
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRobust dicopper(I) μ-boryl complexes supported by a dinucleating naphthyridine-based ligand†
Pablo
Ríos
 a,
Matthew S.
See
a,
Rex C.
Handford
a,
Simon J.
Teat
b and
T. Don
Tilley
*a
a,
Matthew S.
See
a,
Rex C.
Handford
a,
Simon J.
Teat
b and
T. Don
Tilley
*a
aDepartment of Chemistry, University of California, Berkeley, USA
bAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720-1460, USA. E-mail: tdtilley@berkeley.edu
First published on 13th May 2022
Abstract
Copper boryl species have been widely invoked as reactive intermediates in Cu-catalysed C–H borylation reactions, but their isolation and study have been challenging. Use of the robust dinucleating ligand DPFN (2,7-bis(fluoro-di(2-pyridyl)methyl)-1,8-naphthyridine) allowed for the isolation of two very thermally stable dicopper(I) boryl complexes, [(DPFN)Cu2(μ-Bpin)][NTf2] (2) and [(DPFN)Cu2(μ-Bcat)][NTf2] (4) (pin = 2,3-dimethylbutane-2,3-diol; cat = benzene-1,2-diol). These complexes were prepared by cleavage of the corresponding diborane via reaction with the alkoxide [(DPFN)Cu2(μ-OtBu)][NTf2] (3). Reactivity studies illustrated the exceptional stability of these boryl complexes (thermal stability in solution up to 100 °C) and their role in the activation of C(sp)–H bonds. X-ray diffraction and computational studies provide a detailed description of the bonding and electronic structures in these complexes, and suggest that the dinucleating character of the naphthyridine-based ligand is largely responsible for their remarkable stability.
Introduction
Copper-catalysed C–H borylation is a powerful tool in organic chemistry due to the stability and versatility of the resulting organoboron species as synthetic building blocks.1 This catalysis is thought to involve Cu(I) boryl (Cu–BR2) complexes as key reactive intermediates. However, the chemistry of Cu(I) boryls has only recently been observed; while the first proposal of a Cu–BR2 intermediate stems from a 2000 report by Miyaura2 and Hosomi,3 the first well-defined Cu(I) boryl species was described by Sadighi and coworkers in 2005.4 Since then, only a few have been isolated.5 The paucity of examples is likely due to their inherent instability, given that most of those reported decompose in solution at or below room-temperature to elemental Cu.4,5 Notably, these cases involve monodentate N-heterocyclic carbene (NHC) or phosphine supporting ligands. The reported examples stable at room-temperature often make use of apolar solvents (e.g. benzene), such as the ring-expanded NHC-supported (6-Dipp)CuBpin complex reported by Liptrot et al.5h In this example, limited decomposition is observed at 25 °C after several weeks. In other cases, the empty p orbital on boron is stabilised by means of adjacent nitrogen atoms, as in the systems described by Yamashita, Nozaki et al. Their synthetic procedure involves mixing an anionic boryl ligand with copper halides, giving the corresponding boryl complexes, generally in ca. 25–30% yield.5e,fIn 2016, Sadighi reported the first dinuclear boryl, {[(SIPr)Cu]2(μ-Bcat)}{BF4} (Scheme 1, top left, SIPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene), from reaction of {[(SIPr)Cu]2(μ-OSiMe3)}{BF4} with bis(catecholato)diboron (B2cat2) at −35 °C.6 The same dicopper cation was obtained by Kleeberg in 2019 upon treatment of mononuclear (SIPr)CuOtBu with B2cat2 in THF. Interestingly, mononuclear (SIPr)CuBcat was observed when the reaction was carried out in toluene.7 By modifying the boryl and supporting ligands, the Kleeberg group obtained additional examples of neutral dicopper boryls (Scheme 1).5c,8 However, the latter species readily decompose in solution at or below room-temperature and require storage in the solid-state under an inert atmosphere, complicating their characterisation and study as catalytic intermediates.
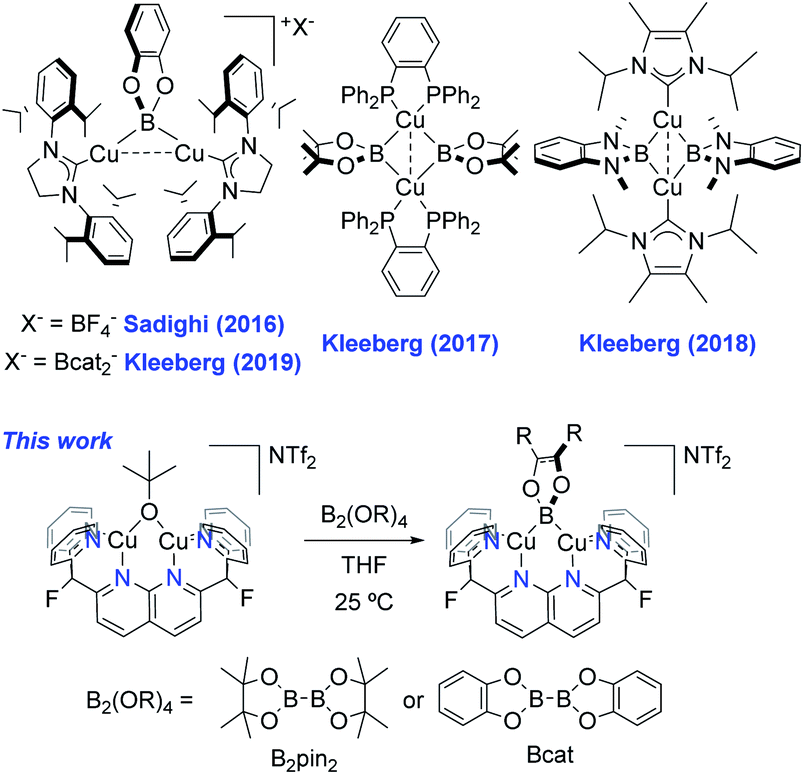 | ||
| Scheme 1 Examples of dicopper(I) μ-boryl complexes. | ||
Here we report the synthesis and characterisation of two dicopper(I) boryl complexes supported by a 1,8-naphthyridine-based dinucleating ligand. These species are persistent in solution and tolerate heating to at least 100 °C. In addition, their role in the activation of C(sp)–H bonds and the origins of their stability are described.
Results and discussion
Previous results from this laboratory established 2,7-bis(fluoro-di(2-pyridyl)methyl)-1,8-naphthyridine (DPFN) as an effective platform for stabilisation of bimetallic units. Dicopper(I) [(DPFN)Cu2]+ complexes, accessed via the convenient starting material [(DPFN)Cu2(μ-NCMe)][NTf2]2 (1),9 exhibit remarkable bimetallic electrophilicity and have allowed for the study of several elusive reactive intermediates such as a dicopper(I,II) nitrenoid,10 pentanuclear metal hydrides,11 and an unprecedented dicopper(I) bridging triazolide (Scheme 2).12–14 Thus, [(DPFN)Cu2]n+ seemed a promising framework for the synthesis and stabilisation of dicopper boryl units.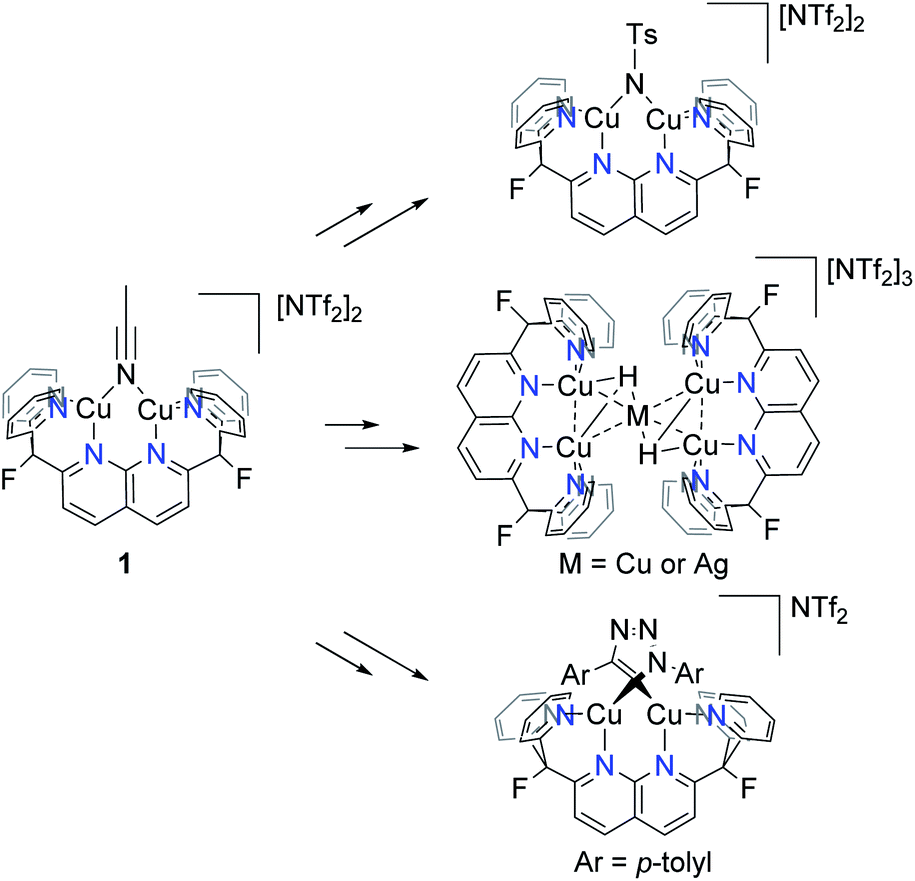 | ||
| Scheme 2 Examples of detected and/or isolated reactive intermediates containing the [(DPFN)Cu2]n+ scaffold. | ||
Due to the electrophilic character of 1, introduction of a boryl fragment requires an anionic borylation reagent. Whereas few synthons for boryl anions are available, a Lewis base adduct of a tetraalkoxy diboron (with e.g., alkoxide, fluoride or NHC) gives an isolable sp2–sp3 anionic diboron compound capable of transferring a boryl group to an electrophile under metal-free conditions.15 To pursue this possibility, one equivalent of K[B2pin2OtBu] was added to complex 1 in ortho-difluorobenzene (o-DFB) at 25 °C to give a color change from orange to dark green after 3 h. A 1H NMR analysis revealed formation of a single new symmetrical species (2) with a set of resonances corresponding to the DPFN ligand. This was corroborated by 19F{1H} NMR spectroscopy, which showed that the singlet at −175.1 ppm for 1 had been replaced by a singlet at −177.8 ppm. The 1H NMR spectrum of the reaction mixture revealed the presence of free MeCN and tBuOBpin, as well as a singlet at 1.44 ppm associated with 2. This assignment is supported by the 11B NMR spectrum containing a single broad resonance for tBuOBpin at 21.7 ppm, in agreement with reported values (Fig. S13†).16 Altogether, these data suggest the formation of boryl complex [(DPFN)Cu2(μ-Bpin)][NTf2] (2, Scheme 3).
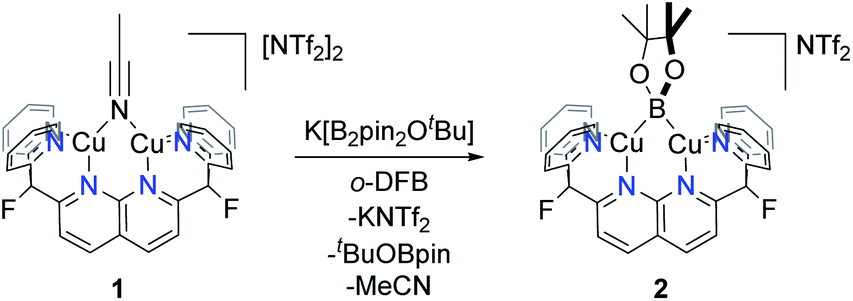 | ||
| Scheme 3 Initial synthesis of complex 2. | ||
Scale-up of the synthesis of 2, or use of THF as solvent, led to formation of several side-products as ascertained by 1H NMR analysis (Fig. S14†). One possible side product could result from the transfer of –OtBu from K[B2pin2OtBu]. To test this hypothesis, one equivalent of KOtBu was added to a solution of 1 in o-DFB at 25 °C, after which the reaction mixture became dark orange (Scheme 4). The 1H NMR spectrum after 1 h exhibited a new set of resonances resulting from the DPFN ligand, along with a new singlet at 1.76 ppm indicating the clean formation of [(DPFN)Cu2(μ-OtBu)][NTf2] (3). The resonances for 3 correspond to those of one of the aforementioned side-products formed in the synthesis of 2, and demonstrates –OtBu transfer to some extent.
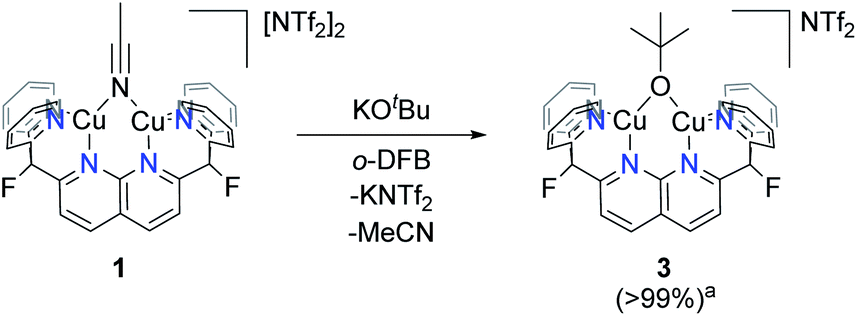 | ||
| Scheme 4 Synthesis of complex 3. aConversion determined by 1H NMR spectroscopy. | ||
Complex 3 was isolated by precipitation from o-DFB or THF with pentane, but removal of volatile components from the resulting powder in vacuo resulted in conversion to an insoluble, intractable material. Nonetheless, dark brown X-ray quality crystals were grown by layer diffusion of O(SiMe3)2 into an o-DFB solution of 3 (generated in solution) after 3 days at 25 °C.17 The solid-state structure, as determined by single crystal diffraction analysis, confirms the proposed assignment (Fig. 1). While the Cu–O bond distances are similar to those observed in previously reported dicopper complexes containing a 1,8-naphthyridine diphosphine ligand (ca. 1.9 Å),18 the Cu⋯Cu distance in 3 is much shorter (2.687(1) Å vs. 2.9–3.0 Å) and the Cu–O–Cu angle is considerably more acute (87.6(2)° vs. 100–108°), indicating a closer metal–metal contact enforced by the dipyridyl (vs. phosphinyl) side-arms. However, the metrical parameters for 3 are in good agreement with previous results from this laboratory on a dicopper(I) DPFN complex with a bridging aryloxide ligand.19
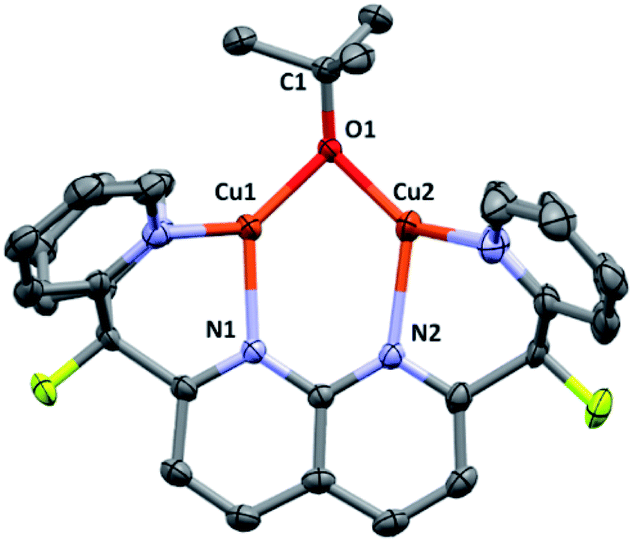 | ||
| Fig. 1 Solid-state molecular structure of 3 (50% probability ellipsoids); H atoms are omitted for clarity. Only the major disorder component of the tert-butoxide fragment is shown. | ||
With a reliable route to complex 3, reactions with diboranes were explored. Addition of B2pin2 to complex 3 in THF (prepared in situ from 1) resulted in a dark green solution after stirring at 25 °C for 15 h. Clean formation of boryl species 2 and tBuOBpin (Scheme 5) was observed by 1H, 19F{1H} and 11B{1H} NMR spectroscopy, and no side-products were observed upon increasing the concentration of the reaction solution. Complex 2 was isolated as green crystals by layering pentane over a o-DFB/THF solution which gave analytically pure 2 in 75% yield. The identity of the complex (Fig. 2, top) was confirmed by single-crystal X-ray diffraction. Similarly, B2cat2 reacted with complex 3 generated in situ to give the corresponding dicopper(I) boryl complex [(DPFN)Cu2(μ-Bcat)][NTf2] (4) after 2 h at 25 °C (Scheme 5), as judged by multinuclear NMR analysis on the resulting purple THF solution. The much shorter reaction time compared to that for the formation of 2 might be due to the higher Lewis acidity of B2cat2 in comparison to B2pin2.20 Similar work-up conditions afforded pure 4 as green X-ray quality crystals in 85% yield (Fig. 2, bottom). Unlike most previously described mono- or dicopper boryl complexes, 2 and 4 were synthesised and isolated at 25 °C, indicating a considerably higher stability. This is also reflected in mass spectra (Fig. S28 and S29†), which indicate persistence of the dinuclear structures under the ionisation conditions.
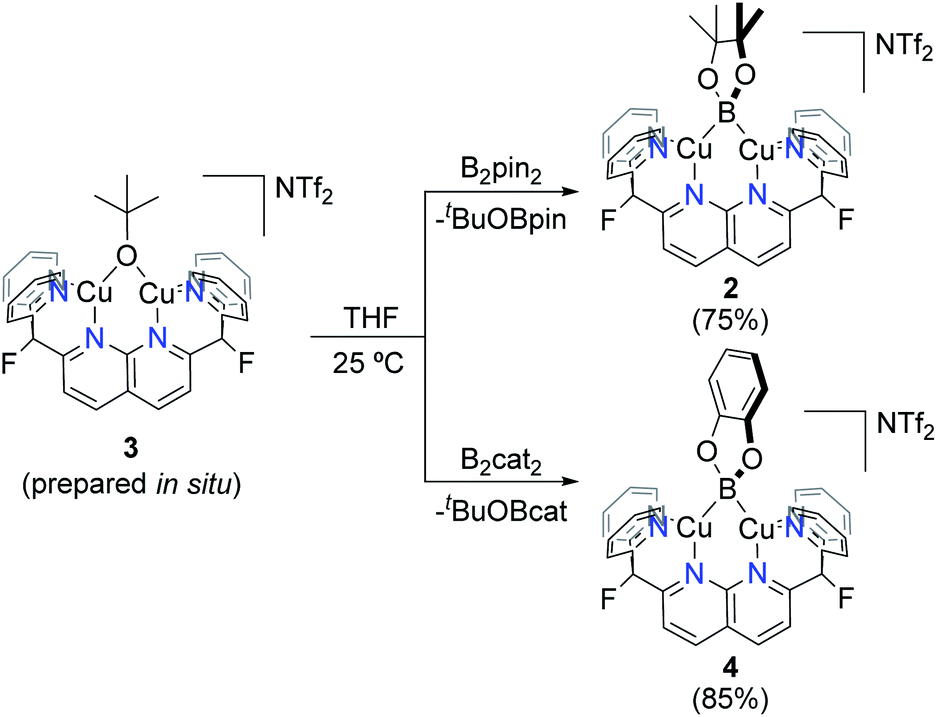 | ||
| Scheme 5 Synthesis of complexes 2 and 4 from complex 3 (generated in situ). Isolated yields in parentheses. | ||
 | ||
| Fig. 2 Solid-state molecular structures (50% probability ellipsoids) of 2 (top) and 4 (bottom); H atoms are omitted for clarity. | ||
The increased stability of these boryl complexes in solution raises interest in their bond metrics compared to those of reported, thermally unstable analogues. However, 2 and 4 possess geometrical parameters that are within the range of those reported for dicopper μ-boryl complexes. The Cu–B bond distances (2.07–2.09 Å) are considerably similar to those in cationic {[(SIPr)Cu]2(μ-Bcat)}{BF4} (2.04–2.05 Å),6 but are substantially shorter than those in the dinuclear neutral complexes synthesised by Kleeberg and co-workers (>2.17 Å).5c,8 Similarly, 2 and 4 feature Cu–B–Cu bond angles (67–68°) that are more acute than that in Sadighi's dinuclear complex (72.1(2)°) but more obtuse than in Kleeberg's complexes (ca. 60°). Finally, the Cu⋯Cu distances (ca. 2.32 Å) are between those of the cationic (2.4083(9) Å)6 and neutral (ca. 2.22–2.27 Å)5c,8 μ-boryl examples previously reported. Nevertheless, all such distances are significantly shorter than the sum of covalent radii for Cu (2.64 Å),21 implying that the short metal–metal distances are due to the bridging boryl ligands and/or cuprophilic interactions.22 Interestingly, the increased stability of 2 and 4 in solution does not seem to be attributable to peculiar binding metrics.
The robust character of these boryl species is also manifested in their reactivity. Both 2 and 4 exhibited no reaction with a number of small molecules including internal alkynes, olefins, silanes, azides or organic molecules possessing weak C–H bonds (e.g. fluorene), over a range of reaction conditions (THF or o-DFB as solvents and temperatures up to 70 or 100 °C, respectively; Table S1/Fig. S15 and S16†). In all cases, these bimetallic complexes coexist in solution with the added reagent even at reaction times of 24 h. In contrast to results reported by Kleeberg et al. for dicopper boryls, 2 and 4 are inert towards 4-iodotoluene.8 Aldehydes (1,3,5-trioxane or mesitylaldehyde) did not react with 2 or 4, in stark contrast to results observed for (IPr)CuBpin (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene),23 suggesting that nuclearity might be one of the reasons behind this difference in reactivity (vide infra). Warming of 2 or 4 to 75 °C in the presence of either CO2 or O2 gave rise to a complex mixture of products, as shown in the NMR spectra of Fig. S17 and S18,† whereas H2 was unreactive. In contrast, HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C(p-CF3–C6H4) cleanly reacted with both bridging boryl complexes to yield [(DPFN)Cu2(μ-CC(C6H4)CF3)][NTf2] (5),19 as summarised in Scheme 6 (Fig. S19–S21†). Concomitant formation of HBpin is observed in the case of complex 2. In the case of 4, HBcat seems to decompose over time to catBOBcat under the reaction conditions. This has been confirmed by warming HBcat in o-DFB at 100 °C (see Fig. S22 and S27† for more information). The increased Lewis basicity of boryl 2 compared to 4 is evident from the reaction conditions necessary to achieve high (>80%) conversions: whereas 2 reacted with 1.5 equiv of alkyne at 70 °C in 21 hours, boryl 4 required more forcing conditions (6 equiv of alkyne and warming for 90 hours at 100 °C). This difference was further confirmed by monitoring reactions of 2 and 4 with a different alkyne, PhCCH, in parallel (by 1H NMR spectroscopy). For this alkyne, both reactions were slower than those observed for p-CF3C6H4CC–H, as expected, and whereas the reaction of 2 with 1.5 equiv of alkyne reached 45% completion at 70 °C after 22 h, the reaction of 4 required a higher concentration of the alkyne (6 equiv) and higher temperature (100 °C) to reach 56% conversion after 24 h.
C(p-CF3–C6H4) cleanly reacted with both bridging boryl complexes to yield [(DPFN)Cu2(μ-CC(C6H4)CF3)][NTf2] (5),19 as summarised in Scheme 6 (Fig. S19–S21†). Concomitant formation of HBpin is observed in the case of complex 2. In the case of 4, HBcat seems to decompose over time to catBOBcat under the reaction conditions. This has been confirmed by warming HBcat in o-DFB at 100 °C (see Fig. S22 and S27† for more information). The increased Lewis basicity of boryl 2 compared to 4 is evident from the reaction conditions necessary to achieve high (>80%) conversions: whereas 2 reacted with 1.5 equiv of alkyne at 70 °C in 21 hours, boryl 4 required more forcing conditions (6 equiv of alkyne and warming for 90 hours at 100 °C). This difference was further confirmed by monitoring reactions of 2 and 4 with a different alkyne, PhCCH, in parallel (by 1H NMR spectroscopy). For this alkyne, both reactions were slower than those observed for p-CF3C6H4CC–H, as expected, and whereas the reaction of 2 with 1.5 equiv of alkyne reached 45% completion at 70 °C after 22 h, the reaction of 4 required a higher concentration of the alkyne (6 equiv) and higher temperature (100 °C) to reach 56% conversion after 24 h.
 | ||
| Scheme 6 Reactivity of complexes 2 and 4 with HCC(p-CF3–C6H4), yielding complex 5, which can further react with B2cat2 to regenerate boryl species 4. | ||
The proposed higher basicity of 2 finds additional support from DFT calculations (see Fig. S40†). Interestingly, bridging alkyne 5 further reacted with B2cat2 (but not with B2pin2) to regenerate Bcat complex 4 (Fig. S21†), and this reactivity trend is consistent with the observed reactions of 3 with diboranes (vide supra). The boron-containing product is not readily identified in the reaction mixture involving 5 and B2cat2, as its NMR resonances are obscured by the solvent peak. Attempts to cleanly isolate and fully characterize this product have not been successful (see ESI†).
The clean deprotonation of HCC(p-CF3–C6H4) to give 5 at high temperatures is surprising, given the potential side-reactions that are possible in the presence of free hydroborane, such as alkyne hydroboration24 or dehydrogenative borylation. Bertrand and coworkers recently reported catalytic C(sp)–H dehydrogenative borylation using carbene-stabilised Cu complexes, presumably by way of a catalytically active σ,π-dicopper acetylide intermediate A (Scheme 7A).25 Unlike species A, the triple bond of bridging alkynyl 5 is not engaged in π-bonding to copper, making it susceptible to hydroboration processes. Control experiments involved the independent synthesis of 5 by treating the previously reported bridging phenyl complex [(DPFN)Cu2(μ-Ph)][NTf2]26 with HCC(p-CF3–C6H4). Addition of HBpin or HBcat to 5 did not result in reaction at 25 or 100 °C over 48 h (Fig. S25 and S26†). This divergence in reactivity from that observed by Bertrand et al. could be due to the different binding mode of the alkyne fragment to the dicopper core (σ,σ vs. σ,π), or to the rigidity imposed by the dinucleating DPFN ligand.
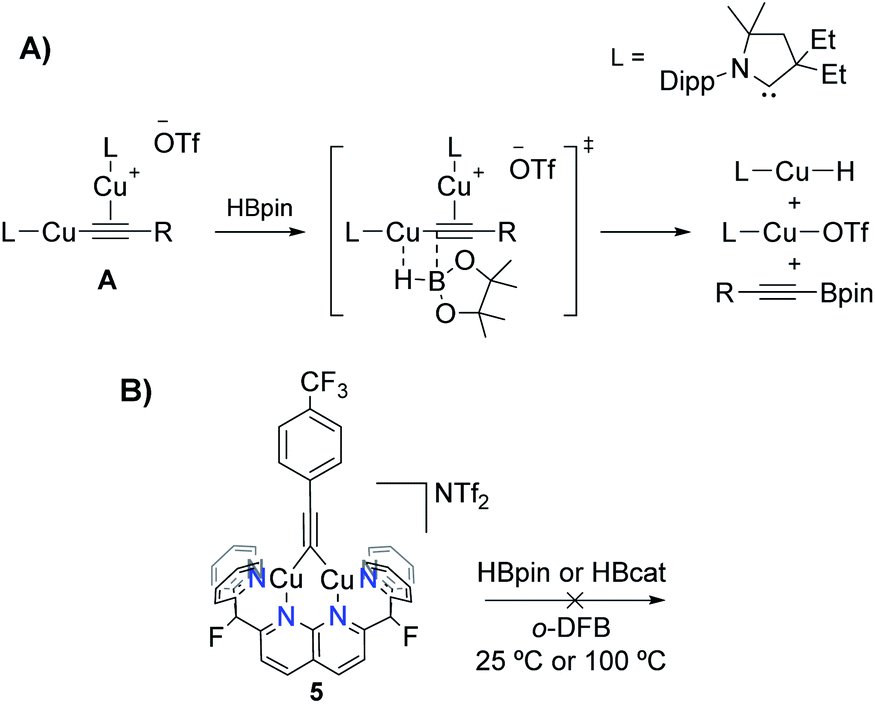 | ||
| Scheme 7 (A) Terminal alkyne dehydrogenative borylation mechanism proposed by Bertrand and coworkers.25 (B) Control experiments using 5 and hydroboranes that rule out alkyne hydroboration processes. | ||
Computational methods were used to gain insights into the unusual robustness of bridging boryls 2 and 4. First, steric congestion around the boron atom was quantified by determining its percent buried volume (%Vbur), described as the volume of a sphere centered on boron that is occupied by the [(DPFN)Cu2]+ scaffold and the substituents on B (Fig. S32–S35†).27,28 As expected, %Vbur values for 2 and 4 are remarkably large (ca. 80%), yet intermediate between values observed for unstable species in solution, namely mononuclear (IPr)CuBpin (%Vbur = 65.3%) and {[(SIPr)Cu]2(μ-Bcat)}{BF4} (%Vbur = 83.4%). In light of this, steric hindrance around boron is not likely to be a strong contributor to the stability.
DFT analyses were performed on 2 and 4 to understand their electronic structures, using the PBE0-D3/6-31g(d,p)/SDD level of theory on the cationic fragments.29 The optimised structures were found to be local minima, with geometries in excellent agreement with metrics observed for the solid state structures (Table S2†). Natural charges30 on Cu and B reflect minimal differences with those of related cationic systems, as displayed in Table 1. Likewise, Wiberg Bond Order analysis exhibited similar values for all Cu–B bonds, albeit the Cu⋯Cu interactions seem to be stronger in the cationic fragments of 2 and 4, as expected based on the observed, short Cu⋯Cu distances. In addition, no bond critical point between the Cu atoms was observed by QTAIM calculations (Fig. S38 and S39†).31,32
Finally, the electron distribution in the Cu–B bonds was investigated, since studies attribute the high nucleophilicity and reactivity exhibited by these complexes to the Cu-boryl σ-bonding electrons. Computational work by Carbó and Fernández in 2012,33 and Sheong and Lin in 2021,34 revealed that the contribution of B-based orbitals to the M–BR2 bond is remarkably high (ca. 70%, with a p/s ratio ≈ 0.9)35 in the case of mononuclear Cu-boryl species, whereas in other examples like Au- or Pd-boryl compounds, the contribution of B is 56% and 53%, respectively. Therefore, the Cu–B bond tends to be substantially more polarised towards the B atom. Natural Localised Molecular Orbital (NLMO)30 analysis of 2 and 4 reveals σ-donation of the boryl ligand to the empty 4s orbitals on the Cu atoms (as previously observed for [(DPFN)Cu2]n+ complexes)9 with negligible back-donation from the metals to boron. This 3c–2e bonding scenario is illustrated in Fig. 3. Surprisingly, the contributions of Cu- and B-based orbitals to the bonding are almost identical to those previously reported for reactive, mononuclear Cu-boryl complexes (ca. 30% Cu and 70% B), with a considerably smaller p/s ratio on B in the case of 4.
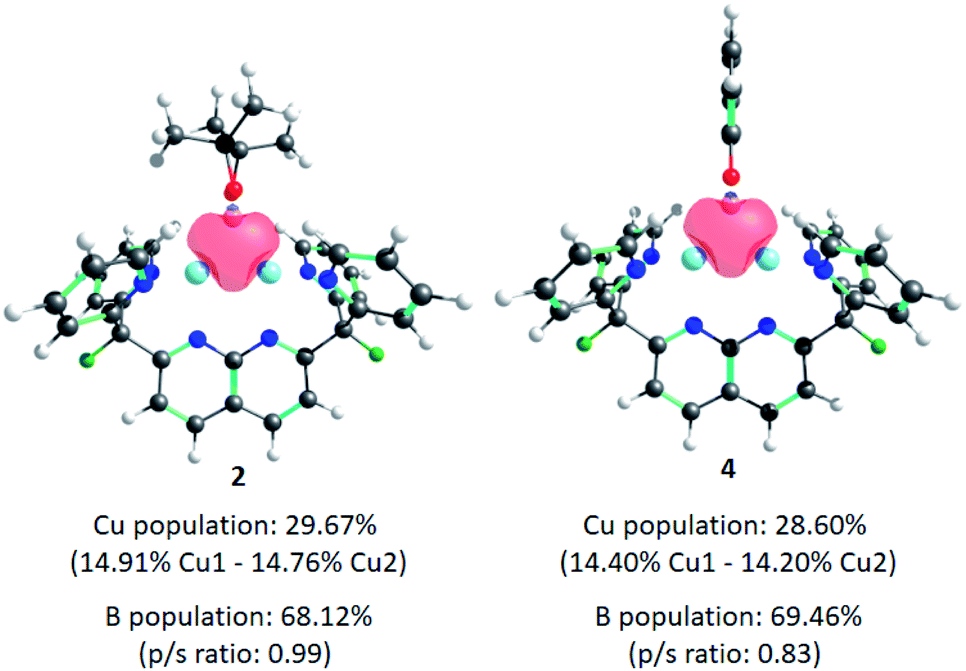 | ||
| Fig. 3 Natural Localised Molecular Orbitals (NLMO) of complexes 2 and 4, depicting σ-donation of the boryl fragment to the dicopper core. | ||
These data indicate that the extreme stability and chemical inertness of boryls 2 and 4 in polar solvents does not stem from peculiar geometrical or electronic factors, since these properties are similar to those of much less stable analogues. Rather, the rigid framework imposed by the dinucleating 1,8-naphthyridine-based ligand seems to be responsible for the observed stability, by precluding dynamic behaviour in solution that might produce an equilibrium concentration of more reactive, monomeric species.4,5,7 This hypothesis finds additional support from a recent study by Ito and coworkers, which concludes that a borylcopper(I) dimer is the dormant species in a catalytic asymmetric borylation process, whereas the mononuclear Cu-boryl complex is the active form of the catalyst.36
Conclusions
The μ-OtBu complex 3 provides a convenient synthetic pathway to a new type of dicopper boryl derivative, by a method that may provide more general access to complexes featuring reactive fragments stabilised by a dicopper core. In reactions of 3 with diboranes B2pin2 and B2cat2, mild conditions lead to the dinuclear boryl compounds 2 and 4 by cleavage of a B–B bond. Striking properties of the new boryl complexes reflect a high stability for the Cu2B cores. This stability appears to relate to strong, delocalised bonding across the bridging interaction supported by an effective, rigid dinucleating ligand that suppresses access to reactive monocopper boryl units. These results are therefore consistent with earlier observations concerning the ability of the [(DPFN)Cu2]n+ platform to stabilise reactive intermediates. Notably, the stability/reactivity balance in μ-boryl dicopper complexes should be tunable by way of modifications to the dinucleating ligand. For example, side arms that enforce a lower coordination number at the metal could lead to higher reactivity, and such possibilities are currently being explored in this laboratory.Data availability
Crystallographic data for complexes 2, 3 and 4 have been deposited at the CCDC under 2130621–2130623Author contributions
P. R. and T. D. T. conceived of the idea. P. R. and M. S. performed the experiments and analysed the data for synthesis, reactivity and characterization. The crystallographic data were analysed by R. C. H. (complexes 2, 3 and 4) and S. J. T. (complex 2). P. R. performed the DFT studies. P. R., M. S., R. C. H. and T. D. T. wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division under Contract no. DE-AC02-05CH11231. This project also received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 841154 through a fellowship for P. R. We acknowledge the National Institutes of Health (NIH) for funding the UC Berkeley CheXray X-ray crystallographic facility under grant no. S10-RR027172, the UC Berkeley College of Chemistry NMR facility under Grants SRR023679A, S10OD024998, and 1S10RR016634-01, and the UC Berkeley Molecular Graphics and Computation Facility under grant no. S10OD023532. We thank Dr Addison N. Desnoyer and T. Alex Wheeler for helpful discussions and Dr Nicholas S. Settineri for crystallographic assistance.Notes and references
- (a) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC; (b) H. Shinokubo, Proc. Jpn. Acad., Ser. B, 2014, 90, 1–11 CrossRef CAS PubMed; (c) H. Yoshida, ACS Catal., 2016, 6, 1799–1811 CrossRef CAS; (d) D. Hemming, R. Fritzmeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC; (e) S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Chem. Soc. Rev., 2020, 49, 4564–4591 RSC.
- K. Takahashi, T. Ishiyama and N. Miyaura, Chem. Lett., 2000, 29, 982–983 CrossRef.
- H. Ito, H. Yamanaka, J.-I. Tateiwa and A. Hosomi, Tetrahedron Lett., 2000, 41, 6821–6825 CrossRef CAS.
- D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed.
- Examples of mononuclear Cu-boryl species unstable in solution: (a) K. Semba, M. Shinomiya, T. Fujihara, J. Terao and Y. Tsuji, Chem.–Eur. J., 2013, 19, 7125–7132 CrossRef CAS PubMed; (b) A. J. Jordan, P. K. Thompson and J. P. Sadighi, Org. Lett., 2018, 20, 5242–5246 CrossRef CAS PubMed; (c) C. Kleeberg and C. Borner, Organometallics, 2018, 37, 4136–4146 CrossRef CAS; (d) W. Drescher, C. Borner and C. Kleeberg, New J. Chem., 2021, 45, 14957–14964 RSC . Examples of stable Cu-boryl complexes:; (e) Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 CrossRef CAS PubMed; (f) T. Kajiwara, T. Terabayashi, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2008, 47, 6606–6610 CrossRef CAS PubMed; (g) C. Borner and C. Kleeberg, Eur. J. Inorg. Chem., 2014, 2486–2489 CrossRef CAS; (h) T. M. H. Downie, R. S. C. Charman, J. W. Hall, M. F. Mahon, J. P. Lowe and D. J. Liptrot, Dalton Trans., 2021, 50, 16336–16342 RSC.
- C. M. Wyss, J. Bitting, J. Bacsa, T. G. Gray and J. P. Sadighi, Organometallics, 2016, 35, 71–74 CrossRef CAS.
- W. Drescher and C. Kleeberg, Inorg. Chem., 2019, 58, 8215–8229 CrossRef PubMed.
- C. Borner, L. Anders, K. Brandhorst and C. Kleeberg, Organometallics, 2017, 36, 4687–4690 CrossRef CAS.
- A. N. Desnoyer, A. Nicolay, P. Rios, M. S. Ziegler and T. D. Tilley, Acc. Chem. Res., 2020, 53, 1944–1956 CrossRef CAS PubMed.
- A. N. Desnoyer, A. Nicolay, M. S. Ziegler, K. V. Lakshmi, T. R. Cundari and T. D. Tilley, J. Am. Chem. Soc., 2021, 143, 7135–7143 CrossRef CAS PubMed.
- A. N. Desnoyer, A. Nicolay, M. S. Ziegler, N. A. Torquato and T. D. Tilley, Angew. Chem., Int. Ed., 2020, 59, 12769–12773 CrossRef CAS.
- M. S. Ziegler, K. V. Lakshmi and T. D. Tilley, J. Am. Chem. Soc., 2017, 139, 5378–5386 CrossRef CAS PubMed.
- B. F. Straub, Chem. Commun., 2007, 3868–3870 RSC.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS.
- Inspiration came from (a) S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem.–Eur. J., 2015, 21, 7082–7099 CrossRef CAS PubMed . Recent reviews on sp2-sp3 anionic diboranes can be found in; (b) R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC; (c) J. J. Carbó and E. Fernández, Chem. Commun., 2021, 57, 11935–11947 RSC.
- (a) E. A. Romero, J. L. Peltier, R. Jazzar and G. Bertrand, Chem. Commun., 2016, 52, 10563–10565 RSC; (b) C. Kim, B. Roh and H. G. Lee, Chem. Sci., 2021, 12, 3668–3673 RSC.
- The formation of an insoluble material after exposing complex 3 to vacuum prevented the synthesis of analytically pure solid samples of this compound..
- E. Kounalis, M. Lutz and D. L. J. Broere, Chem.–Eur. J., 2019, 25, 13280–13284 CrossRef CAS PubMed.
- M. S. Ziegler, N. A. Torquato, D. S. Levine, A. Nicolay, H. Celik and T. D. Tilley, Organometallics, 2018, 37, 2807–2823 CrossRef CAS.
- L. Dang, H. Zhao, Z. Lin and T. B. Marder, Organometallics, 2008, 27, 1178–1186 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Álvarez, Dalton Trans., 2008, 2832–2838 RSC.
- N. V. S. Harisomayajula, S. Makovetskyi and Y.-C. Tsai, Chem.–Eur. J., 2019, 25, 8936–8954 CrossRef CAS PubMed.
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 128, 11036–11037 CrossRef CAS PubMed.
- S. K. Bose, L. Mao, L. Kuehn, U. Radius, J. Nekvinda, W. L. Santos, S. A. Westcott, P. G. Steel and T. B. Marder, Chem. Rev., 2021, 121, 13238–13341 CrossRef CAS PubMed.
- (a) E. A. Romero, R. Jazzar and G. Bertrand, Chem. Sci., 2017, 8, 165–168 RSC . Species A was previously isolated, see; (b) L. Jin, D. R. Tolentino, M. Melaimi and G. Bertrand, Sci. Adv., 2015, 1, e1500304 CrossRef PubMed.
- M. S. Ziegler, D. S. Levine, K. V. Lakshmi and T. D. Tilley, J. Am. Chem. Soc., 2016, 138, 6484–6491 CrossRef CAS PubMed.
- A. Poater, F. Ragone, S. Giudice, C. Costabile, R. Dorta, S. P. Nolan and L. Cavallo, Organometallics, 2018, 27, 2679–2681 CrossRef.
- SambVca 2.1 was used to calculate %Vbur: L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- The calculations were performed with the Gaussian 16 program: Gaussian 16, Revision A.03, Gaussian, Inc. Wallingford CT, 2016. The full Gaussian citation can be found in the ESI.†.
- E. D. Glendening, A. E. Reed, J. E. Carpenter, and F. Weinhold, NBO Version 3.1 Search PubMed.
- R. F. W. Bader, Atom in Molecules: A Quantum Theory; Oxford University Press, Oxford, U.K., 1995 Search PubMed.
- Previous work in this laboratory demonstrated the presence of a Bond Critical Point (BCP) between the Cu atoms in a complex with a longer Cu···Cu distance (2.4457(4) Å), suggesting a stronger cuprophilic interaction. See T. C. Davenport and T. D. Tilley, Angew. Chem., Int. Ed., 2011, 50, 12205–12208 CrossRef CAS PubMed.
- J. Cid, J. J. Carbó and E. Fernández, Chem.–Eur. J., 2012, 18, 12794–12802 CrossRef CAS PubMed.
- X. Guo, T. Yang, F. K. Sheong and Z. Lin, ACS Catal., 2021, 11, 5061–5068 CrossRef CAS.
- The p/s ratio also gives an idea of the nucleophilicity of a M-boryl bond. A boryl ligand with more p character on B will donate more electron density to the metal, making the M-B bond stronger. On the contrary, a boryl ligand with more s character on B will polarise the M-B bond towards B. For more information, see J. Zhu, Z. Lin and T. B. Marder, Inorg. Chem., 2005, 44, 9384–9390 CrossRef CAS PubMed 33 or .
- H. Iwamoto, Y. Ozawa, Y. Takenouchi, T. Imamoto and H. Ito, J. Am. Chem. Soc., 2021, 143, 6413–6422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available CCDC 2130621–2130623. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00848c |
| This journal is © The Royal Society of Chemistry 2022 |