
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advancing homogeneous catalysis for parahydrogen-derived hyperpolarisation and its NMR applications†
Ben. J.
Tickner
 *ab and
Vladimir V.
Zhivonitko
*a
*ab and
Vladimir V.
Zhivonitko
*a
aNMR Research Unit, Faculty of Science, University of Oulu, P.O. Box 3000, Oulu, 90014, Finland. E-mail: ben.tickner@alumni.york.ac.uk; vladimir.zhivonitko@oulu.fi
bDepartment of Chemical and Biological Physics, Faculty of Chemistry, Weizmann Institute of Science, Rehovot, 7610001, Israel
First published on 22nd March 2022
Abstract
Parahydrogen-induced polarisation (PHIP) is a nuclear spin hyperpolarisation technique employed to enhance NMR signals for a wide range of molecules. This is achieved by exploiting the chemical reactions of parahydrogen (para-H2), the spin-0 isomer of H2. These reactions break the molecular symmetry of para-H2 in a way that can produce dramatically enhanced NMR signals for reaction products, and are usually catalysed by a transition metal complex. In this review, we discuss recent advances in novel homogeneous catalysts that can produce hyperpolarised products upon reaction with para-H2. We also discuss hyperpolarisation attained in reversible reactions (termed signal amplification by reversible exchange, SABRE) and focus on catalyst developments in recent years that have allowed hyperpolarisation of a wider range of target molecules. In particular, recent examples of novel ruthenium catalysts for trans and geminal hydrogenation, metal-free catalysts, iridium sulfoxide-containing SABRE systems, and cobalt complexes for PHIP and SABRE are reviewed. Advances in this catalysis have expanded the types of molecules amenable to hyperpolarisation using PHIP and SABRE, and their applications in NMR reaction monitoring, mechanistic elucidation, biomedical imaging, and many other areas, are increasing.
 Ben. J. Tickner | Ben. J. Tickner did his PhD research (2017–2020) in the group of Simon Duckett at the Centre for Hyperpolarisation in Magnetic Resonance, University of York. His research was focused on developing new iridium SABRE catalysts to hyperpolarise target molecules such as pyruvate for applications in biomedical imaging, chemosensing, and reaction monitoring. Since then he has worked with Ville-Veikko Telkki and Vladimir Zhivonitko on hyperpolarised ultrafast Laplace NMR as a postdoctoral researcher at the University of Oulu, Finland (2020–2021). He is currently continuing work on hyperpolarisation with Lucio Frydman at the Weizmann Institute of Science, Israel. |
 Vladimir V. Zhivonitko | Associate Professor Vladimir V. Zhivonitko studied coordination and organic chemistry at the Novosibirsk State University (2000–2005). He received his PhD degree in 2008 in the group of Professor Igor V. Koptyug at the International Tomography Center, Novosibirsk, for studies of nonlinear catalytic processes using NMR imaging. He continued at ITC as a postdoc, researcher, and senior researcher in the field of nuclear spin hyperpolarization and catalysis. Since 2017 he has worked at the NMR Research Unit of the University of Oulu. His research interests include signal enhancement in NMR, applications of NMR and MRI in catalysis and materials science, Laplace and ultrafast NMR methods. |
1. Introduction
Nuclear Magnetic Resonance (NMR) spectroscopy and imaging are widely known methods that examine nuclear spins within molecules and materials. Consequently, they provide important structural, dynamic, and spatial information about matter without using ionising radiation that can often destroy the samples under examination. Moreover, they can be applied to study any state of matter: gas, liquid, or solid. For these reasons, NMR-based techniques have become a method-of-choice for many scientists to interrogate the structure and dynamic properties of small molecules, proteins, catalytic and functional materials, tissues and many other systems.1 Despite these advantages, they are considered relatively insensitive as concentrated samples (≥mM concentrations) are often required to get strong enough signals to draw any conclusions. This insensitivity stems from the small population differences across closely spaced nuclear spin energy levels which is dictated by the Boltzmann distribution under standard thermal equilibrium conditions. This is evident from the tiny fraction of nuclear spins (e.g., 1 in every 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 1H nuclei at 9.4 T) that effectively contribute to a detected NMR signal. The sensitivity issue becomes even more pronounced for nuclei that possess a smaller gyromagnetic ratio (such as 13C, 15N, 19F, 31P and many others) and at lower magnetic fields, as nuclear spin states become closer in energy. This is usually mitigated by using concentrated samples and/or time-consuming signal averaging to generate spectra of sufficient signal-to-noise ratio (SNR).
000 1H nuclei at 9.4 T) that effectively contribute to a detected NMR signal. The sensitivity issue becomes even more pronounced for nuclei that possess a smaller gyromagnetic ratio (such as 13C, 15N, 19F, 31P and many others) and at lower magnetic fields, as nuclear spin states become closer in energy. This is usually mitigated by using concentrated samples and/or time-consuming signal averaging to generate spectra of sufficient signal-to-noise ratio (SNR).
Great effort is focussed towards instrumental and methodological developments that improve the sensitivity of NMR.2,3 As a result, approaches collectively known as hyperpolarisation have arisen from these efforts. Hyperpolarisation refers to creation of a non-equilibrium nuclear spin state with dramatically larger population differences between the energy levels relative to those for thermally polarised samples (i.e., those coming from a Boltzmann distribution). NMR signals can be orders of magnitude more intense as a consequence.3 Hyperpolarisation is an active topic of research as it can significantly reduce experiment times since large numbers of repetitions are no longer required to generate discernible NMR signals. The enhanced NMR signals provided by hyperpolarisation have advantages for many applications such as detecting reaction intermediates and reaction monitoring, mechanistic elucidations, analysis of low concentration mixtures, and metabolic imaging.4–7
Several hyperpolarisation techniques are known to derive strong non-equilibrium nuclear spin polarisation via various sources.4 For instance, Spin Exchange Optical Pumping (SEOP)8 and dissolution Dynamic Nuclear Polarisation (DNP)9 utilize circularly polarised light and polarisation of electron spins, respectively, to hyperpolarise nuclear spins. To a great extent, these methods are underpinned by advanced physics and have both been known for at least the last 65 years.4 In this review, we focus on hyperpolarisation techniques that are much more chemical by nature since they utilise catalytic transformations of parahydrogen (para-H2), the spin-0 isomer of H2. Para-H2 serves as both a chemical reagent and the source of hyperpolarisation at the same time. Methods that exploit para-H2 spin order as a source of hyperpolarisation are collectively termed as ParaHydrogen-Induced Polarisation (PHIP) and have been developed since the 1980s.6,10–12 They provide a low-cost route to hyperpolarise target molecules because expensive equipment is not required and the para-H2 feedstock is cheap and easy to produce.13
The para-H2 molecule is NMR silent (i.e. it does not give any NMR signal). However, breaking the symmetry of para-H2 in a chemical reaction can lead to a dramatic enhancement of NMR signals of the reaction products.14,15 This chemistry underpins the PHIP effect and can be hugely beneficial for the detection of low concentration (≪1 mM) or short-lived molecules (<1 minute) that might be challenging to observe using conventional NMR with thermal polarisation. Herein, we discuss various types of chemical reactions that can break para-H2 symmetry and give rise to PHIP. These reactions are often catalysed by transition metal complexes, and we discuss the role these species play in achieving PHIP. In general, many researchers are working to increase the substrate scope, efficiency, conversion rates, lifetime of hyperpolarised states, solvent tolerability, and many other properties of these catalysts which are directly linked to the NMR signal enhancements that can be produced. Therefore, we focus on recent advances in homogeneous catalyst design which have allowed the hyperpolarisation of a wider range of molecules with greater efficiency using PHIP and related SABRE16,17 effects. We also give some key examples of the tremendous value these novel catalysts have in the areas of mixture analysis, mechanistic elucidation, reaction monitoring, and biomedical imaging. In addition to metal complexes, recent emerging metal-free catalysts are also discussed.
2. Para-H2: properties, production and PHIP
In this review we describe the properties and production of para-H2 only briefly and refer the interested reader to other works6,17–19 for specific details. Generally, at room temperature dihydrogen gas (H2) exists as a mixture of ortho and para nuclear spin isomers quite accurately in a 3:1 ratio, and conversion between these isomers is slow (on the order of days and even longer) due to the symmetry disallowed nature of this transition.18 H2 can be enriched in the lower energy para isomer (para-H2) via interactions with a metal centre or a paramagnetic material (catalyst) at low temperatures.20–22 Para-H2 enrichments of up to 50% can occur at liquid nitrogen temperatures (77 K) and even higher values of 98% can be achieved at 28 K.5 This enrichment survives removal of the spin interconversion catalyst and return to ambient temperature,13,20,22 and it is important since analogous PHIP reactions with hydrogen gas thermally equilibrated at or near room temperature does not typically produce NMR signal enhancements.
Para-H2 has only one nuclear spin manifold and its wavefunction is denoted as (|αβ〉 − |βα〉)/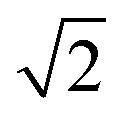 or |S0〉 (nuclear spin singlet). Ortho-H2 has a nuclear spin degeneracy of three in the absence of a magnetic field, and these states can be described as |αα〉, (|αβ〉 + |βα〉)/
or |S0〉 (nuclear spin singlet). Ortho-H2 has a nuclear spin degeneracy of three in the absence of a magnetic field, and these states can be described as |αα〉, (|αβ〉 + |βα〉)/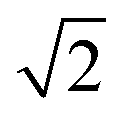 , and |ββ〉, or by using the notation |T+1〉, |T0〉, and |T−1〉 (nuclear spin triplet), respectively. In this notation, α and β are spin-up and spin-down states of individual nuclei in H2 molecules. As para-H2 has a nuclear spin quantum number of 0, it does not respond with a signal to the radiofrequency excitation applied during NMR pulse sequences and is often referred to as being ‘NMR silent’. The symmetry of para-H2 must be broken to allow the pair of 1H nuclei to become observable using 1H NMR. This can be achieved in a catalytic process in which, e.g., the 1H nuclei of the para-H2 molecule are incorporated into a target molecule (Fig. 1a).9,24,25 These newly introduced protons can exhibit dramatically enhanced 1H NMR signals. The nuclear hyperpolarisation, however, is typically short-lived and decays reaching the thermal equilibrium level due to nuclear spin relaxation which is typically characterised by the time taken to establish thermal longitudinal magnetisation, T1.
, and |ββ〉, or by using the notation |T+1〉, |T0〉, and |T−1〉 (nuclear spin triplet), respectively. In this notation, α and β are spin-up and spin-down states of individual nuclei in H2 molecules. As para-H2 has a nuclear spin quantum number of 0, it does not respond with a signal to the radiofrequency excitation applied during NMR pulse sequences and is often referred to as being ‘NMR silent’. The symmetry of para-H2 must be broken to allow the pair of 1H nuclei to become observable using 1H NMR. This can be achieved in a catalytic process in which, e.g., the 1H nuclei of the para-H2 molecule are incorporated into a target molecule (Fig. 1a).9,24,25 These newly introduced protons can exhibit dramatically enhanced 1H NMR signals. The nuclear hyperpolarisation, however, is typically short-lived and decays reaching the thermal equilibrium level due to nuclear spin relaxation which is typically characterised by the time taken to establish thermal longitudinal magnetisation, T1.
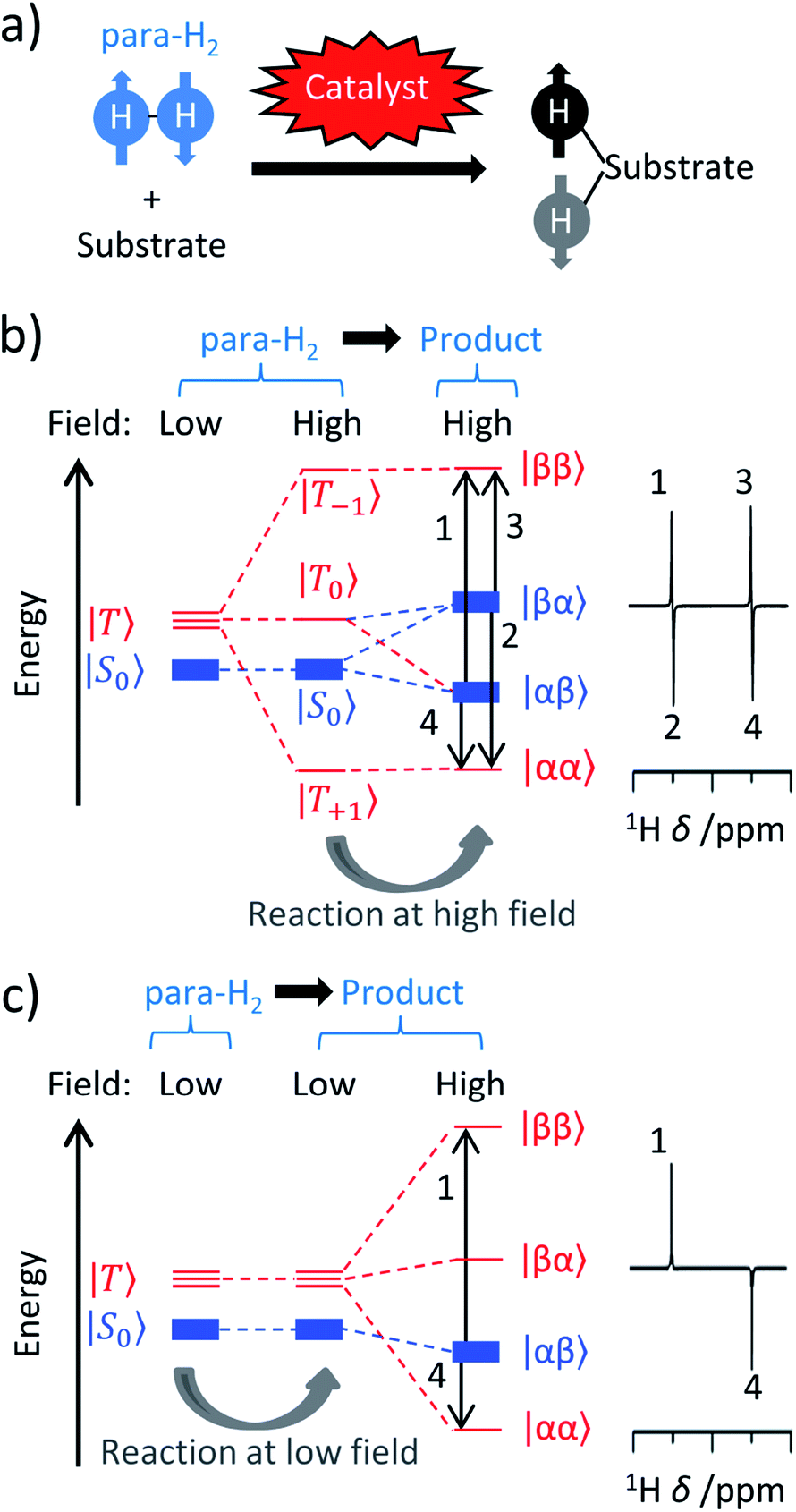 | ||
| Fig. 1 (a) Parahydrogen is ‘NMR silent’ and can only be observed by NMR if its symmetry can be broken in a pairwise reaction, often catalysed by a metal centre. Products containing 1H nuclei that were originally located in para-H2 can exhibit NMR signals orders-of-magnitude more intense than would be recorded under ‘thermal’ (i.e. Boltzmann) controlled conditions. (b) Under PASADENA conditions, the para-H2 symmetry-breaking process in a chemical reaction occurs at the spectrometer (high) field and results in population of both the |αβ〉 and |βα〉 nuclear spin energy levels of the product. (c) Under ALTADENA conditions the reaction occurs at Earth's (low) field outside the spectrometer before insertion into the spectrometer for NMR detection. As a result, only one of the |αβ〉 or |βα〉 levels of the product are populated. In this depiction the |αβ〉 state is shown lower in energy and populated. Note that the two protons in the product are assumed to form a weakly coupled AX spin system and the energies are not shown to scale. NMR spectra may show a different appearance when recorded using different flip angles. In these depictions, the population of each state is indicated by the thickness of the line. | ||
A typical requirement for such reactions to yield strong PHIP effects is that catalysed incorporation of para-H2 into the target substrate must occur in a pairwise manner. In other words, both introduced 1H nuclei should originate from the same para-H2 molecule for them to transfer the two-spin correlation (nuclear spin order) inherited from para-H2 to the substrate. In cases of non-pairwise addition, the spin correlation is not transferred to the substrate and, as a consequence, PHIP effects will not be observed. At the same time, PHIP can be observed for catalytic intermediates if initial para-H2 activation by the catalyst active centre is pairwise, although the full cycle can be non-pairwise. Based on this possibility, effects such as oneH-PHIP23–28 can be observed wherein only one of the para-H2 originating protons ends up in the hyperpolarised product. There are also reactions in which molecules can become hyperpolarised without chemical incorporation of para-H2 into the target structures, as takes place in SABRE.16,17,29 These processes still contain a pairwise symmetry-breaking step in the catalytic cycle and rely on other means (e.g., level anticrossings, rf pulsing) to transfer the polarisation or the spin order to the target sites. Therefore, an understanding of the catalysis underpinning these reactions is clearly vital to rationalise and account for these differing effects.
The appearance of PHIP NMR spectra can differ if the catalytic reaction takes place directly at the high magnetic field of the spectrometer (while the sample is inside the magnet) or outside the magnet at the Earth's magnetic field (sometimes referred to as ‘low’ field) before the sample is transferred to the spectrometer for ‘high’ field NMR detection.14,18,30,31 For instance, Fig. 1b and c shows schematically the influence of these two different (spectrometer and Earth's) fields on the populations of the nuclear spin states of para-H2 and the resultant reaction product(s) in the case when the para-H2 originating protons form a weakly coupled AX spin system at high field. These regimes have been termed Parahydrogen And Synthesis Allow Dramatically Enhanced Nuclear Alignment (PASADENA)14 for symmetry-breaking at high field and Adiabatic Longitudinal Transport After Dissociation Engenders Nuclear Alignment (ALTADENA)32 for symmetry-breaking at low field. Hyperpolarisation created under ALTADENA and PASADENA conditions may display different 1H NMR signal shapes (Fig. 1b and c), although both approaches can be described more generally as PHIP.
3. Hyperpolarised metal dihydrides: ligation and oxidative addition of para-H2
There are different types of chemical reactions that can break the symmetry of para-H2 in a pairwise fashion. Perhaps the simplest of these is ligation of para-H2 to a metal centre to form an η2-H2 complex. The implications of this for catalysis are that the improvement in NMR sensitivity provided by PHIP can make detection of η2-H2 complexes feasible. An excellent example is the indirect detection of a [Ni(η2-H2)(L-κ2P,P')2]2+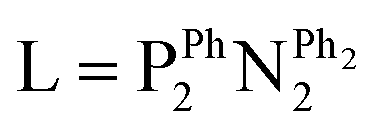 complex that is impossible to discern using standard thermally-polarised 1H NMR measurements.33 The short-lived nature of η2-H2 species usually requires advanced NMR methods (such as Chemical Exchange Saturation Transfer (CEST) and partially negative line effect) to detect them indirectly.33–35
complex that is impossible to discern using standard thermally-polarised 1H NMR measurements.33 The short-lived nature of η2-H2 species usually requires advanced NMR methods (such as Chemical Exchange Saturation Transfer (CEST) and partially negative line effect) to detect them indirectly.33–35
Further reaction of η2-H2 complexes to form metal dihydrides can result in PHIP effects as oxidative addition of para-H2 to a metal centre also breaks para-H2 symmetry.11,36–39 Activation of dihydrogen by transition metal centres has been known since the 1960s.40,41 This typically occurs via a concerted mechanism where the newly introduced groups are located in a cis relationship.9,10 These examples provide a fertile playground for PHIP if this activation is pairwise and faster than nuclear spin relaxation. Early examples in the late 1980s were dihydride complexes of Rh, Ir and Ru.14,15,30,42 Many of these are short-lived intermediates in catalytic processes27,43–45 (such as hydrogenation or hydroformylation), whereas others are stable hydride species whose ligand exchange processes may be reversible or even light-activated.36,46,47 For example, one of the first reported examples of PHIP was the activation of para-H2 by [RhCl(PPh3)3] to form [RhCl(H)2(PPh3)3] in which the 1H NMR signals of its hydride ligands were significantly enhanced (Fig. 2a).14 Related examples around this time showed hyperpolarised 1H NMR signals for the hydride ligands of [IrBr(H)2CO(PPh2(CH2)2PPh2)], which forms upon oxidative addition of para-H2 to [IrBrCO(PPh2(CH2)2PPh2)] and is the first step in the hydrogenation of alkynes to alkenes using this catalyst (Fig. 2b).31 In these contexts, the enhanced 1H NMR signals for the hydride sites of transition metal complexes are extremely useful for enabling their detection, particularly in cases where the metal complex may be at low concentration. Namely, hydride NMR signals for catalytic intermediates such as [Rh(H)2Cl(PPh3)2(styrene)], which is formed following phosphine loss from [RhCl(H)2(PPh3)3] and subsequent alkene coordination, can be made visible using PHIP.45 This intermediate is involved in the hydrogenation of alkenes and the detection of such species can be important in determining reaction mechanisms.7,10,11
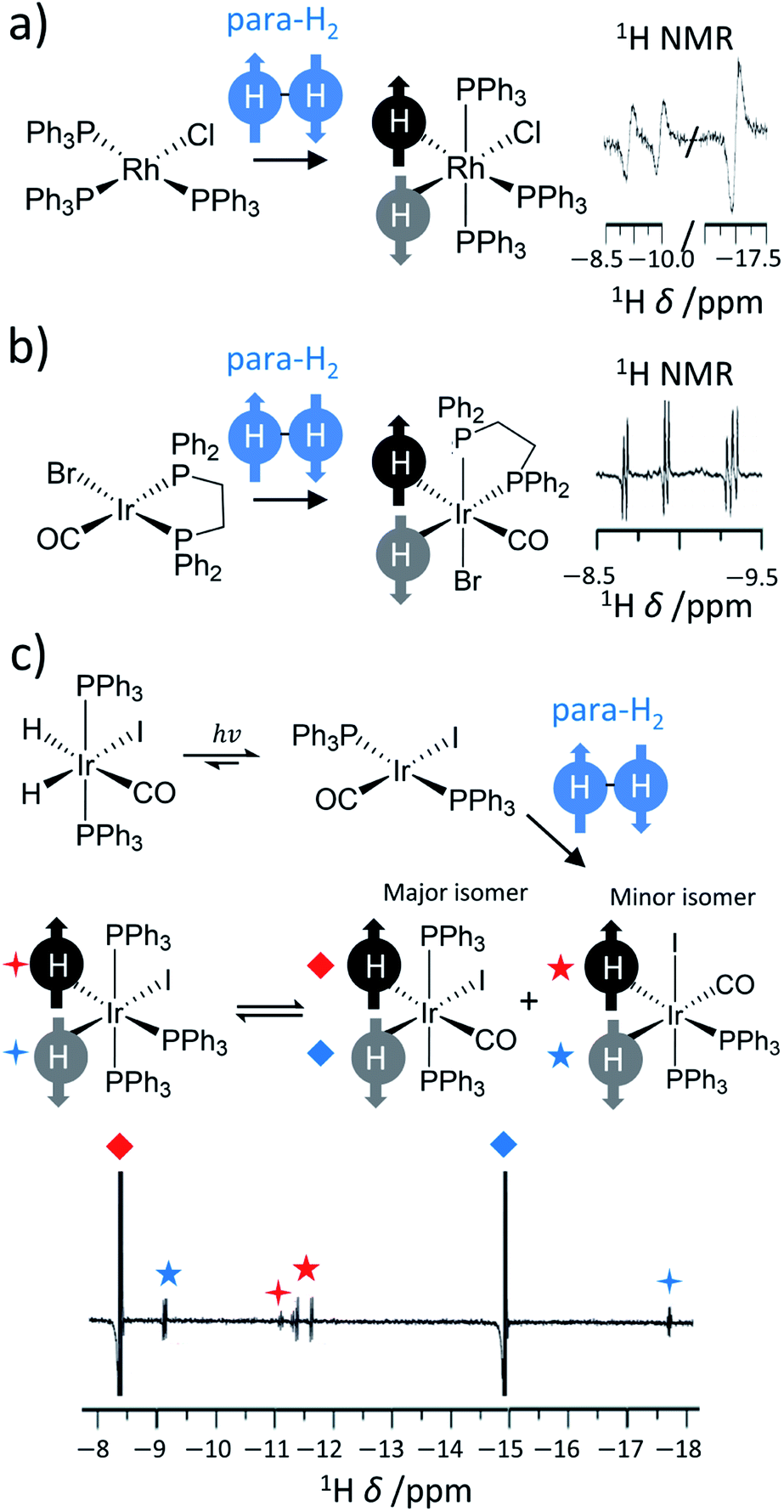 | ||
| Fig. 2 Examples of symmetry-breaking oxidative addition reactions. Formation of hyperpolarised (a) [RhCl(H)2(PPh3)3] upon para-H2 addition to [RhCl(PPh3)3], (b) [IrBr(H)2CO(PPh2(CH2)2PPh2)] following para-H2 addition to [IrBrCO(PPh2(CH2)2PPh2)] and (c) [IrCO(H)2I(PPh3)2] upon para-H2 addition to [IrCOI(PPh3)2]. Enhanced hydride NMR signals for a minor geometric isomer of [IrCO(H)2I(PPh3)2] are also visible as para-H2 addition can occur to both symmetry axes of [IrCOI(PPh3)2]. Hyperpolarised hydride NMR signals for [Ir(H)2I(PPh3)3], which is formed from CO loss from [IrCO(H)2I(PPh3)2], are also discerned. Adapted from (a) ref. 14 (b) ref. 31 and (c) ref. 36. | ||
In many of these mechanisms, activation of para-H2 is irreversible and, as a consequence, PHIP effects can only be observed over a narrow time window in which reactivity is occurring and nuclear spin relaxation is yet to dominate. Many catalysts, particularly those based on iridium complexes, have been shown to reversibly activate para-H2 and therefore enhanced hydride signals can be observed at will upon reaction with fresh para-H2.29,35,48 This is particularly useful in mechanistic studies involving iridium dihydride complexes38 and the detection of low concentrations of metal complexes for which enhanced hydride resonances may be diagnostic of particular ligands in complex mixtures.38,49–51 Reactivity of this type is exploited in SABRE as enhanced hydride polarisation can be transferred to other sites in the catalyst (this is discussed in more detail in Section 7).29
Other catalysts utilise photochemistry to promote ligand loss pathways and produce enhanced hydride NMR signals.52 For example, a [IrCOI(PPh3)2] complex can be formed from the light-induced reductive elimination of hydrogen from [IrCO(H)2I(PPh3)2]. The original [IrCO(H)2I(PPh3)2] can be reformed from [IrCOI(PPh3)2], albeit in a hyperpolarised state, by oxidative addition of para-H2 (Fig. 2c). In this example, PHIP enables the formation of metal hydrides over millisecond timescales to be monitored and kinetic rate constants for this process to be determined: feats that are impossible to achieve using thermal NMR polarisation due to the need to perform time consuming signal averaging.52
As the systems that undergo homolytic dihydrogen splitting have increased rapidly in recent years, an analogous increase in PHIP catalysts has accompanied this. Nowadays, metal catalysts based on Pd,47,53–56 Co26,27,57–59 and Pt23 have all been reported to break para-H2 symmetry via oxidative addition. We also note that metal clusters11 and heterogeneous supported catalysts60 have emerged as valuable catalysts that can activate para-H2 giving rise to PHIP and SABRE effects. The heterogeneous systems have been described in detail elsewhere.5,61,62
4. Metal-free PHIP: heterolytic para-H2 splitting and beyond
Over recent years, many metal-free catalysts have been developed to activate para-H2. Most of these systems are based on the use of frustrated Lewis pairs (FLPs)63 and can split para-H2 heterolytically in a way that also produces enhanced NMR signals for sites within the FLP.64–69 The heterolytic activation occurs in a pairwise manner for hyperpolarised reaction products to be observed and can provide valuable insight into the mechanism of H2 activation by FLPs. These effects were first demonstrated using ansa-aminoborane FLPs such as 1-{2-[bis(pentafluorophenyl)boryl]benzyl}-2,2,4,7-tetramethyl-1,2,3,4-tetrahydroquinoline (QCAT) (Fig. 3a).66 Activation of para-H2 by QCAT leads to formation of a QCAT-H2 adduct in which the 1H NMR signals for those nuclei originating from para-H2 are enhanced by up to 30-fold. Polarisation transfer from these sites to 11B nuclei was achieved using an INEPT-type pulse sequence and yielded a 10-fold enhancement in 11B NMR signal intensity. Since then, a wider range of ansa-aminoboranes have been used for metal-free PHIP, including those activating para-H2 under ambient temperatures in a reversible fashion.65 Spontaneous polarisation transfer from para-H2 derived 1H nuclei to 11B and 15N sites in a 7 T spectrometer is also possible with significant 350-fold signal enhancements recorded for 15N sites of the FLP.64 Activation of para-H2 using FLPs based on P–C68 and P–Sn70 have also been documented.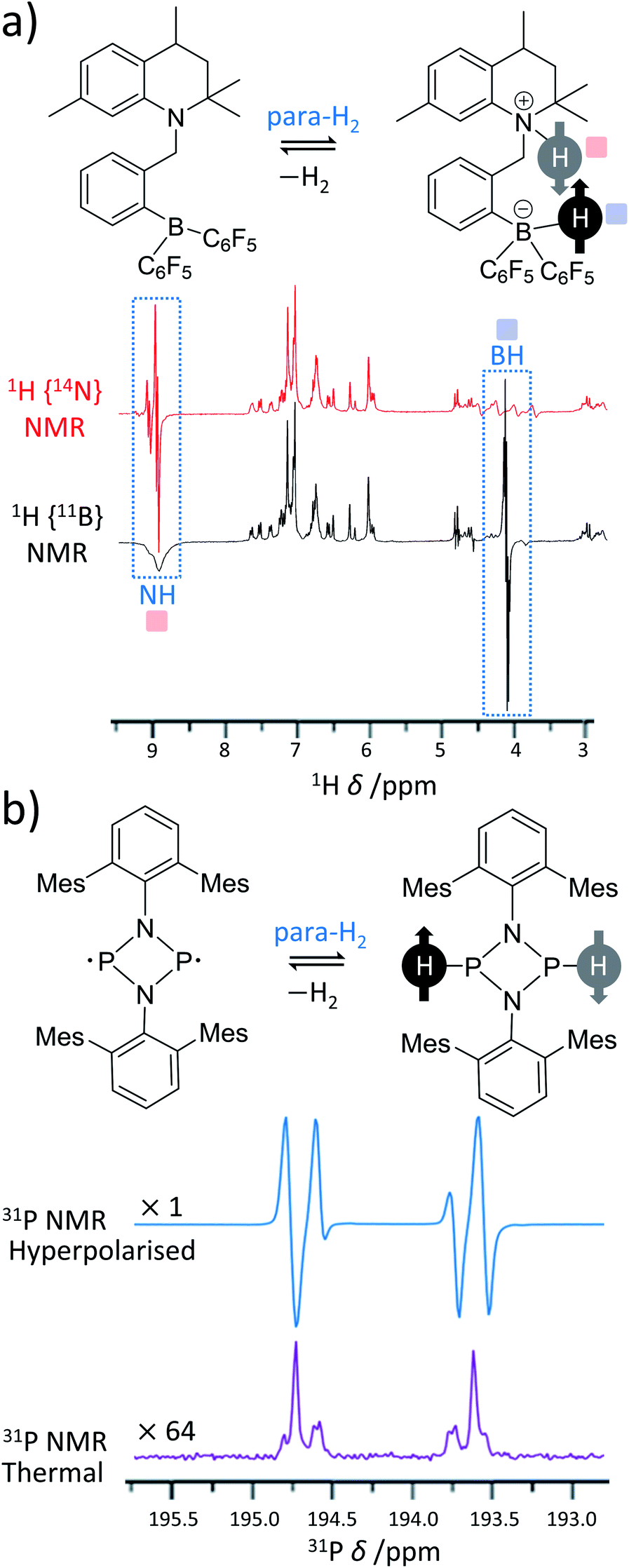 | ||
| Fig. 3 Examples of PHIP in metal-free para-H2 activations. (a) ansa-Aminoboranes can activate para-H2 using a N–B frustrated Lewis pair centre. For example, 14N- and 11B-decoupled 1H NMR spectra acquired using QCAT ansa-aminoborane are presented. (b) Pnictogen biradicaloids can also activate para-H2 and produce PHIP. For instance, a P–P biradicaloid pair where Mes = 2,4,6-trimethylphenyl gives the presented 31P NMR spectrum. For comparison, the thermal 31P spectrum expanded vertically by a factor of 64 is also shown in the figure. Adapted from (a) ref. 66 and (b) ref. 67. | ||
In addition to FLPs, P–P67,69 and As–P69 biradicaloids, an alternative class of metal-free para-H2 activators, have also been used in PHIP with up to 2000-fold signal enhancements for 31P sites at 9.4 T. In this case, the activation is homolytic in nature leading to the formation of hyperpolarised biradicaloid-H2 adducts. In addition to enhanced para-H2-derived 1H sites, the hyperpolarisation of 31P sites could be observed either spontaneously or by using rf pulsing to transfer polarisation from 1H to 31P, depending on the structure of the biradicaloid (Fig. 3b). Generally, P–P, As–P and As–As biradicaloids containing four- and five-membered rings were shown to activate para-H2, in many cases reversibly.69 This reversibility allowed the observation of hyperpolarised starting para-H2 metal-free activators (FLPs64 or biradicaloids69) without actual modification of their structures and demonstrates features of a metal-free SABRE effect. In these cases, PHIP has been used as a great mechanistic tool to study para-H2 activation by such systems.
While current examples of metal-free systems for para-H2 activation have been reported in organic solvents such as toluene-d8,66,69 dichloromethane-d2,64,65 and bromobenzene-d5,68 they may in the future provide a promising route for PHIP in aqueous solvents without the use of toxic metal catalysts. Generally, metal-free PHIP using FLPs offers an alternative route to break para-H2 symmetry that does not rely on a transition metal complex for traditional oxidative addition. Homolytic H2 activation typically involves complex orbital overlap effects.11 Repulsive interactions between filled orbitals on the metal centre and the H2 ligand can be a barrier to oxidative addition that must be overcome. For some metal complexes, this barrier can be so large that heterolytic splitting of H2 becomes feasible.71 Therefore, examples of metal-catalysed heterolytic para-H2 addition34,71,72 to generate enhanced NMR signals for reaction products may emerge in the future.
5. Hydrogenation reactions
5.1 Transition metal catalysed alkyne and alkene hydrogenation
Many catalytic processes involve H2 activation as an initial step, including metal-catalysed hydrogenation or hydroformylation. Early PHIP studies that involved para-H2 activation by square planar group 9 precatalysts of rhodium30,42 and iridium31,73,74 were accompanied by transfer of these newly introduced hydrides to an alkyne or alkene (Fig. 4a). If this reaction occurs rapidly relative to the relaxation rate of the hyperpolarised metal dihydride then enhanced polarisation may survive transfer into the product.14,30,31,45 Early PHIP studies involved hydrogenation of simple alkenes and alkynes such as styrene and phenylacetylene,15,30,75 although in more recent years PHIP has been extended to significantly more complex molecules including amino acid derivatives76 and even complex synthetic oligopeptides which have been of use to probe protein–ligand interactions.77–82 Heteronuclear polarisation within the transition metal hydrogenation catalysts, or hydrogenated reaction products, can also be achieved following either dedicated pulse sequences,74,76,83–85 spontaneous low field transfer,86 or field cycling.87–89 | ||
| Fig. 4 (a) Depiction of the general process of a pairwise hydrogenation reaction. Oxidative addition is the primary step that breaks the symmetry of para-H2. Subsequent steps such as substrate coordination, hydride migration, and product elimination, yield a hydrogenated product with enhanced 1H NMR signals. (b) Examples of catalysts reported for early PHIP studies14,30,31,42,45 with commercial catalysts commonly used for PHIP today. (c) Example hydrogenation of vinyl acetate to form ethyl acetate, which was used to study the effect of Rh catalyst on factors such as 13C polarisation level and conversion to the product. Adapted from ref. 90. (d) Example water-soluble catalysts reported to produce PHIP in aqueous solutions.44,98 (e) Representative examples of ruthenium,15 palladium47,54 and cobalt59 catalysts used for PHIP. | ||
The most commonly used catalysts for these PHIP hydrogenation reactions remain based on rhodium, and to a lesser extent iridium (Fig. 4b), although there were early examples of less well-defined systems using ruthenium15 to achieve hyperpolarised signals for hydrogenated products. Rapid hydrogenation is required to observe PHIP effects before nuclear spin relaxation destroys the hyperpolarisation. Therefore, optimisation of these catalysts is essential as the level of hyperpolarisation achieved strongly depends on factors such as hydrogenation rate.90 Significant efforts have been directed at the optimisation of catalysts for standard hydrogenation reactions, which usually focus on improving the catalyst turnover, reactivity and selectivity. Such efforts are not typically applied to the optimisation of hydrogenation catalysts for PHIP reactions, which often use commercially available Rh catalysts (such as [Rh(diene)(dppb)][BF4], diene = norbornadiene or 1,5-cyclooctadiene) that are sometimes incapable of completely hydrogenating a target molecule in just a few seconds without the use of high H2 pressure.91 In PHIP reactions, the presence of paramagnetic impurities, catalyst degradation products, resting states, or transient intermediates are likely to relax hyperpolarised molecules and destroy the PHIP effect. As soon as para-H2 is ligated to a metal centre its symmetry is broken and the spin order can then be lost before further transfer of the para-H2 originating protons. Therefore, unwanted polarisation loss will depend on relaxation and the lifetimes of catalyst-bound para-H2 species, of which there may be many in the catalytic cycle. These factors will play a role in the polarisation transfer efficiency of PHIP and are additional factors that are not typically considered in traditional hydrogenation catalysis with thermally equilibrated H2.
Therefore, the roles of catalyst structure and concentration on hyperpolarisation efficiency were studied for [Rh(COD)(BINAP)][BF4] and [Rh(COD)(dppb)][BF4] catalysts in butyl acrylate hydrogenation using para-H2.92 The authors defined a parameter called polarisation transfer efficiency (PTE) which quantified the ratio of the experimental and the idealised hyperpolarisation that could be achieved for the product (butyl propanoate) in an ensemble of product molecules; it depended on measurables such as relaxation and hydrogenation rate. For [Rh(COD)(BINAP)][BF4], PTE increased as the catalyst concentration is lowered, and generally increased as reaction time is increased, which was related to a drop in hydrogen pressure as the reaction proceeds. The authors suggested that this effect is likely related to a shortened residence time of the para-H2 molecule in the coordinated state on the metal centre when the alkene/hydrogen ratio decreases.92 The shorter residence can favour high PTE since the time for relaxation of the para-H2 spin order is reduced. The PHIP efficiency was stronger at higher magnetic fields (14 T compared to 7 T)92 where the T1 relaxation time of dihydrides is typically longer.93 The change of PTE as a function of hydrogenation reaction time appeared different demonstrating rise and fall behavior when the [Rh(COD)(dppb)][BF4] catalyst was used which highlights the importance the catalyst can have on the efficiency of the PHIP process. The reaction time, magnetic field, catalyst identity and concentration, all influence the efficiency of PHIP and must be carefully considered in conjunction with the rate, turnover and yield usually addressed in studies of traditional hydrogenation systems.
Some studies have sought to improve the Rh complexes used in PHIP by variation of ligands in the coordination sphere. A series of [Rh(norbornadiene)(bisphosphine)][BF4] catalysts (1–4) were used to hydrogenate the example substrate vinyl acetate.90 A variety of different bisphosphines ligands were screened and their effect on hydrogenation kinetics and the magnitude of PHIP NMR signal enhancements were recorded (Fig. 4c). This study demonstrated that highly electron-donating bisphosphines with dicyclohexylphosphine groups (2–4) gave higher catalytic activity with 1.8–2.0 times higher turnover frequencies compared to the standard commercially available [Rh(norbornadiene)(dppb)][BF4] (1).90 The initial precatalyst activation involves the loss of the norbornadiene ligand and its replacement with two solvent molecules (in this case, methanol). This activation step was faster using 2–4 as the rate of norbornadiene ligand loss was 3.3–4.8 times faster than the standard Rh system.90 Similarly, hydrogenation was also faster in the case of the modified catalysts which in turn gave rise to higher NMR signal enhancements in PHIP experiments. The reason for this higher catalytic activity is yet unconfirmed with the mechanism of this transformation not yet completely understood. The active [Rh(solvent)2(diphosphine)][BF4] catalyst can coordinate substrate before irreversible oxidative addition of hydrogen.94 However, routes in which hydrogen activation occurs before substrate coordination have also been proposed.95 Further studies of this nature are likely to discover catalysts that can produce greater PHIP than those currently used. Investigation into hydrogenation catalytic cycles focussed on PHIP are likely to stimulate new and improved systems that retain the para-H2 spin state more efficiently during catalytic transformations, and give rise to larger NMR signal enhancements for hydrogenated products.
PHIP catalysis performed in coordinating solvents including methanol, ethanol or acetone can give high hydrogenation efficiencies and high NMR signal enhancements.96,97 This has been linked to the relatively high para-H2 solubility in these solvents, and is also closely related to the role of metal-solvent adducts in catalysis. Displacement of norbornadiene in [Rh(norbornadiene)(dppb)][BF4] to form active [Rh(solvent)2(diphosphine)][BF4] can be favoured in such solvents. Readily-coordinating solvents also favour rapid elimination of hydrogenated products from the metal coordination sphere, which limits the destructive influence of nuclear spin relaxation on PHIP of the products while bound to the metal centre.96 Commonly-used PHIP catalysts are typically most soluble in organic solvents (such as methanol, ethanol, acetone, chloroform, dichloromethane, toluene, etc.) but exhibit limited solubility in aqueous solutions. Many have sought to increase catalyst solubility in water by modification of spectator ligands (Fig. 4d).44,98 For example, [Rh(norbornadiene)(dppb)][BF4]-derived complexes in which the phenyl groups of the 1,4-bis(diphenylphosphino)butane ligand are modified to contain OMe and/or SO3H groups have been used to perform PHIP of dimethyl maleate in D2O.44 Related [Rh(norbornadiene)(THP)2][BF4] catalysts that contain monodentate tris(hydroxymethyl)phosphine (THP) ligands are also water-soluble and have been used to hydrogenate 2-hydroxyethyl 1-13C-acrylate-d2,3,3 yielding hyperpolarised 2-hydroxyethyl 1-13C-propionate-d2,3,3.98 Also ionic liquids based on rhodium salts have been used as hydrogenation catalysts to achieve PHIP in aqueous solvents.73 Further development of highly active water-soluble PHIP catalysts are likely to facilitate hyperpolarisation of biologically relevant molecules, which may have applications in NMR imaging.4,6
The use of cheaper and more readily available metals as catalysts in symmetry-breaking hydrogenation reactions has also been explored (Fig. 4e). Consequently, PHIP has been used to study the hydrogenation of molecules such as phenylacetylene53 or diphenylacetylene47,54 by palladium phosphine47,54 or palladium bisimino53 complexes. In the last few years catalysts based on cobalt such as [Co(bis(mesityl-benzimidazol-2-ylidene)phenyl)(pyridine)] have been used to hydrogenate ethyl acrylate with 13C signal enhancements of ca. 1130-fold at 11.7 T for the terminal site of the hydrogenated product in acetone-d6.59 This is significantly larger than the ca. 250-fold achieved under analogous conditions using a traditional [Rh(COD)(dppb)][BF4] catalyst.59 The scope of PHIP studies will expand as catalysts are improved to optimise their functional group tolerance, performance, speed, and many other factors. We also note that other metals such as V, Zr, Ta, Ce and Mo have been used to demonstrate PHIP with heterogeneous catalysts,99–101 and therefore the list of possible metal-based homogeneous catalysts may be extended significantly in the future.
5.2 Ruthenium catalysts for trans and geminal alkene hydrogenations
Most transition metal-catalysed alkyne hydrogenations involve the formation of Z (cis) alkenes and is a consequence of concerted hydrogen activation in which the introduced hydride ligands are located in a cis relationship.40,102 This typically restrains the types of molecules that can be produced from the homogeneous hydrogenation of alkynes in a hyperpolarised state to Z alkenes. E (trans) alkenes can form from homogeneous alkyne hydrogenation, although in many cases this process is actually a cis hydrogenation followed by isomerisation from the Z to E alkene mediated by the metal hydrogenation catalyst.103 Such trans hydrogenations can occur when heterogeneous solid surface catalysts are employed. In recent years, examples of homogeneous water-soluble ruthenium pentamethylcyclopentadiene (Cp*) catalysts have arisen that are able to trans-hydrogenate internal alkynes104 in a process that typically involves carbene intermediates (Fig. 5a).105,106 These [Ru(Cp*)(L)n] systems have been reported to react with alkyne and H2 to form a [Ru(Cp*)(H)2(alkyne)] intermediate that breaks para-H2 symmetry by oxidative addition.105 Subsequent hydride migration and rearrangement can form a ruthenium carbene intermediate which can react either to give cis or trans hydrogenated products depending on the geometry of this intermediate. Hydrogenation in a trans fashion is driven by steric factors in these intermediates105 and can be favoured by parameters such as temperature and solvent.104,107 These novel catalysts have expanded the scope of PHIP by allowing the preparation of E-alkenes in a hyperpolarised state (Fig. 5b). The formation of E-alkenes from alkynes via ruthenium carbene intermediates can also produce geminal hydrogenated products in which both 1H nuclei of the starting para-H2 are transferred to a single (rather than adjacent) carbon atom (Fig. 5a).105,106,108 While the production of hyperpolarised CH2 groups via geminal PHIP is possible (Fig. 5b), a challenge of this approach is the slower reaction times compared to traditional PHIP.109 Fast relaxation of the newly introduced CH2 protons can limit signal enhancements of these sites. Nevertheless, an appropriate resonant radiofrequency field can preserve the singlet spin order within the CH2 group which decays with the relaxation time constant for the long-lived state, TLLS, which can be longer than T1.109 In these systems, trans and geminal hydrogenations can both occur as competing pathways.53,109 Examples of hyperpolarised products resulting from both trans and geminal PHIP catalysis are expected to increase in the future as more catalytic systems that can perform such chemistry are developed. This has been particularly important for the production of hyperpolarised fumarate which can be used as a metabolic imaging probe and is discussed in more detail in Section 8.1.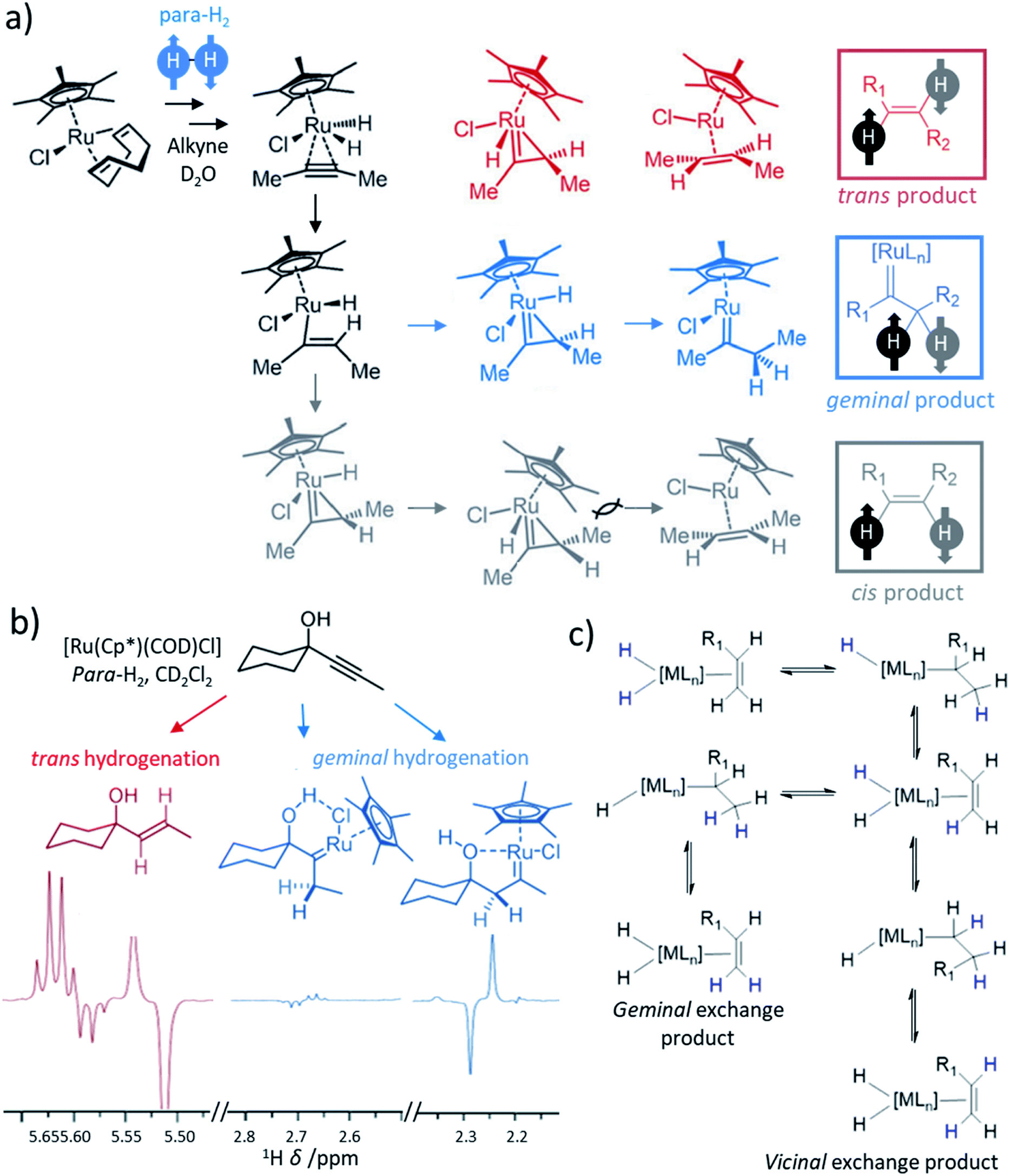 | ||
| Fig. 5 (a) Proposed mechanism for the hydrogenation of alkynes, in this example 2-butyne, by ruthenium cyclopentadienyl complexes yielding products that can be hyperpolarised according to traditional PHIP, trans, or geminal PHIP. Adapted from ref. 105. Similar mechanisms are given in ref. 106 and 107, although binuclear mechanisms involving two ruthenium centres have also been proposed.104 (b) Example partial 1H NMR spectra showing enhanced signals following trans hydrogenation of the indicated alkyne using a ruthenium catalyst. Enhanced 1H NMR signals for ruthenium carbenes result from geminal hydrogenation. Adapted from ref. 106. (c) Depiction of metal-catalysed vicinal and geminal exchange in which proton sites on an alkene are exchanged with a metal dihydride. If a dihydride has formed from activation of para-H2, then this exchange can lead to enhanced 1H NMR signals for the exchanged sites in the alkene. | ||
5.3 Metal-catalysed vicinal and geminal proton exchange leading to PHIP
Exchange of vicinal and geminal alkene protons is often a feature of metal-catalysed alkyne or alkene hydrogenation.53,110–112 In this process, protons on an alkene can become scrambled via metal hydride intermediates. In the context of PHIP experiments, these kinds of metal-catalysed proton exchange events provide a route to introduce parahydrogen-derived protons into alkenes (Fig. 5c). This can occur following a series of hydride migration steps, with bond rotation in the intermediate facilitating the scrambling of these sites. Such reactions can typically be revealed by analogous reaction of D2 gas which will exchange with proton sites on the alkene to form H2 gas and deuterated alkene.53 Similarly, PHIP NMR experiments utilising para-H2 can also reveal the occurrence of these reactions from the appearance of hyperpolarised 1H NMR spectra. For example, reaction of styrene and para-H2 or D2 with Rh and Pd catalysts result in scrambling of the para-H2 or D2 labels with the geminal alkene protons.53 Similar exchange of para-H2 protons with vicinal or geminal alkene protons has been reported for Rh systems.112 Consequently, 1H NMR signals for these sites can become enhanced using PHIP. It is important to note that vicinal or geminal exchange of a single H or D by itself is usually insufficient to give rise to the PHIP effect as both para-H2 atoms must be exchanged for the transfer to be pairwise.53 The catalysis underlying these NMR signal enhancements does not necessarily require hydrogenation of the alkene, or formation of an alkene from alkyne hydrogenation. Therefore, while geminal and vicinal exchange often occur in parallel to hydrogenation chemistry, they are reversible and do not necessitate any net changes to the chemical structure of hyperpolarised molecules. Examples of this reactivity in PHIP studies is unusual, likely due to competing hydrogenation that occurs via the same metal intermediates. Nevertheless, they provide unforeseen opportunities for hyperpolarisation at CC double-bonds and analysis of hyperpolarised line shapes can give valuable information on these processes, which may often go unidentified.1125.4 Homogeneous hydrogenation of CN groups
The hydrogenation of unsaturated CC bonds, such as alkynes and alkenes, represents the most common example of substrates used in PHIP studies. However, the hydrogenation of other unsaturated functionalities with para-H2 has also been reported, although examples of these are rare. The hydrogenation of nitriles to imines and primary amines using a cobalt bis(carbene) pincer catalyst has been demonstrated (Fig. 6).58 This system involves reduction of the cobalt precursor with NaHBEt3 before the hydrogenation of CN triple bonds via a Co(I/III) redox process that includes an oxidative addition step.58 This process is facilitated by the presence of a Lewis acid (such as BEt3) which promotes the η2 coordination of the nitrile important for initial transfer of para-H2 to form imine. In these examples, hyperpolarised NMR signals for the imine are observed which suggest it is formed from nitrile hydrogenation in a pairwise fashion. However, no hyperpolarised signals for the amine products are observed which is indicative either of a non-pairwise hydrogenation of imine to form amine, or that the pairwise formation of amine is slow compared to the rate of relaxation. These catalysts have been used to hydrogenate a wide range of nitriles58 (>20) and indicates that a greater variety of functionalities may become amenable to hyperpolarisation using para-H2 as new catalysts to perform these transformations are developed.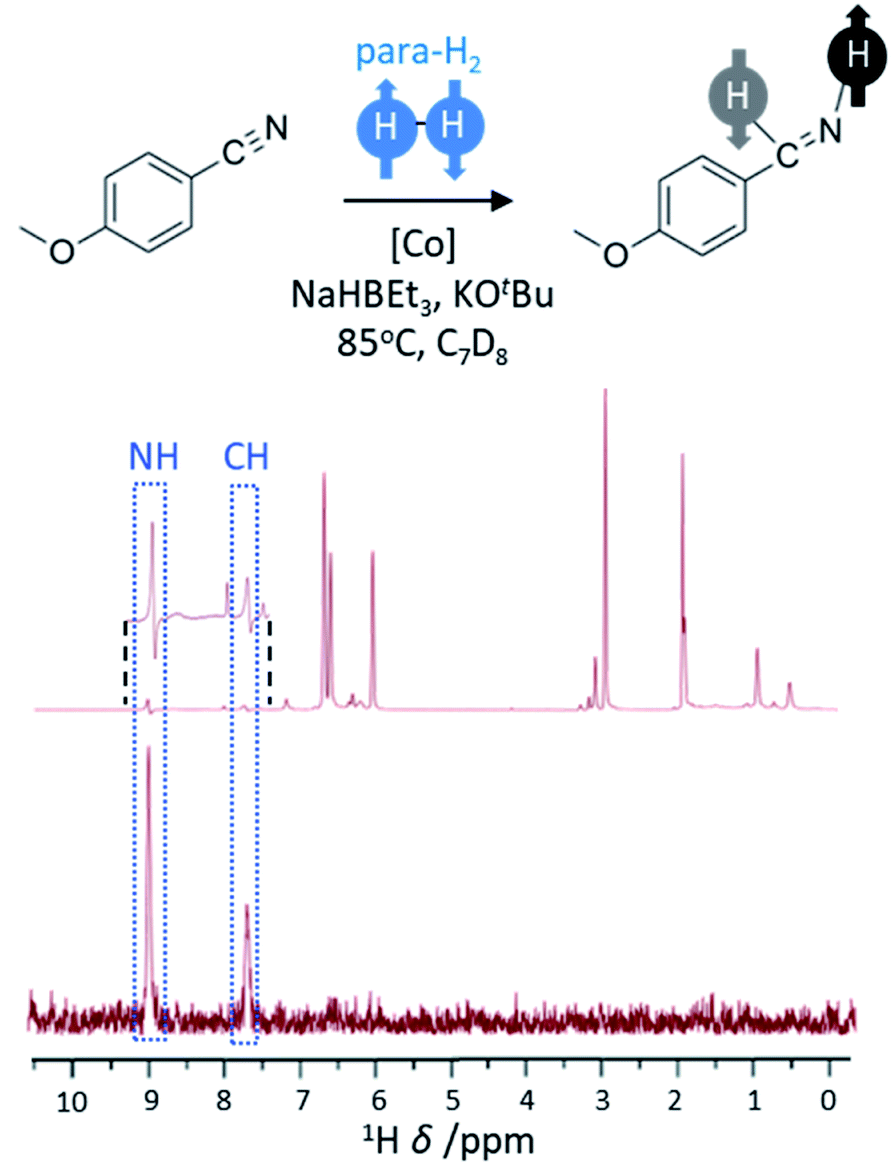 | ||
| Fig. 6 PHIP effects can be observed when nitriles are hydrogenated using para-H2. The example 1H NMR spectrum (600 MHz) has been recorded for the reaction mixture shown using a 45° pulse (above) with enhanced imine signals (inset) and a 1H-OPSY sequence (below) that confirms these protons are derived from para-H2. Adapted from ref. 58. | ||
5.5 Metal-free alkyne hydrogenation
As the range of metal-based systems for PHIP is increasing, so too are examples of metal-free systems. Very recently, ansa-aminoboranes have been shown to hydrogenate alkynes with para-H2 leading to hyperpolarisation of alkene products.28 In the reported examples, a frustrated Lewis pair (HCAT) reacts with an alkyne to form a HCAT-alkyne adduct (Fig. 7a). This adduct performs heterolytic splitting of para-H2 as the key step in the catalytic cycle to form a HCAT-alkyne-H2 species. Hyperpolarised 1H, 15N and 11B NMR signals for the NH and BH sites are observed which is consistent with similar para-H2 activation by FLPs (vide supra). However, HCAT-alkyne-H2 species can eliminate alkene products with enhanced NMR signals (Fig. 7b). This is the first example of a metal-free catalytic system capable of producing hyperpolarised products other than para-H2 activation adducts. Interestingly, only one proton originating from para-H2 is transferred into the alkene product, with the other remaining within the HCAT catalyst. This highlights that in some cases both protons originating from the same para-H2 molecule need not end up in the same product molecule. Similar effects have been reported for reversible iridium-catalysed exchange of para-H2 and H2O,113 and metal-catalysed hydroformylation reactions (oneH-PHIP as discussed in the next section), although the catalysis and/or polarisation mechanisms involved in these processes are different in each case.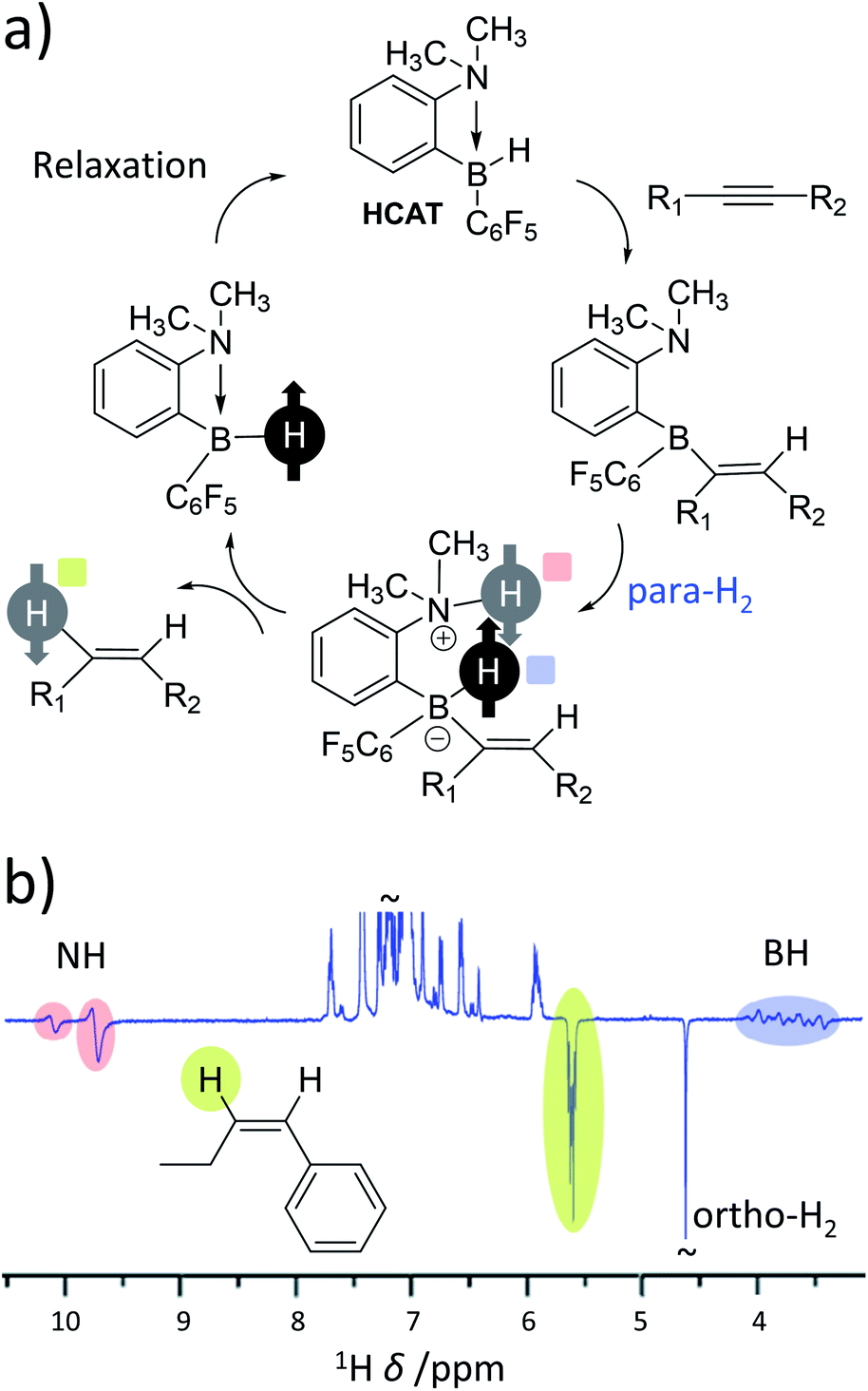 | ||
| Fig. 7 (a) Mechanism of metal-free hydrogenation and PHIP of alkynes using the ansa-aminoborane HCAT. Notably, para-H2 protons are split heterolytically and only one of these protons is incorporated into the alkene product. (b) Example 1H NMR spectrum for an alkene product displaying enhanced 1H NMR signals for a single proton originating from the para-H2 feedstock in addition to the NH and BH sites of catalytic intermediates. Adapted from ref. 28. | ||
6. Hydroformylation reactions
Hydroformylation reactions, in which alkenes are converted into aldehydes, present a different type of reaction that can break para-H2 symmetry leading to PHIP. These reactions lead to the so-called oneH-PHIP effect as only a single 1H nucleus originating from para-H2 ends up in the aldehyde product but nonetheless can exhibit a net hyperpolarisation.23 Despite the fact that the full catalytic cycle in this case is non-pairwise, a pairwise process must occur for PHIP effects to be observed at initial reaction steps. This is achieved by an initial oxidative addition reaction in which both 1H nuclei within para-H2 are transferred to an intermediary metal dihydride complex, before only one of these hydrides is incorporated into the final product (Fig. 8). One of the first examples of oneH-PHIP was in the hydroformylation of ethene in C6D6 using [Ir(H)(CO)2(dppe)].23 Upon reaction of this precursor with 3 bar para-H2 at 80 °C, [Ir(COEt)(H)2(CO)(dppe)] is formed in which the two 1H nuclei from para-H2 are strongly coupled and thereby can gain a significant level of single-spin net polarisations of opposite signs. Reductive elimination of this acyl dihydride intermediate yields propanal with an enhanced aldehyde CHO proton due to oneH-PHIP. Hyperpolarised signals for the metal product of reductive elimination, [Ir(H)(CO)(dppe)], were not observed which was attributed to either short relaxation times for this intermediate, or a short lifetime due to rapid formation of [Ir(H)(CO)2(dppe)].23 Hyperpolarised signals for a [IrH3(CO)(dppe)] species are also observed which presumably forms from activation of para-H2 by [Ir(H)(CO)(dppe)].23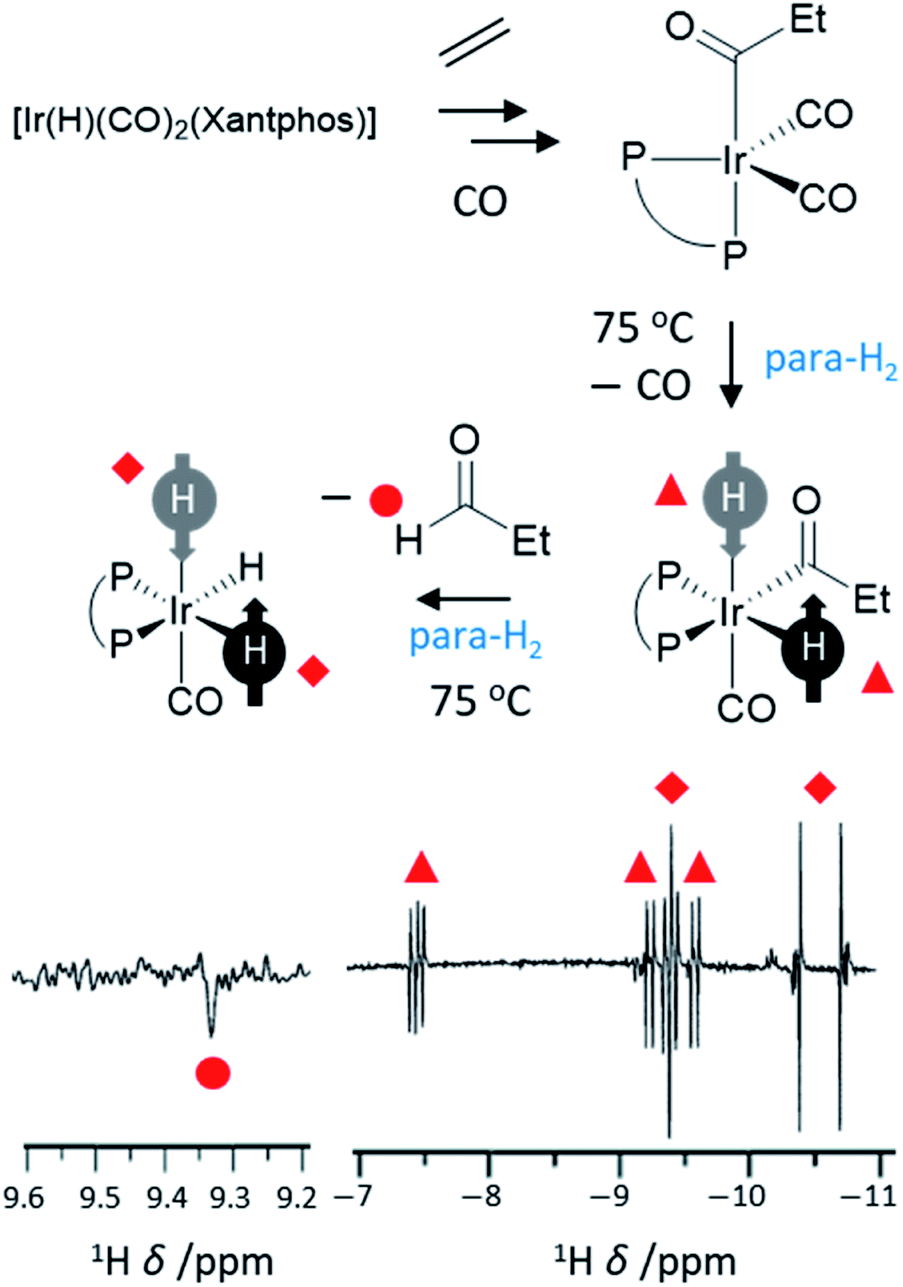 | ||
| Fig. 8 Example oneH-PHIP 1H NMR spectrum showing polarisation for propanal (lower left) and for iridium hydride intermediates formed during the hydroformylation process (lower right). Xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene adapted from ref. 101. | ||
These initial studies also utilised Pt-based systems and since then the range of catalysts used for oneH-PHIP have increased. For example, [Ir(CO)(PPh3)2(η3-C3H5)]25,114 or [IrI(CO)2(xantphos)]24 hydroformylation catalysts have been employed and consequently, several PHIP-hyperpolarised iridium acyl and alkyl dihydride intermediates could be detected using NMR. The use of less well-defined cobalt systems to perform such oneH-PHIP reactions has also been studied.26,27 A wider range of carbonylation reactions, in which CO is incorporated into a substrate, are also accessible to oneH-PHIP. For example, a palladium bis-phosphine complex was demonstrated to convert diphenylacetylene into methyl-2,3-diphenyl acrylate in the presence of CO and H2.55 This transformation was accompanied by the formation of cis- and trans-stilbene and the 1H NMR signals for all three products were enhanced when this process was performed using para-H2. In the case of stilbene, enhanced signals are due to hydrogenative PHIP, although in methyl-2,3-diphenyl acrylate the enhanced 1H NMR signals were attributed to a oneH-PHIP effect in which a single proton from para-H2 was incorporated into the substrate molecule.55
The 1H NMR signal enhancements observed for aldehyde protons using oneH-PHIP are significantly lower than can be achieved for alkene and alkanes produced using standard PHIP hydrogenation reactions. Nevertheless, strongly enhanced NMR signals for metal hydride intermediates has been of great use in mapping hydroformylation reaction pathways.23–27,114 OneH-PHIP has expanded the types of reactions that can be examined using para-H2 and has been of tremendous value in probing inorganic reaction mechanisms.
7. Reversible reactions with para-H2 and polarisation transfer to other molecules (SABRE)
The reactions discussed so far all result in functionalisation of a to-be-hyperpolarised molecule with protons previously located in the para-H2 feedstock. It was first reported in 2009 that target substrates can become hyperpolarised without incorporation of para-H2 originating atoms.29 The method has been called signal amplification by reversible exchange (SABRE) and it works on the basis of reversible interactions of both para-H2 and a to-be-hyperpolarised molecule with a metal catalyst.16,17,29 The symmetry of para-H2 is initially broken in a reversible oxidative addition reaction to form an iridium dihydride. The difference is that polarisation is then transferred from the hydride ligands to other 1H sites or even heteronuclei of ligands bound transiently within the metal complex. Dissociation of the active SABRE catalyst generates hyperpolarised ligand free in solution without hydrogenation of the ligand (Fig. 9). A key advantage of SABRE compared to hydrogenative PHIP is that substrates are not chemically altered. Therefore, SABRE removes the requirement for target molecules to contain unsaturated functionality, although they must now contain motifs that can ligate to the SABRE catalyst. An added benefit is that SABRE is reversible: once the hyperpolarised signals have decayed due to relaxation, they can be regained by shaking or bubbling with fresh para-H2. This is in contrast to hydrogenative PHIP which is only visible if the unsaturated substrate is not consumed completely in the reaction.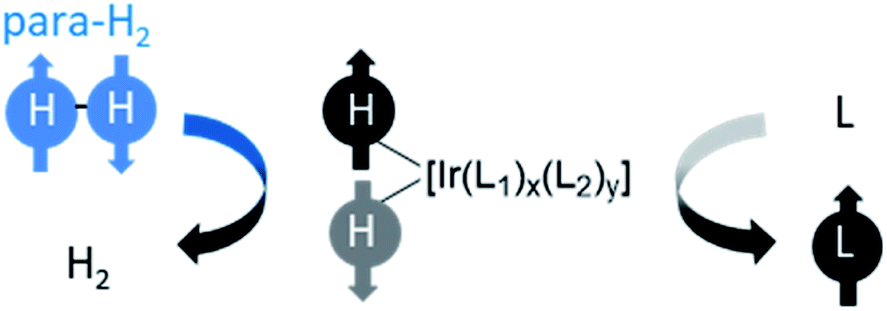 | ||
| Fig. 9 Generic depiction of reversible polarisation transfer using SABRE. The symmetry of para-H2 is broken using a reversible oxidative addition reaction to form a hyperpolarised metal dihydride complex. This catalyses spontaneous polarisation transfer to other ligands (L1) bound within the complex at low magnetic field (>milliTesla). Auxiliary ligands (L2), which can be non-exchanging, often play a role in this process where x + y = 4. The iridium complex may be neutral or positively charged depending on the identity of L1 and L2 and is often formed in situ. | ||
The spin dynamics of polarisation transfer within SABRE catalysts can be complex and there have been several excellent treatments of these effects.17,19,115,116 Briefly, polarisation transfer from the para-H2 originating proton pair to target sites is enabled by spin–spin couplings connecting the two hydrides to each other and to the target ligands in the SABRE complex.17,19,116,117 Typically, most efficient transfer to 1H sites is achieved at 0–10 mT magnetic fields118,119 which are often produced in the stray fields of strong NMR magnets (>7 T). It is possible to transfer magnetisation from hydride ligands directly to heteronuclei including 15N,120–12413C,125–12719F,128,12931P,39,130119Sn131 and 31Si131,132 in a target molecule. Heteronuclear SABRE follows the same principles as 1H-SABRE but must be performed at much lower magnetic fields (0.5–20 μT) in order to meet resonance conditions.39,121 Polarisation transfer within the SABRE catalyst is spontaneous at low magnetic fields (ALTADENA conditions), but it can also occur via radiofrequency excitation at high field (Tesla) (PASADENA conditions).133–138 In addition, feasible, but less efficient, polarisation transfer can occur spontaneously at high magnetic fields, giving rise to high-field SABRE effects.133
7.1 Ligand exchange pathways important for SABRE catalysis
The catalysts involved in SABRE are typically of the form [Ir(H)2(L)(substrate)3]+ where L is an auxiliary ligand. Such complexes are formed in situ by the reaction of 16-electron square planar precatalysts such as [Ir(COD)(L)(MeCN)][BF4] or [IrCl(COD)(L)] (where COD is cis,cis-1,5-cyclooctadiene) with para-H2 and an excess of the substrate of interest.29 These reactions typically proceed via 16-electron intermediates of the form [Ir(COD)(L)(substrate)]+, in which a labile ligand within the precatalyst is replaced by the substrate.48,139 Addition of H2 to square planar [IrCl(COD)(L)] initially forms the octahedral 18-electron [Ir(H)2(COD)(L)(substrate)]+. This must undergo hydride migration to form the SABRE-active [Ir(H)2(L)(substrate)3]+ (Fig. 10).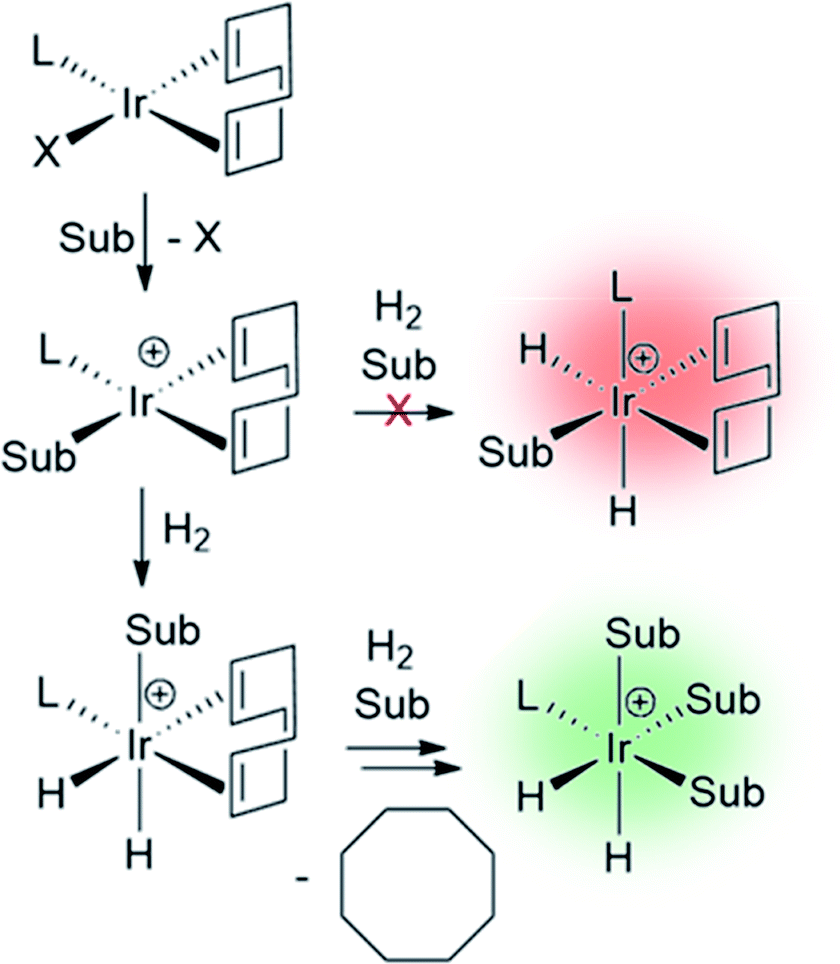 | ||
| Fig. 10 Formation of SABRE-active polarisation transfer catalysts from a 16-electron air-stable precatalyst. In this case L represents an auxiliary ligand and Sub a substrate of interest. The charges of the complexes shown in this scheme will be neutral or positive depending on the identity of X. | ||
Studies of ligand exchange within these complexes using exchange spectroscopy (EXSY) has revealed that substrate ligands located trans to hydride are typically in reversible exchange with their counterpart free in solution.140 This reversible exchange allows significant hyperpolarisation for the substrate free in solution to be built up constantly. Mechanistic studies have shown that substrate loss from [Ir(H)2(L)(substrate)3]Cl to form the 5-coordinate 16-electron intermediate [Ir(H)2(L)(substrate)2]Cl is a key step in the catalytic SABRE process.38,141 The formation of this intermediate (via substrate dissociation) is the dominant pathway for both hydrogen and substrate exchange. For example, such transient intermediates can react with para-H2 to form [Ir(H)2(η2-para-H2)(L)(substrate)2]Cl which undergoes rapid rearrangement to eliminate H2 (Fig. 11). The 5-coordinate [Ir(H)2(L)(substrate)2]Cl exists in a square-based pyramidal geometry immediately after substrate dissociation.141 However, formation of a trigonal bipyramidal structure is possible if the complex has a sufficiently long lifetime for ligand reorientation. Isomerisation between different square-based pyramidal structures via a trigonal bipyramidal form, or reaction of a trigonal bipyramidal intermediate with either para-H2 or substrate on either face, is responsible for scrambling of the hydride ligand sites (Fig. 11).142 This becomes more important for complexes that contain inequivalent hydride ligands.38
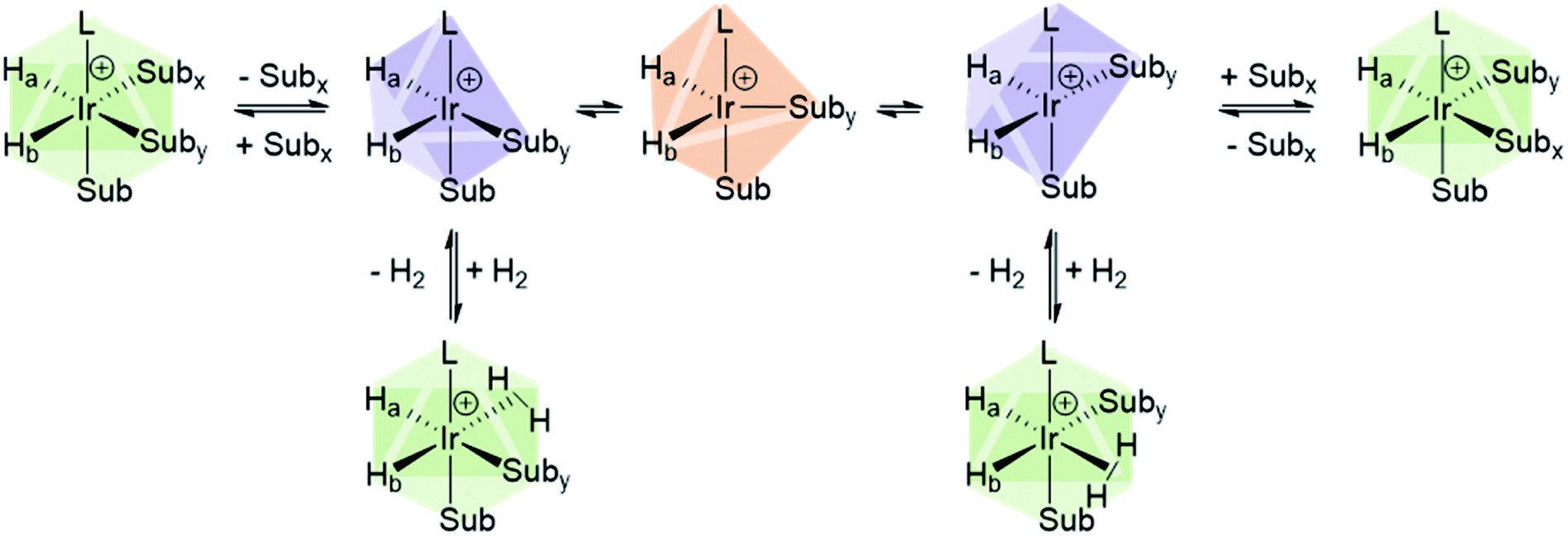 | ||
| Fig. 11 Depiction of isomerisation pathways in SABRE complexes via the five coordinate intermediate [Ir(H)2(IMes)(Sub)2]+. | ||
For catalytic polarisation transfer to be observed, [Ir(H)2(η2-para-H2)(L)(substrate)2]Cl must be short-lived to limit the conversion of para-H2 into ortho-H2.143 Instead, [Ir(H)2(η2-para-H2)(L)(substrate)2]Cl serves to refresh the para-H2-derived singlet spin order within the iridium dihydride catalyst. The rate of H2 loss typically decreases as the substrate concentration is increased, but increases as hydrogen pressure is increased.38,48,141 These observations have been used to deduce that hydrogen and substrate exchange occur through a common pathway in which both hydrogen and substrate bind competitively to intermediates such as [Ir(H)2(L)(substrate)2]Cl. In other words, pathways involving hydrogen loss directly from [Ir(H)2(L)(pyridine)3]Cl to form [Ir(L)(pyridine)3]Cl with changes in oxidation state are unlikely.38,141 The transient intermediates involved in SABRE catalysis are often too short-lived to be observed directly by NMR methods, although Density Functional Theory (DFT) calculations have provided supporting evidence for these mechanistic studies.38,141,144 These studies have confirmed that direct reductive elimination of hydrogen is accompanied with a large energy barrier, favouring a pathway involving species with constant oxidation states.
7.2 Variation of the auxiliary ligand to fine tune substrate exchange and relaxation
The SABRE catalyst plays a crucial role in determining the substrates that can become hyperpolarised, and the magnitude of the NMR signal enhancements that can be achieved. The lifetime of the active SABRE catalyst is of vital importance: if it is too short then ligand exchange will occur before there has been sufficient time for a substrate to become hyperpolarised, if it is too long then the hyperpolarisation of substrate will decay due to the enhanced spin relaxation while bound to the metal centre. The active catalyst lifetime is closely linked to the substrate exchange rate, which can be altered by variation of auxiliary ligands on the catalyst, although factors such as temperature122,145,146 or even solvent will also influence this.142,147 The steric and electronic properties of the auxiliary ligand can also influence the types of substrates that can bind to the SABRE catalyst. Therefore, careful catalyst optimisation can play a significant role in tuning ligand exchange which in turn provides a valuable route to improving the substrate scope and hyperpolarisation levels achieved using SABRE. Novel pulse sequences have also been used as a route to achieve high polarisation by compensating for suboptimal ligand exchange processes.148Catalysts involved in early SABRE studies contained an auxiliary phosphine ligand and were of the form [Ir(H)2(PCy3)(substrate)3][BF4], where Cy is cyclohexyl. It was soon noted that the identity of the auxiliary phosphine ligand was one of many important factors that determine the efficiency of SABRE catalysis and the resulting NMR signal enhancements that can be achieved. For example, catalysts containing the phosphine ligand PCy2Ph yielded higher 1H NMR enhancements for pyridine compared to the use of catalysts derived from other phosphines including Pcy3 and PPh3.149 Sterically large electron-rich phosphines are preferred which is attributed to more favourable substrate dissociation.
Since then, a variety of more complex phosphine ligands have been used that contain mixed P and N donor sites. Namely, iridium pincer complexes containing a κ3-N,P,P auxiliary ligand have been used as polarisation transfer catalysts (Fig. 12a).142 Related iridium κ2-N,P systems that use a 2-(2-(diphenylphosphanyl)phenyl)-4,5-dihydrooxazole (Phox) ligand have also been reported.150 This catalyst achieved much lower SABRE efficiency for pyridine (2-fold ortho1H NMR signal enhancement) compared to using [Ir(H)2(PCy3)(pyridine)3]PF4 (100-fold ortho1H NMR signal enhancement).150 However, the significant advantage of these Phox-containing systems is that they are able to hyperpolarise sterically larger substrates. In particular, pyridines with substituents at the ortho position such as 2-methylpyridine, 2-fluoropyridine, and 2-ethylpyridine exhibit 1H NMR signal enhancements for their aromatic ortho sites of 132-, 140-, and 25-fold respectively150 and could not be hyperpolarised using [Ir(H)2(PCy3)(substrate)3]PF4.
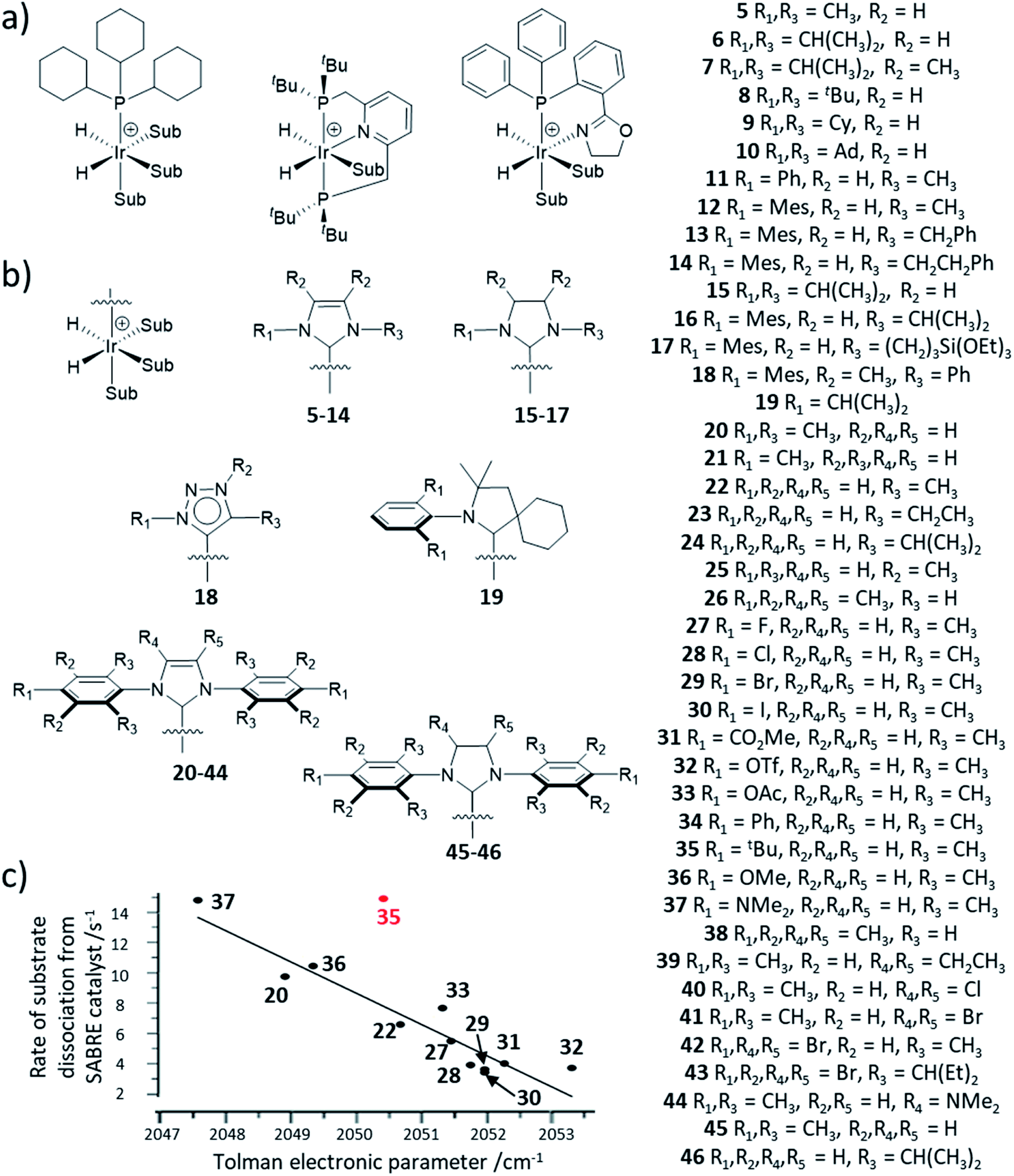 | ||
| Fig. 12 (a) Example iridium phosphine complexes used for catalytic polarisation transfer from para-H2. Reported examples are based on traditional monodentate κ1-P phosphine ligands11,150,151 (left), κ3-N,P,P pincer ligands154 (middle) or κ2-N,P phox ligands156 (right). (b) SABRE catalysts containing different auxiliary NHC ligands. (c) Effect of para substituted NHC ligands as a function of their TEP on the rate of substrate dissociation from [Ir(H)2(NHC)(sub)3]Cl at 298 K where substrate is methyl-4,6-d2-nicotinate. The outlier, 35 (shown in red), is attributed to steric effects of the bulky tBu group. Adapted from ref. 153. | ||
It was soon discovered that the use of carbene ligands, instead of phosphines, has a large effect in increasing the efficiency of SABRE catalysis. Their greater SABRE efficiency has been rationalised in terms of optimised substrate exchange within the active catalytic cycle. The rate constant of pyridine dissociation from [Ir(H)2(IMes)(pyridine)3]Cl was determined by EXSY to be 23 s−1 at 300 K (ref. 48) which is ca. > 50 times faster than pyridine dissociation from [Ir(H)2(Pcy3)(pyridine)3]+ (0.45 s−1 at 295 K).149 As a consequence, 1H NMR signal enhancements of 8100-fold could be achieved for pyridine in methanol-d4 at 3 T using an [Ir(H)2(IMes)(pyridine)3]Cl active catalyst141 which is much higher than those achieved using phosphine-based catalysts (<100-fold at 9.4 T).149
The NHC ligand used to make [IrCl(COD)(NHC)] SABRE precatalysts can easily be modified to exhibit a variety of steric, and to a lesser extent, electronic properties.151,152 Consequently, a large variety of SABRE catalysts with different NHC auxiliary ligands have been reported (Fig. 13b, also see ESI Table S1†) and their effect on SABRE efficiency has been investigated.48,153–156 Generally, it is those catalysts containing an NHC ligand based on IMes that provide some of the highest NMR signal gains for substrates of interest using SABRE. When comparing IMes-derivatives with variation in the substituent at the para position, R1 (20–44), there is a good correlation between electron-rich carbenes (low TEP) and fast rates of substrate dissociation from [Ir(H)2NHC(substrate)3]Cl (Fig. 12c).153 This is generally beneficial for refreshing para-H2 spin order and facilitating catalytic build-up of substrate polarisation, although rationalisation of catalyst efficiencies can be challenging. Relaxation within the active catalyst is also an important factor that determines the magnitude of hyperpolarised NMR signals that SABRE can produce. Therefore, selected 1H sites within the NHC ligand can be replaced with deuterium (2H) labels.153 This reduces any wastage of para-H2 derived spin order by preventing unwanted transfer to 1H sites of the auxiliary ligand. It also serves to reduce relaxation of the substrate when bound to the iridium catalyst and therefore substrate relaxation times in the presence of the catalyst are often longer when deuterium-labelled catalysts are used compared to their protium counterparts.153
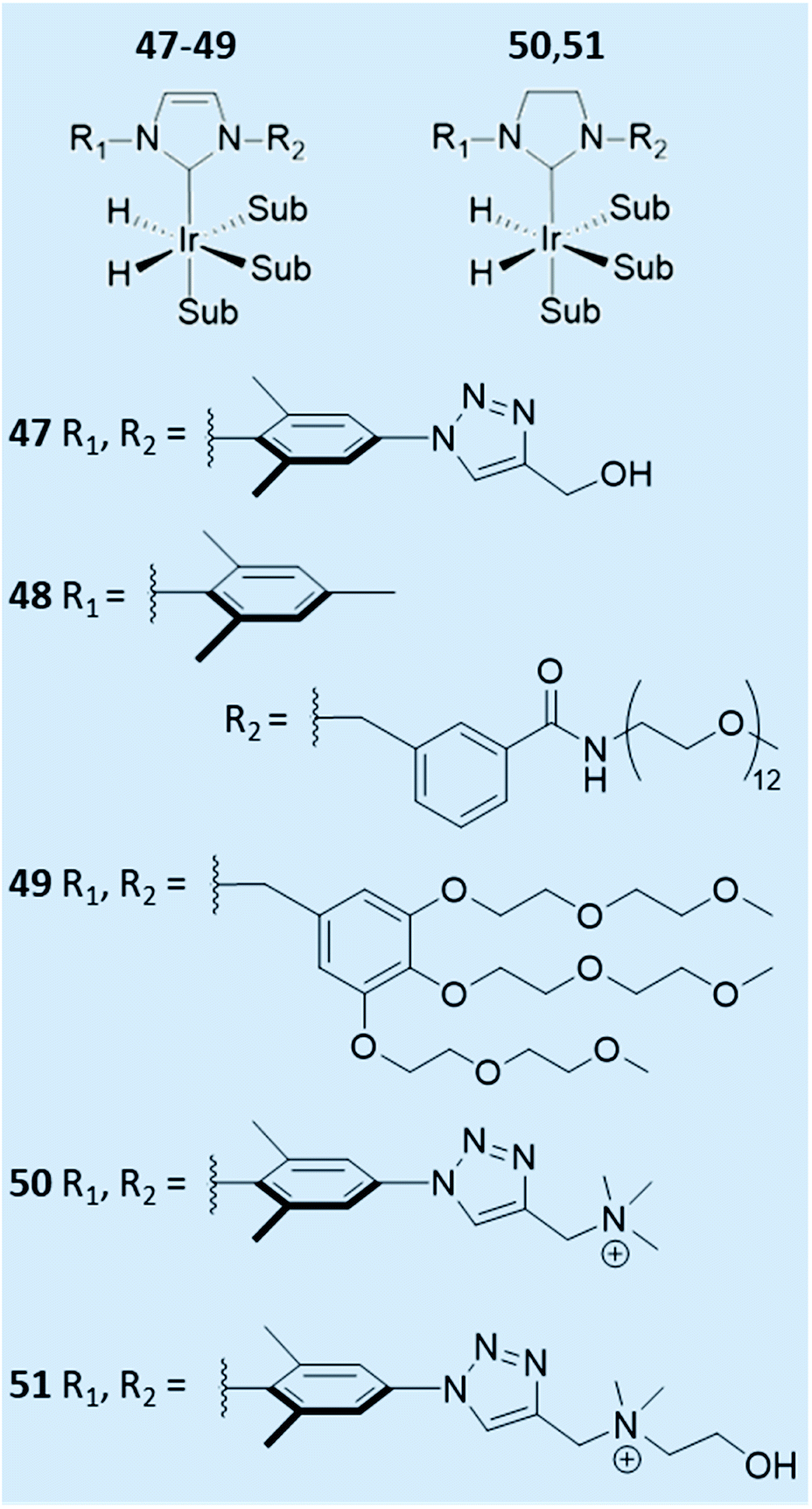 | ||
| Fig. 13 Reported examples of water-soluble SABRE catalysts. | ||
7.3 Variation of the auxiliary ligand for water-soluble catalysts
The effect of the auxiliary ligand can also extend beyond subtle steric, electronic, and relaxation effects. For example, catalyst solubility can be altered by changing the auxiliary ligand. Ir–NHC catalysts are typically highly soluble in organic solvents and therefore polarisation transfer reactions are typically performed in methanol-d4 (ref. 29, 38, 146, 157 and 158) or even dichloromethane-d2,37,38 chloroform-d159 or ethanol-d6.146 The efficiency of SABRE catalysis in other solvents can be improved by alteration of the auxiliary NHC ligand. For example, a neutral iridium carbene complex where the NHC was 3-(2-methylene-4-nitrophenolate)-1-(2,4,6-trimethylphenyl)imidazolylidene, which contained a pendant alkoxide ligand, has been utilised in low polarity solvents including benzene-d6, and THF-d8.160Water-soluble SABRE catalysts allow the entire hyperpolarisation process to be performed in aqueous solvents. Specifically, auxiliary PPh3 ligands were functionalised with SO2Na or SO3K groups to produce water-soluble catalysts.161 A variety of water-soluble IMes-derived ligands have also been developed (Fig. 13).161–163 Catalyst 51 was able to efficiently catalyse SABRE polarisation transfer to pyridine in methanol-d4 giving total 1H NMR signal enhancements of 750-fold.161 However, when this was repeated in 70:30 D2O:ethanol-d6 this value dropped to just 22-fold. Even though 47, 50–51 were soluble in water they failed to yield appreciable NMR signal enhancements for pyridine in water which was attributed to low para-H2 solubility.161 In contrast, 48 could achieve 1H NMR signal enhancements of ca. 30-fold for pyridine in water.162 Similar signal enhancements were observed for pyridine using 49, with up to 42-fold 1H NMR signal enhancement for the ortho site of nicotinamide also reported.163 These studies have shown that variation of the auxiliary ligand can alter catalyst solubility and they have been successful in allowing reversible polarisation transfer reactions to be performed directly in aqueous solvent. Nevertheless, they have not yet been reported to achieve NMR signal enhancements comparable to those that can be attained in organic solvents. Further alterations of the auxiliary ligand may yield higher-performing water-soluble catalysts that can give more significant NMR signal enhancements in the future.
7.4 Coligand-supported SABRE catalysts to hyperpolarise sterically large or weakly donating substrates
For bulky substrates, formation of [Ir(H)2(IMes)(substrate)3]Cl may be prevented by steric effects.118,150,158 The use of catalysts with sterically smaller auxiliary ligands, such as phosphines,150 asymmetric carbenes,164 or bidentate carbenes165 can be used to form SABRE catalysts with bulkier substrate ligands. A more effective way to create stable SABRE catalysts in cases where [Ir(H)2(NHC)(substrate)3]Cl is not formed due to sterically large, or weakly-ligating, substrates is the use of coligand-supported catalysts.158,166 For example, coordination of pyridine-derived substrates with functionality in the ortho position that hinders iridium ligation can be facilitated by a coligand.150,166 To demonstrate, 1H NMR signal enhancements up to 1442 ± 84-fold were recorded for 2,5-lutidine using catalysts of the form [IrCl(H)2(NHC)(sulfoxide)(substrate)]. This approach is notable as substrates of this type are not amenable to SABRE using [Ir(H)2(IMes)(substrate)3]Cl due to steric constraints. These catalysts could also improve upon NMR signal enhancements for molecules such as 2-picoline compared to those achieved using typical [Ir(H)2(IMes)(substrate)3]Cl systems.166 The inclusion of a coligand can in some cases have a beneficial effect on increasing substrate signals enhancements for systems in which [Ir(H)2(NHC)(substrate)3]Cl is readily formed.123,167–170 This is possible if relaxation or ligand exchange is more favourable for [Ir(H)2(IMes)(substrate)2(coligand)]Cl compared to [Ir(H)2(IMes)(substrate)3]Cl.Careful selection of coligands around the metal centre can create SABRE catalysts in which weakly-ligating substrates can bind and become hyperpolarised. Particularly, O-donor molecules such as carboxylic acids and ketoacids do not form the [Ir(H)2(NHC)(substrate)3]Cl typically necessary for polarisation transfer in reversible reactions. However, novel SABRE catalysts containing stabilising coligands have been developed to catalyse polarisation transfer to O-donor molecules. For example, 13C NMR enhancements of 108-fold for 13C-acetate were achieved using [Ir(H)2(IMes)(acetate)(pyridine)2].126 More notably, sulfoxides have emerged as a versatile class of SABRE coligand38,125,146,147,157 and as a result, novel SABRE catalysts of the form [Ir(H)2(κ2-O,O-pyruvate)(NHC)(sulfoxide)]Cl or [Ir(H)2(κ2-O,O-ketoisokaproate)(NHC)(sulfoxide)]Cl can deliver up to 2135 and 985-fold 13C NMR signal enhancements for sodium pyruvate-1,2-[13C2]146 and sodium ketoisocaproate-1-[13C]157 in methanol-d4, respectively (Fig. 14b and c). Since these initial studies, the efficiency of these catalysts have been improved further by variation of temperature to optimise ligand exchange processes and ca. 11% 13C polarisation for sodium pyruvate-1-[13C] has been reported.171
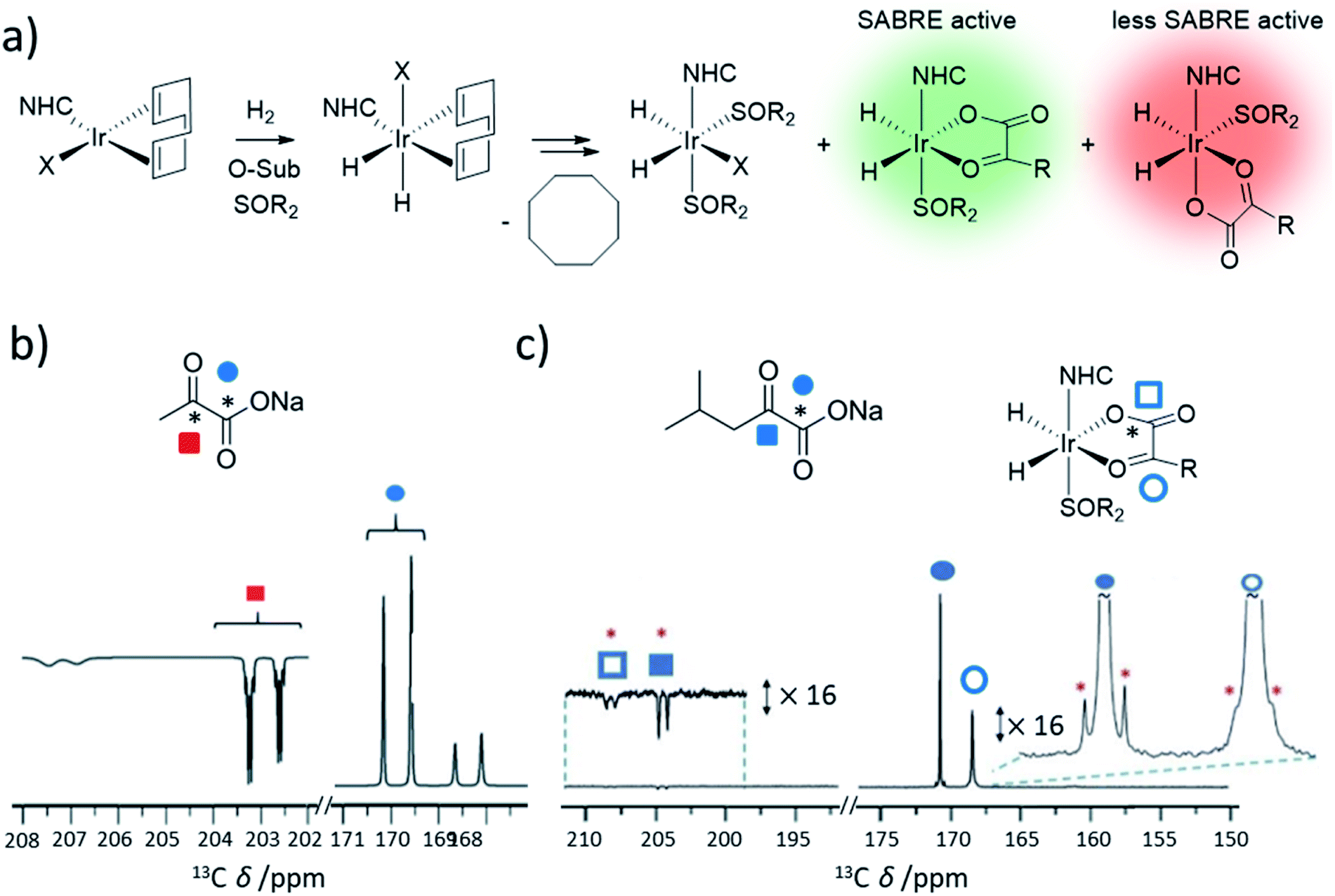 | ||
| Fig. 14 (a) Formation of sulfoxide-containing SABRE catalysts that can hyperpolarise O-donor ketoacids. (b) Example use of these catalysts to achieve hyperpolarisation of pyruvate. Partial hyperpolarised 13C NMR spectra for the keto region (left) and carbonyl region (right) recorded after [IrCl(COD)(IMes)] (5 mM), methylphenylsulfoxide (50 mM), and sodium pyruvate-1,2-[13C2] (30 mM) are shaken in methanol-d4 (0.6 mL) with 3 bar para-H2 for 30 seconds in a mu-metal shield. (c) Example use of these catalysts to achieve hyperpolarisation of ketoisocaproate. Partial hyperpolarised 13C NMR spectra when [IrCl(COD)(IMes)] (5 mM) and ketoisocaproate-1-[13C] are shaken with dimethylsulfoxide (50 mM) and para-H2 (3 bar) in methanol-d4 (0.6 mL) for 10 seconds in a mu-metal shield at ca. 1 μT. The signals marked by a red asterisk denote singlet magnetisation of naturally abundant ketoisocaproate-1,2-[13C2]. Adapted from (b) ref. 146 and (c) ref. 157. | ||
The mechanism of formation, and action, of these sulfoxide-containing catalysts contains several key differences from that of traditional [Ir(H)2(NHC)(substrate)3]Cl-based systems. There is no evidence of displacement of chloride from [IrCl(COD)(NHC)] by either O-donor substrate or sulfoxide.147 As a consequence, hydrogen addition occurs directly to [IrCl(COD)(NHC)] to form [IrCl(H)2(COD)(NHC)] (Fig. 14).147,172 Hydrogenation of the COD ligand then occurs to form [IrCl(H)2(sulfoxide)2(NHC)] which exists in equilibrium with [Ir(H)2(κ2-O,O-substrate)(NHC)(sulfoxide)].125 Unusually, [Ir(H)2(κ2-O,O-substrate)(NHC)(sulfoxide)] exhibits relatively slow ligand exchange on the NMR timescale.147 There are other species also present in solution and those of the form [IrCl(H)2(sulfoxide)2(NHC)] are known to undergo fast para-H2 exchange.147 They are therefore likely to play a role in refreshing para-H2 spin order within the catalytic system. The role of these species is also highlighted by the fact that the identity of the ligand, X, in [IrX(H)2(sulfoxide)2(NHC)] can have a large effect on the substrate NMR signals enhancements of O-donor substrates.147 Catalysis can be further complicated in these examples by the existence of two isomers of [Ir(H)2(κ2-O,O-substrate)(NHC)(sulfoxide)].125,146,171 It is those species containing substrate ligated in the same plane as the hydride ligands that have a spin topology most appropriate for efficient polarisation transfer to substrate.125 The proportion of active isomer can be maximised by alteration of the steric properties of the NHC.146 Such systems have allowed the polarisation of previously inaccessible molecules: continued development of novel classes of coligands with unique reactivity will unlock the ability to hyperpolarise an ever-greater range of molecules using SABRE.
7.5 Role of low concentration adducts on SABRE catalysis
Some of the most efficient SABRE catalysts have been highlighted. It is worth noting that there are many reports of low concentration adducts formed in solution that are not efficient SABRE catalysts and their presence can often have a detrimental effect on SABRE efficiency.35,173 SABRE catalysis is most commonly performed in methanol-d4, which can act as a coordinating solvent. Therefore, the trapping of short-lived 5-coordinate reaction intermediates, such as [Ir(H)2(NHC)(substrate)2]Cl, with methanol to form [Ir(H)2(NHC)(substrate)2(CD3OD)]Cl48,173 can occur. These adducts are typically of low concentration and are short-lived due to the weak binding of solvent methanol. The role that these adducts play can be revealed by recording SABRE efficiency as a function of polarisation transfer field (PTF).173 These PTF plots can include multiple maxima and this has been rationalised in terms of different optimal magnetic fields for polarisation transfer to substrate catalysed by [Ir(H)2(NHC)(substrate)2(CD3OD)]Cl compared to [Ir(H)2(NHC)(substrate)3]Cl.173 These differences are a result of the different J coupling spin topology formed in the solvent adduct compared to the traditional tris-substrate catalyst. These studies have shown that the role of undiscerned species in SABRE catalysis can be challenging to determine and may be important for those seeking to model polarisation transfer during SABRE.19,115,116,174,175Other adducts such as [IrCl(H)2(NHC)(substrate)2]Cl have been reported35,173 and they are typically detected via distinctive hydride resonances or indirect methods based on CEST.35 [IrCl(H)2(NHC)(substrate)2] has been reported to have a detrimental effect on SABRE efficiency as it results in a loss of para-H2 spin order. This loss of correlation, or singlet–triplet leakage, is typically larger at higher magnetic field and it can be reduced by performing SABRE experiments at lower fields176 or by applying a spin lock pulse during para-H2 bubbling to increase the lifetime of para-H2 spin order.35
7.6 Inefficient Ir–NHC SABRE catalysts: multinuclear decomposition products
The SABRE-active catalysts discussed so far are all formed over differing activation time periods which can range from several minutes146 to several days.160 While SABRE can easily be repeated multiple times by adding fresh para-H2 and repeating the polarisation step, this cannot occur indefinitely and, as with all catalytic processes, there is catalyst degradation over longer timescales.131,157 Different systems will remain active for different time windows, which are usually sufficient to allow many repeated hyperpolarisation measurements to take place. For some catalysts, activity is observed for several hours after catalyst formation. For others, deactivation can be more rapid.146 Formation of catalyst decomposition products, which are usually dimers or higher order oligomers, can be responsible for a drop in SABRE efficiency at longer reaction times.146,177 The formation of a wide range of dimer or trimer products, which can be revealed using X-ray crystallography, have been reported (Fig. 15).131,147,157,159,172,177 These often contain iridium–iridium single bonds146,157,159,172 and may also contain bridging hydride,157,159,172 chloride,159 substrate,131,147 or in the case of sulfoxide-containing catalysts, bridging S2− or SR− ligands.146,157,172 These species are typically no longer able to undergo the ligand exchange processes necessary for reversible polarisation transfer and therefore do not act as SABRE catalysts.177 Hence, their formation should be minimised by modification of the substrate, auxiliary ligand or coligand (if applicable).146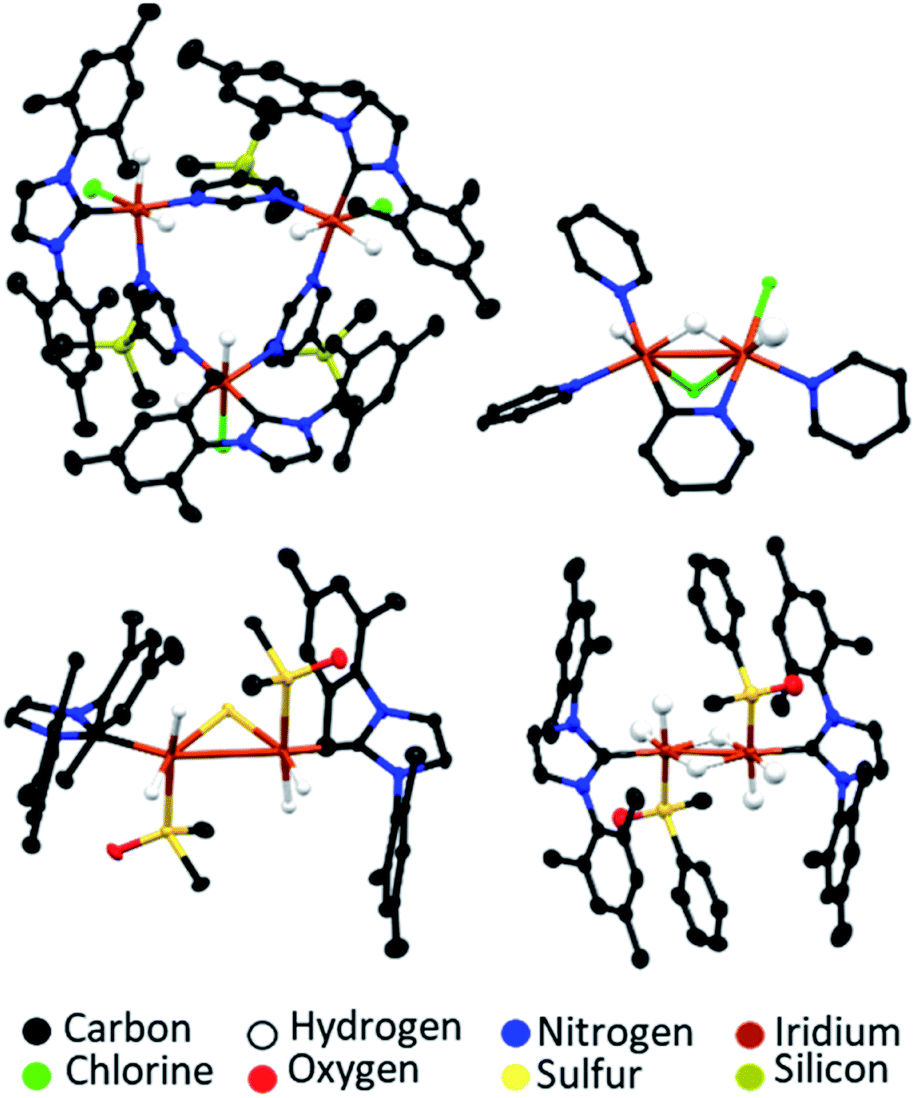 | ||
| Fig. 15 Example X-ray crystal structures of catalyst decomposition products reported in ref. 131 (upper left), ref. 159 (upper right) and ref. 157 (lower). Note that thermal ellipsoids are shown at 50% probability and all non-hydride hydrogen atoms and solvent of crystallisation have been omitted for clarity. | ||
7.7 Novel catalysts based on cobalt for SABRE-like hyperpolarisation
Almost all currently reported SABRE catalysts are based on iridium. In the future, completely new systems that are not based on Ir-phosphine or Ir–NHC catalysts are likely to become available that may be more efficient than the current state-of-the-art catalysts. This includes discovery of catalysts that are based on other transition metals. Rhodium complexes have been the focus of attempts to create non-Ir SABRE catalysts178 as it also resides in group 9 of the periodic table, exhibits similar chemistry to iridium, and is used for hydrogenative PHIP. Despite this, no significant breakthroughs in the development of such Rh-based systems have yet been reported. However, in recent years a novel cobalt dihydride system containing a pincer bis(carbene) ligand has been developed that is capable of hydrogenating alkenes.59,103,179 When these reactions were studied using PHIP, NMR signal enhancements for the hydrogenated products were observed, as is typical of the PHIP effect. However, enhanced signals for the alkene reagents were also visible which were attributed to a reversible SABRE-like effect. This is possible due to reaction of [Co(MesCCC)(N2)(PR3)] (where MesCCC = bis(mesityl-benzimidazol-2-ylidene)phenyl) with para-H2 to form [Co(MesCCC)(H2)(PR3)] (Fig. 16).179 Subsequent phosphine displacement by reaction with an alkene yields [Co(MesCCC)(H2)(alkene)]. It is this species that performs a symmetry-breaking oxidative addition to form [Co(MesCCC)(H)2(alkene)]. It is worth noting that in these examples [Co(MesCCC)(H2)(PR3)] does not appear able to undergo the analogous reaction (i.e. [Co(MesCCC)(H)2(PR3)] does not form). The active species [Co(MesCCC)(H)2(alkene)] can hydrogenate the alkene to form an alkane, which exists in a hyperpolarised state due to PHIP. As alkene binding must be reversible, it can dissociate chemically unchanged. It also exists in a non-Boltzmann spin state which is a consequence of SABRE-like polarisation transfer within [Co(MesCCC)(H)2(alkene)] (Fig. 16).179 Recently, these effects have been extended to a wider range of substrates (>15) including styrenes, terpenes, acrylates, and pentenoates with 1H and 13C signals enhanced by up to 150-fold.57 Other mechanisms for these observations, such as insertion–elimination (PHIP-IE) of the alkene to effectively exchange its protons with those derived from para-H2, have also been proposed (Fig. 16).57 In these cobalt systems, substrates can undergo competing PHIP or PHIP-IE/SABRE103 and additional studies are anticipated to further understand the different polarisation transfer mechanisms in action in these systems. Nevertheless, these ongoing studies have utilised a more widely available, cheaper, transition metal (compared to iridium or rhodium), and have demonstrated that reversible polarisation transfer is likely not exclusive to iridium catalysts. It is also expected that these breakthroughs will stimulate further design of novel systems that may exhibit vastly different catalysis to currently used systems.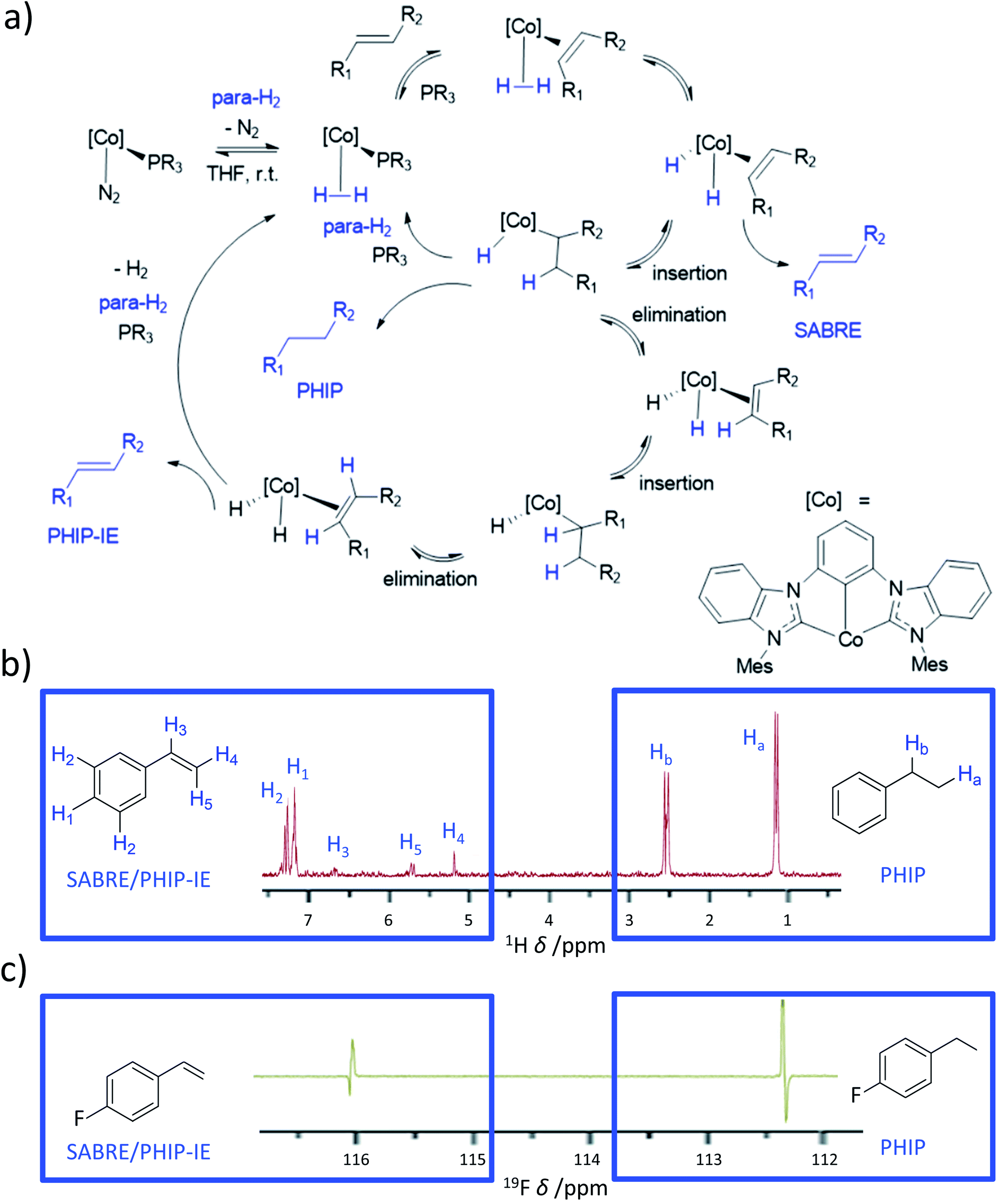 | ||
| Fig. 16 Cobalt-based system used to achieve both PHIP and SABRE-like hyperpolarisation. (a) Simplified mechanism showing ways in which alkenes can become hydrogenated using para-H2 to give PHIP for alkane products, or the starting alkene can become hyperpolarised in a SABRE or PHIP-IE pathway. (b) Example 1H OPSY spectrum showing enhanced 1H NMR signals due to both PHIP and SABRE/PHIP-IE for phenylethane and phenylpropene respectively. (c) 19F NMR Spectrum for fluorophenylpropene and fluorophenylpropane hyperpolarised using these cobalt systems. Adapted from (b) ref. 179 and (c) ref. 57. | ||
8. Applications of novel PHIP catalysts in hyperpolarisation
In this final section we highlight just two recent applications of PHIP hyperpolarisation in NMR that would not have been possible without the development of novel catalysts that can break para-H2 symmetry.8.1 Water-soluble ruthenium catalysts for trans hydrogenation: in vivo molecular imaging of hyperpolarised fumarate
The NMR signal gains that can be provided by hyperpolarisation have provided exciting opportunities for biomolecular imaging.4,6,180 MRI is currently used for routine diagnosis of tissue structure abnormalities, but its low sensitivity necessitates detecting a signal for bulk water at tens of molar concentrations in the body. Boosting MRI signal intensity allows molecules at much lower (i.e., millimolar) concentrations such as disease markers, drugs, and many other biologically relevant molecules, to be detected in vivo. This dramatically improves the diagnostic value of MRI as it can now give information about tissue biochemistry that are not available in thermally polarised MRI measurements. There are many key metabolic intermediates that are the focus of hyperpolarisation studies as their injection and imaging allows specific metabolic processes to be interrogated in vivo in real time.9 One of these molecules is fumarate, a key intermediate of the Krebs cycle. Hyperpolarised [1,4-13C2]-fumarate can be used to probe necrotic cell death by observation of enhanced signals for [1-13C] and [4-13C]-malate products (Fig. 17).107,181 The advent of water-soluble ruthenium catalysts that can perform trans hydrogenation reactions unlock the potential for PHIP to produce fumarate in a hyperpolarised state (Fig. 17a).181,182 Transfer of polarisation from introduced protons to a 13C-labelled site can occur using a magnetic field cycling (MFC) approach and was reported to yield 13C signals for [1,4-13C2]-fumarate with a polarisation level of up to 24%.182 It has since been reported that this has been increased to 30–45% by optimisation of the equipment, reaction conditions, and purification procedure (precipitation as a solid before redissolving).183,184 The great benefit of this approach is that it is highly compatible with biological imaging studies as it can produce aqueous solutions of hyperpolarised fumarate without contaminant side products or toxic catalyst.107,183 Production and injection of PHIP-hyperpolarised [1-13C]-fumarate into mice containing acetaminophen-induced hepatitis has allowed fumarate biochemistry to be examined in vivo using 2D chemical shift imaging at 1.5 T (Fig. 17c).107 This feat would not be possible without development of catalytic systems that can hydrogenate starting unsaturated precursors in a trans fashion. Notably, 13C polarisation levels for fumarate are comparable to those achieved using alternative hyperpolarisation techniques such as dissolution dynamic nuclear polarisation (d-DNP).185,186 However, PHIP provides several advantages compared to d-DNP as it is rapid (seconds-minutes) and does not require the technically demanding equipment associated with d-DNP.9,187 Further advances in catalyst design and agent purification steps are expected to increase signal enhancements, biocompatibility, and consequently, further progress in this direction is expected. Optimisation of current catalysts is expected to improve the NMR signal enhancements that can be achieved, which in turn facilitates MRI detection of molecules at lower concentrations. Continued development of novel catalysts that can break the symmetry of para-H2 is likely to stimulate hyperpolarisation of a greater variety of metabolic imaging probes for disease diagnosis.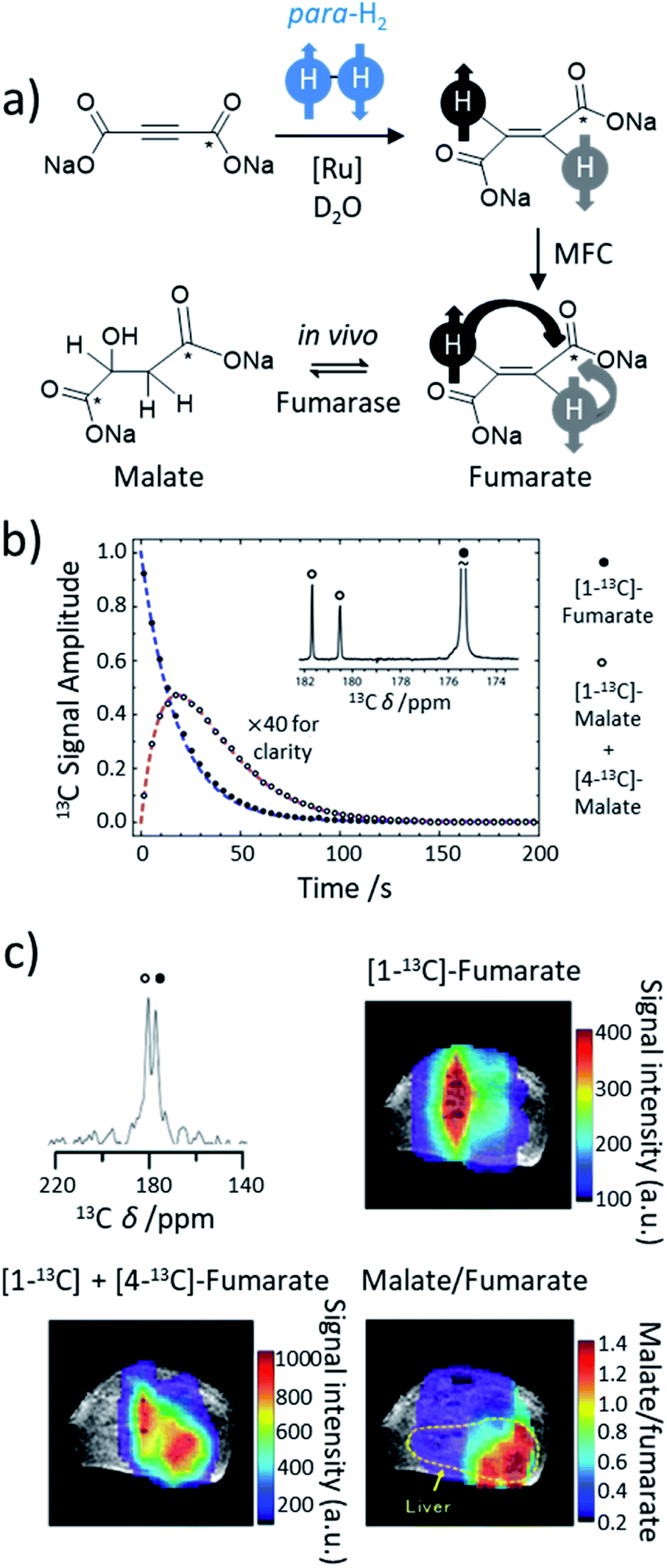 | ||
| Fig. 17 (a) Reaction scheme showing the formation of PHIP-hyperpolarised [1-13C]-fumarate from trans hydrogenation of an unsaturated [1-13C]acetylene dicarboxylate precursor. Proton magnetisation is transferred to the 13C site by magnetic field cycling (MFC). Metabolic conversion following in vivo injection can give rise to [1-13C]-malate and [4-13C]-malate products. Note that * represents a 13C labelled site and the nuclei represented in black circles are hyperpolarised. (b) Time course of hyperpolarised fumarate and malate in a suspension of lysed EL-4 tumour at 14.1 T with an example single acquisition (inset). Adapted from ref. 182. (c) In vivo13C chemical shift imaging of PHIP-hyperpolarised [1-13C]-fumarate and its metabolic products in an acetaminophen-induced hepatitis mouse at 1.5 T. An example single voxel 13C NMR spectrum is shown in the upper left. Maps of hyperpolarised 13C signal intensity for [1-13C]-fumarate (upper right) and both [1-13C] and [4-13C]malate (lower left) are shown with a parametric map of the malate/fumarate ratio (lower right). Adapted from ref. 107. | ||
8.2 Hyperpolarised metal dihydride catalysts for coligand sensing: applications for mixture analysis by NMR
A selection of PHIP catalysts have been engineered with reporter hydride NMR signals designed to act as an indirect marker of a target analyte bound to the metal centre. For example, [IrCl(CO)(PPh3)2] can activate para-H2 to form [IrCl(H)2(CO)(PPh3)2] which can be detected with enhanced hydride NMR signals.46 Reaction of this catalyst with the analytes pyridine, benzimidazole, purine, or adenine, forms complexes of the form [IrCl(H)2(PPh3)2(analyte)] which also display enhanced hydride NMR resonances with distinctive chemical shifts that depend on the identity of the analyte. The improved NMR sensitivity provided by PHIP allows picomolar concentrations of these analytes to be detected via hyperpolarised hydride signals.Similar effects can be achieved using SABRE catalysts, which also display hydride NMR chemical shifts sensitive to the identity of the substrate molecule trans to them.38,51 In particular, catalysts with inequivalent hydride ligands have been highly beneficial as their NMR signals display a wider chemical shift dispersion in a spectrally uncrowded region.51 The enhanced hydride signals of these [Ir(H)2(IMes)(Nsub1)(Nsub2)(Nsub3)]Cl catalysts have been used to detect specific analytes present in complex mixtures and has been applied to study metabolomics and other systems that contain a great variety of molecules.50,51 In these examples, peak overlap, even within the hydride region of 1H NMR spectra, can often become a problem due to the large number of SABRE-amenable analytes in the mixture. It is therefore often advantageous to use 2D NMR methods such as COSY,188,189 DOSY190,191 or others49,51 to provide increased chemical resolution between analytes.
A further way to increase chemical shift resolution of these reporter complexes is through novel catalyst design. Recently, the enhanced hydride signals of [Ir(H)2(IMes)(κ2-N,O-imine)(sub)] catalysts have been reported as sensitive markers of particular analytes.38 They can achieve much wider chemical shift dispersion for their hydride signals than [Ir(H)2(IMes)(Nsub1)(Nsub2)(Nsub3)]Cl systems. The chemical inequivalence of these hydrides, which are located trans to the O-donor site of the imine and trans to the analyte which can bind through an N-, S- or even C-donor site (Fig. 18a) is advantageous in preventing NMR signal overlap.37,38 As a consequence, the hydride resonance trans to the analyte can display signals over a 15.5 ppm chemical shift range depending on the identity of the analyte bound within the catalyst.38 Aliphatic and aromatic NMR signals of the analyte itself can become enhanced via SABRE, but for complex mixtures these signals are poorly resolved. The enhanced hydride signals of closely related [Ir(H)2(IMes)(κ2-N,O-amino acids)(pyridine)] catalysts have been used to distinguish different amino acids (Fig. 18b) in complex bio-mixtures.49 The applications of these catalysts in NMR mixture analysis and biofluid analysis has been clearly demonstrated and further examples are expected in the years ahead.
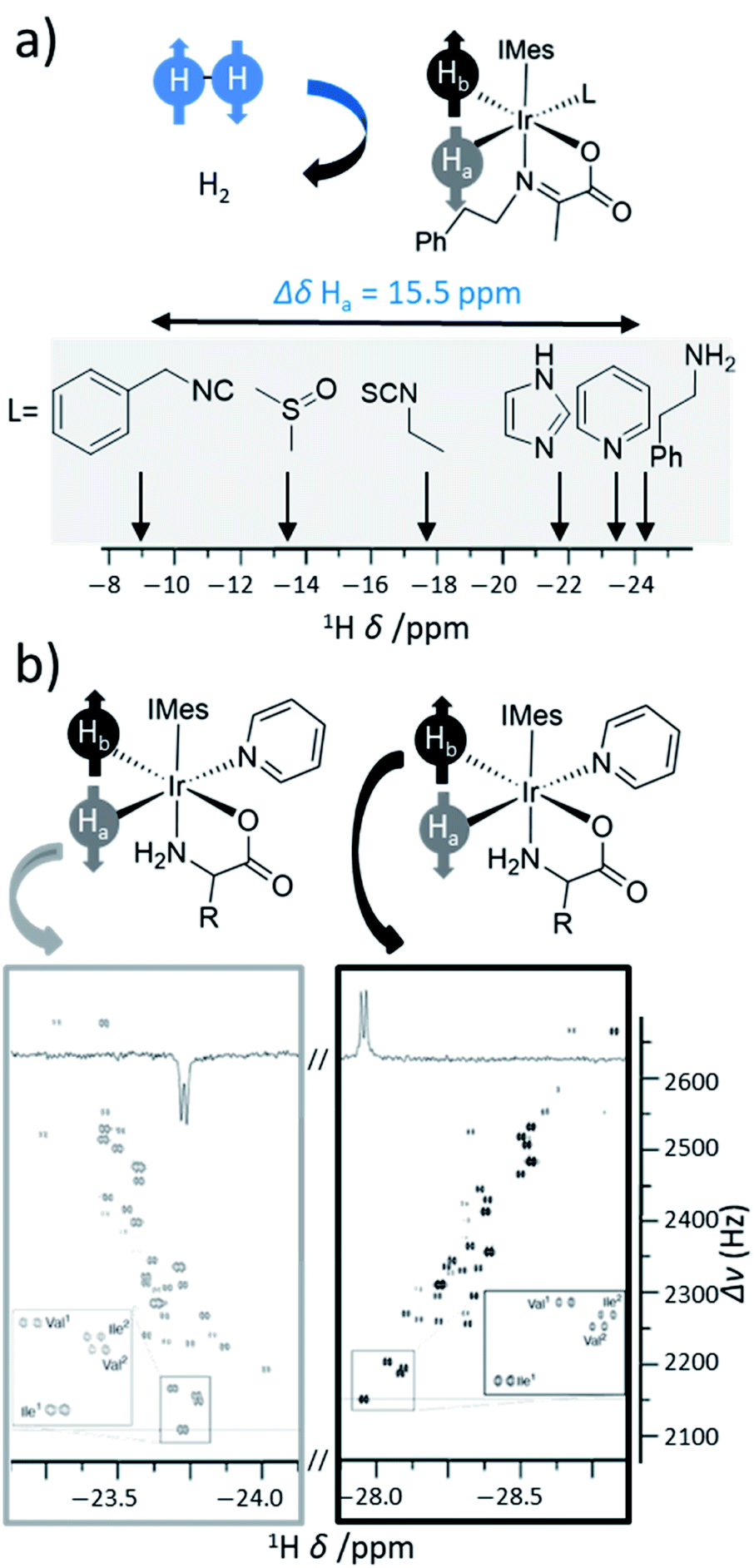 | ||
| Fig. 18 (a) The enhanced hydride NMR signals of PHIP and SABRE catalysts can provide significant chemical shift resolution which is of great use for the indirect detection of analytes in mixtures.38 (b) Example 2D NMR measurements that utilise PHIP hyperpolarisation of metal dihydride catalysts for sensing and identification of complex mixtures of amino acids. Adapted from ref. 49. | ||
9. Conclusions
The use of para-H2 to enhance NMR signal intensity is a versatile platform that allows the hyperpolarisation of a range of different functionalities including metal complexes,14,30,36,38,45 alkenes,14,30,57 alkanes,14,30 esters,88 aldehydes,23–27,114 carboxylic acids,126,192 N-heterocycles,17,29 amines,58,193 diazirines,124,194,195 nitriles,167 silanes,132 sulfoxides,147,172 α-ketoacids,125,146,147,157,196 amino acids,197 peptides78,80,82,198 and many others. Enhanced NMR signals can be achieved most commonly for 1H, 13C and 15N sites, although PHIP and SABRE-enhanced 2H,199,20011B,28,64,6619F,85,128,129,20129Si,131,13231P,39,69,74,130 and 119Sn131 NMR signals have also been reported. The preparation of these molecules in a hyperpolarised state allows exciting applications in the areas of mechanistic elucidations,11 reaction monitoring,139,196,202 mixture analysis,51,188,189,203,204 biomedical imaging,6,107,182,205,206 and many others. In recent years this list of applications has even grown to include novel uses in magnetooptics,207 RASER physics,208 micron-scale NMR,209 SQUID-based NMR,210 and zero-field NMR.211,212Development of novel catalyst systems has spurred some of the greatest advances in this area of science by expanding the scope of molecules that can be hyperpolarised. Many other advances in hyperpolarisation are occurring simultaneously: proton exchange effects have allowed polarisation to be relayed to molecules that might not contain any unsaturated (PHIP-X)213 or iridium ligating (SABRE-Relay)214 sites such as alcohols,215 sugars,216 silanols132 and many others.213,214,217 It is expected that further catalyst development will be a large driving force behind further extension of substrate scope, increased efficiency, and enhanced biocompatibility. In particular, novel metal-free,28,64–67trans hydrogenation,106,109,181,182 sulfoxide-containing SABRE catalysts125,146,147,157 and cobalt-based SABRE-like systems59,179 are likely to lead to exciting new results in this area in the years ahead.
Abbreviations
| ALTADENA | Adiabatic longitudinal transport after dissociation engenders nuclear alignment |
| BINAP | 2,20-Bis(diphenylphosphino)-1,10-binaphthyl |
| CEST | Chemical exchange saturation transfer |
| COD | cis,cis-1,5-Cyclooctadiene |
| COSY | Correlation spectroscopy |
| Cp* | 1,2,3,4,5-Pentamethylcyclopentadiene |
| Cy | Cyclohexyl |
| dppb | 1,4-Bis(diphenylphosphino)butane |
| dppe | (Diphenylphosphino)ethane |
| d-DNP | Dissolution dynamic nuclear polarisation |
| DFT | Density functional theory |
| DOSY | Diffusion ordered spectroscopy |
| EL-4 | A cancer cell line |
| Et | Ethyl |
| EXSY | Exchange spectroscopy |
| FLP | Frustrated Lewis pair |
| gem | Geminal |
| HCAT | Hydroborane catalyst N,N-dimethyl-2-[(pentafluorophenyl)boryl]aniline |
| HP | Hyperpolarised |
| I | Nuclear spin quantum number |
| IMes | 1,3-Bis(2,4,6-trimethylphenyl)imidazole-2-ylidene |
| INEPT | Insensitive nuclei enhancement by polarisation transfer |
| J | J coupling constant |
| Me | Methyl |
| Mes | Mesityl |
| MesCCC | Bis(mesityl-benzimidazol-2-ylidene)phenyl |
| MFC | Magnetic field cycling |
| NHC | N-Heterocyclic carbene |
| NMR | Nuclear magnetic resonance |
| O-Sub | O-donor substrate |
| OneH | One proton |
| OPSY | Only parahydrogen spectroscopy |
| Ortho-H2 | Orthohydrogen |
| Para-H2 | Parahydrogen |
| PASADENA | Parahydrogen and synthesis allow dramatically enhanced nuclear alignment |
| Ph | Phenyl |
| PHIP | Parahydrogen induced polarisation |
| PHIP-IE | PHIP insertion elimination |
| PHIP-X | PHIP exchange |
| Phox | 2-(2-(Diphenylphosphanyl)phenyl)-4,5-dihydrooxazole |
| ppm | Parts per million |
| PTF | Polarisation transfer field |
| Py | Pyridine |
| QCAT | 1-{2-[Bis(pentafluorophenyl)boryl]benzyl}-2,2,4,7-tetramethyl-1,2,3,4-tetrahydroquinoline |
| RASER | Radio-frequency amplification by stimulated emission of radiation |
| RF | Radiofrequency |
| SABRE | Signal amplification by reversible exchange |
| SEOP | Spin exchange optical pumping |
| SQUID | Superconducting quantum interference device |
| TEP | Tolman electronic parameter |
| Tf | Triflate |
| THF | Tetrahydrofuran |
| THP | Tris(hydroxymethyl)phosphine |
| T 1 | Longitudinal relaxation time |
| t Bu | Tertiary butyl |
| T LLS | Relaxation time of long lived singlet order |
| Xantphos | 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene |
Author contributions
BJT: conceptualisation, writing – original draft, review and editing. VVZ: conceptualisation, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
VVZ acknowledges funding from the Academy of Finland (grant no. 323480), the European Research Council under Horizon 2020 (grant agreement no. 772110), and the University of Oulu (Kvantum Institute).References
- J. Keeler, Understanding NMR Spectroscopy, John Wiley & Sons, Chichester, 2010 Search PubMed.
- Hyperpolarization Methods in NMR Spectroscopy, Topics in Current Chemistry, ed. L. T. Kuhn, Springer, Berlin, 2013, vol. 338 Search PubMed.
- J. Ardenkjaer-Larsen, G. S. Boebinger, A. Comment, S. Duckett, A. S. Edison, F. Engelke, C. Griesinger, R. G. Griffin, C. Hilty, H. Maeda, G. Parigi, T. Prisner, E. Ravera, J. van Bentum, S. Vega, A. Webb, C. Luchinat, H. Schwalbe and L. Frydman, Angew. Chem., Int. Ed., 2015, 54, 9162–9185 CrossRef CAS PubMed.
- P. Nikolaou, B. M. Goodson and E. Y. Chekmenev, Chem.–Eur. J., 2015, 21, 3156–3166 CrossRef CAS PubMed.
- K. V. Kovtunov, E. V. Pokochueva, O. G. Salnikov, S. F. Cousin, D. Kurzbach, B. Vuichoud, S. Jannin, E. Y. Chekmenev, B. M. Goodson and D. A. Barskiy, Chem.–Asian J., 2018, 13, 1857–1871 CrossRef CAS PubMed.
- J. Hövener, A. N. Pravdivtsev, B. Kidd, C. R. Bowers, S. Glöggler, K. V. Kovtunov, M. Plaumann, R. Katz-Brull, K. Buckenmaier, A. Jerschow, F. Reineri, T. Theis, R. V. Shchepin, S. Wagner, P. Bhattacharya, N. M. Zacharias and E. Y. Chekmenev, Angew. Chem., Int. Ed., 2018, 57, 11140–11162 CrossRef PubMed.
- S. B. Duckett and N. J. Wood, Coord. Chem. Rev., 2008, 252, 2278–2291 CrossRef CAS.
- T. G. Walker and W. Happer, Rev. Mod. Phys., 1997, 69, 629 CrossRef CAS.
- K. R. Keshari and D. M. Wilson, Chem. Soc. Rev., 2014, 43, 1627–1659 RSC.
- R. Eisenberg, Acc. Chem. Res., 1991, 24, 110–116 CrossRef CAS.
- S. B. Duckett and D. Blazina, Eur. J. Inorg. Chem., 2003, 2003, 2901–2912 CrossRef.
- S. B. Duckett and R. E. Mewis, Acc. Chem. Res., 2012, 45, 1247–1257 CrossRef CAS PubMed.
- A. B. Schmidt, C. R. Bowers, K. Buckenmaier, E. Y. Chekmenev, H. de Maissin, J. Eills, F. Ellermann, S. Glöggler, J. W. Gordon, S. Knecht, I. V. Koptyug, J. Kuhn, A. N. Pravdivtsev, F. Reineri, T. Theis, K. Therm and J.-B. Hövener, Anal. Chem., 2022, 94, 479–502 CrossRef CAS PubMed.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- R. U. Kirss, T. C. Eisenschmid and R. Eisenberg, J. Am. Chem. Soc., 1988, 110, 8564–8566 CrossRef CAS.
- P. J. Rayner and S. Duckett, Angew. Chem., Int. Ed., 2018, 57, 6742–6753 CrossRef CAS PubMed.
- D. A. Barskiy, S. Knecht, A. V. Yurkovskaya and K. L. Ivanov, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 33–70 CrossRef CAS PubMed.
- D. Canet, C. Aroulanda, P. Mutzenhardt, S. Aime, R. Gobetto and F. Reineri, Concepts Magn. Reson., Part A, 2006, 28, 321–330 CrossRef.
- R. W. Adams, S. B. Duckett, R. A. Green, D. C. Williamson and G. G. R. Green, J. Chem. Phys., 2009, 131, 194505 CrossRef PubMed.
- J. R. Birchall, A. M. Coffey, B. M. Goodson and E. Y. Chekmenev, Anal. Chem., 2020, 92, 15280–15284 CrossRef CAS PubMed.
- B. Feng, A. M. Coffey, R. D. Colon, E. Y. Chekmenev and K. W. Waddell, J. Magn. Reson., 2012, 214, 258–262 CrossRef CAS PubMed.
- F. Ellermann, A. Pravdivtsev and J.-B. Hövener, Magn. Reson., 2021, 2, 49–62 CrossRef CAS.
- A. B. Permin and R. Eisenberg, J. Am. Chem. Soc., 2002, 124, 12406–12407 CrossRef CAS PubMed.
- D. J. Fox, S. B. Duckett, C. Flaschenriem, W. W. Brennessel, J. Schneider, A. Gunay and R. Eisenberg, Inorg. Chem., 2006, 45, 7197–7209 CrossRef CAS PubMed.
- D. Guan, C. Godard, S. M. Polas, R. P. Tooze, A. C. Whitwood and S. B. Duckett, Dalton Trans., 2019, 48, 2664–2675 RSC.
- C. Godard, S. B. Duckett, S. Polas, R. Tooze and A. C. Whitwood, Dalton Trans., 2009, 2496–2509 RSC.
- C. Godard, S. B. Duckett, S. Polas, R. Tooze and A. C. Whitwood, J. Am. Chem. Soc., 2005, 127, 4994–4995 CrossRef CAS PubMed.
- D. O. Zakharov, K. Chernichenko, K. Sorochkina, S. Yang, V.-V. Telkki, T. Repo and V. V. Zhivonitko, Chem.–Eur. J., 2021, 28, e2021103501 Search PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645 CrossRef CAS PubMed.
- T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft, R. Eisenberg, J. Bargon, R. G. Lawler and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 8089–8091 CrossRef CAS.
- M. G. Pravica and D. P. Weitekamp, Chem. Phys. Lett., 1988, 145, 255–258 CrossRef CAS.
- N. V. Kireev, A. S. Kiryutin, A. A. Pavlov, A. V. Yurkovskaya, E. I. Musina, A. A. Karasik, E. S. Shubina, K. L. Ivanov and N. V. Belkova, Eur. J. Inorg. Chem., 2021, 41, 4265–4272 CrossRef.
- A. S. Kiryutin, G. Sauer, A. V. Yurkovskaya, H.-H. Limbach, K. L. Ivanov and G. Buntkowsky, J. Phys. Chem. C, 2017, 121, 9879–9888 CrossRef CAS.
- S. Knecht, S. Hadjiali, D. A. Barskiy, A. Pines, G. Sauer, A. S. Kiryutin, K. L. Ivanov, A. V. Yurkovskaya and G. Buntkowsky, J. Phys. Chem. C, 2019, 123, 16288–16293 CrossRef CAS.
- B. Procacci, P. M. Aguiar, M. E. Halse, R. N. Perutz and S. B. Duckett, Chem. Sci., 2016, 7, 7087–7093 RSC.
- B. J. Tickner, W. Iali, S. S. Roy, A. C. Whitwood and S. B. Duckett, ChemPhysChem, 2019, 20, 241–245 CrossRef CAS PubMed.
- B. J. Tickner, R. O. John, S. S. Roy, S. J. Hart, A. C. Whitwood and S. B. Duckett, Chem. Sci., 2019, 10, 5235–5245 RSC.
- V. V. Zhivonitko, I. V. Skovpin and I. V. Koptyug, Chem. Commun., 2015, 51, 2506–2509 RSC.
- P. B. Chock and J. Halpern, J. Am. Chem. Soc., 1966, 88, 3511–3514 CrossRef CAS.
- J. Halpern, J. Phys. Chem., 1959, 63, 398–403 CrossRef CAS.
- R. U. Kirss and R. Eisenberg, J. Organomet. Chem., 1989, 359, C22–C26 CrossRef CAS.
- J. Bargon, in The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley, Weinheim, 2006, pp. 313–358 Search PubMed.
- M. Ahlquist, M. Gustafsson, M. Karlsson, M. Thaning, O. Axelsson and O. F. Wendt, Inorg. Chim. Acta, 2007, 360, 1621–1627 CrossRef CAS.
- S. B. Duckett, C. L. Newell and R. Eisenberg, J. Am. Chem. Soc., 1994, 116, 10548–10556 CrossRef CAS.
- N. J. Wood, J. A. Brannigan, S. B. Duckett, S. L. Heath and J. Wagstaff, J. Am. Chem. Soc., 2007, 129, 11012–11013 CrossRef CAS PubMed.
- J. López-Serrano, S. B. Duckett and A. Lledós, J. Am. Chem. Soc., 2006, 128, 9596–9597 CrossRef PubMed.
- L. S. Lloyd, A. Asghar, M. J. Burns, A. Charlton, S. Coombes, M. J. Cowley, G. J. Dear, S. B. Duckett, G. R. Genov and G. G. R. Green, Catal. Sci. Technol., 2014, 4, 3544–3554 RSC.
- L. Sellies, R. Aspers, M. C. Feiters, F. Rutjes and M. Tessari, Angew. Chem., 2021, 133, 27160–27165 CrossRef.
- N. Reimets, K. Ausmees, S. Vija and I. Reile, Anal. Chem., 2021, 93, 9480–9485 CrossRef PubMed.
- L. Sellies, I. Reile, R. L. E. G. Aspers, M. C. Feiters, F. P. J. T. Rutjes and M. Tessari, Chem. Commun., 2019, 55, 7235–7238 RSC.
- B. Procacci, P. M. Aguiar, M. E. Halse, R. N. Perutz and S. B. Duckett, Chem. Sci., 2016, 7, 7087–7093 RSC.
- A. Harthun, R. Giernoth, C. J. Elsevier and J. Bargon, Chem. Commun., 1996, 2483–2484 RSC.
- J. López-Serrano, S. B. Duckett, J. P. Dunne, C. Godard and A. C. Whitwood, Dalton Trans., 2008, 4270–4281 RSC.
- D. Guan, A. J. Holmes, J. López-Serrano and S. B. Duckett, Catal. Sci. Technol., 2017, 7, 2101–2109 RSC.
- J. McCormick, A. M. Grunfeld, Y. N. Ertas, A. N. Biswas, K. L. Marsh, S. Wagner, S. Glöggler and L.-S. Bouchard, Anal. Chem., 2017, 89, 7190–7194 CrossRef CAS PubMed.
- S. R. Muhammad, R. B. Greer, S. B. Ramirez, B. M. Goodson and A. R. Fout, ACS Catal., 2021, 11, 2011–2020 CrossRef CAS.
- K. Tokmic, B. J. Jackson, A. Salazar, T. J. Woods and A. R. Fout, J. Am. Chem. Soc., 2017, 139, 13554–13561 CrossRef CAS PubMed.
- K. Tokmic, R. B. Greer, L. Zhu and A. R. Fout, J. Am. Chem. Soc., 2018, 140, 14844–14850 CrossRef CAS PubMed.
- I. V. Koptyug, K. V. Kovtunov, S. R. Burt, M. S. Anwar, C. Hilty, S.-I. Han, A. Pines and R. Z. Sagdeev, J. Am. Chem. Soc., 2007, 129, 5580–5586 CrossRef CAS PubMed.
- E. V. Pokochueva, D. B. Burueva, O. G. Salnikov and I. V. Koptyug, ChemPhysChem, 2021, 22, 1421–1440 CrossRef CAS PubMed.
- K. V. Kovtunov, O. G. Salnikov, I. V. Skovpin, N. V. Chukanov, D. B. Burueva and I. V. Koptyug, Pure Appl. Chem., 2020, 92, 1029–1046 CrossRef CAS.
- Frustrated Lewis Pairs I: Uncovering and Understanding, Topics in Current Chemistry, ed. G. Erker and D. W. Stephan, Springer, Berlin, 2013, vol. 332 Search PubMed.
- K. Sorochkina, V. V. Zhivonitko, K. Chernichenko, V.-V. Telkki, T. Repo and I. V. Koptyug, J. Phys. Chem. Lett., 2018, 9, 903–907 CrossRef CAS PubMed.
- V. V. Zhivonitko, K. Sorochkina, K. Chernichenko, B. Kótai, T. Földes, I. Pápai, V.-V. Telkki, T. Repo and I. Koptyug, Phys. Chem. Chem. Phys., 2016, 18, 27784–27795 RSC.
- V. V. Zhivonitko, V.-V. Telkki, K. Chernichenko, T. Repo, M. Leskelä, V. Sumerin and I. V. Koptyug, J. Am. Chem. Soc., 2014, 136, 598–601 CrossRef CAS PubMed.
- V. V. Zhivonitko, J. Bresien, A. Schulz and I. V. Koptyug, Phys. Chem. Chem. Phys., 2019, 21, 5890–5893 RSC.
- L. E. Longobardi, C. A. Russell, M. Green, N. S. Townsend, K. Wang, A. J. Holmes, S. B. Duckett, J. E. McGrady and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 13453–13457 CrossRef CAS PubMed.
- V. V. Zhivonitko, H. Beer, D. O. Zakharov, J. Bresien and A. Schulz, ChemPhysChem, 2021, 22, 813–817 CrossRef CAS PubMed.
- P. Holtkamp, J. Schwabedissen, B. Neumann, G. Stammler, I. Koptyug, V. Zhivonitko and N. W. Mitzel, Chem.–Eur. J., 2020, 26, 17381–17385 CrossRef CAS PubMed.
- A. Dedieu, S. Humbel, C. Elsevier and C. Grauffel, Theor. Chem. Acc., 2004, 112, 305–312 Search PubMed.
- P. J. Albietz, J. F. Houlis and R. Eisenberg, Inorg. Chem., 2002, 41, 2001–2003 CrossRef CAS PubMed.
- G. Papp, H. Horváth and F. Joó, ChemCatChem, 2019, 11, 3000–3003 CrossRef CAS.
- T. C. Eisenschmid, J. McDonald, R. Eisenberg and R. G. Lawler, J. Am. Chem. Soc., 1989, 111, 7267–7269 CrossRef CAS.
- R. Giernoth, P. Huebler and J. Bargon, Angew. Chem., Int. Ed., 1998, 37, 2473–2475 CrossRef CAS PubMed.
- S. Glöggler, S. Wagner and L.-S. Bouchard, Chem. Sci., 2015, 6, 4261–4266 RSC.
- M. Körner, G. Sauer, A. Heil, D. Nasu, M. Empting, D. Tietze, S. Voigt, H. Weidler, T. Gutmann and O. Avrutina, Chem. Commun., 2013, 49, 7839–7841 RSC.
- G. Sauer, D. Nasu, D. Tietze, T. Gutmann, S. Englert, O. Avrutina, H. Kolmar and G. Buntkowsky, Angew. Chem., Int. Ed., 2014, 53, 12941–12945 CrossRef CAS PubMed.
- A. S. Kiryutin, G. Sauer, D. Tietze, M. Brodrecht, S. Knecht, A. V. Yurkovskaya, K. L. Ivanov, O. Avrutina, H. Kolmar and G. Buntkowsky, Chem.–Eur. J., 2019, 25, 4025–4030 CrossRef CAS PubMed.
- F. Gruppi, X. Xu, B. Zhang, J. A. Tang, A. Jerschow and J. W. Canary, Angew. Chem., 2012, 124, 11957–11960 CrossRef.
- R. Mandal, P. Pham and C. Hilty, Chem. Sci., 2021, 12, 12950–12958 RSC.
- T. Ratajczyk, G. Buntkowsky, T. Gutmann, B. Fedorczyk, A. Mames, M. Pietrzak, P. Szkudlarek and Z. Puzio, ChemBioChem, 2021, 22, 855–860 CrossRef CAS PubMed.
- S. B. Duckett, C. L. Newell and R. Eisenberg, J. Am. Chem. Soc., 1993, 115, 1156–1157 CrossRef CAS.
- V. P. Kozinenko, A. S. Kiryutin, A. V. Yurkovskaya and K. L. Ivanov, J. Magn. Reson., 2019, 309, 106594 CrossRef CAS PubMed.
- L. T. Kuhn, U. Bommerich and J. Bargon, J. Phys. Chem. A, 2006, 110, 3521–3526 CrossRef CAS.
- M. Stephan, O. Kohlmann, H. G. Niessen, A. Eichhorn and J. Bargon, Magn. Reson. Chem., 2002, 40, 157–160 CrossRef CAS.
- H. Jóhannesson, O. Axelsson and M. Karlsson, C. R. Phys., 2004, 5, 315–324 CrossRef.
- E. Cavallari, C. Carrera, T. Boi, S. Aime and F. Reineri, J. Phys. Chem. B, 2015, 119, 10035–10041 CrossRef CAS PubMed.
- B. Joalland, A. B. Schmidt, M. S. H. Kabir, N. V. Chukanov, K. V. Kovtunov, I. V. Koptyug, J. Hennig, J.-B. Hövener and E. Y. Chekmenev, Anal. Chem., 2019, 92, 1340–1345 CrossRef PubMed.
- M. Itoda, Y. Naganawa, M. Ito, H. Nonaka and S. Sando, RSC Adv., 2019, 9, 18183–18190 RSC.
- R. V. Shchepin, D. A. Barskiy, A. M. Coffey, I. V. Manzanera Esteve and E. Y. Chekmenev, Angew. Chem., 2016, 128, 6175–6178 CrossRef.
- M. Emondts, J. F. P. Colell, B. Blümich and P. P. M. Schleker, Phys. Chem. Chem. Phys., 2017, 19, 21933–21937 RSC.
- P. G. Jessop and R. H. Morris, Coord. Chem. Rev., 1992, 121, 155–284 CrossRef CAS.
- R. A. Sánchez-Delgado and M. Rosales, Coord. Chem. Rev., 2000, 196, 249–280 CrossRef.
- I. D. Gridnev, N. Higashi, K. Asakura and T. Imamoto, J. Am. Chem. Soc., 2000, 122, 7183–7194 CrossRef CAS.
- O. Bondar, E. Cavallari, C. Carrera, S. Aime and F. Reineri, Catal. Today, 2021 DOI:10.1016/j.cattod.2021.11.030.
- E. Cavallari, C. Carrera, S. Aime and F. Reineri, J. Magn. Reson., 2018, 289, 12–17 CrossRef CAS PubMed.
- R. V. Shchepin, A. M. Coffey, K. W. Waddell and E. Y. Chekmenev, J. Phys. Chem. Lett., 2012, 3, 3281–3285 CrossRef CAS PubMed.
- V. V. Zhivonitko, I. V. Skovpin, K. C. Szeto, M. Taoufik and I. V. Koptyug, J. Phys. Chem. C, 2018, 122, 4891–4900 CrossRef CAS PubMed.
- D. B. Burueva, A. A. Smirnov, O. A. Bulavchenko, I. P. Prosvirin, E. Y. Gerasimov, V. A. Yakovlev, K. V. Kovtunov and I. V. Koptyug, Top. Catal., 2020, 63, 2–11 CrossRef CAS.
- E. W. Zhao, H. Zheng, R. Zhou, H. E. Hagelin-Weaver and C. R. Bowers, Angew. Chem., 2015, 127, 14478–14483 CrossRef.
- C. E. Johnson and R. Eisenberg, J. Am. Chem. Soc., 1985, 107, 3148–3160 CrossRef CAS.
- K. Tokmic and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 13700–13705 CrossRef CAS PubMed.
- D. Schleyer, H. G. Niessen and J. Bargon, New J. Chem., 2001, 25, 423–426 RSC.
- M. Leutzsch, L. M. Wolf, P. Gupta, M. Fuchs, W. Thiel, C. Farès and A. Fürstner, Angew. Chem., 2015, 127, 12608–12613 CrossRef PubMed.
- A. Guthertz, M. Leutzsch, L. M. Wolf, P. Gupta, S. M. Rummelt, R. Goddard, C. Farès, W. Thiel and A. Fürstner, J. Am. Chem. Soc., 2018, 140, 3156–3169 CrossRef CAS PubMed.
- N. J. Stewart, H. Nakano, S. Sugai, M. Tomohiro, Y. Kase, Y. Uchio, T. Yamaguchi, Y. Matsuo, T. Naganuma, N. Takeda, I. Nishimura, H. Hirata, T. Hashimoto and S. Matsumoto, ChemPhysChem, 2021, 22, 915–923 CrossRef CAS PubMed.
- T. Biberger, S. N. Hess, M. Leutzsch and A. Fürstner, Angew. Chem., 2022, 134, e202113827 CrossRef.
- L. Dagys, B. Ripka, M. Leutzsch, G. A. I. Moustafa, J. Eills, J. F. P. Colell and M. H. Levitt, Magn. Reson., 2020, 1, 175–186 CrossRef.
- A. Harthun, R. Selke and J. Bargon, Angew. Chem., Int. Ed., 1996, 35, 2505–2507 CrossRef CAS.
- I. V. Skovpin, V. V. Zhivonitko and I. V. Koptyug, Appl. Magn. Reson., 2011, 41, 393–410 CrossRef CAS.
- A. N. Pravdivtsev, A. Brahms, S. Kienitz, F. D. Sönnichsen, J. Hövener and R. Herges, ChemPhysChem, 2021, 22, 370–377 CrossRef CAS PubMed.
- S. Lehmkuhl, M. Emondts, L. Schubert, P. Spannring, J. Klankermayer, B. Bluemich and P. Schleker, ChemPhysChem, 2017, 18, 2426–2429 CrossRef CAS PubMed.
- C. Godard, S. B. Duckett, C. Henry, S. Polas, R. Toose and A. C. Whitwood, Chem. Commun., 2004, 1826–1827 RSC.
- A. N. Pravdivtsev, K. L. Ivanov, A. V. Yurkovskaya, P. A. Petrov, H.-H. Limbach, R. Kaptein and H.-M. Vieth, J. Magn. Reson., 2015, 261, 73–82 CrossRef CAS PubMed.
- A. N. Pravdivtsev, A. V. Yurkovskaya, H. Vieth, K. L. Ivanov and R. Kaptein, ChemPhysChem, 2013, 14, 3327–3331 CrossRef CAS PubMed.
- N. Eshuis, R. L. E. G. Aspers, B. J. A. van Weerdenburg, M. C. Feiters, F. P. J. T. Rutjes, S. S. Wijmenga and M. Tessari, J. Magn. Reson., 2016, 265, 59–66 CrossRef CAS PubMed.
- E. B. Dücker, L. T. Kuhn, K. Münnemann and C. Griesinger, J. Magn. Reson., 2012, 214, 159–165 CrossRef PubMed.
- A. S. Kiryutin, A. V. Yurkovskaya, H. Zimmermann, H. Vieth and K. L. Ivanov, Magn. Reson. Chem., 2018, 56, 651–662 CrossRef CAS PubMed.
- R. V. Shchepin, D. A. Barskiy, A. M. Coffey, T. Theis, F. Shi, W. S. Warren, B. M. Goodson and E. Y. Chekmenev, ACS Sens., 2016, 1, 640–644 CrossRef CAS PubMed.
- T. Theis, M. L. Truong, A. M. Coffey, R. V. Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Am. Chem. Soc., 2015, 137, 1404–1407 CrossRef CAS PubMed.
- M. L. Truong, T. Theis, A. M. Coffey, R. V. Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Phys. Chem. C, 2015, 119, 8786–8797 CrossRef CAS PubMed.
- M. Fekete, F. Ahwal and S. B. Duckett, J. Phys. Chem. B, 2020, 124, 4573–4580 CrossRef CAS PubMed.
- K. Shen, A. W. J. Logan, J. F. P. Colell, J. Bae, G. X. Ortiz Jr, T. Theis, W. S. Warren, S. J. Malcolmson and Q. Wang, Angew. Chem., 2017, 129, 12280–12284 CrossRef.
- W. Iali, S. S. Roy, B. J. Tickner, F. Ahwal, A. J. Kennerley and S. B. Duckett, Angew. Chem., 2019, 131, 10377–10381 CrossRef.
- M. E. Gemeinhardt, M. N. Limbach, T. R. Gebhardt, C. W. Eriksson, S. L. Eriksson, J. R. Lindale, E. A. Goodson, W. S. Warren, E. Y. Chekmenev and B. M. Goodson, Angew. Chem., 2020, 132, 426–431 CrossRef.
- Z. Zhou, J. Yu, J. F. P. Colell, R. Laasner, A. Logan, D. A. Barskiy, R. V. Shchepin, E. Y. Chekmenev, V. Blum and W. S. Warren, J. Phys. Chem. Lett., 2017, 8, 3008–3014 CrossRef CAS PubMed.
- R. V. Shchepin, B. M. Goodson, T. Theis, W. S. Warren and E. Y. Chekmenev, ChemPhysChem, 2017, 18, 1961–1965 CrossRef CAS PubMed.
- A. M. Olaru, T. B. R. Robertson, J. S. Lewis, A. Antony, W. Iali, R. E. Mewis and S. B. Duckett, ChemistryOpen, 2018, 7, 97–105 CrossRef CAS PubMed.
- M. J. Burns, P. J. Rayner, G. G. R. Green, L. A. R. Highton, R. E. Mewis and S. B. Duckett, J. Phys. Chem. B, 2015, 119, 5020–5027 CrossRef CAS PubMed.
- A. M. Olaru, A. Burt, P. J. Rayner, S. J. Hart, A. C. Whitwood, G. G. R. Green and S. B. Duckett, Chem. Commun., 2016, 52, 14482–14485 RSC.
- P. J. Rayner, P. M. Richardson and S. B. Duckett, Angew. Chem., 2020, 132, 2732–2736 CrossRef.
- D. A. Barskiy, K. V. Kovtunov, I. V. Koptyug, P. He, K. A. Groome, Q. A. Best, F. Shi, B. M. Goodson, R. V Shchepin and A. M. Coffey, J. Am. Chem. Soc., 2014, 136, 3322–3325 CrossRef CAS PubMed.
- T. Theis, M. Truong, A. M. Coffey, E. Y. Chekmenev and W. S. Warren, J. Magn. Reson., 2014, 248, 23–26 CrossRef CAS PubMed.
- J. R. Lindale, C. P. N. Tanner, S. L. Eriksson and W. S. Warren, J. Magn. Reson., 2019, 307, 106577 CrossRef CAS PubMed.
- A. N. Pravdivtsev, A. V. Yurkovskaya, H.-M. Vieth and K. L. Ivanov, J. Phys. Chem. B, 2015, 119, 13619–13629 CrossRef CAS PubMed.
- S. Knecht, A. S. Kiryutin, A. V. Yurkovskaya and K. L. Ivanov, J. Magn. Reson., 2018, 287, 10–14 CrossRef CAS PubMed.
- S. Knecht, A. S. Kiryutin, A. V. Yurkovskaya and K. L. Ivanov, Mol. Phys., 2019, 117, 2762–2771 CrossRef CAS.
- O. Semenova, P. M. Richardson, A. J. Parrott, A. Nordon, M. E. Halse and S. B. Duckett, Anal. Chem., 2019, 91, 6695–6701 CrossRef CAS PubMed.
- K. D. Atkinson, M. J. Cowley, S. B. Duckett, P. I. P. Elliott, G. G. R. Green, J. López-Serrano, I. G. Khazal and A. C. Whitwood, Inorg. Chem., 2009, 48, 663–670 CrossRef CAS PubMed.
- M. J. Cowley, R. W. Adams, K. D. Atkinson, M. C. R. Cockett, S. B. Duckett, G. G. R. Green, J. A. B. Lohman, R. Kerssebaum, D. Kilgour and R. E. Mewis, J. Am. Chem. Soc., 2011, 133, 6134–6137 CrossRef CAS PubMed.
- A. J. Holmes, P. J. Rayner, M. J. Cowley, G. G. R. Green, A. C. Whitwood and S. B. Duckett, Dalton Trans., 2015, 44, 1077–1083 RSC.
- A. Thomas, M. Haake, F. Grevels and J. Bargon, Angew. Chem., Int. Ed., 1994, 33, 755–757 CrossRef.
- K. Lin, P. TomHon, S. Lehmkuhl, R. Laasner, T. Theis and V. Blum, ChemPhysChem, 2021, 22, 1937–1938 CrossRef CAS PubMed.
- H. Zeng, J. Xu, J. Gillen, M. T. McMahon, D. Artemov, J. Tyburn, J. A. B. Lohman, R. E. Mewis, K. D. Atkinson, G. G. R. Green, S. B. Duckett and P. C. M. van Zijl, J. Magn. Reson. Imag., 2013, 237, 73–78 CrossRef CAS PubMed.
- B. J. Tickner, O. Semenova, W. Iali, P. J. Rayner, A. C. Whitwood and S. B. Duckett, Catal. Sci. Technol., 2020, 10, 1343–1355 RSC.
- B. J. Tickner, J. S. Lewis, R. O. John, A. C. Whitwood and S. B. Duckett, Dalton Trans., 2019, 48, 15198–15206 RSC.
- S. L. Eriksson, J. R. Lindale, X. Li and W. S. Warren, Sci. Adv., 2022, 8, eabl3708 CrossRef PubMed.
- K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, J. Lopez-Serrano and A. C. Whitwood, J. Am. Chem. Soc., 2009, 131, 13362–13368 CrossRef CAS PubMed.
- J. Colell, A. W. J. Logan, Z. Zhou, J. R. Lindale, R. Laasner, R. Shchepin, E. Chekmenev, V. Blum, W. S. Warren and S. J. Malcolmson, Chem. Commun., 2020, 56, 9336–9339 RSC.
- D. G. Gusev, Organometallics, 2009, 28, 6458–6461 CrossRef CAS.
- R. A. Kelly III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef.
- P. J. Rayner, P. Norcott, K. M. Appleby, W. Iali, R. O. John, S. J. Hart, A. C. Whitwood and S. B. Duckett, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed.
- B. J. A. van Weerdenburg, N. Eshuis, M. Tessari, F. P. J. T. Rutjes and M. C. Feiters, Dalton Trans., 2015, 44, 15387–15390 RSC.
- B. J. A. van Weerdenburg, S. Glöggler, N. Eshuis, A. H. J. T. Engwerda, J. M. M. Smits, R. de Gelder, S. Appelt, S. S. Wymenga, M. Tessari, M. C. Feiters, B. Blümich and F. P. J. T. Rutjes, Chem. Commun., 2013, 49, 7388–7390 RSC.
- S. Hadjiali, R. Savka, M. Plaumann, U. Bommerich, S. Bothe, T. Gutmann, T. Ratajczyk, J. Bernarding, H.-H. Limbach and H. Plenio, Appl. Magn. Reson., 2019, 50, 895–902 CrossRef CAS.
- B. J. Tickner, F. Ahwal, A. C. Whitwood and S. B. Duckett, ChemPhysChem, 2021, 22, 13–17 CrossRef CAS PubMed.
- B. J. Tickner, Y. Borozdina, S. B. Duckett and G. Angelovski, Dalton Trans., 2021, 50, 2448–2461 RSC.
- W. Iali, G. G. R. Green, S. J. Hart, A. C. Whitwood and S. B. Duckett, Inorg. Chem., 2016, 55, 11639–11643 CrossRef CAS PubMed.
- A. J. Ruddlesden, R. E. Mewis, G. G. R. Green, A. C. Whitwood and S. B. Duckett, Organometallics, 2015, 34, 2997–3006 CrossRef CAS PubMed.
- M. Fekete, C. Gibard, G. J. Dear, G. G. R. Green, A. J. J. Hooper, D. Roberts, F. Cisnetti and S. B. Duckett, Dalton Trans., 2015, 44, 7870–7880 RSC.
- F. Shi, P. He, Q. A. Best, K. Groome, M. L. Truong, A. M. Coffey, G. Zimay, R. V. Shchepin, K. W. Waddell and E. Y. Chekmenev, J. Phys. Chem. C, 2016, 120, 12149–12156 CrossRef CAS PubMed.
- P. Spannring, I. Reile, M. Emondts, P. P. M. Schleker, N. K. J. Hermkens, N. G. J. van der Zwaluw, B. J. A. van Weerdenburg, P. Tinnemans, M. Tessari and B. Blümich, Chem.–Eur. J., 2016, 22, 9277–9282 CrossRef CAS PubMed.
- C. M. Wong, M. Fekete, R. Nelson-Forde, M. R. D. Gatus, P. J. Rayner, A. C. Whitwood, S. B. Duckett and B. A. Messerle, Catal. Sci. Technol., 2018, 8, 4925–4933 RSC.
- P. Pham and C. Hilty, Chem. Commun., 2020, 56, 15466–15469 RSC.
- P. J. Rayner, J. P. Gillions, V. D. Hannibal, R. O. John and S. B. Duckett, Chem. Sci., 2021, 12, 5910–5917 RSC.
- R. E. Mewis, R. A. Green, M. C. R. Cockett, M. J. Cowley, S. B. Duckett, G. G. R. Green, R. O. John, P. J. Rayner and D. C. Williamson, J. Phys. Chem. B, 2015, 119, 1416–1424 CrossRef CAS PubMed.
- R. Mandal, P. Pham and C. Hilty, ChemPhysChem, 2020, 21, 2166–2172 CrossRef CAS PubMed.
- F. F. Diaz-Rullo, F. Zamberlan, R. E. Mewis, M. Fekete, L. Broche, L. A. Cheyne, S. DallAngelo, S. B. Duckett, D. Dawson and M. Zanda, Bioorg. Med. Chem., 2017, 25, 2730–2742 CrossRef PubMed.
- J. F. P. Colell, A. W. J. Logan, Z. Zhou, R. V. Shchepin, D. A. Barskiy, G. X. Ortiz Jr, Q. Wang, S. J. Malcolmson, E. Y. Chekmenev and W. S. Warren, J. Phys. Chem. C, 2017, 121, 6626–6634 CrossRef CAS PubMed.
- P. TomHon, M. Abdulmojeed, I. Adelabu, S. Nantogma, M. S. H. Kabir, S. Lehmkuhl, E. Y. Chekmenev and T. Theis, J. Am. Chem. Soc., 2022, 144, 282–287 CrossRef CAS PubMed.
- B. J. Tickner, R. R. Parker, A. C. Whitwood and S. B. Duckett, Organometallics, 2019, 38, 4377–4382 CrossRef CAS PubMed.
- M. Fekete, S. S. Roy and S. B. Duckett, Phys. Chem. Chem. Phys., 2020, 22, 5033–5037 RSC.
- D. A. Barskiy, A. N. Pravdivtsev, K. L. Ivanov, K. V. Kovtunov and I. V. Koptyug, Phys. Chem. Chem. Phys., 2016, 18, 89–93 RSC.
- S. Knecht, D. A. Barskiy, G. Buntkowsky and K. L. Ivanov, J. Chem. Phys., 2020, 153, 164106 CrossRef CAS PubMed.
- T. Theis, N. M. Ariyasingha, R. V. Shchepin, J. R. Lindale, W. S. Warren and E. Y. Chekmenev, J. Phys. Chem. Lett., 2018, 9, 6136–6142 CrossRef CAS PubMed.
- M. L. Truong, F. Shi, P. He, B. Yuan, K. N. Plunkett, A. N. Coffey, R. V. Shchepin, D. A. Barskiy, K. V. Kovtunov, I. V. Koptyug, K. W. Waddell, B. M. Goodson and E. Y. Chekmenev, J. Phys. Chem. B, 2014, 118, 13882–13889 CrossRef CAS PubMed.
- T. B. Robertson, PhD thesis, Manchester Metropolitan University, U.K., 2019.
- K. Tokmic, C. R. Markus, L. Zhu and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 11907–11913 CrossRef CAS PubMed.
- F. Reineri, E. Cavallari, C. Carrera and S. Aime, Magn. Reson. Mater. Phys., Biol. Med., 2021, 34, 25–47 CrossRef CAS PubMed.
- B. Ripka, J. Eills, H. Kouřilová, M. Leutzsch, M. H. Levitt and K. Münnemann, Chem. Commun., 2018, 54, 12246–12249 RSC.
- J. Eills, E. Cavallari, C. Carrera, D. Budker, S. Aime and F. Reineri, J. Am. Chem. Soc., 2019, 141, 20209–20214 CrossRef CAS PubMed.
- S. Knecht, J. W. Blanchard, D. Barskiy, E. Cavallari, L. Dagys, E. Van Dyke, M. Tsukanov, B. Bliemel, K. Münnemann, S. Aime, F. Reineri, M. H. Levitt, G. Buntkowsky, A. Pines, P. Blumler, D. Budker and J. Eills, Proc. Natl. Acad. Sci., 2021, 118, e2025383118 CrossRef CAS PubMed.
- L. Wienands, F. Theiß, J. Eills, L. Rösler, S. Knecht and G. Buntkowsky, Appl. Magn. Reson., 2021, 1–20 Search PubMed.
- T. Witney, M. Kettunen, D. Hu, F. Gallagher, S. Bohndiek, R. Napolitano and K. Brindle, Br. J. Cancer, 2010, 103, 1400–1406 CrossRef CAS PubMed.
- F. A. Gallagher, M. I. Kettunen, D.-E. Hu, P. R. Jensen, M. Karlsson, A. Gisselsson, S. K. Nelson, T. H. Witney, S. E. Bohndiek and G. Hansson, Proc. Natl. Acad. Sci., 2009, 106, 19801–19806 CrossRef CAS PubMed.
- J. H. Ardenkjær-Larsen, S. Bowen, J. R. Petersen, O. Rybalko, M. S. Vinding, M. Ullisch and N. C. Nielsen, Magn. Reson. Med., 2019, 81, 2184–2194 CrossRef PubMed.
- L. S. Lloyd, R. W. Adams, M. Bernstein, S. Coombes, S. B. Duckett, G. G. R. Green, R. J. Lewis, R. E. Mewis and C. J. Sleigh, J. Am. Chem. Soc., 2012, 134, 12904–12907 CrossRef CAS PubMed.
- V. Daniele, F. Legrand, P. Berthault, J. Dumez and G. Huber, ChemPhysChem, 2015, 16, 3413–3417 CrossRef CAS PubMed.
- I. Reile, R. L. E. G. Aspers, J. Tyburn, J. G. Kempf, M. C. Feiters, F. P. J. T. Rutjes and M. Tessari, Angew. Chem., 2017, 129, 9302–9305 CrossRef.
- L. Guduff, P. Berthault, C. Van Heijenoort, J. Dumez and G. Huber, ChemPhysChem, 2019, 20, 392–398 CrossRef CAS PubMed.
- E. Cavallari, C. Carrera, S. Aime and F. Reineri, Chem.–Eur. J., 2017, 23, 1200–1204 CrossRef CAS PubMed.
- W. Iali, P. J. Rayner, A. Alshehri, A. J. Holmes, A. J. Ruddlesden and S. B. Duckett, Chem. Sci., 2018, 9, 3677–3684 RSC.
- B. Procacci, S. S. Roy, P. Norcott, N. Turner and S. B. Duckett, J. Am. Chem. Soc., 2018, 140, 16855–16864 CrossRef CAS PubMed.
- T. Theis, G. X. Ortiz, A. W. J. Logan, K. E. Claytor, Y. Feng, W. P. Huhn, V. Blum, S. J. Malcolmson, E. Y. Chekmenev and Q. Wang, Sci. Adv., 2016, 2, e1501438 CrossRef PubMed.
- B. J. Tickner, P. J. Rayner and S. B. Duckett, Anal. Chem., 2020, 92, 9095–9103 CrossRef CAS PubMed.
- A. N. Pravdivtsev, G. Buntkowsky, S. B. Duckett, I. V. Koptyug and J.-B. Hövener, Angew. Chem., Int. Ed., 2021, 60, 23496–23507 CrossRef CAS PubMed.
- S. Glöggler, R. Müller, J. Colell, M. Emondts, M. Dabrowski, B. Blümich and S. Appelt, Phys. Chem. Chem. Phys., 2011, 13, 13759–13764 RSC.
- S. Aime, R. Gobetto, F. Reineri and D. Canet, J. Chem. Phys., 2003, 119, 8890–8896 CrossRef CAS.
- V. P. Kozinenko, A. S. Kiryutin, S. Knecht, G. Buntkowsky, H.-M. Vieth, A. V. Yurkovskaya and K. L. Ivanov, J. Chem. Phys., 2020, 153, 114202 CrossRef CAS PubMed.
- N. V. Chukanov, O. G. Salnikov, R. V. Shchepin, A. Svyatova, K. V. Kovtunov, I. V. Koptyug and E. Y. Chekmenev, J. Phys. Chem. C, 2018, 122, 23002–23010 CrossRef CAS PubMed.
- H. Chae, S. Min, H. J. Jeong, S. K. Namgoong, S. Oh, K. Kim and K. Jeong, Anal. Chem., 2020, 92, 10902–10907 CrossRef CAS PubMed.
- N. Eshuis, R. L. E. G. Aspers, B. J. A. van Weerdenburg, M. C. Feiters, F. P. J. T. Rutjes, S. S. Wijmenga and M. Tessari, Angew. Chem., Int. Ed., 2015, 54, 14527–14530 CrossRef CAS PubMed.
- N. Eshuis, N. Hermkens, B. J. A. van Weerdenburg, M. C. Feiters, F. P. J. T. Rutjes, S. S. Wijmenga and M. Tessari, J. Am. Chem. Soc., 2014, 136, 2695–2698 CrossRef CAS PubMed.
- E. Cavallari, C. Carrera, S. Aime and F. Reineri, ChemPhysChem, 2019, 20, 318–325 CrossRef CAS PubMed.
- P. Bhattacharya, E. Y. Chekmenev, W. H. Perman, K. C. Harris, A. P. Lin, V. A. Norton, C. T. Tan, B. D. Ross and D. P. Weitekamp, J. Magn. Reson., 2007, 186, 150–155 CrossRef CAS PubMed.
- P. Štěpánek and A. M. Kantola, J. Phys. Chem. Lett., 2019, 10, 5458–5462 CrossRef PubMed.
- S. Appelt, S. Lehmkuhl, S. Fleischer, B. Joalland, N. M. Ariyasingha, E. Y. Chekmenev and T. Theis, J. Magn. Reson., 2021, 322, 106815 CrossRef CAS PubMed.
- N. Arunkumar, D. B. Bucher, M. J. Turner, P. TomHon, D. Glenn, S. Lehmkuhl, M. D. Lukin, H. Park, M. S. Rosen and T. Theis, PRX Quantum, 2021, 2, 010305 CrossRef.
- K. Buckenmaier, M. Rudolph, C. Back, T. Misztal, U. Bommerich, P. Fehling, D. Koelle, R. Kleiner, H. A. Mayer and K. Scheffler, Sci. Rep., 2017, 7, 1–9 CrossRef CAS PubMed.
- T. Theis, P. Ganssle, G. Kervern, S. Knappe, J. Kitching, M. P. Ledbetter, D. Budker and A. Pines, Nat. Phys., 2011, 7, 571–575 Search PubMed.
- T. Theis, M. P. Ledbetter, G. Kervern, J. W. Blanchard, P. J. Ganssle, M. C. Butler, H. D. Shin, D. Budker and A. Pines, J. Am. Chem. Soc., 2012, 134, 3987–3990 CrossRef CAS PubMed.
- K. Them, F. Ellermann, A. N. Pravdivtsev, O. G. Salnikov, I. V. Skovpin, I. V. Koptyug, R. Herges and J.-B. Hövener, J. Am. Chem. Soc., 2021, 143, 13694–13700 CrossRef CAS PubMed.
- W. Iali, P. J. Rayner and S. B. Duckett, Sci. Adv., 2018, 4, eaao6250 CrossRef PubMed.
- P. J. Rayner, B. J. Tickner, W. Iali, M. Fekete, A. D. Robinson and S. B. Duckett, Chem. Sci., 2019, 10, 7709–7717 RSC.
- P. M. Richardson, W. Iali, S. S. Roy, P. J. Rayner, M. E. Halse and S. B. Duckett, Chem. Sci., 2019, 10, 10607–10619 RSC.
- E. Vaneeckhaute, J.-M. Tyburn, D. Kilgour, J. G. Kempf, F. Taulelle, J. A. Martens and E. Breynaert, J. Phys. Chem. C, 2020, 124, 14541–14549 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00737a |
| This journal is © The Royal Society of Chemistry 2022 |