
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical 1,2,3-tricarbofunctionalization of α-vinyl-β-ketoesters enabled by a carbon shift from an all-carbon quaternary center†
Qi
Zhang
a,
Mong-Feng
Chiou
a,
Changqing
Ye
a,
Xiaobin
Yuan
a,
Yajun
Li
*ab and
Hongli
Bao
 *ac
*ac
aKey Laboratory of Coal to Ethylene Glycol and Its Related Technology, State Key Laboratory of Structural Chemistry, Center for Excellence in Molecular Synthesis, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, 155 Yangqiao Road West, Fuzhou, Fujian 350002, P. R. China. E-mail: liyajun@fjirsm.ac.cn; hlbao@fjirsm.ac.cn
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Lingling Road 345, Shanghai 200032, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 2nd May 2022
Abstract
Herein, we report an intermolecular, radical 1,2,3-tricarbofunctionalization of α-vinyl-β-ketoesters to achieve the goal of building molecular complexity via the one-pot multifunctionalization of alkenes. This reaction allows the expansion of the carbon ring by a carbon shift from an all-carbon quaternary center, and enables further C–C bond formation on the tertiary carbon intermediate with the aim of reconstructing a new all-carbon quaternary center. The good functional group compatibility ensures diverse synthetic transformations of this method. Experimental and theoretical studies reveal that the excellent diastereoselectivity should be attributed to the hydrogen bonding between the substrates and solvent.
A leading motive for the impressive achievements in the area of assembling molecular complexity is the transformation of simple feedstock chemicals into complex molecular skeletons with superior bioactive properties. In this respect, the direct functionalization of alkenes has been demonstrated as one of the most effective and simple strategies to meet this criterion at a high level. While the difunctionalization of alkenes in a one-pot process is the major theme of considerable interest in this field,1 the multifunctionalization of alkenes,2 for example, a 1,2,3-trifunctionalization of alkenes, has the power to simultaneously incorporate multifunctional groups. Therefore, this multifunctionalization reaction model can be regarded as an efficient and novel strategy to afford molecules with high structural diversity and complexity. However, such methods are elusive.
During the last decades, radical alkene functionalizations have been revealed to be a powerful tool for building complex molecular frameworks by employing a radical initiator, a transition metal catalyst, or a photocatalyst.1f–i However, only several successful methods for the radical multifunctionalization of alkenes have been achieved. For example, the Studer group reported an elegant 1,2-boryl shift-enabled radical 1,2,3-trifunctionalization of allylboronic esters using AIBN as the radical initiator (Fig. 1a).3 Shi et al. disclosed an excellent photocatalytic perfluoroalkylation of a vinyl-substituted all-carbon quaternary center through 1,2-aryl migration (Fig. 1b).4 Herein, we report a new one-pot protocol to realize an intermolecular, radical 1,2,3-tricarbofunctionalization of α-vinyl-β-ketoesters through a cascade process of deconstruction–reconstruction of the all-carbon quaternary center (Fig. 1c).5
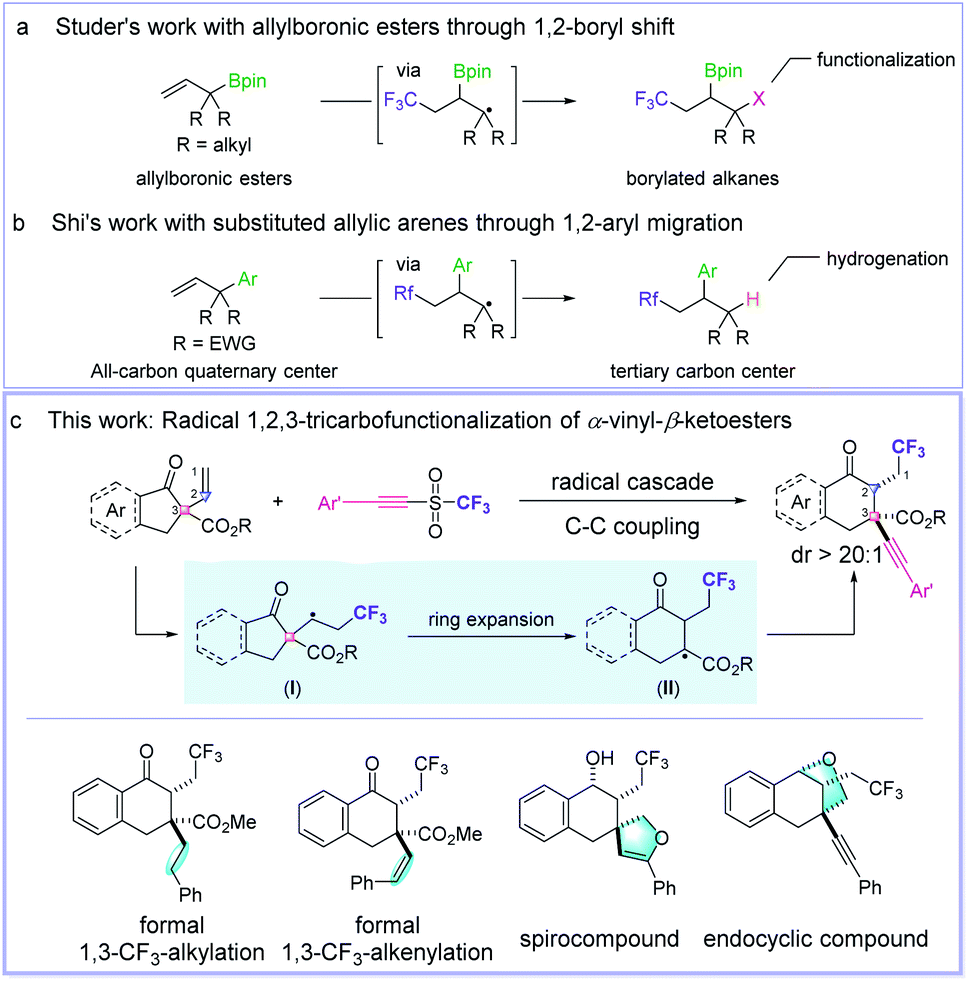 | ||
| Fig. 1 Radical 1,2,3-trifunctionalization of alkenes. (a) Studer's work; (b) Shi's work; (c) This work. | ||
The direct incorporation of a fluorine atom or fluorinated moieties into organic compounds has been extensively investigated and proved to be an significant synthetic strategy in the field of discovering new pharmaceuticals.6 Recently, we are interested in the radical functionalization of alkenes with fluoroalkyl groups,7 and we envisioned that, different from the typical Dowd–Beckwith8 ring expansion reaction,9 the addition of a fluoroalkyl radical to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond would generate an adduct radical species I, which will transform into the radical intermediate II upon ring expansion (Fig. 1c). Finally, the cascade C–C coupling affords the product with a reconstructed all-carbon quaternary center. However, there are several challenging issues that need to be addressed: (1) the carbon shift from an all-carbon quaternary center to afford a tertiary carbon center which is bulkier than the tertiary carbon center formed in a typical Dowd–Beckwith ring expansion reaction; (2) the reconstruction of all-carbon quaternary center from tertiary carbon radical II will meet the associated conformational restriction and steric congestion; (3) side reactions, such as 1,2-radical addition to the alkenyl group, homolytic couplings of the carbon radical intermediates I and II, and direct H-atom abstraction;10 (4) how to control the diastereomeric ratio of the products. To meet these challenges, we developed a novel method for the 1,2,3-trifunctionalization of alkenes using alkynyl triflones as both the CF3 (ref. 6) and alkynyl sources, providing the ring-expanded cyclic β-ketoesters with excellent diastereoselectivity and functional group diversity. In addition, good functional group compatibility of this method was observed, which ensures the diverse synthetic transformations. Moreover, hydrogen bonding between the substrates and 2,2,2-trifluoroethanol solvent was revealed to be the key factor for the excellent diastereoselectivity obtained in this reaction, and this result was confirmed by both experimental and theoretical studies.
C double bond would generate an adduct radical species I, which will transform into the radical intermediate II upon ring expansion (Fig. 1c). Finally, the cascade C–C coupling affords the product with a reconstructed all-carbon quaternary center. However, there are several challenging issues that need to be addressed: (1) the carbon shift from an all-carbon quaternary center to afford a tertiary carbon center which is bulkier than the tertiary carbon center formed in a typical Dowd–Beckwith ring expansion reaction; (2) the reconstruction of all-carbon quaternary center from tertiary carbon radical II will meet the associated conformational restriction and steric congestion; (3) side reactions, such as 1,2-radical addition to the alkenyl group, homolytic couplings of the carbon radical intermediates I and II, and direct H-atom abstraction;10 (4) how to control the diastereomeric ratio of the products. To meet these challenges, we developed a novel method for the 1,2,3-trifunctionalization of alkenes using alkynyl triflones as both the CF3 (ref. 6) and alkynyl sources, providing the ring-expanded cyclic β-ketoesters with excellent diastereoselectivity and functional group diversity. In addition, good functional group compatibility of this method was observed, which ensures the diverse synthetic transformations. Moreover, hydrogen bonding between the substrates and 2,2,2-trifluoroethanol solvent was revealed to be the key factor for the excellent diastereoselectivity obtained in this reaction, and this result was confirmed by both experimental and theoretical studies.
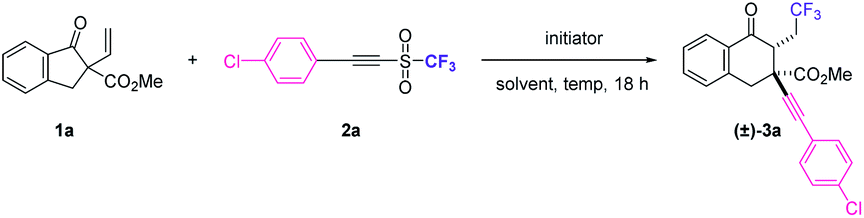
This study began by surveying radical initiators for 1,2,3-tricarbofunctionalizing α-vinyl-β-ketoester 1a with alkynyl triflone 2a11 (Table 1). Although dibenzoyl peroxide (BPO) and lauroyl peroxide (LPO) have been shown as good radical initiators for this radical reaction, the purification of the desired product (±)-3a turned out to be a great challenge12 (Table 1, entries 1 and 2). When azodiisobutyronitrile (AIBN) was used as the radical initiator, the complexity of the reaction decreased and product (±)-3a could be obtained in 63% 19F NMR yield with a diastereomeric ratio (dr) of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 3). The screening of solvents indicates that non-polar solvents such as methyl tert-butyl ether (MTBE) and dichloroethane (DCE) cannot improve the performance of the reaction (Table 1, entries 4 and 5). However, the reaction failed when another non-polar solvent, toluene, was used as the solvent, presumably due to the generation of trifluoromethylated toluene (Table 1, entry 6). When the reaction was performed in an aprotic polar solvent DMF or a protic polar solvent MeOH, only a trace amount of the desired product (±)-3a was detected (Table 1, entries 7 and 8). Interestingly, the use of 2,2,2-trifluoroethanol (TFE)13 dramatically increased the diastereoselectivity and (±)-3a could be obtained in an identical yield with an even higher dr value (dr > 20:1) (Table 1, entry 9). Increasing the reaction temperature resulted in a slight loss of yield (Table 1, entries 10 and 11), while increasing the loading amount of AIBN increased the yield of (±)-3a (Table 1, entry 12). Furthermore, by changing the loading model, the best 19F NMR yield of (±)-3a was obtained when alkynyl triflone 2a and AIBN were added as two equal portions with an interval of 9 h (Table 1, entries 13 vs. 14).14 Without the addition of a radical initiator, a reaction did not happen (Table 1, entry 15). It is worth mentioning that some amounts of the 1,2-alkynyltrifluoromethylation product (without ring-expansion) were detected in all the cases during the reaction optimization. Under optimal conditions, the desired 1,3-alkynyltrifluoromethylation product (±)-3a could be isolated in 65% yield as only syn diastereoisomers.
:1 (Table 1, entry 3). The screening of solvents indicates that non-polar solvents such as methyl tert-butyl ether (MTBE) and dichloroethane (DCE) cannot improve the performance of the reaction (Table 1, entries 4 and 5). However, the reaction failed when another non-polar solvent, toluene, was used as the solvent, presumably due to the generation of trifluoromethylated toluene (Table 1, entry 6). When the reaction was performed in an aprotic polar solvent DMF or a protic polar solvent MeOH, only a trace amount of the desired product (±)-3a was detected (Table 1, entries 7 and 8). Interestingly, the use of 2,2,2-trifluoroethanol (TFE)13 dramatically increased the diastereoselectivity and (±)-3a could be obtained in an identical yield with an even higher dr value (dr > 20:1) (Table 1, entry 9). Increasing the reaction temperature resulted in a slight loss of yield (Table 1, entries 10 and 11), while increasing the loading amount of AIBN increased the yield of (±)-3a (Table 1, entry 12). Furthermore, by changing the loading model, the best 19F NMR yield of (±)-3a was obtained when alkynyl triflone 2a and AIBN were added as two equal portions with an interval of 9 h (Table 1, entries 13 vs. 14).14 Without the addition of a radical initiator, a reaction did not happen (Table 1, entry 15). It is worth mentioning that some amounts of the 1,2-alkynyltrifluoromethylation product (without ring-expansion) were detected in all the cases during the reaction optimization. Under optimal conditions, the desired 1,3-alkynyltrifluoromethylation product (±)-3a could be isolated in 65% yield as only syn diastereoisomers.
|
|
||
|---|---|---|
| Entry | Solvent | Yieldb (%) |
| a Reaction conditions: alkene 1a (0.2 mmol, 1 equiv.), 2a (0.6 mmol, 3.0 equiv.), and AIBN (0.3 equiv.) in 3 mL of solvent at 85 °C for 18 h in a sealed tube under a nitrogen atmosphere. b Crude yield and crude diastereomeric ratio were determined by 19F NMR. c LPO was used as the initiator. d BPO was used as the initiator. e The reaction was performed at 100 °C. f The reaction was performed at 120 °C. g AIBN (60 mol%) was used. h 2a (3.0 equiv.) and AIBN (60 mol%) were added as two equal portions with an interval of 9 h. i Isolated yield in parentheses. j 2a (3.0 equiv.) and AIBN (60 mol%) were added as three equal portions with an interval of 6 h. | ||
| 1 | EA | 60 (dr = 13:1)c |
| 2 | EA | 55 (dr = 11:1)d |
| 3 | EA | 63 (dr = 12:1) |
| 4 | MTBE | 45 (dr = 10:1) |
| 5 | DCE | 63 (dr = 15:1) |
| 6 | Toluene | Trace |
| 7 | DMF | Trace |
| 8 | MeOH | Trace |
| 9 | TFE | 63 (dr > 20:1) |
| 10e | TFE | 60 (dr > 20:1) |
| 11f | TFE | 56 (dr > 20:1) |
| 12g | TFE | 70 (dr > 20:1) |
| 13h | TFE | 76 (65)i (dr > 20:1) |
| 14j | TFE | 71 (dr > 20:1) |
| 15 | TFE | Trace |
Under optimal conditions, a diverse array of α-vinyl-β-ketoesters serve as substrates in this metal-free deconstruction–construction of all-carbon quaternary centers for the synthesis of carbon-ring expanded cyclic β-ketoesters (Fig. 2). In most of the cases, excellent diastereoselectivities (dr > 20:1) were observed by crude 19F NMR analysis. Substrates with the substituents at the 5- or 6-position of the α-vinyl-β-ketoesters generally produced the corresponding product (±)-3 in higher yields than those with the substituents at the 4-position. Apart from the carbonyl group and the ester group, functional groups such as chloride ((±)-3b and (±)-3f), fluoride ((±)-3c), a methoxyl group ((±)-3d and (±)-3h), a methyl group ((±)-3e and (±)-3g) and a phenyl group ((±)-3i) can be tolerated under the reaction conditions. Notably, the phenyl ring of the core structure with two substituents reacted smoothly to afford the corresponding products ((±)-3j and (±)-3k). When substrate 1l that lacks the fused benzene ring was used for this carbon-ring expansion reaction, a dramatical loss of diastereoselectivity was detected, presumably because of the feasible interconversion of the boat and chair conformations of the intermediate. Substrates with an ethyl ester or a benzyl ester group, as opposed to a methyl ester group, delivered the corresponding products ((±)-3m and (±)-3n) with moderate yields and excellent diastereoselectivity. When the CH2 unit of the six membered-ring was replaced by a CMe2 group, only a trace amount of the desired product (±)-3o was detected. A reaction with the purpose of realizing an extension from the six-membered ring was also carried out and (±)-3p was obtained, although with a low yield and low diastereoselectivity. Notably, the diastereochemistries of products (±)-3e and (±)-3h have been confirmed by X-ray crystallography.
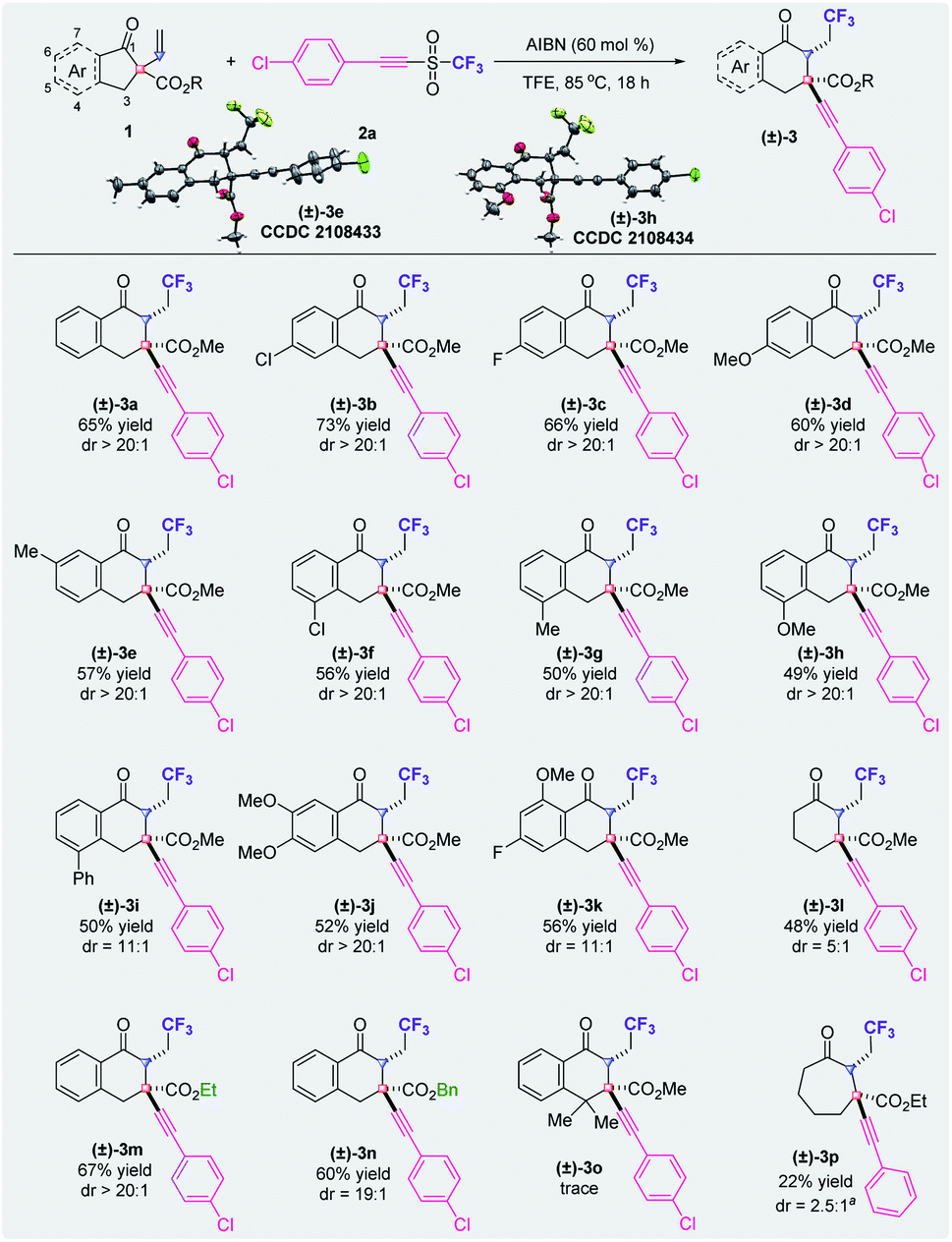 | ||
| Fig. 2 Substrate scope of α-vinyl-β-ketoesters. aThe reaction was performed with 1p and 2b. | ||
The scope with respect to the alkynyl triflones was also investigated and the results are summarized in Fig. 3. Generally, substituents on the phenyl ring of the arylethynyl moiety have little impact on the yields of the corresponding products. The functional groups at the para-, meta-, or ortho-position of the phenyl ring produced the desired products ((±)-4a–(±)-4k) with excellent diastereoselectivities. Furthermore, the method is compatible with alkynyl triflones that have a thienyl group or a perfluorobutyl group and the reactions afforded the product ((±)-4l or (±)-4m) with an excellent dr value, respectively. However, when the arylethynyl moiety was replaced by an alkylethynyl or a silylethynyl part, the reaction failed to produce the targeted tricarbofunctionalization product ((±)-4n or (±)-4o).15 Moreover, when triflic azide or (Z)-TolCHCHSO2CF3 was used in place of the alkynyl triflone, the desired product was not obtained and most of the starting material was recovered. Notably, the diastereochemistry of product (±)-4a has been confirmed by X-ray crystallography.
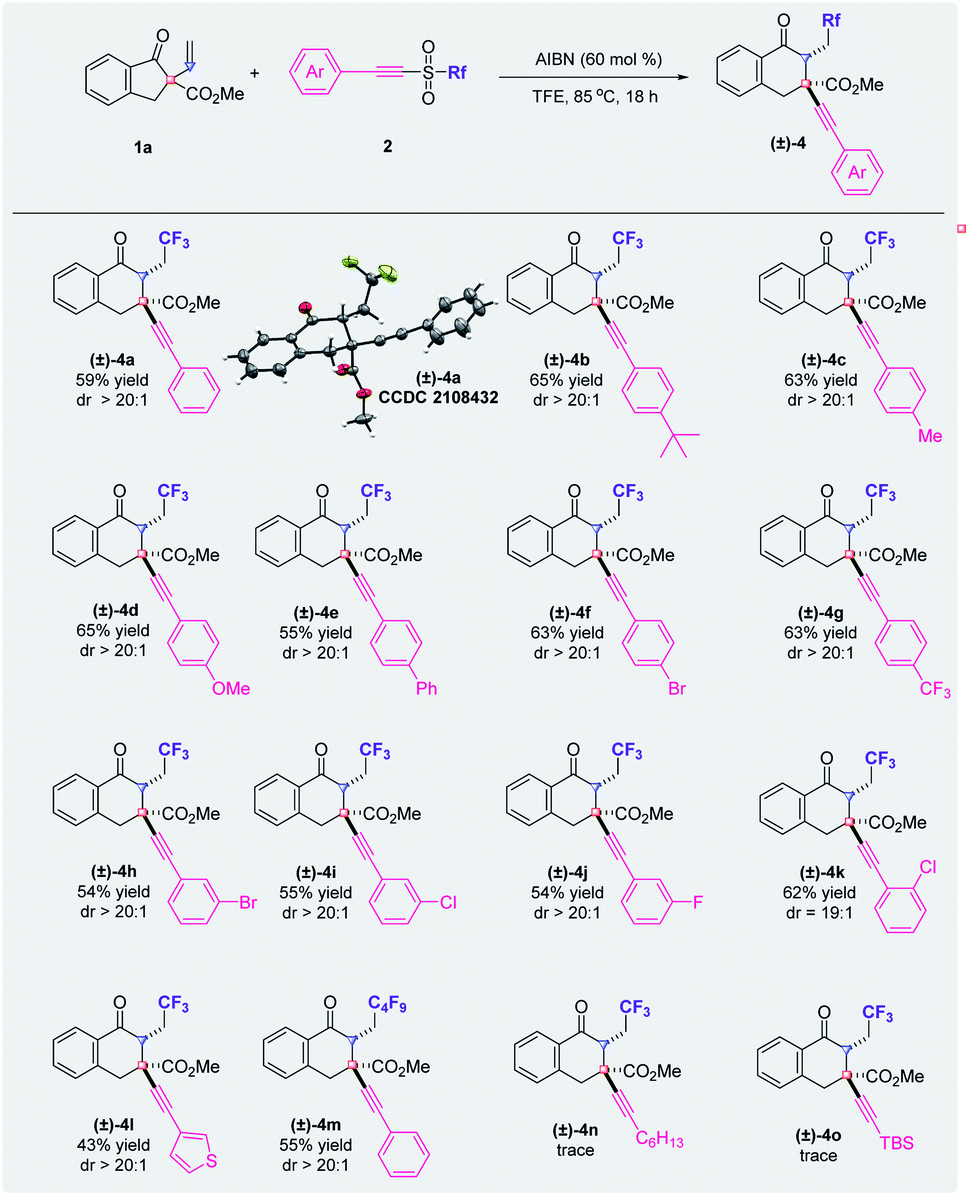 | ||
| Fig. 3 Substrate scope of alkynyl triflones. | ||
This 1,2,3-trifunctionalization reaction not only allows the deconstruction and reconstruction of all-carbon quaternary centers, but features good functional group tolerance and excellent diastereoselectivity. Regarding the diverse reactivities of these functional groups, many valuable synthetic transformations have been successfully achieved (Fig. 4). For example, the C–C triple bond of (±)-4a can be completely reduced to a CH2CH2 unit ((±)-5) in the presence of hydrogen and a Pd/C catalyst,16 while the selective reduction of (±)-4a gives rise to a Z-alkene (±)-6 when quinoline is added as an additive for the Lindlar reduction.17 The diastereochemistry of (±)-6 has been confirmed by X-ray crystallography. The selective reducing methods afford formal approaches for radical 1,3-trifluoromethylalkylation and 1,3-trifluoromethylalkenylation of α-vinyl-β-ketoesters, respectively, to produce the corresponding products which are otherwise difficult to obtain. In addition, the C–C triple bond can be oxidized under oxidative conditions with RuCl3/NaIO4, and (±)-4a can be smoothly transformed into the trifluoromethylated triketone (±)-7 in 65% yield.18 With a large excess amount of reducing agent LiAlH4, the carbonyl group and the ester group, together with the C–C triple bond, can be unexpectedly reduced simultaneously, affording the alkenyl diol (±)-8 in excellent regioselectivity. The hydrolysis process under basic conditions provided a reliable method for access to a free carboxylic acid (±)-9. Interestingly, when the reaction was performed under milder conditions compared to those for the synthesis of (±)-8, (±)-4a was successfully converted into an alkynyl diol (±)-10, which can be cyclized into a spiro compound (±)-11 (ref. 19) and an endocyclic compound (±)-12,20 respectively. Notably, in the majority of these cases, the excellent diastereoselectivity was reserved. These synthetic applications can demonstrate the significant value of this method.
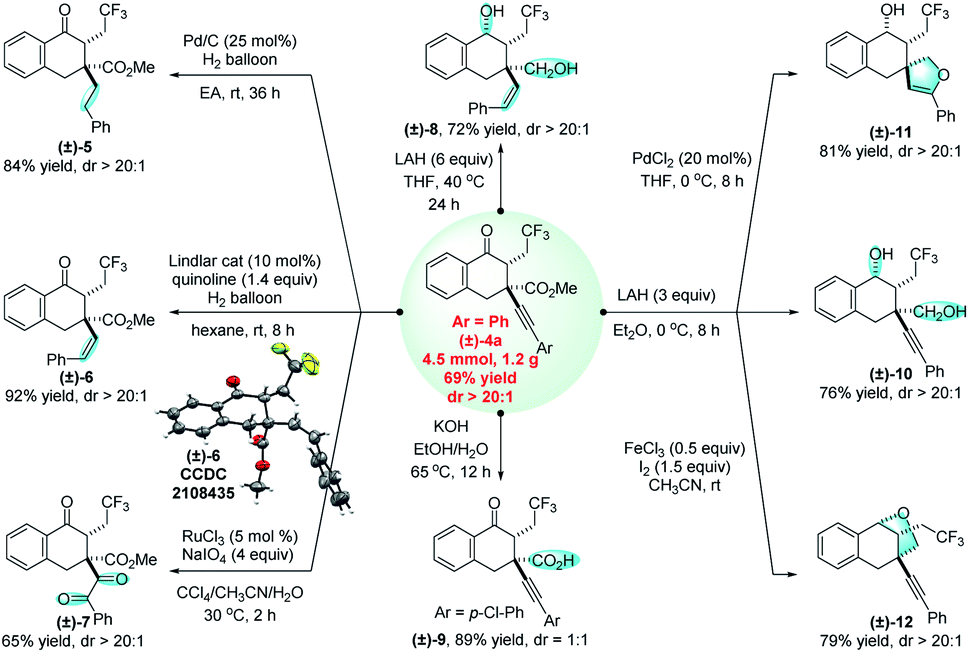 | ||
| Fig. 4 Synthetic transformations. | ||
In order to gain some mechanistic insights into this radical cascade reaction, subsequent efforts have been made (Fig. 5). First, the detection of trifluoromethylated toluene (with toluene as the solvent, Table 1, entry 6), TEMPO–CMe2(CN) adduct (with TEMPO additive), and a 1,2-trifluoromethylalkynylation product of 1,1-diphenylethylene (with 1,1-diphenylethylene additive) by GC-MS analysis suggested that the reaction proceeded through a radical pathway (Fig. 5a, see ESI† for details). Second, we were curious about the excellent diastereoselectivity associated with the use of TFE as the solvent. As can be seen in Fig. 5b, 1H NMR titration of 1a with increasing amounts of TFE showed a chemical shift of the resonance signal corresponding to protons. The 2D NOESY spectrum indicates the existence of an interaction between 1a and TFE (Fig. 5c). Moreover, Job plot studies by both 1H NMR and 19F NMR imply a 1:1.5 stoichiometry of the complex adduct resulting from 1a and TFE (Fig. 5d). These mechanistic studies strongly suggest that the excellent diastereoselectivity of this reaction might be attributed to the hydrogen bonding between TFE and the α-vinyl-β-ketoester.
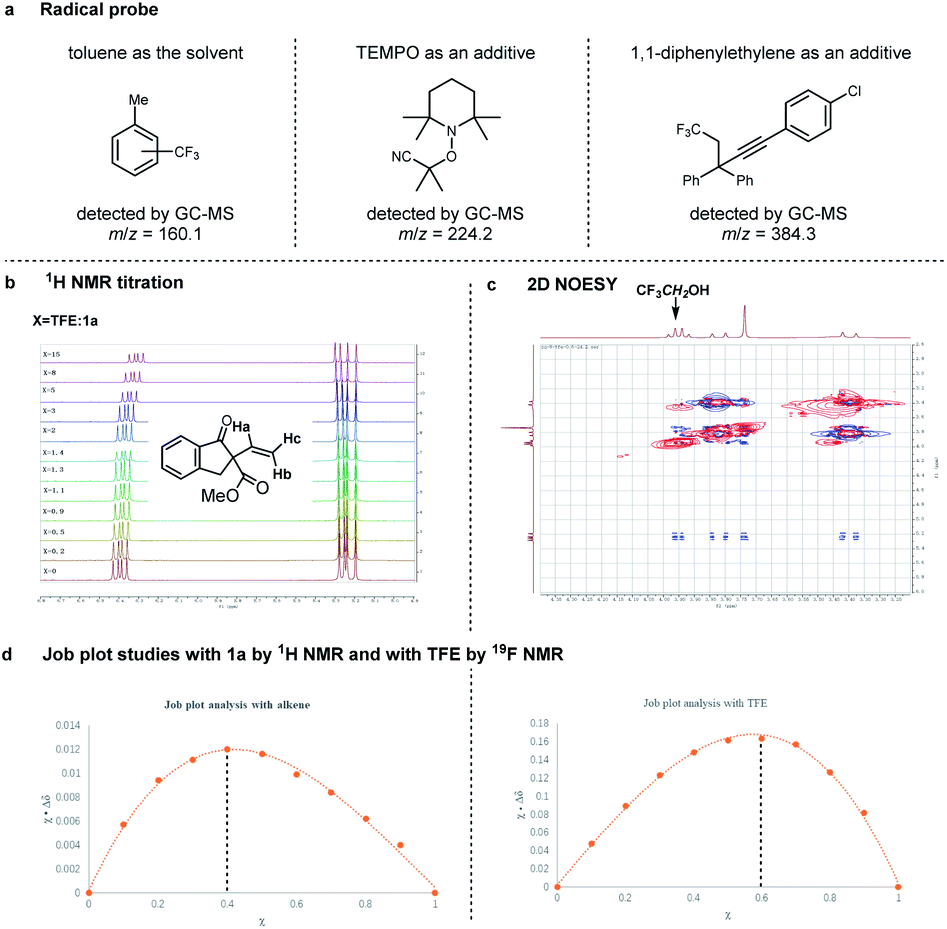 | ||
| Fig. 5 Mechanism studies. (a) Radical probe; (b) 1H NMR titration; (c) 2D NOESY; (d) Job plot studies. | ||
On the other hand, density functional theory (DFT) calculations have also been performed at the B3LYP-D3(SMD)/Def2-TZVP//B3LYP-D3(SMD)/Def2-SVP level of theory in the TFE solvent model to further investigate the reaction pathways (Fig. 6). On the basis of the experimental results, herein, the radical pathway was considered. Initially, the CF3 radical addition onto 1a was calculated, and a transition state, TS1, was located with a free energy barrier of 10.9 kcal mol−1 to deliver the radical intermediate int1 with an exergonicity of 20.5 kcal mol−1. Then, a bicyclic transition state, TS2,21 with a barrier of 11.0 kcal mol−1 through a concerted 1,2-shift route was found to be the lower barrier TS for int2 formation than that of the addition to 2b for the byproduct (see Fig. S5 in ESI†), which is consistent with the experimental results of the mainly hexacyclic products. Moreover, the intrinsic reaction coordinate (IRC) calculations and the root mean square (RMS) gradient of the potential energy surface from TS2 suggested that no transition state for the formation of the previously proposed strained alkoxyl radical was found. Next, the radical intermediate int2 attacking 2b was calculated. To understand the diastereoselectivity of this step, the transition states of the addition of 2b onto the Re and Si faces of C3 in int2 were located with barriers of 12.5 and 17.4 kcal mol−1 (TS3 and TS3′), respectively. It is noteworthy that the torsion angle of C1–C2–C3–C4 in TS3′ is −62.3°, larger than that of −40.9° in int2 and −49.0° in TS3, indicating that the distortion factor in TS3′ is large due to the steric effect from the trifluoroethyl group in int2 and, therefore, increases the barrier. The transition states of 2b addition were also optimized in solvents DCE and EA, and the free energy barrier differences between TS3 and TS3′ [ΔG‡ = G‡(TS3′) − G‡(TS3)] are 3.6 and 3.0 kcal mol−1, respectively, in agreement with the experimental observations. Finally, dissociation of a SO2 molecule with a CF3 radical from int3 to deliver the product was conducted, and a transition state TS4 with a much lower barrier of only 7.1 kcal mol−1 was located, which led to the major product (±)-4a with a relative free enthalpy of −51.6 kcal mol−1.
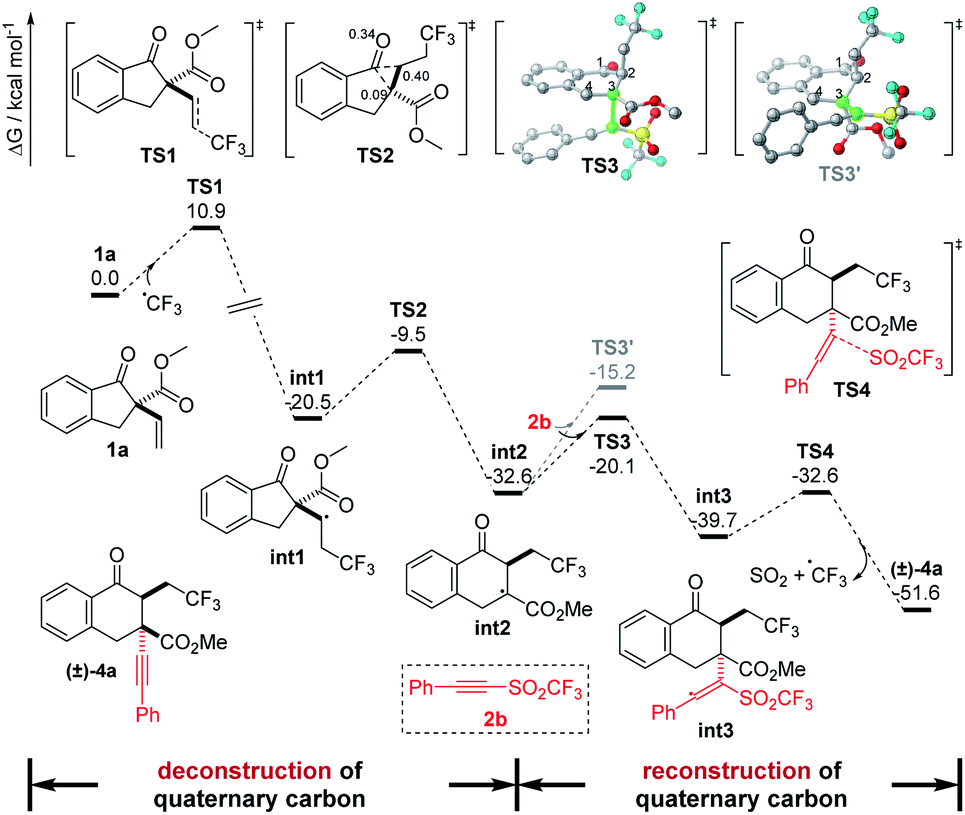 | ||
| Fig. 6 Gibbs free energy profile for the synthesis of 4a in the TFE solvent model. | ||
Conclusions
In conclusion, we have developed an intermolecular 1,2,3-tricarbofunctionalization of α-vinyl-β-ketoesters through a radical process of deconstruction and construction of all-carbon quaternary centers. This one-pot reaction allows the expansion of the carbon ring by a carbon shift from the all-carbon quaternary center and enables further C–C bond formation on the tertiary carbon intermediate with the aim to reconstruct a new all-carbon quaternary center. The high level of functional group compatibility ensures the diverse synthetic transformations of this method. The experimental and theoretical studies reveal that the hydrogen bonding between the substrates and trifluoroethanol solvent is beneficial to obtaining the excellent diastereoselectivity.Data availability
All experimental and characterization data, as well as DFT calculation data are available in the ESI.†Author contributions
H. Bao and Y. Li directed the investigations. Y. Li prepared the manuscript. Q. Zhang, C. Ye, and X. Yuan performed the synthetic experiments and analyzed the experimental data. M.-F. Chiou cunducted the theoretical calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (Grant No. 22001251, 21871258, 21922112), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000), and the National Key R&D Program of China (Grant No. 2017YFA0700103).Notes and references
- (a) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083–4088 RSC; (b) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed; (c) R. M. Romero, T. H. Woste and K. Muniz, Chem.–Asian J., 2014, 9, 972–983 CrossRef CAS PubMed; (d) T. Besset, T. Poisson and X. Pannecoucke, Eur. J. Org. Chem., 2015, 2015, 2765–2789 CrossRef CAS; (e) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413–2423 CrossRef CAS PubMed; (f) X.-W. Lan, N.-X. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 2017, 5821–5851 CrossRef CAS; (g) Y. Tian, S. Chen, Q.-S. Gu, J.-S. Lin and X.-Y. Liu, Tetrahedron Lett., 2018, 59, 203–215 CrossRef CAS; (h) F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS PubMed; (i) Z. L. Li, G. C. Fang, Q. S. Gu and X. Y. Liu, Chem. Soc. Rev., 2020, 49, 32–48 RSC.
- (a) Y. Li, D. Qiu, R. Gu, J. Wang, J. Shi and Y. Li, J. Am. Chem. Soc., 2016, 138, 10814–10817 CrossRef CAS PubMed; (b) W. Zhao and J. Montgomery, J. Am. Chem. Soc., 2016, 138, 9763–9766 CrossRef CAS PubMed.
- (a) K. Jana, A. Bhunia and A. Studer, Chem, 2020, 6, 512–522 CrossRef CAS; (b) K. Jana and A. Studer, Org. Lett., 2022, 24, 1100–1104 CrossRef CAS PubMed.
- Z. Li, M. Wang and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 186–190 CrossRef CAS PubMed.
- (a) S. F. Martin, Tetrahedron, 1980, 36, 419–460 CrossRef CAS; (b) E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388–401 CrossRef; (c) K. Fuji, Chem. Rev., 2002, 93, 2037–2066 CrossRef; (d) B. M. Trost and C. Jiang, Synthesis, 2006, 369–396 CrossRef CAS; (e) M. Shimizu, Angew. Chem., Int. Ed., 2011, 50, 5998–6000 CrossRef CAS PubMed; (f) B. Wang and Y. Q. Tu, Acc. Chem. Res., 2011, 44, 1207–1222 CrossRef CAS PubMed; (g) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed; (h) X. P. Zeng, Z. Y. Cao, Y. H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS PubMed; (i) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS PubMed; (j) Y. Li and S. Xu, Chem.–Eur. J., 2018, 24, 16218–16245 CrossRef CAS PubMed; (k) K.-F. Zhang, K.-J. Bian, C. Li, J. Sheng, Y. Li and X.-S. Wang, Angew. Chem., Int. Ed., 2019, 58, 5069–5074 CrossRef CAS PubMed; (l) Y. e. You and S. Ge, Angew. Chem., Int. Ed., 2021, 60, 12046–12052 CrossRef CAS PubMed; (m) C. Wang and S. Ge, J. Am. Chem. Soc., 2018, 140, 10687–10690 CrossRef CAS PubMed.
- (a) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS PubMed; (b) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC; (c) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765–825 CrossRef CAS PubMed; (d) S. Barata-Vallejo, M. V. Cooke and A. Postigo, ACS Catal., 2018, 8, 7287–7307 CrossRef CAS; (e) H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC.
- (a) W. Deng, W. Feng, Y. Li and H. Bao, Org. Lett., 2018, 20, 4245–4249 CrossRef CAS PubMed; (b) H. Xiong, N. Ramkumar, M.-F. Chiou, W. Jian, Y. Li, J.-H. Su, X. Zhang and H. Bao, Nat. Commun., 2019, 10, 122 CrossRef PubMed; (c) X. Zhu, W. Deng, M.-F. Chiou, C. Ye, W. Jian, Y. Zeng, Y. Jiao, L. Ge, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2019, 141, 548–559 CrossRef CAS PubMed; (d) L. Ge, H. Zhou, M.-F. Chiou, H. Jiang, W. Jian, C. Ye, X. Li, X. Zhu, H. Xiong, Y. Li, L. Song, X. Zhang and H. Bao, Nat. Catal., 2020, 4, 28–35 CrossRef; (e) M. Israr, H. Xiong, Y. Li and H. Bao, Adv. Synth. Catal., 2020, 362, 2211–2215 CrossRef CAS; (f) M. Taj Muhammad, Y. Jiao, C. Ye, M.-F. Chiou, M. Israr, X. Zhu, Y. Li, Z. Wen, A. Studer and H. Bao, Nat. Commun., 2020, 11, 416 CrossRef CAS PubMed; (g) Q. Zhang, M. T. Muhammad, M.-F. Chiou, Y. Jiao, H. Bao and Y. Li, Org. Lett., 2020, 22, 5261–5265 CrossRef CAS PubMed.
- (a) A. L. J. Beckwith, D. M. O'Shea, S. Gerba and S. W. Westwood, J. Chem. Soc., Chem. Commun., 1987, 666–667 RSC; (b) P. Dowd and S.-C. Choi, J. Am. Chem. Soc., 1987, 109, 3493–3494 CrossRef CAS; (c) P. Dowd and S.-C. Choi, J. Am. Chem. Soc., 1987, 109, 6548–6549 CrossRef CAS; (d) A. L. J. Beckwith, D. M. O'Shea and S. W. Westwood, J. Am. Chem. Soc., 1988, 110, 2565–2575 CrossRef CAS.
- (a) W. Zhang and P. Dowd, Chem. Rev., 1993, 93, 2091–2115 CrossRef; (b) L. Yet, Tetrahedron, 1999, 55, 9349–9403 CrossRef CAS.
- L. Chen, L. N. Guo, S. Liu, L. Liu and X. H. Duan, Chem. Sci., 2020, 12, 1791–1795 RSC.
- (a) L. Wang, Y. Xia, K. Bergander and A. Studer, Org. Lett., 2018, 20, 5817–5820 CrossRef CAS PubMed; (b) F. Le Vaillant and J. Waser, Chem. Sci., 2019, 10, 8909–8923 RSC; (c) Y. Xia and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9836–9840 CrossRef CAS PubMed; (d) X.-Q. Chu, D. Ge, Y.-Y. Cui, Z.-L. Shen and C.-J. Li, Chem. Rev., 2021, 121, 12548–12680 CrossRef CAS PubMed; (e) Y. Yang, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 2145–2148 CrossRef CAS PubMed.
- The desired product was always obtained with some amount of impurities.
- (a) J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS PubMed; (b) K. Ebisawa, K. Izumi, Y. Ooka, H. Kato, S. Kanazawa, S. Komatsu, E. Nishi and H. Shigehisa, J. Am. Chem. Soc., 2020, 142, 13481–13490 CrossRef CAS PubMed; (c) X. L. Lai, X. M. Shu, J. Song and H. C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS PubMed; (d) T. Sheridan, H. G. Yayla, Y. Lian, J. Genovino, N. Monck and J. W. Burton, Eur. J. Org. Chem., 2020, 2020, 2766–2770 CrossRef CAS.
- L. Ge, Y. Li and H. Bao, Org. Lett., 2019, 21, 256–260 CrossRef CAS PubMed.
- Z. Xiong, F. Zhang, Y. Yu, Z. Tan and G. Zhu, Org. Lett., 2020, 22, 4088–4092 CrossRef CAS PubMed.
- M. Q. Tian, Z. Y. Shen, X. Zhao, P. J. Walsh and X. H. Hu, Angew. Chem., Int. Ed., 2021, 60, 9706–9711 CrossRef CAS PubMed.
- X. Gao, R. Cheng, Y.-L. Xiao, X.-L. Wan and X. Zhang, Chem, 2019, 5, 2987–2999 CAS.
- X. Y. Dong, J. T. Cheng, Y. F. Zhang, Z. L. Li, T. Y. Zhan, J. J. Chen, F. L. Wang, N. Y. Yang, L. Ye, Q. S. Gu and X. Y. Liu, J. Am. Chem. Soc., 2020, 142, 9501–9509 CrossRef CAS PubMed.
- T. Saito, T. Suzuki, M. Morimoto, C. Akiyama, T. Ochiai, K. Takeuchi, T. Matsumoto and K. Suzuki, J. Am. Chem. Soc., 1998, 120, 11633–11644 CrossRef CAS.
- L. Su, C.-Y. Lei, W.-Y. Fan and L.-X. Liu, Synth. Commun., 2011, 41, 1200–1207 CrossRef CAS.
- (a) D. Ardura and T. L. Sordo, Tetrahedron Lett., 2004, 45, 8691–8694 CrossRef CAS; (b) D. Ardura and T. L. Sordo, J. Org. Chem., 2005, 70, 9417–9423 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2108432–2108435. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00902a |
| This journal is © The Royal Society of Chemistry 2022 |