
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegiocontrolled allylic functionalization of internal alkene via selenium-π-acid catalysis guided by boron substitution†
Ling
Yang
,
Yuan
Liu
,
Wen-Xin
Fan
,
Dong-Hang
Tan
,
Qingjiang
Li
 and
Honggen
Wang
*
and
Honggen
Wang
*
Guangdong Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, People's Republic of China. E-mail: wanghg3@mail.sysu.edu.cn
First published on 22nd April 2022
Abstract
The selenium-π-acid-catalysis has received increasing attention as a powerful tool for olefin functionalization, but the regioselectivity is often problematic. Reported herein is a selenium-catalyzed regiocontrolled olefin transpositional chlorination and imidation reaction. The reaction outcome benefits from an allylic B(MIDA) substitution. And the stabilization of α-anion from a hemilabile B(MIDA) moiety was believed to be the key factor for selectivity. Broad substrate scope, good functional group tolerance and generally good yields were observed. The formed products were demonstrated to be valuable precursors for the synthesis of a wide variety of structurally complex organoborons.
Introduction
The past few years have witnessed considerable progress in electrophilic chalcogen-catalysis.1 In this research realm, the selenium-π-acid catalysis,2 wherein a selenium catalyst chemoselectively activates an alkene or alkyne, is particularly interesting by offering unique reactivities which could be complementary to those of transition metal catalysts3 (e.g., gold,4 platinum5 and palladium6 complexes). The high carbophilicity of selenium catalyst offers additional advantages such as good functional group tolerance, simplicity of synthetic operation and mild conditions. However, the utility of selenium-π-acid catalysis may be plagued by the intrinsically challenging regioselectivity when nonpolarized internal alkenes are used as reaction partners (Scheme 1a).2c In a typical reaction manifold, an initial coordination of selenium ion onto olefinic π-bond leads to the formation of a highly electrophilic seleniranium intermediate A. This intermediate could be stereoselectively, but not regioselectively, trapped by a nucleophile to form two sets of selenide adducts (B and C). Depending on the reaction parameters (for examples, substituents on the double bond and the oxidant), both adducts could be potentially converted to either allylic, vinylic or 1,2-difunctionalization product.1 This was reflected by the seminal work of Sharpless,7 wherein the use of unsymmetrical internal alkenes produced the unselective chlorination mixtures (Scheme 1b). | ||
| Scheme 1 Selenium-π-acid catalysis and our work. | ||
To address the regioselectivity issue, the most commonly used and fruitful strategy is to run the process intramolecularly by introducing a pendent nucleophile to the substrates.8 Also effective is the employment of tri-substituted alkenes as substrates, which usually leads to clean tertiary functionalized products as guided resided at the allylic position can function as a directing group by the Markovnikov's rule.9 A recent study from Zhao suggests that the hydroxyl group (via precoordination with the selenium cation) to enable a selective anti-Markovnikov aza-Wacker imidation reaction (Scheme 1c).10 A more general solution is by electronic perturbation via the installation of an electron-withdrawing group (EWG) at the allylic position, as firstly reported by Tunge (Scheme 1d).11 Thus, by virtue of the reversibility of the chloroselenylation and oxidation processes (A → C, Scheme 1a),12 a rate-limiting dehydrodeselenylative elimination eventually leads to an allylic chloride product. The acidity of the proton α to the EWG accounts for the profound regioselectivity by facilitating the irreversible deprotonation process. Linear alkenes bearing no allylic EWGs result in complex product mixtures. Similar strategy was later found to be applicable to the elegant selenium-π-acid catalytic amination,13 oxygenation9b,14 and fluorination9c,15 reactions.
On the other hand, the protection of sp2-B organoboron into the pyramidalized sp3-B MIDA (N-methyliminodiacetic acid) boronates brings about tremendously improved stability, which thereby offers new opportunities to the late-stage modification of organoborons.16 In this context, previous studies have showcased that in certain cases, the MIDA boron moiety is by no means a bystander, but could confer interesting reactivity to the substrates.17 For instance, independent work from Yudin18 and us19 uncovered an intriguing directing effect of MIDA boron in metal-catalyzed functionalized of alkenes and alkynes, respectively. This effect is attributed to a neighboring stereoelectronic stabilizing donation of electron density to boron, thanks to the hemilabile nature of the MIDA B–N dative bond.20 In line with the selenium-π-acid catalysis, we reason the similar neighboring group effect would offer a chance for a regiocontrolled allylic functionalization reactions. Herein, we report our realization of a selenium-catalyzed double bond transpositional allylic chlorination and imidation of allyl MIDA boronates (Scheme 1e). The reaction leads to the facile synthesis of multifunctional amphoteric building blocks with both the olefin and boron functionality being preserved as valuable handles for further chemical elaboration.
Results and discussion
To start, we examined the feasibility of MIDA boron-directed allylic chlorination. The reaction of allyl MIDA boronate 1a with stoichiometric amount of PhSeCl as both the oxidant and chlorine source was firstly investigated. A brief survey revealed that mixing 1a with PhSeCl (2.5 equiv.) in THF in the presence of 4 Å molecular sieve provided cleanly a single allylic chlorination product 2a in 75% yield (eqn (1)). The double bond was selectively transpositioned towards the boron moiety and only the E geometric isomer was formed as expected. We also turned our attention to the catalytic version of this reaction. And was found that with 10 mol% of PhSeCl catalyst and N-chlorosuccinimide (NCS, 1.1 equiv.) in MeCN, a comparable yield of 78% was obtained. Considering the potential inhibition effect of NCS as observed by Tunge,11 the slow addition of NCS via a syringe pump gives an improved yield of 84% (eqn (2)). | (1) |
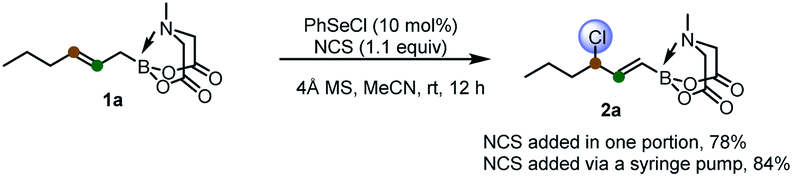 | (2) |
Encouraged by these results and considering the importance of allyl amines, we further attempted to explore the applicability of this concept to allylic functionalization imidation reactions. With N-fluorobenzenesulfonimide (NFSI, 1.0 equiv.) as oxidant and diphenyl diselane (5 mol%) as catalyst in THF at room temperature, an allylic imidation product (25% yield) was indeed formed with most of the staring materials being untouched (Table 1, entry 1). The site and stereoselectivity were in accordance with the above chlorination protocol, confirming the versatility of boron-directing effect. A higher yield was obtained by changing the solvent to MeCN (entries 2 and 3). Further improvements were achieved by introducing the molecular sieve (entry 4), increasing the oxidant dosage (entry 5), and slightly raising the reaction temperature (entry 6). And finally, the use of DCE as solvent in associated with 10 mol% of catalyst were able to reduce the oxidant (to 1.5 equiv.) while enhancing the yield to 68% (entries 7–9).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | x | y | Solvent | Temp. | 4 Å MS | % yield |
| a Reaction condition: 1a (0.2 mmol), NFSI (x equiv.), (PhSeSePh, y mol%), 4 Å MS (mg), temp. (°C), solvent, under Ar, 12 h. | ||||||
| 1 | 1 | 5 | THF | 25 | 0 | 25 |
| 2 | 1 | 5 | 1,4-Dioxane | 25 | 0 | 21 |
| 3 | 1 | 5 | MeCN | 25 | 0 | 29 |
| 4 | 2 | 5 | MeCN | 25 | 10 | 32 |
| 5 | 2 | 5 | MeCN | 25 | 20 | 43 |
| 6 | 2 | 5 | MeCN | 35 | 20 | 55 |
| 7 | 2 | 5 | DCE | 35 | 20 | 67 |
| 8 | 1.5 | 5 | DCE | 35 | 20 | 61 |
| 9 | 1.5 | 10 | DCE | 35 | 20 | 68 |
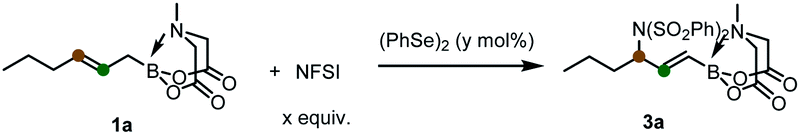
With the optimized conditions in hand, the substrate scopes for both the oxidation allylic chlorination and imidation reactions were explored. As shown in Scheme 2, a wide variety of alkyl-substituted allyl MIDA boronates were well applicable, generating the γ-chlorinated/imidized alkenyl boronates both as single regio- and E-geometric isomers in moderate to good yields. Some commonly encountered functional groups, such as ester (1f, 1l, 1m), phenoxy (1g), alkoxy (1h), acetal (1i), amide (1j), carbamate (1k), halogen (1n) were all well tolerated, indicating the mildness of the protocols. Secondary allyl MIDA boronates showed no reactivity in this reaction, probably for steric reasons.
 | ||
| Scheme 2 Substrate scope. Chlorination: 1a (0.2 mmol), NCS (1.1 equiv.), PhSeCl (10 mol%), 4 Å MS, MeCN, rt. aNCS (dissolved in MeCN) was added via syringe pump. Imidation: 1a (0.2 mmol), NFSI (1.5 equiv.), PhSeSePh (10 mol%), 4 Å MS, DCE, 35 °C, under Ar, 12 h. | ||
Substrate 1o with a Z geometry also successfully delivered the desired chlorination product 2o with E configuration in good yield (eqn (3)). However, the imidation product 3o was formed in low yield, probably due to the greater steric hindrance of nitrogen-centered nucleophile. The reaction of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1d and Z-1d produced E product 2d stereo-convergently (eqn (4)).
:1 mixture of 1d and Z-1d produced E product 2d stereo-convergently (eqn (4)).
 | (3) |
 | (4) |
The utility of the formed products was then investigated (Scheme 3). The allylic C–Cl bond in 2a provided the valuable handle for follow-up functional group manipulations. For examples, subjecting 2a to palladium-catalyzed Tsuji–Trost reactions using malonate and sulfinate as nucleophiles delivered the corresponding allylic alkylation (4) and sulfonylation (6) products, respectively, in good yields. A copper-catalyzed displacement of chloride furnished an allyl fluoride 7 without difficulty.21 In addition, the iodofluorination of the double bond in 2a following our previous protocol22 produced the α-iodo-β-fluoro-γ-chloroalkyl MIDA boronate 8 (confirmed by X-ray single crystal diffraction analysis)23 with good regio- and stereoselectivity. And the epoxidation reaction gave efficiently a highly functionalized amphoteric epoxide 9. Finally, the transesterification reaction of the B(MIDA) moiety with pinacol allowed the efficient synthesis of alkenyl Bpin 10. And under a slow-release reaction conditions, the palladium-catalyzed Suzuki–Miyaura coupling with aryl iodide was successful to provide the styrenyl product 5.
 | ||
| Scheme 3 Synthetic derivatizations of 2a. Reaction condition: [a] dimethyl malonate, NaH, Pd(OAc)2, PPh3, THF, rt.; [b] Pd(OAc)2, Sphos, p-iodoanisole, 1 M NaOH, THF, rt.; [c] sodium benzenesulfinate, Pd(PPh3)4, THF/DMSO, 50 °C; [d] AgF, CuBr, MeCN, rt.; [e] DIH, Et3N·HF, DCM, rt.; [f] mCPBA, DCM, 30 °C; [g] pinacol, 2 M H2SO4, THF, rt., 1H NMR yield; [h] isolated yield. For more details, see the ESI.† DIH = 1,3-diiodo-5,5-dimethylhydantoin. | ||
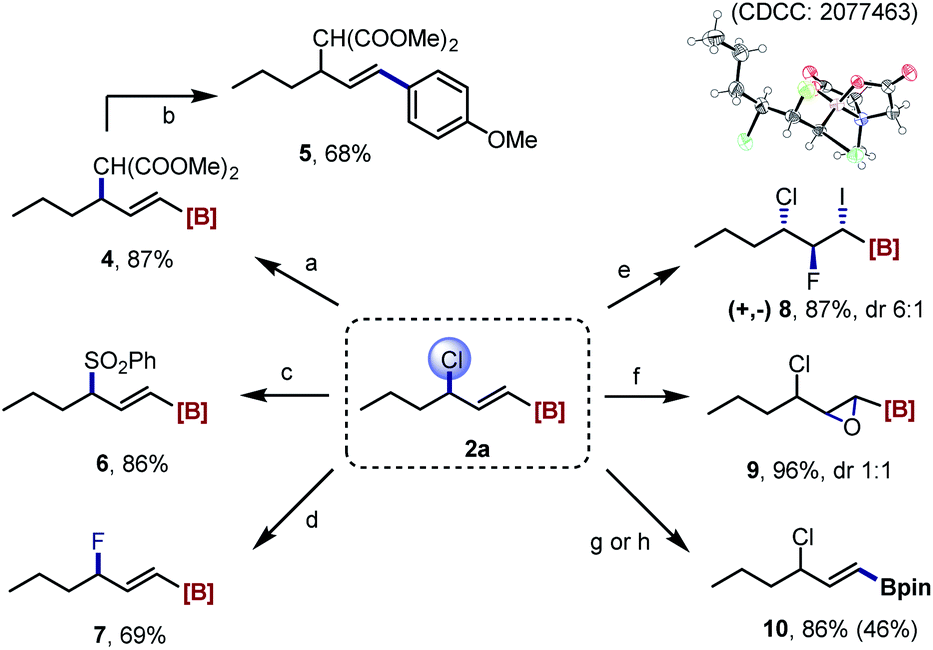
Interestingly, reacting the imidation product 3a with NaN3 provided the γ-azidated alkenyl MIDA boronate 11, which can then be applicable to the iodofluorination reaction as well (12). A E2 elimination occurred by heating 3a with NaI under microwave irradiation to give a boryl-substituted diene 13 (Scheme 4).
 | ||
| Scheme 4 Synthetic derivatizations of 3a. Reaction condition: [a] NaN3, NaI, DMF, 100 °C; [b] DIH, Et3N·HF, DCM, rt.; [c] NaI, DMF, 140 °C (microwave). For more details, see the ESI.† | ||
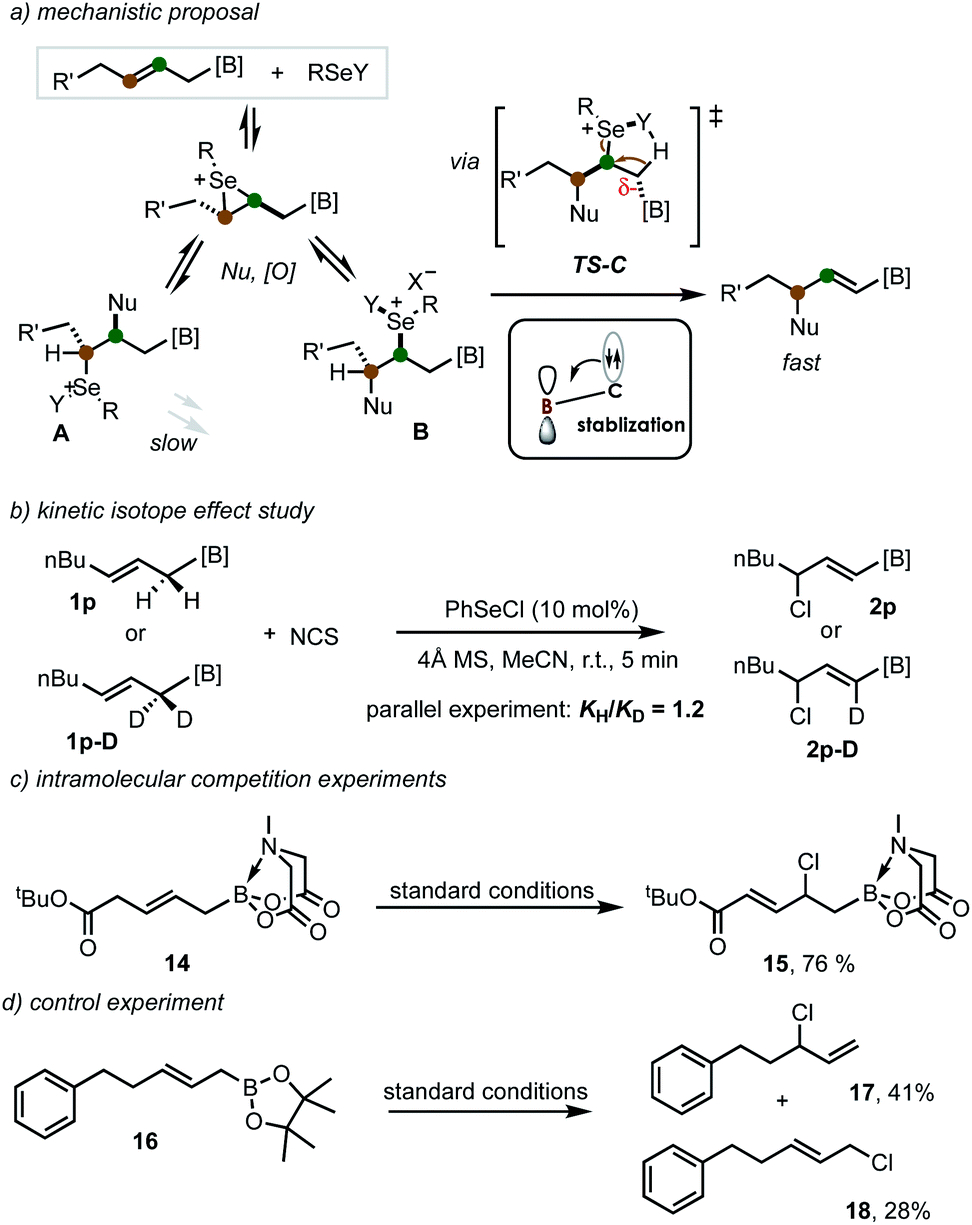
Based on the previous observations on the electron-withdrawing group (EWG)-directed allylic functionalization11–14,15b and the notion on the capacity of B(MIDA) to accept adjacent electron density,18–20 a possible reaction mechanism was proposed (Scheme 5a). The reversible olefin coordination/nucleophilic substitution/selenide oxidation forms two regioisomeric intermediates A and B. Both are prone to undergo dehydrodeselenylation or nucleophilic displacement of the selenium moiety. However, the activation effect from B(MIDA) by stabilizing the developing negative charge in the transition state TS-C would facilitate a regioselective syn-dehydrodeselenenylation, thereby shifting the equilibrium towards the alkenyl MIDA boronate formation.24 Interestingly, previous evidence from Tunge has suggested that the dehydrodeselenylative elimination is the rate-limiting step.25 However, kinetic isotope effect studies in our case by using deuterated allyl MIDA boronate revealed a small KIE value of 1.2 (Scheme 5b). This result indicates that the C–H cleavage is no longer involved in the rate-limiting step, in accordance with an activation effect of B(MIDA) (by lowering the energy of TS-C) in this reaction. Interestingly, allyl MIDA boronate 14 with an electron-withdrawing ester group on the other side produced α,β-unsaturated ester 15, suggesting the ester group showed a greater directing ability (Scheme 5c). And the use of pinacol boronate only led to the formation of protodeborylation products (69% yield as a mixture, Scheme 5d).
 | ||
| Scheme 5 Proposed mechanism and mechanistic study. | ||
Conclusions
In summary, a selenium-π-acid-catalyzed regiocontrolled olefin transpositional allylic chlorination and imidation were developed. The reaction outcome benefits from a B(MIDA) substitution. And the stabilization of α-anion from a hemilabile B(MIDA) moiety was believed to be the key factor for selectivity, which facilitates the irreversible selenium-mediated syn-H elimination. The protocol led to the synthesis of a wide array of multifunctional amphoteric building blocks with both the olefin and boron functionality being preserved as valuable handles for further chemical elaboration. The utilities of the products were demonstrated. The unique effect of B(MIDA) revealed in this protocol may inspire more interesting boron chemistry from the synthetic community.Data availability
Experimental data has been provided as ESI.†Author contributions
L. Y. discovered the reaction, performed the experiments and analysed the data. H. W. conceived and directed the project. H. W. and L. Y. wrote the manuscript. Y. L. and W. F. synthesized some raw materials. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22022114, 21971261), the Guang-dong Basic and Applied Basic Research Foundation (2020A1515010624), the Fundamental Research Funds for the Central Universities (20ykzd12) and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y093).Notes and references
- L. Bayeh, P. Q. Le and U. K. Tambar, Nature, 2017, 547, 196 CrossRef CAS PubMed.
- (a) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS PubMed; (b) A. Breder and S. Ortgies, Tetrahedron Lett., 2015, 56, 2843 CrossRef CAS; (c) S. Ortgies and A. Breder, ACS Catal., 2017, 7, 5828 CrossRef CAS; (d) R. Guo, L. Liao and X. Zhao, Molecules, 2017, 22, 835 CrossRef PubMed; (e) V. Rathore, C. Jose and S. Kumar, New J. Chem., 2019, 43, 8852 RSC; (f) L. Shao, Y. Li, J. Lu and X. Jiang, Org. Chem. Front., 2019, 6, 2999 RSC; (g) F. V. Singh and T. Wirth, Catal. Sci. Technol., 2019, 9, 1073 RSC; (h) L. Liao and X. Zhao, Synlett, 2021, 32, 1262 CrossRef CAS.
- (a) Y. Yamamoto, J. Org. Chem., 2007, 72, 7817 CrossRef CAS PubMed; (b) F. Guerra, A. García-Cabeza, F. Moreno-Dorado and M. Ortega, Synthesis, 2016, 48, 2323 CrossRef; (c) S. Muthusamy, N. Kumarswamyreddy, V. Kesavan and S. Chandrasekaran, Tetrahedron Lett., 2016, 57, 5551 CrossRef CAS.
- (a) A. Corma, A. Leyva-Perez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (b) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed; (c) A. Furstner, Angew. Chem., Int. Ed., 2018, 57, 4215 ( Angew. Chem. , 2018 , 130 , 4289 ) CrossRef PubMed.
- A. Furstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 ( Angew. Chem. , 2007 , 46 , 3410 ) CrossRef PubMed.
- (a) I. I. Moiseev and M. N. Vargaftik, Coord. Chem. Rev., 2004, 248, 2381 CrossRef CAS; (b) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed; (c) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS PubMed.
- (a) T. Hori and K. B. Sharpless, J. Org. Chem., 2002, 44, 4204 CrossRef; (b) T. Hori and K. B. Sharpless, J. Org. Chem., 2002, 44, 4208 CrossRef.
- (a) D. M. Browne, O. Niyomura and T. Wirth, Org. Lett., 2007, 9, 3169 CrossRef CAS PubMed; (b) F. V. Singh and T. Wirth, Org. Lett., 2011, 13, 6504 CrossRef CAS PubMed; (c) R. Guo, J. Huang, H. Huang and X. Zhao, Org. Lett., 2016, 18, 504 CrossRef CAS PubMed; (d) Y. Kawamata, T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2016, 138, 5206 CrossRef CAS PubMed; (e) S. Ortgies, R. Rieger, K. Rode, K. Koszinowski, J. Kind, C. M. Thiele, J. Rehbein and A. Breder, ACS Catal., 2017, 7, 7578 CrossRef CAS; (f) A. Breder, K. Rode, M. Palomba, S. Ortgies and R. Rieger, Synthesis, 2018, 50, 3875 CrossRef.
- (a) S. Raucher, Tetrahedron Lett., 1977, 18, 3909 CrossRef; (b) S. Torii, K. Uneyama, M. Ono and T. Bannou, J. Am. Chem. Soc., 1981, 103, 4606 CrossRef CAS; (c) S. Bloom, J. L. Knippel, M. G. Holl, R. Barber and T. Lectka, Tetrahedron Lett., 2014, 55, 4576 CrossRef CAS; (d) M. Ke, Z. Liu, K. Zhang, S. Zuo and F. Chen, Green Synth. Catal., 2021, 2, 228 CrossRef.
- Z. Deng, J. Wei, L. Liao, H. Huang and X. Zhao, Org. Lett., 2015, 17, 1834 CrossRef CAS PubMed.
- J. A. Tunge and S. R. Mellegaard, Org. Lett., 2004, 6, 1205 CrossRef CAS PubMed.
- (a) D. L. J. Clive, C. G. Russell, G. Chittattu and A. Singh, Tetrahedron, 1980, 36, 1399 CrossRef CAS; (b) S. E. Denmark and M. G. Edwards, J. Org. Chem., 2006, 71, 7293 CrossRef CAS PubMed.
- J. Trenner, C. Depken, T. Weber and A. Breder, Angew. Chem., Int. Ed., 2013, 52, 8952 CrossRef CAS PubMed.
- (a) M. Tiecco, L. Testaferri, M. Tingoli, L. Bagnoli and C. Santi, J. Chem. Soc., Chem. Commun., 1993, 637 RSC; (b) S. Ortgies, C. Depken and A. Breder, Org. Lett., 2016, 18, 2856 CrossRef CAS PubMed; (c) C. Depken, F. Kratzschmar, R. Rieger, K. Rode and A. Breder, Angew. Chem., Int. Ed., 2018, 57, 2459 CrossRef CAS PubMed.
- (a) K. Uneyama, H. Asai, Y. Dan-oh and H. Matta, Electrochim. Acta, 1997, 42, 2005 CrossRef CAS; (b) R. Guo, J. Huang and X. Zhao, ACS Catal., 2018, 8, 926 CrossRef CAS.
- (a) T. Mancilla, R. Contreras and B. Wrackmeyer, J. Organomet. Chem., 1986, 307, 1 CrossRef CAS; (b) Z. He and A. K. Yudin, J. Am. Chem. Soc., 2011, 133, 13770 CrossRef CAS PubMed; (c) J. Li and M. D. Burke, J. Am. Chem. Soc., 2011, 133, 13774 CrossRef CAS PubMed; (d) Z. He, P. Trinchera, S. Adachi, J. D. St Denis and A. K. Yudin, Angew. Chem., Int. Ed., 2012, 51, 11092 ( Angew. Chem. , 2012 , 124 , 11254 ) CrossRef CAS PubMed; (e) L. Xu, S. Ding and P. Li, Angew. Chem., Int. Ed., 2014, 53, 1822 ( Angew. Chem. , 2014 , 126 , 1853 ) CrossRef CAS PubMed; (f) J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221 CrossRef CAS PubMed; (g) J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297 CrossRef CAS PubMed; (h) J. Taguchi, T. Ikeda, R. Takahashi, I. Sasaki, Y. Ogasawara, T. Dairi, N. Kato, Y. Yamamoto, J. W. Bode and H. Ito, Angew. Chem., Int. Ed., 2017, 56, 13847 ( Angew. Chem. , 2017 , 129 , 14035 ) CrossRef CAS PubMed.
- (a) Y. F. Zeng, W. W. Ji, W. X. Lv, Y. Chen, D. H. Tan, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 14707 ( Angew. Chem. , 2017 , 129 , 14899 ) CrossRef CAS PubMed; (b) C. F. Lee, D. B. Diaz, A. Holownia, S. J. Kaldas, S. K. Liew, G. E. Garrett, T. Dudding and A. K. Yudin, Nat. Chem., 2018, 10, 1062 CrossRef CAS PubMed; (c) W. X. Lv, Q. Li, J. L. Li, Z. Li, E. Lin, D. H. Tan, Y. H. Cai, W. X. Fan and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 16544 ( Angew. Chem. , 2018 , 130 , 16782 ) CrossRef CAS PubMed; (d) D. H. Tan, Y. H. Cai, Y. F. Zeng, W. X. Lv, L. Yang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 13784 ( Angew. Chem. , 2019 , 131 , 13922 ) CrossRef CAS PubMed; (e) Y. F. Zeng, X. G. Liu, D. H. Tan, W. X. Fan, Y. N. Li, Y. Guo and H. Wang, Chem. Commun., 2020, 56, 4332 RSC; (f) L. Yang, D. H. Tan, W. X. Fan, X. G. Liu, J. Q. Wu, Z. S. Huang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 3454 ( Angew. Chem. , 2021 , 133 , 3496 ) CrossRef CAS PubMed.
- V. B. Corless, A. Holownia, H. Foy, R. Mendoza-Sanchez, S. Adachi, T. Dudding and A. K. Yudin, Org. Lett., 2018, 20, 5300 CrossRef CAS PubMed.
- E. E. Lin, J.-Q. Wu, F. Schäfers, X.-X. Su, K.-F. Wang, J.-L. Li, Y. Chen, X. Zhao, H. Ti, Q. Li, T.-M. Ou, F. Glorius and H. Wang, Commun. Chem., 2019, 2, 34 CrossRef.
- Y. Liu, W. Luo, J. Wu, Y. Fang, Y. Li, X. Jin, L. Zhang, Z. Zhang, F. Xu and C. Du, Org. Chem. Front., 2020, 7, 1588 RSC.
- Z. Zhang, F. Wang, X. Mu, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2013, 52, 7549 ( Angew. Chem. , 2013 , 125 , 7697 ) CrossRef CAS PubMed.
- W. X. Fan, J. L. Li, W. X. Lv, L. Yang, Q. Li and H. Wang, Chem. Commun., 2020, 56, 82 RSC.
- L. Yang, CCDC 227635: CSD Communication, 2021, DOI:10.5517/ccdc.csd.cc27qryp.
- We can not exclude that the B(MIDA) group may directly interact with the selenium unit, thereby leading to the observed regioselectivity.
- S. R. Mellegaard-Waetzig, C. Wang and J. A. Tunge, Tetrahedron, 2006, 62, 7191 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00954d |
| This journal is © The Royal Society of Chemistry 2022 |