
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric synthesis of enantioenriched α-deuterated pyrrolidine derivatives†
Xin
Chang‡
ab,
Xiang
Cheng‡
a and
Chun-Jiang
Wang
 *ab
*ab
aEngineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, China. E-mail: cjwang@whu.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin, 300071, China
First published on 17th March 2022
Abstract
The recent promising applications of deuterium-labeled pharmaceutical compounds have led to an urgent need for the efficient synthetic methodologies that site-specifically incorporate a deuterium atom into bioactive molecules. Nevertheless, precisely building a deuterium-containing stereogenic center, which meets the requirement for optimizing the absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of chiral drug candidates, remains a significant challenge in organic synthesis. Herein, a catalytic asymmetric strategy combining H/D exchange (H/D-Ex) and azomethine ylide-involved 1,3-dipolar cycloaddition (1,3-DC) was developed for the construction of biologically important enantioenriched α-deuterated pyrrolidine derivatives in good yields with excellent stereoselectivities and uniformly high levels of deuterium incorporation. Directly converting glycine-derived aldimine esters into the deuterated counterparts with D2O via Cu(I)-catalyzed H/D-Ex, and the subsequent thermodynamically/kinetically favored cleavage of the α-C–H bond rather than the α-C–D bond to generate the key N-metallated α-deuterated azomethine ylide species for the ensuing 1,3-DC are crucial to the success of α-deuterated chiral pyrrolidine synthesis. The current protocol exhibits remarkable features, such as readily available substrates, inexpensive and safe deuterium source, mild reaction conditions, and easy manipulation. Notably, the synthetic utility of a reversed 1,3-DC/[H/D-Ex] protocol has been demonstrated by catalytic asymmetric synthesis of deuterium-labelled MDM2 antagonist idasanutlin (RG7388) with high deuterium incorporation.
Introduction
Deuterium as one of the readily accessible and nonradioactive atoms plays crucial roles in life science because of its ability to promote the discovery and development of drugs,1–4 to study the reaction mechanisms,5,6 and to be as an ideal internal standard for mass spectrometry.7 Particularly, incorporating a deuterium atom into the stereogenic center of a chiral drug candidate can not only stabilize its structure8–12 but also improve the metabolic profile.13,14 For example, the deuterated analogue of (−)-CC-122, a compound currently in human clinical trials for hematological cancers and solid tumors, exhibits higher in vitro anti-inflammatory and in vivo antitumorigenic effects with the enhanced configuration stability.8,9 To suppress the disfavored epimerization of telaprevir without losing activity against the hepatitis C virus NS3-4A protease, the proton at the easily-epimerized stereogenic center was replaced with deuterium.10 The incorporation of a deuterium-containing stereogenic center into erythromycin B led to a remarkable reduction of acid-promoted enol ether formation without compromising the antibacterial effect.11 The enantioenriched 5-deuterated (R)-pioglitazone proves to be a better therapeutic agent than non-deuterated pioglitazone and racemic mixtures of deuterated counterparts (Fig. 1).12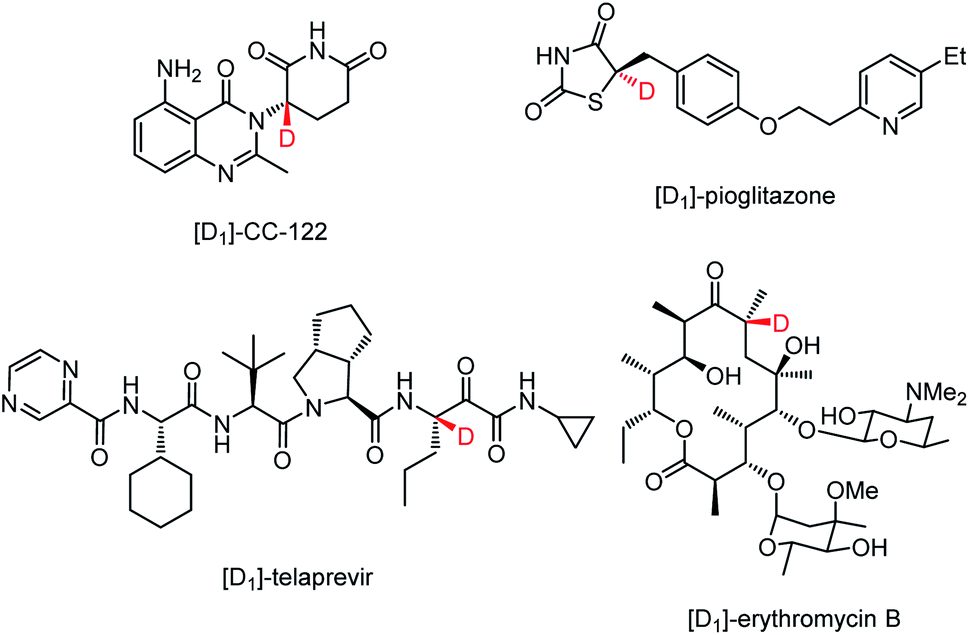 | ||
| Fig. 1 Representative drug molecules bearing a deuterium-containing stereogenic center. | ||
Accordingly, much attention has been paid to exploit various methods for the incorporation of a deuterium atom into the stereogenic center of chiral pharmaceutical molecules or their key building blocks.15–21 Catalytic asymmetric synthesis, as one of the most straightforward approaches towards this goal, mainly focuses on asymmetric reduction and asymmetric deuteration of prochiral compounds22–30 (Scheme 1a). In contrast, catalytic asymmetric 1,3-dipolar cycloaddition reactions,31–35 occupying an important position in the field of organic synthesis and medicinal chemistry, have never been documented to install a deuterium-containing stereogenic center in heterocyclic adducts presumably due to the difficulty and unavailability of generating the corresponding deuterium-containing starting materials or key intermediates. Therefore, developing a novel synthetic strategy capable of precisely incorporating a deuterium-containing stereogenic center into biologically active and therapeutically-relevant heterocyclic scaffolds via a catalytic asymmetric cycloaddition reaction is of great significance and urgent demand.
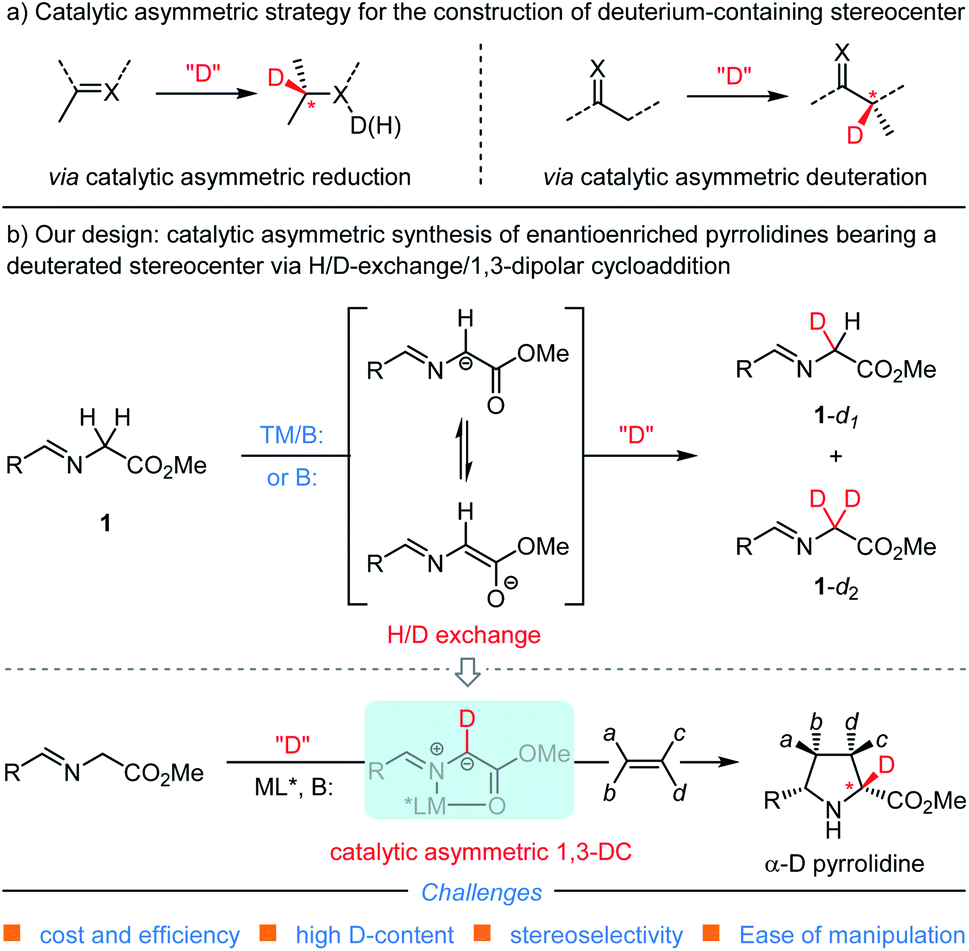 | ||
| Scheme 1 Strategies for catalytic asymmetric construction of a deuterium-containing stereocenter. | ||
On the other hand, optically active pyrrolidines are prevalent core structures found in many biologically active natural products and pharmaceutical molecules.36–38 However, only sporadic studies on the preparation of deuterated counterparts have been reported so far.39,40 In 1994, Beak and co-workers disclosed a sequential asymmetric deprotonation–deuteration of N-Boc-pyrrolidine in the presence of excess amount of (−)-sparteine, affording enantioenriched α-deuterated N-Boc-pyrrolidine using MeOD as the electrophile and deuterium source.39 In another example, based on the modified Pieters's protocol,21 the team of Roche documented that stereoretentive α-deuteration of L-proline could be achieved using ruthenium on a carbon catalyst with deuterium oxide as the deuterium reagent.40 Despite these advances, such methods suffered from harsh reaction conditions, non-catalytic asymmetric process or extremely narrow substrate scope, which would limit their practical application.
In view of the importance of deuterium-labelled chiral N-heterocycles and our continuing interest in the catalytic asymmetric construction of enantioenriched pyrrolidine derivatives with azomethine ylide,31,41,42 we envisioned that merging the H/D-exchange (H/D-Ex) protocol15 with the 1,3-dipolar cycloaddition reaction would meet this great challenge in a catalytic asymmetric manner and exactly cater for the ever-growing demand of deuterated bioactive heterocyclic molecules in the pharmaceutical industry.43 As shown in Scheme 1b, we proposed that base-promoted and/or transition metal-catalyzed H/D-exchange of readily available glycine-derived aldimine ester with certain deuterium reagent would deliver the corresponding deuterated intermediate 1-dn (n = 1 or 2);44,45 the deuterated aldimine ester could coordinate with the transition-metal cation to form the key N-metallated azomethine ylide bearing a deuterium atom at the α-position. Subsequently, catalyst-controlled asymmetric cycloaddition of the generated D-azomethine ylide with dipolarophiles provides α-deuterated enantioenriched pyrrolidine derivatives. While this reaction design is conceptually straightforward, there are still some significant challenges associated with this proposal: (1) driving keto–enol tautomerization towards the deuterated intermediate 1-dn could be theoretically achieved by the formation of a slightly more stable C–D bond versus the C–H bond and by adding a large excess of certain polar deuterium reagent,46 nonetheless, we cognized that such a thermodynamic process could be complicated by the level of deuterium-enrichment of aldimine esters and the ensuing de-deuteration or de-protonation to form the key N-metallated D-azomethine ylides with metal cations; (2) whether or not the employed polar deuterium reagent deteriorates the efficiency and diastereo-/enantioselectivity control of transition metal-catalyzed asymmetric 1,3-dipolar cycloaddition, since less polar and anhydrous solvents, such as dichloromethane, toluene or tetrahydrofuran, were commonly used in the established catalytic systems.
Herein, we report the development of an efficient synthesis of α-deuterated enantioenriched pyrrolidine derivatives via the strategy of combining H/D-exchange with catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides. This protocol enables us to directly convert glycine derived aldimine esters to the deuterated counterparts employing relatively safe, low-cost and operationally convenient D2O as the deuterium source under mild reaction conditions, and therefore provides a facile access to α-deuterated chiral pyrrolidines with high functionality, multiple stereocenters and uniformly high levels of deuterium incorporation for the first time.
Results and discussion
Reaction development and optimization
We initiated our investigations into the H/D-exchange performance of glycine-derived aldimine ester. In order to get insight into the H/D exchange process of aldimine ester 1a, a series of 1H NMR experiments were carried out with inexpensive and easy to handle D2O as the deuterium reagent in the presence of a catalytic amount of chiral metal complex Cu(I)/(S)-TF-BiphamPhos as the catalyst and Et3N as the base.42 As shown in Fig. 2A, along with the addition of the increased amount of D2O (monitored with 1H NMR at the indicated time intervals with 3, 5, 7, 14, 28 and 56 equiv. of D2O, respectively, see the ESI† for details), the starting material aldimine ester 1a was fast consumed and converted into two deuterated intermediates 1a-d1 and 1a-d2, and the content of intermediate 1a-d1 showed a fast increase and then a slow decrease in this H/D exchange process. The whole deuterium enrichment of aldimine ester 1a improved gradually with the increased excess of D2O. When the amount of D2O was increased up to 28 equivalents, the ratio of the two species 1a-d1 and 1a-d2 tends to be stable. However, the complete α-deuteration of 1a cannot be reached even with 56 equivalents of D2O. The ratio of 1a and two deuterated species 1a-d1 and 1a-d2 did not show any remarkable changes when the amount of D2O was increased from 28 equiv. to 56 equiv., and the resulting mixture still contained a considerable amount of 1a-d1 and a small amount of 1a.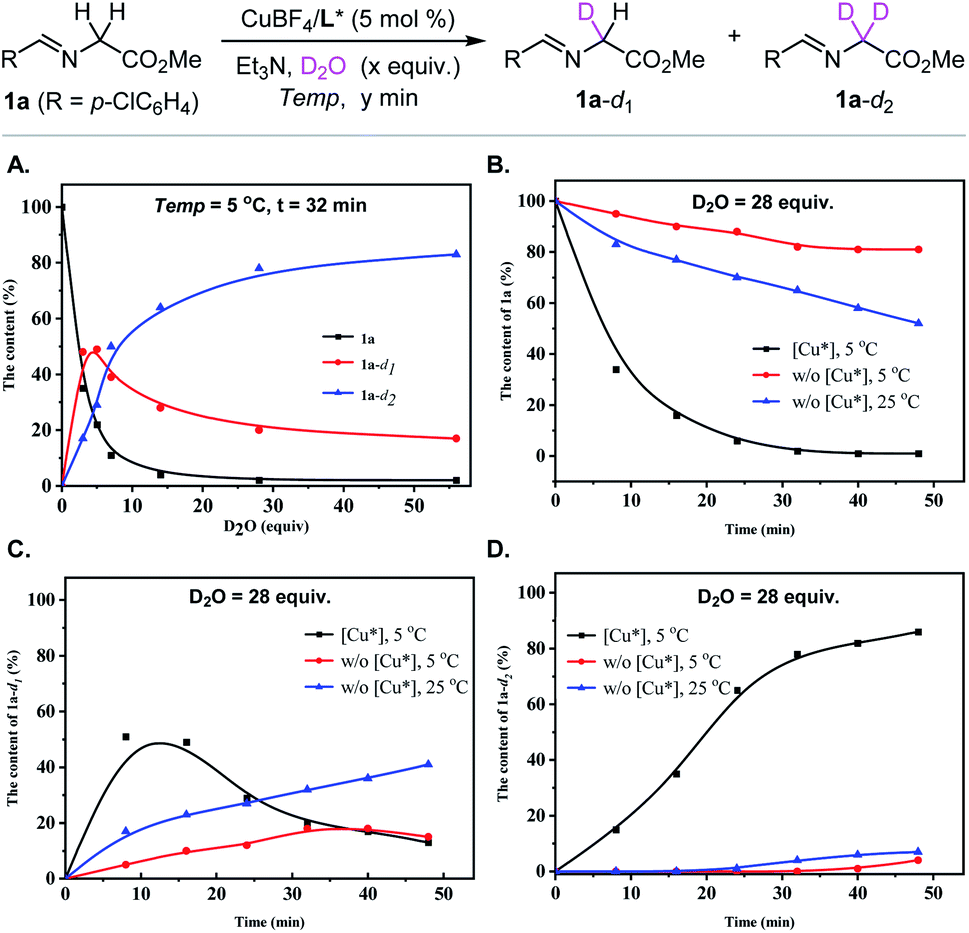 | ||
| Fig. 2 1H NMR studies on the level of deuterium incorporation of aldimine ester 1a with D2O. (A) Monitoring the content of aldimine ester 1a, 1a-d1 and 1a-d2 when the different amount of D2O was used. (B) Studying the effect of reaction parameters such as chiral copper complex, reaction time and temperature on the content of aldimine ester 1a. (C) Studying the effect of reaction parameters such as chiral copper complex, reaction time and temperature on the content of intermediate 1a-d1. (D) Studying the effect of reaction parameters such as chiral copper complex, reaction time and temperature on the content of intermediate 1a-d2. | ||
Furthermore, the effect of other reaction parameters, such as chiral copper complex, temperature and time, on the H/D exchange performance was also investigated with 28 equiv. of D2O being added. It is worth noting that metal complex Cu(I)/(S)-TF-BiphamPhos significantly promotes the H/D exchange of aldimine ester 1a. As shown in Fig. 2B–D, removing the chiral copper complex from the above condition seriously decayed the rate of H/D-exchange. A comparative H/D-exchange level could not be reached even with a prolonged reaction time or at an elevated reaction temperature in the absence of the copper complex. These control experimental results revealed that the chiral copper complex served as a crucial factor to promote the H/D-exchange of aldimine ester 1a. When the process of H/D-exchange was monitored over time (8 min, 16 min, 24 min, 32 min, 40 min, and 48 min), the kinetic curve of 1a, 1a-d1, and 1a-d2 gradually became steady at 32 min, which suggested that the mixed system reached an equilibrium and thus introducing dipolarophiles at this time point would be suitable for the ensuing construction of enantioenriched α-deuterated pyrrolidines via 1,3-dipolar cycloaddition. Accordingly, the amount of 28 equiv. of D2O might be the compromise choice for H/D-exchange of aldimine ester, although leaving an uncertainty on whether a high level of deuterium incorporation could be achieved or not in the final pyrrolidine products, which to a great extent depends on the in situ formation of the deuterated azomethine ylide species via the selectivity of de-deuteration or de-protonation of aldimine ester 1a-d1.
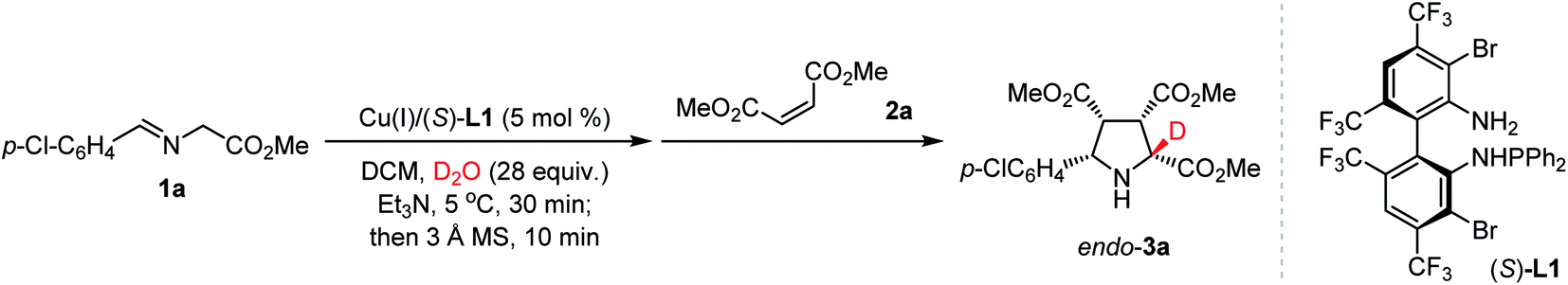
Since the complete α-deuteration of aldimine ester 1a into 1a-d2 could not be reached with 28 equiv. of D2O, the subsequent investigation commenced with the hope that a slightly more stable C–D bond would thermodynamically and kinetically favor the cleavage of the α-C–H bond rather than the α-C–D bond to form the key N-metallated α-deuterated azomethine ylide with a synthetically useful level of deuterium-enrichment. To explore the selectivity issue of de-deuteration or de-protonation, we started to investigate the model reaction of aldimine ester 1a and dimethyl maleate 2a for optimization of reaction conditions.
As shown in Table 1, aldimine ester 1a was pre-stirred in a mixture of dichloromethane and D2O (28 equiv.) for 30 min in the presence of Cu(I)/(S)-TF-BiphamPhos (5 mol%) and Et3N (1 equiv.) at 5 °C. To eliminate the disfavored interference of polar deuterium solvent in the stereoselectivity of cycloaddition, 3 Å MS (200 mg) was added to remove the remaining D2O/DHO from the system, and then dimethyl maleate 2a was introduced at room temperature. The reaction finished smoothly to give cycloadduct endo-3a in 92% yield with an exclusive diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr), excellent enantioselectivity (99% ee) and 96% deuterium incorporation (Table 1, entry 1). This excellent deuterium enrichment confirmed that the cleavage of α-C–H in 1-d1 is kinetically more favored than that of the C–D bond even in the presence of a chiral copper catalyst. Further the control experiment revealed that removing the remaining D2O/DHO is crucial to enhancing the reactivity and diastereoselectivity (entry 2). Using a lower amount of D2O (7 or 14 equiv.) led to a significantly reduced deuterium-incorporation level (entries 3 and 4). Increasing the amount of D2O to 56 equiv. could not improve the deuterium-incorporation level further (entry 5), which is fully consistent with the observation in 1H NMR experiments (vide supra). When D2O was replaced with another commonly-used deuterium reagent MeOD, a high level of deuterium-incorporation could still be achieved albeit with unsatisfactory yield and diastereoselectivity, which is probably caused by the incompatibility of the polar solvent in the catalytic asymmetric cycloaddition step (entries 6 and 7). Therefore, D2O was chosen as the optimal deuterium-donor due to the inexpensiveness, safety and convenient operation of this material.
:1 dr), excellent enantioselectivity (99% ee) and 96% deuterium incorporation (Table 1, entry 1). This excellent deuterium enrichment confirmed that the cleavage of α-C–H in 1-d1 is kinetically more favored than that of the C–D bond even in the presence of a chiral copper catalyst. Further the control experiment revealed that removing the remaining D2O/DHO is crucial to enhancing the reactivity and diastereoselectivity (entry 2). Using a lower amount of D2O (7 or 14 equiv.) led to a significantly reduced deuterium-incorporation level (entries 3 and 4). Increasing the amount of D2O to 56 equiv. could not improve the deuterium-incorporation level further (entry 5), which is fully consistent with the observation in 1H NMR experiments (vide supra). When D2O was replaced with another commonly-used deuterium reagent MeOD, a high level of deuterium-incorporation could still be achieved albeit with unsatisfactory yield and diastereoselectivity, which is probably caused by the incompatibility of the polar solvent in the catalytic asymmetric cycloaddition step (entries 6 and 7). Therefore, D2O was chosen as the optimal deuterium-donor due to the inexpensiveness, safety and convenient operation of this material.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Derivation from standard conditions | Yieldb (%) | drc (endo:exo) |
Dc (%) | eec (%) |
| a All reactions were carried out with 0.2 mmol of 1a, 0.3 mmol of 2a, and 0.2 mmol of Et3N in 2 mL of DCM and 0.1 mL of D2O for 3–6 h, see the ESI for details. b Yields refer to isolated yields of deuterated products. c The dr value was determined by crude 1H NMR. D refers to D-incorporation percentages based on the calculations described in the ESI. The ee value was determined by HPLC analysis. d Without 3 Å MS. | |||||
| 1 | None | 92 | >20:1 |
96 | 99 |
| 2 | Without 3 Å MS | 82 | 15:1 |
96 | 99 |
| 3 | D2O (7 equiv.) instead of D2O (28 equiv.) | 94 | >20:1 |
78 | 99 |
| 4 | D2O (14 equiv.) instead of D2O (28 equiv.) | 93 | >20:1 |
91 | 99 |
| 5 | D2O (56 equiv.) instead of D2O (28 equiv.) | 92 | >20:1 |
97 | 99 |
| 6d | MeOD as solvent | 85 | 3:1 |
96 | 96 |
| 7d |
V
DCM:VMeOD = 10:1 as the solvent |
80 | 17:1 |
95 | 99 |
Substrate scope
With the optimized reaction conditions established, we turned our attention to investigating the scope and generality of the current catalytic system with regard to aldimine esters. The representative results are shown in Table 2. A range of para- and meta-substituted aryl aldimine esters bearing electron-deficient (1a, 1c, 1d, and 1e), electron neutral (1f), or electron-rich (1g, 1i, and 1j) groups on the arene ring performed well to afford the corresponding α-deuterated products endo-3a–3j in high yield (57–95%) with excellent stereoselectivity (13:1–20:1 dr, 97–99% ee) and uniformly high levels of (94–96%) deuterium incorporation (Table 2, entries 1, 3–7, 9 and 10). Notably, sterically hindered ortho-methyl and ortho-chloro substituted aldimine esters 1b and 1h were well compatible with this catalytic system, leading to the desired α-deuterated cycloadducts 3b and 3h in good to high yields with perfect diastereo-/enantioselectivities and excellent levels of deuterium incorporation (entries 2 and 8). Aldimine esters with fused aromatic rings (1k and 1l) or heteroaryl group (1m) are also well tolerated in this sequential H/D-exchange/1,3-dipolar cycloaddition process. Remarkably, the challenging and less reactive alkyl aldimine ester 1n derived from cyclohexanecarbaldehyde is a viable substrate in this transformation, furnishing the desired α-deuterated endo-product (3n) with a high enantioselectivity (95%) and excellent deuterium incorporation (92%) with a moderate yield (45%) (entry 14). Comparable reactivity, stereoselectivity and deuterium-enrichment level were maintained when the reaction was carried out at 1.0 mmol scale (entry 15).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | 3 | Yieldb (%) | drc (endo:exo) |
Dc (%) | eec (%) |
| a All reactions were carried out with 0.2 mmol of 1, 0.3 mmol of 2, 0.01 mmol of Cu(I)/(S)-L1, and 0.2 mmol of Et3N in 2 mL of DCM and 0.1 mL of D2O for 3–6 h, see the ESI for details. b Yields refer to isolated yields of deuterated products. c The dr value was determined by crude 1H NMR. D refers to D-incorporation percentages based on the calculations described in the ESI. The ee value was determined by HPLC analysis. d 1.0 mmol scale. | ||||||
| 1 | p-ClC6H4 | 3a | 92 | >20:1 |
96 | 99 |
| 2 | o-ClC6H4 | 3b | 74 | 14:1 |
95 | 99 |
| 3 | m-ClC6H4 | 3c | 67 | >20:1 |
95 | 99 |
| 4 | p-BrC6H4 | 3d | 95 | 17:1 |
96 | 99 |
| 5 | p-FC6H4 | 3e | 90 | 13:1 |
96 | 99 |
| 6 | Ph | 3f | 89 | 19:1 |
94 | 99 |
| 7 | p-MeC6H4 | 3g | 88 | 17:1 |
96 | 99 |
| 8 | o-MeC6H4 | 3h | 57 | 19:1 |
96 | 99 |
| 9 | m-MeC6H4 | 3i | 71 | 13:1 |
96 | 97 |
| 10 | p-MeOC6H4 | 3j | 90 | 13:1 |
94 | 99 |
| 11 | 1-Naphthyl | 3k | 63 | 13:1 |
93 | 99 |
| 12 | 2-Naphthyl | 3l | 56 | 17:1 |
95 | 99 |
| 13 | 2-Furyl | 3m | 66 | 9:1 |
96 | 98 |
| 14 | Cyclohexyl | 3n | 45 | >20:1 |
92 | 95 |
| 15d | p-ClC6H4 | 3a | 95 | >20:1 |
96 | 99 |

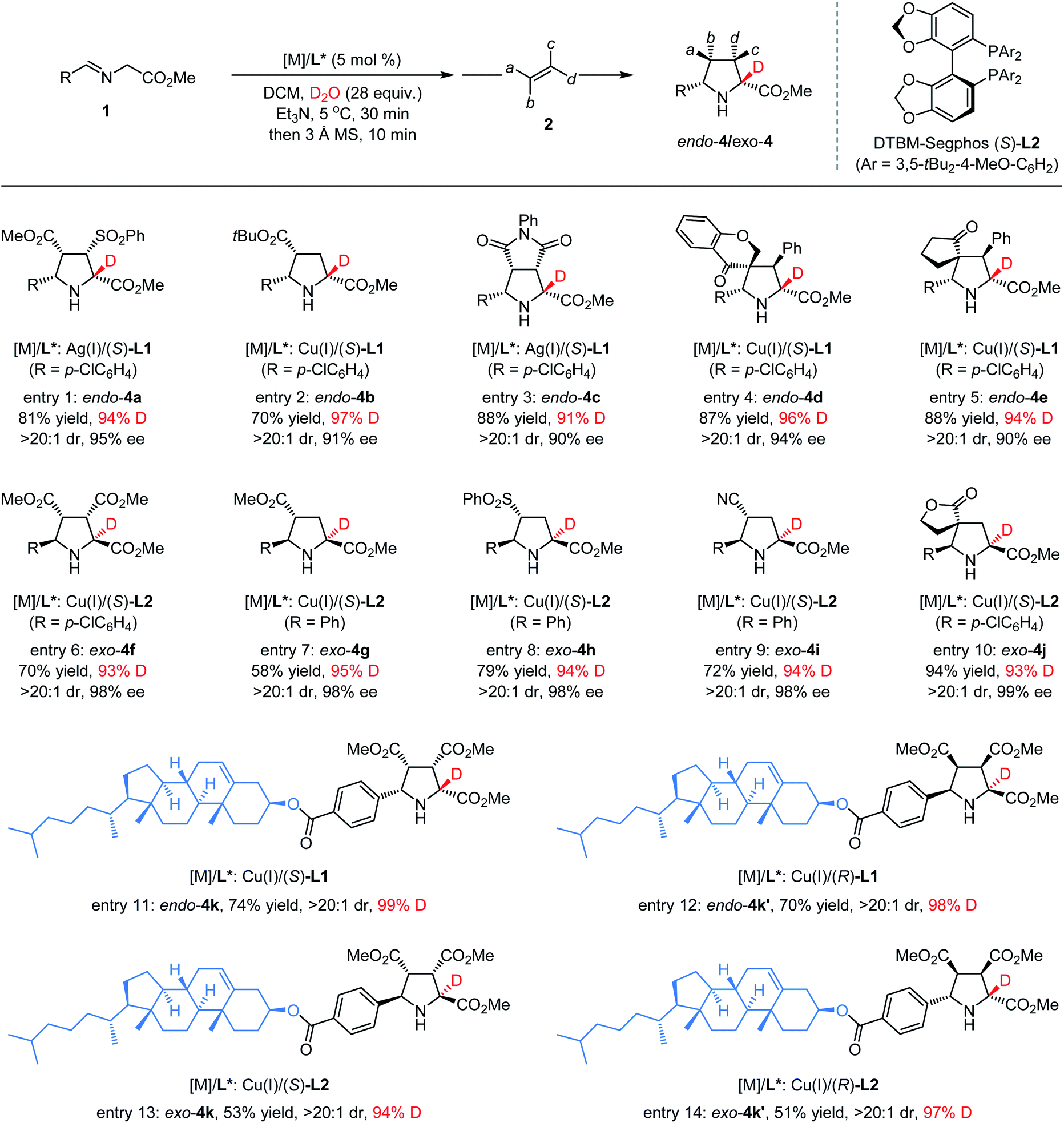
Motivated by the excellent performance of various aldimine esters in the synthesis of α-deuterated chiral pyrrolidines, we were interested in exploring the feasibility of various electron-deficient alkenes such as dipolarophiles in this catalytic asymmetric deuteration system. As depicted in Table 3, employing (Z)-β-sulfonyl acrylate 2b as the dipolarophile and aldimine ester 1a as the ylide precursor with the AgOAc/(S)-L1 complex as the chiral catalyst under otherwise identical reaction conditions,31 the desired cycloadduct α-deuterated endo-4a was isolated exclusively in 81% yield with 95% ee and 94% deuterium incorporation (entry 1). Subjecting tert-butyl acrylate 2c to the standard reaction conditions,42 the corresponding α-deuterated endo-adduct 4b was obtained in good yield with excellent stereoselectivity and a high level of deuterium incorporation (97%) (entry 2). Moreover, good yield and high enantioselectivity with 91% deuterium incorporation could be achieved when N-phenyl maleimide 2d was employed as the reactant partner with AgOAc/(S)-L1 as the metal complex (entry 3). When introducing some trisubstituted cyclic electron-deficient alkenes into the current cycloaddition reaction, highly α-deuterated endo-4d and endo-4e bearing a unique spiro quaternary carbon center were formed in good yield with excellent selectivity, respectively (entries 4 and 5). When using Cu(I)/(S)-DTBM-Segphos-L2 as the chiral catalyst and dimethyl maleate 2a as the dipolarophile,47,48 the corresponding α-deuterated exo-selective cycloadduct 4f could be separated in 70% yield with 98% ee and 93% deuterium incorporation (entry 6). In addition, this exo-selective catalytic system was also highly compatible with a series of olefines bearing a single substitute such as 2g, 2h and 2i, leading to formation of the desired α-deuterated cycloadducts exo-4g, exo-4h and exo-4i in good yield (58–79%) with excellent ee values (97–98%) and deuterium incorporation (94–95%) (entries 7–9). Similarly, highly α-deuterated spirocyclic exo-4j could be obtained in good yield with excellent stereoselectivity control (entry 10). To further demonstrate the scope of this methodology, we introduced a structurally complex molecular unit with biological activity into α-deuterated chiral pyrrolidines. When employing Cu(I)/(S)-L1 as the metal complex, the reaction of cholesterol derived aldimine ester 1k with 2a under the standard reaction conditions, the corresponding endo-selective α-deuterated product 4k could be separated in a good yield (74%) with an excellent diastereoselectivity (>20:1) and high deuterium incorporation (98%) (entry 11). Furthermore, by selecting suitable chiral ligands ((R)-L1, (S)-L2, or (R)-L2) instead of (S)-L1, controllable syntheses of the other three deuterated diastereoisomers (endo-4k′, exo-4k, and exo-4k′) could be facilely accessed with satisfactory results (entries 12–14), which demonstrated that the local stereochemical information on the imine moiety of aldimine esters results in negligible effects on the H/D-exchange and asymmetric induction in the ensuing cycloaddition reaction.
| a All reactions were carried out with 0.2 mmol of 1, 0.3 mmol of 2, 0.01 mmol of [M]/L, and 0.2 mmol of Et3N in 2 mL of DCM and 0.1 mL of D2O for 3–6 h, see the ESI for details. Yields refer to isolated yields of deuterated products. The dr value was determined by crude 1H NMR. D refers to D-incorporation percentages based on the calculations described in the ESI. The ee value was determined by HPLC analysis. |
|---|
|
Subsequently, we turned our attention to whether the above catalyst-controlled protocol for diastereodivergent synthesis of endo-/exo-isomers of α-deuterated pyrrolidines could be further extended to the precise construction of up to eight deuterated endo-/exo-stereoisomers. As shown in Scheme 2, the reaction of (S)-(−)-lactate derived aldimine ester 5 with 2a was performed with appropriate copper(I)/chiral ligand complexes under the standard reaction conditions, and four desired α-deuterated diastereoisomers (endo-6, endo-6′, exo-6, and endo-6′) were successfully prepared in good yield (81–86%) with excellent diastereoselectivity (>20:1 dr) and high deuterium incorporation (93–94% D). As expected, using (R)-(−)-lactate derived imine ester ent-5 as the starting material, the remaining four complementary α-deuterated diastereomers (ent-endo-6, ent-endo-6′, ent-exo-6, and ent-endo-6′) could be readily achieved with similar results via the same protocol.
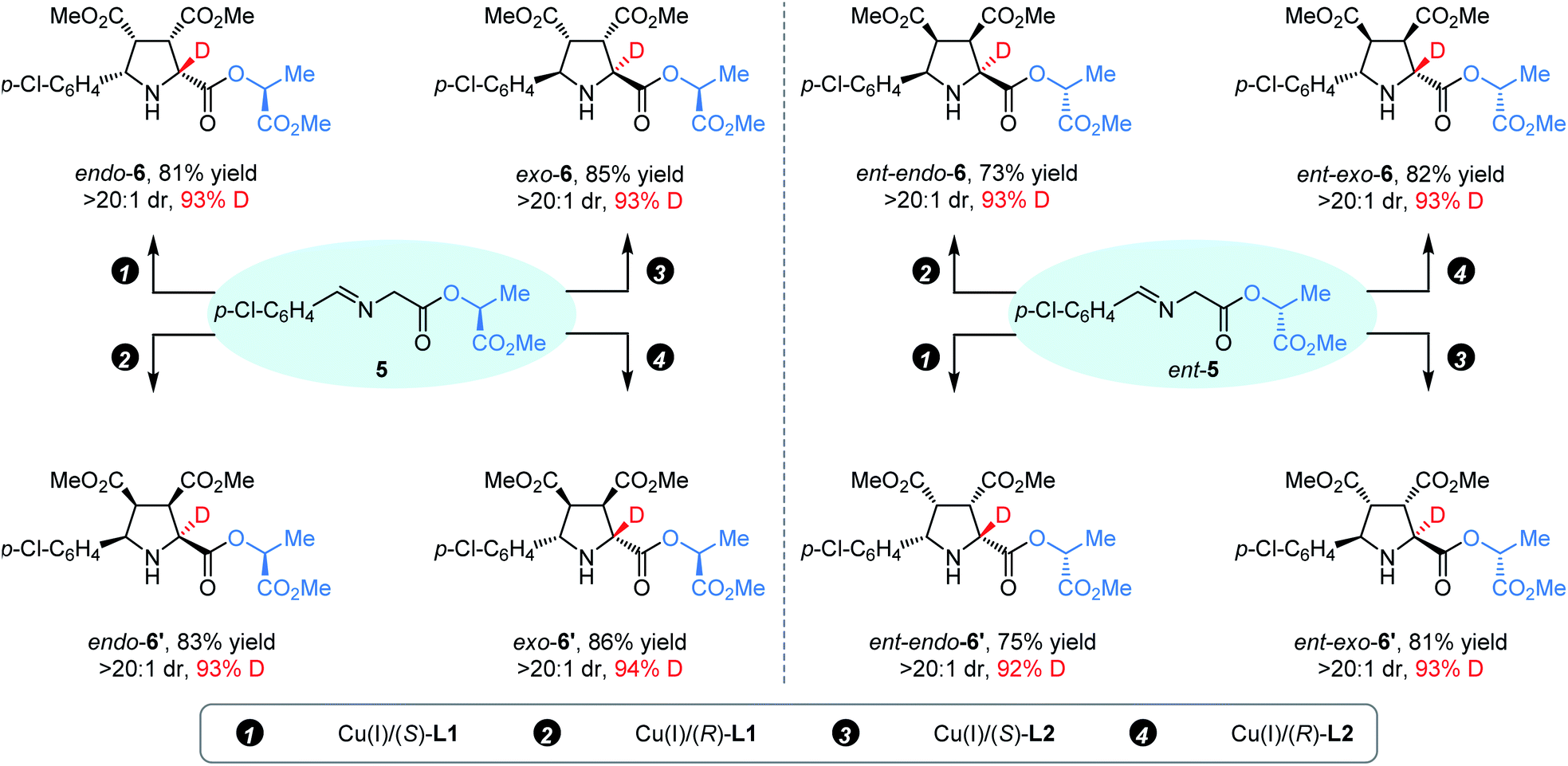 | ||
| Scheme 2 Access to enantioenriched pyrrolidines endo-6, endo-6′, exo-6, and exo-6′, and the enantiomers. | ||
Synthetic applications
Encouraged by the above investigations on the asymmetric synthesis of enantioenriched α-deuterated pyrrolidines via sequential H/D-exchange/1,3-dipolar cycloaddition process, we questioned whether such protocol could be utilized for the preparation of deuterium-labelled bioactive pharmaceutical molecules. For example, MDM2 antagonist idasanutlin (RG7388), discovered by Roche, is a chiral pyrrolidine carboxamide in phase III clinical trials.49 As shown in Scheme 3a, through integrating the H/D-exchange strategy into Cu(OAc)2/(R)-BINAP-catalyzed asymmetric 1,3-dipolar cycloaddition of nitrile olefin 7 and aldimine amide 8,50 the corresponding α-deuterated pyrrolidine carboxamide D-exo-9 could be isolated in 63% yield with 18:1 diastereoselectivity and 95% ee albeit with unsatisfactory 42% deuterium incorporation at the α-position of ester. However, further isomerization of compound D-exo-9 with aqueous sodium hydroxide through a base-promoted ring-opening/[H/D-Ex]/cyclization pathway51 led to idasanutlin 12 without any α-deuterium being detected albeit with retention of the configuration at the α-position of the pyrrolidine ring, and the key keto–enol tautomerization resulted in the complete loss of deuterium in a regular protic solvent. In consideration of the correlation between the deuteration of unlabeled compounds in deuterium solvents and the reverse de-deuteration of labelled compounds in common solvents52 and the previous 1H NMR experimental results of the α-hydrogen atom of aldimine esters could be partially deuterated with D2O in the presence of weak organic base Et3N (vide supra, Fig. 2), we reasoned that using a stronger inorganic base (aqueous sodium hydroxide), it would be possible to install a deuterium atom onto the α-position of idasanutlin with a synthetically useful deuteration level via a reversed 1,3-DC/[H/D-Ex] protocol. As shown in Scheme 3b, Cu(I)/(R,Rp)-L3-catalyzed 1,3-dipolar cycloaddition between nitrile olefin 7 and aldimine amide 8 proceeded smoothly in THF to provide the regular compound exo-9 in 88% yield with an exclusive diastereoselectivity (>20:1 dr) and 96% ee.47 The ensuing isomerization was successfully carried out in a mixed solution of NaOH/D2O (10 M), leading to the α-deuterated idasanutlin D-12 (D-RG7388) in 75% yield with a high level of deuteration incorporation (95% D) and excellent stereoselectivity (>20:1 dr, 95% ee).
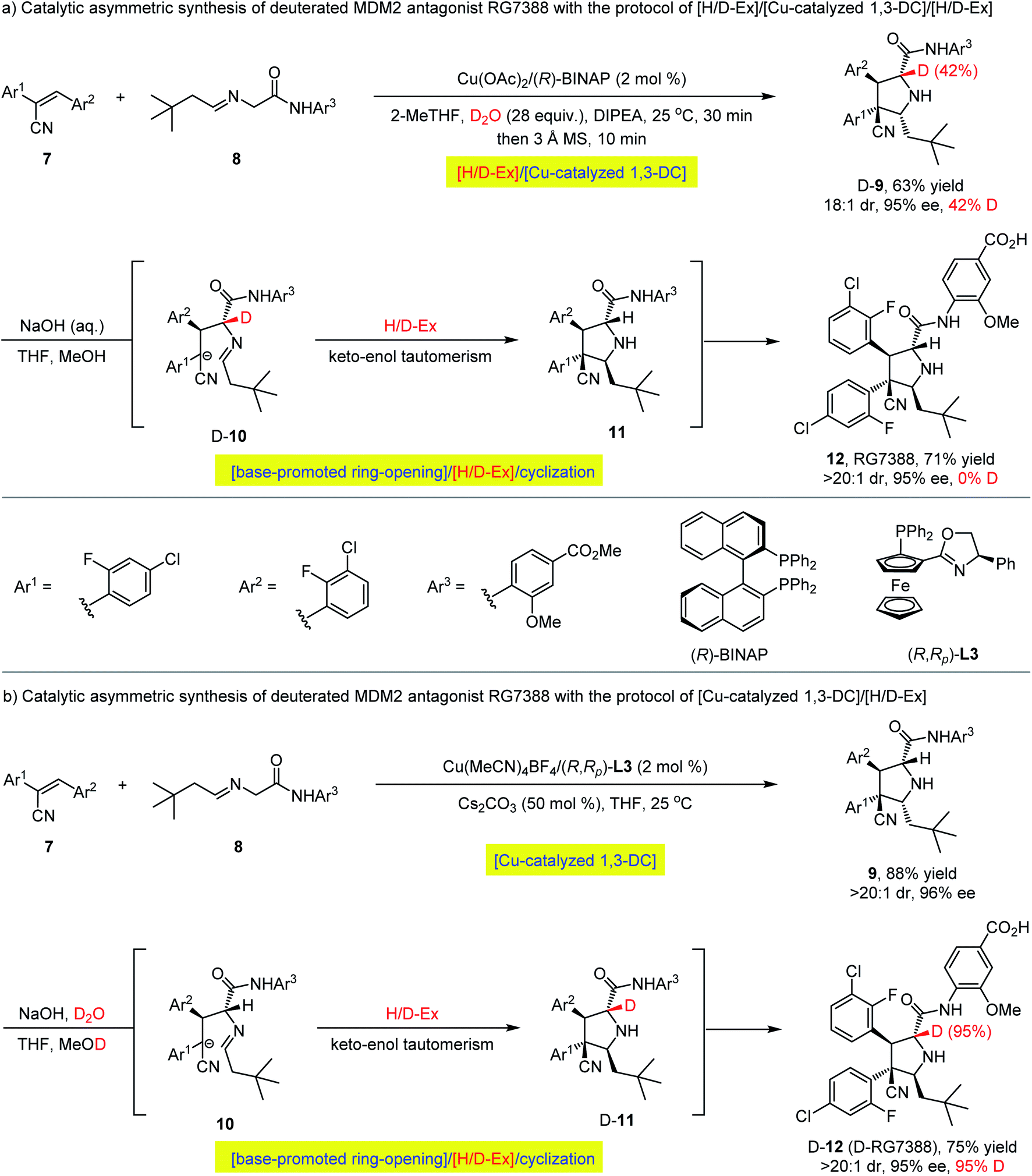 | ||
| Scheme 3 The catalytic asymmetric construction of deuterated MDM2 antagonist RG7388. | ||
Conclusions
In summary, a wide range of enantioenriched pyrrolidine derivatives with a high level of deuteration incorporation have been efficiently synthesized for the first time via the strategy of merging H/D exchange with the transition-metal catalyzed 1,3-dipolar cycloaddition reaction. Directly converting glycine-derived aldimine esters into the deuterated counterparts with D2O by virtue of Cu(I)-catalyzed H/D-exchange and subsequent thermodynamically/kinetically favored cleavage of the α-C–H bond rather than the α-C–D bond are crucial to generate the key N-metallated α-deuterated azomethine ylide species, which enables the success of α-deuterated chiral pyrrolidine synthesis with a high level of deuterium incorporation. The cost-effective and operationally simple protocol not only precisely installs a unique deuterium-containing stereogenic center but also makes contributions to the development of deuterated heterocycle chemistry. The application of such a strategy was further demonstrated through a catalytic asymmetric synthesis of deuterium-labelled MDM2 antagonist idasanutlin (RG7388) via a reversed 1,3-DC/[H/D-exchange] protocol. We anticipate that the current method will be helpful in the construction of deuterated chiral heterocycles to enable new drug discovery.Data availability
All experimental and characterization data in this manuscript are available in the ESI.†Author contributions
C.-J. W. conceived and designed the research. X. C. and X. C. performed the research. C. J. W. and X. C. co-wrote the paper. All the authors analysed the data, discussed the results, and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSFC (220711186, and 22101216), the Hubei Province Natural Science Foundation (2020CFA036), and China Postdoctoral Science Foundation (2021M702514). Support by the Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated.Notes and references
- T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed.
- T. G. Gant, J. Med. Chem., 2014, 57, 3595–3611 CrossRef CAS PubMed.
- S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Chem. Rev., 2022 DOI:10.1021/acs.chemrev.1c00795.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- K. B. Wiberg, Chem. Rev., 1955, 55, 713–743 CrossRef CAS.
- E. Stokvis, H. Rosing and J. H. Beijnen, Rapid Commun. Mass Spectrom., 2005, 19, 401–407 CrossRef CAS PubMed.
- V. Jacques, A. W. Czarnik, T. M. Judge, L. H. T. Van der Ploeg and S. H. DeWitt, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1471–E1479 CrossRef CAS PubMed.
- G. W. Muller, P. H. Schafer, H.-W. Man, L.-H. Zhang, A. Gandhi and R. Chopra, US Pat., 20120230983A1, 2012.
- F. Maltais, Y. C. Jung, M. Chen, J. Tanoury, R. B. Perni, N. Mani, L. Laitinen, H. Huang, S. Liao, H. Gao, H. Tsao, E. Block, C. Ma, R. S. Shawgo, C. Town, C. L. Brummel, D. Howe, S. Pazhanisamy, S. Raybuck, M. Namchuk and Y. L. Bennani, J. Med. Chem., 2009, 52, 7993–8001 CrossRef CAS PubMed.
- P. K. Bhadra, A. Hassanzadeh, B. Arsic, D. G. Allison, G. A. Morris and J. Barber, Org. Biomol. Chem., 2016, 14, 6289–6296 RSC.
- S. Dewitt, V. Jacques and L. H. T. Van der Ploeg, 2016153948A1, PTC Int., 2016.
- A. Katsnelson, Nat. Med., 2013, 19, 656 CrossRef CAS PubMed.
- J. Helfenbein, C. Lartigue, E. Noirault, E. Azim, J. Legailliard, M. J. Galmier and J. C. Madelmont, J. Med. Chem., 2002, 45, 5806–5808 CrossRef CAS PubMed.
- J. Bigeleisen, Science, 1965, 147, 463–471 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed.
- P. Ji, Y. Zhang, Y. Dong, H. Huang, Y. Wei and W. Wang, Org. Lett., 2020, 22, 1557–1562 CrossRef CAS PubMed.
- W. N. Palmer and P. J. Chirik, ACS Catal., 2017, 7, 5674–5678 CrossRef CAS PubMed.
- L. V. A. Hale and N. K. Szymczak, J. Am. Chem. Soc., 2016, 138, 13489–13492 CrossRef CAS PubMed.
- C. Taglang, L. M. Martínez-Prieto, I. del Rosal, L. Maron, R. Poteau, K. Philippot, B. Chaudret, S. Perato, A. Sam Lone, C. Puente, C. Dugave, B. Rousseau and G. Pieters, Angew. Chem., Int. Ed., 2015, 54, 10474–10477 CrossRef CAS PubMed.
- J. S. Rowbotham, H. A. Reeve and K. A. Vincent, ACS Catal., 2021, 11, 2596–2604 CrossRef CAS PubMed.
- S. Guo, X. Wang and J. S. Zhou, Org. Lett., 2020, 22, 1204–1207 CrossRef CAS PubMed.
- T. Sakamoto, K. Mori and T. Akiyama, Org. Lett., 2012, 14, 3312–3315 CrossRef CAS PubMed.
- E. J. Corey and J. O. Link, Tetrahedron Lett., 1989, 30, 6275–6278 CrossRef CAS.
- H. Mizutani, R. Kawanishi and K. Shibatomi, Chem. Commun., 2021, 57, 6676–6679 RSC.
- T. Shao, Y. Li, N. Ma, C. Li, G. Chai, X. Zhao, B. Qiao and Z. Jiang, iScience, 2019, 16, 410–419 CrossRef CAS PubMed.
- K. Moozeh, S. M. So and J. Chin, Angew. Chem., Int. Ed., 2015, 54, 9381–9385 CrossRef CAS PubMed.
- Y. Zhao, X. Lim, Y. Pan, L. Zong, W. Feng, C.-H. Tan and K.-W. Huang, Chem. Commun., 2012, 48, 5479–5481 RSC.
- J.-S. Oh, K. I. Kim and C. E. Song, Org. Biomol. Chem., 2011, 9, 7983–7985 RSC.
- L. Wei, X. Chang and C.-J. Wang, Acc. Chem. Res., 2020, 53, 1084–1100 CrossRef CAS PubMed.
- X. Fang and C.-J. Wang, Org. Biomol. Chem., 2018, 16, 2591–2601 RSC.
- J. Adrio and J. C. Carretero, Chem. Commun., 2019, 55, 11979–11991 RSC.
- T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed.
- J. M. Longmire, B. Wang and X. Zhang, J. Am. Chem. Soc., 2002, 124, 13400–13401 CrossRef CAS PubMed.
- M. Kuhnert, A. Blum, H. Steuber and W. E. Diederich, J. Med. Chem., 2015, 58, 4845–4850 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- Y. Zhao, A. Aguilar, D. Bernard and S. Wang, J. Med. Chem., 2015, 58, 1038–1052 CrossRef CAS PubMed.
- P. Beak, S. T. Kerrick, S. Wu and J. Chu, J. Am. Chem. Soc., 1994, 116, 3231–3239 CrossRef CAS.
- A. Michelotti, F. Rodrigues and M. Roche, Org. Process Res. Dev., 2017, 21, 1741–1744 CrossRef CAS.
- X. Chang, Y. Yang, C. Shen, K.-S. Xue, Z.-F. Wang, H. Cong, H.-Y. Tao, L. W. Chung and C.-J. Wang, J. Am. Chem. Soc., 2021, 143, 3519–3535 CrossRef CAS PubMed.
- C.-J. Wang, G. Liang, Z.-Y. Xue and F. Gao, J. Am. Chem. Soc., 2008, 130, 17250–17251 CrossRef CAS PubMed.
- P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, J. Med. Chem., 2021, 64, 2339–2381 CrossRef CAS PubMed.
- B. Lygo and L. D. Humphreys, Tetrahedron Lett., 2002, 43, 6677–6679 CrossRef CAS.
- T. Yamada, M. Kuwata, R. Takakura, Y. Monguchi, H. Sajiki and Y. Sawama, Adv. Synth. Catal., 2018, 360, 637–641 CrossRef CAS.
- H. Geng, X. Chen, J. Gui, Y. Zhang, Z. Shen, P. Qian, J. Chen, S. Zhang and W. Wang, Nat. Catal., 2019, 2, 1071–1077 CrossRef CAS PubMed.
- W. Gao, X. Zhang and M. Raghunath, Org. Lett., 2005, 7, 4241–4244 CrossRef CAS PubMed.
- G. S. Caleffi, O. Larrañaga, M. Ferrándiz-Saperas, P. R. R. Costa, C. Nájera, A. de Cózar, F. P. Cossío and J. M. Sansano, J. Org. Chem., 2019, 84, 10593–10605 CrossRef CAS PubMed.
- Q. Ding, Z. Zhang, J.-J. Liu, N. Jiang, J. Zhang, T. M. Ross, X.-J. Chu, D. Bartkovitz, F. Podlaski, C. Janson, C. Tovar, Z. M. Filipovic, B. Higgins, K. Glenn, K. Packman, L. T. Vassilev and B. Graves, J. Med. Chem., 2013, 56, 5979–5983 CrossRef CAS PubMed.
- L. Shu, C. Gu, D. Fishlock and Z. Li, Org. Process Res. Dev., 2016, 20, 2050–2056 CrossRef CAS.
- G. Rimmler, A. Alker, M. Bosco, R. Diodone, D. Fishlock, S. Hildbrand, B. Kuhn, C. Moessner, C. Peters, P. D. Rege and M. Schantz, Org. Process Res. Dev., 2016, 20, 2057–2066 CrossRef CAS.
- A. Di Giuseppe, R. Castarlenas and L. A. Oro, C. R. Chim., 2015, 18, 713–741 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00826b |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |