. Chem. Res., 2015, 48, 388–398 CrossRef CAS PubMed; (b) H. Osakabe, S. Saito, M. Miyagawa, T. Suga, T. Uchikura and T. Akiyama, Org. Lett., 2020, 22, 2225–2229 CrossRef CAS PubMed.
- (a) S.-L. You, Chem.–Asian J., 2007, 2, 820–870 CrossRef CAS PubMed; (b) C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC; (c) M. Rueping, E. Sugiono and F. R. Schoepke, Synlett, 2010, 852–865 CrossRef CAS.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2108959 and 2116827. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00636g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |