
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyridine dicarbanion-bonded Ag13 organometallic nanoclusters: synthesis and on-surface oxidative coupling reaction†
Cui-Cui
Li
,
Siqi
Zhang
,
Jian
Tang
,
Ruijun
Jian
,
Yu
Xia
and
Liang
Zhao
 *
*
Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: zhaolchem@mail.tsinghua.edu.cn
First published on 14th June 2022
Abstract
Highly reactive organometallic nanoclusters in situ generated in metal-catalyzed reactions are pivotal in the comprehension of catalytic mechanisms. Herein, we develop a two-step synthetic method to achieve three unprecedented aryl dicarbanion-bonded Ag13 nanoclusters by using protective macrocyclic ligands. Firstly, various aryl dicarbanion–Ag4 cluster intermediates are acquired via a silver-mediated annulation reaction within a macrocyclic ligand. These Ag4 cluster precursors are released from the surrounding macrocycle by protonation, and further undergo an inter-cluster coupling to generate bipyridine products and low-valence silver atoms. The remaining resurgent diide–Ag4 clusters assemble with low-valence silver atoms to yield a series of organometallic Ag13 nanoclusters. These Ag13 nanoclusters feature a unique open-shell electronic structure as well as a chiral cluster architecture due to the asymmetric arrangements of surrounding aryl dianion ligands. Furthermore, the pyridyl diide ligands on the surface of the nanocluster further experience an intra-cluster oxidative coupling to produce bipyridine coupling products and large nanoparticles. The coupling reaction-driven cluster-to-cluster transformation is comprehensively tracked by high resolution mass spectroscopy. This work is not only reminiscent of the detailed evolution of cluster species upon the occurrence of coupling reactions, but also reproduces novel inter- and intra-cluster coupling steps at different reaction stages.
Introduction
The study of metal subnano- or nano-clusters in situ generated in transition metal-catalyzed reactions has become a fascinating and substantial topic in both homogeneous and heterogeneous catalyses.1 These kinds of metal nanoclusters, which are mostly formed through the reduction of metal complexes by reaction substrates, coordinative ligands or additives,1f,2 largely reshape the perception regarding real catalytic species and detailed catalytic mechanisms. They act as either catalytically active species1d,f,3 or alternative reservoirs of active metal complexes,4 and prevent devitalization of the catalytic species by suppressing the formation of insoluble metal species. Structural investigations on the in situ generated metal nanocluster intermediates, especially carbon-bonded organometallic nanoclusters, enable chemists to gain insight into the structural details of privileged intermediates and promote the comprehension of catalytic mechanisms. However, although organometallic clusters have been identified as intermediate species in many metal-catalyzed organic transformations by mass spectrometry,3d,5 electron microscopy2a,3a and X-ray absorption spectroscopy,3b,c their synthesis and structural characterization are still a challenging task due to their transient and highly reactive nature. Accordingly, the transformations of organic ligands attaching on these organometallic nanoclusters are rarely investigated.6The Ag(0/I)-based single-electron redox cycle has been extensively proposed in many silver-catalyzed coupling reactions.7 The presence of low valence silver atoms in the Ag(0/I) redox process facilitates the in situ formation of silver nanoclusters, which have been demonstrated to exhibit excellent catalytic activity in organic transformations.8 However, in-depth structural characterization and reactivity studies of these in situ generated silver nanoclusters suffer from the lack of appropriate synthetic methods and stabilizing means as labile silver–carbon bonds prefer a rapid homolysis disproportionation to generate metallic silver and finally cause decomposition.9 In view of the previously reported successful encapsulation and stabilization of reactive organometallic species within supramolecular cages,10 we envision that the initial encaging of organosilver species within a closed macrocyclic environment followed by a steerable organometallic transformation may provide a viable pathway to acquire organosilver nanoclusters (Scheme 1).
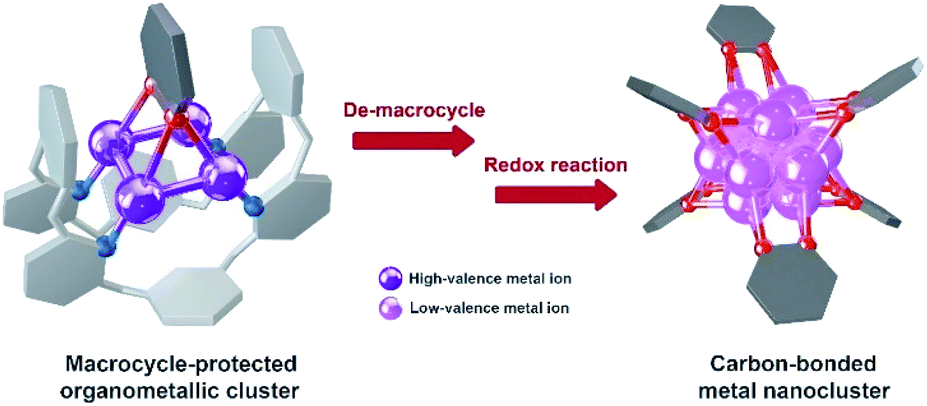 | ||
| Scheme 1 Synthesis of carbon-bonded metal nanoclusters via the transformation of macrocycle-protected organometallic clusters. | ||
In this work, we develop a two-step synthetic method to achieve a series of unprecedented aryl vicinal dicarbanion bonded Ag13 nanoclusters ([Ag13(RPy–H)6](CF3SO3)6, R = Me, n-Pr, Ph). At the initial step, differently substituted pyridine dicarbanion-bonded Ag4 cluster intermediates are acquired via a silver-mediated cyclization reaction within a macrocyclic ligand. After removing the protective macrocycle by protonation, the dicarbanion–Ag4 cluster precursors undergo an inter-cluster oxidative coupling between two pyridine carbanions to in situ generate low-valence silver atoms. The remaining released dicarbanion–Ag4 clusters assemble with a low-valence silver atom to yield vicinal dicarbanion bonded Ag13 nanoclusters. These organometallic nanoclusters feature a unique open-shell electronic structure as well as a chiral cluster architecture as a result of asymmetric arrangement of surrounding pyridyl dianion ligands. Furthermore, the pyridyl dicarbanion ligands on the surface of a Ag13 nanocluster can undergo an intra-cluster oxidative coupling to generate the bipyridine coupling product and large nanoparticle species, reminiscent of the evolution of cluster species in many metal-catalyzed reactions. This work not only reveals novel dicarbanion-bonded metal nanoclusters for the first time, but also provides a new perspective for the dynamic evolution of organometallic species in metal catalysis, from isolated single atom species through low-nuclear clusters to nanoclusters along with the occurrence of intra- and inter-cluster coupling reactions.
Results and discussion
Pyridyl dicarbanion bonded Ag13 nanoclusters
In this work, an attempt was made to generate a gem-dimetallic organometallic species as a representative intermediate motif in metal-catalyzed reactions.11 To achieve this goal, a silver(I)-mediated intermolecular annulation reaction of ketones and propargylamine12 was performed in the presence of excessive silver salts and a polydentate macrocyclic ligand octamethylazacalix[8]pyridine (Py[8]).13 As shown in Fig. 1, various methyl ketones together with propargylamine undergo a continuous multi-step transformation of condensation, imine–enamine isomerization, 6-endo-dig cyclization and oxidative aromatization to yield three differently substituted pyridyl dicarbanion bonded Ag4 clusters (abbreviated as MePyAg5, n-PrPyAg5 and PhPyAg5) in moderate yields (23–61%). The composition and structural features of these Ag4 clusters were confirmed by high-resolution electrospray ionization mass spectrometry (Fig. S1–S3†) and NMR (Fig. S4–S7†).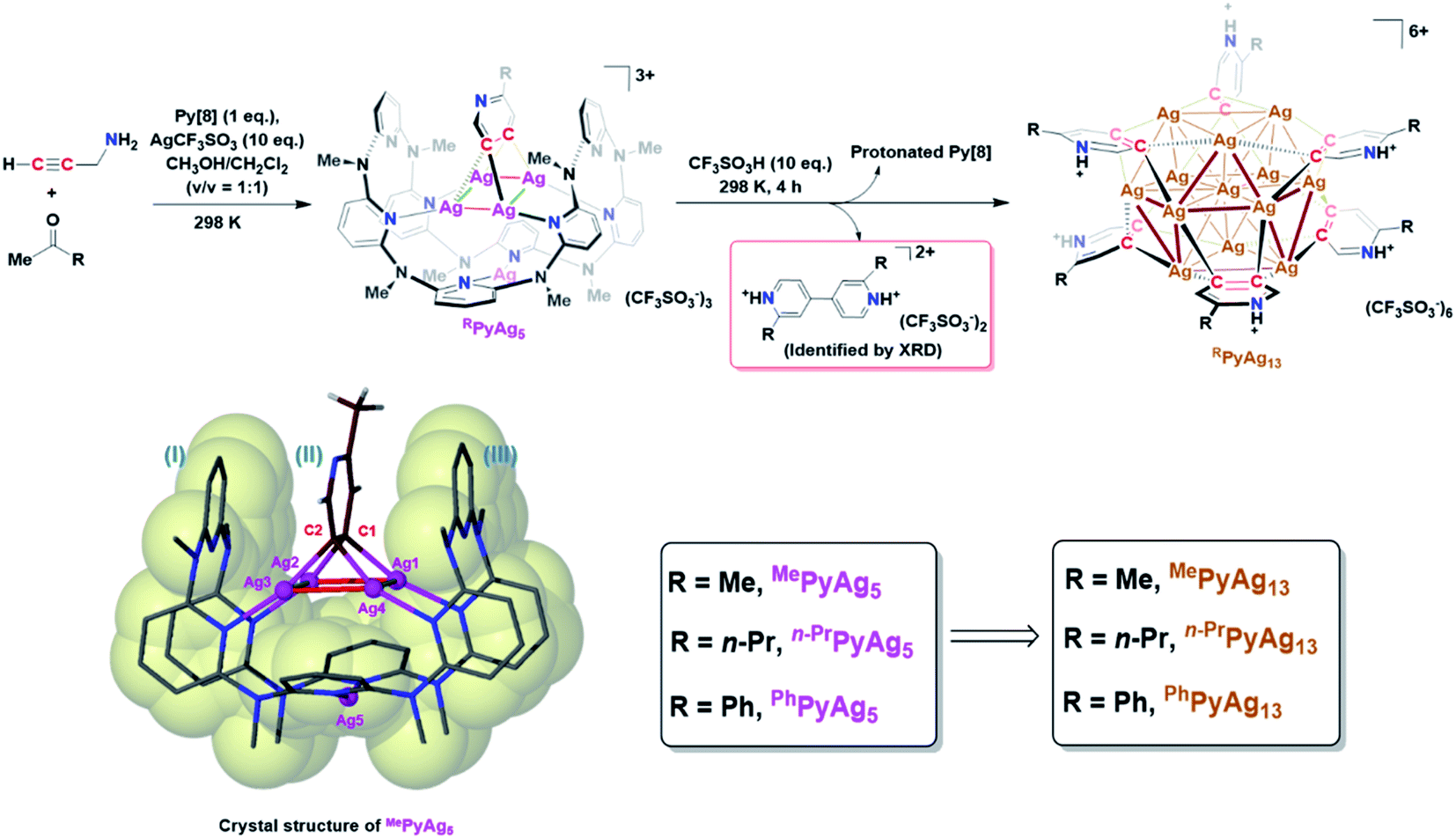 | ||
| Fig. 1 Synthetic procedures for macrocycle-protected Ag4 cluster complexes and aryl vicinal dicarbanion bonded Ag13 nanoclusters. The crystal structure of MePyAg5 is shown as an inset with anions and solvent molecules omitted for clarity. | ||
Crystalline solids of these organosilver cluster compounds are quite stable in the dark even upon exposure to air and moisture for months. The 1H NMR monitoring on the acetone solution of MePyAg5 shows negligible change for up to seven days (Fig. S8†), suggesting its superior stability in the solution state. Single-crystal X-ray analysis reveals a highly isostructural pyridyl diide–Ag4 cluster core in MePyAg5 (Fig. 1), n-PrPyAg5 (Fig. S9†) and PhPyAg5 (Fig. S10†). Taking MePyAg5 for instance, two vicinal carbon atoms C1 and C2 of a pyridyl ring are negatively charged and stabilized by a coplanar argentophilic Ag4 rectangle via four-fold Ag–C σ bonds in the range of 2.148(9)–2.181(9) Å, which are comparable to the CAg2 species in reported arylsilver(I) complexes.10e,14 The Ag4 rectangle is composed of two short Ag–Ag edges (Ag1–Ag2: 2.724(1) Å, Ag3–Ag4: 2.683(1) Å) and two long ones (Ag1–Ag4: 3.164(1) Å, Ag2–Ag3: 3.246(2) Å). The whole pyridyl diide–Ag4 cluster motif is enclosed by a semi-open bowl-shaped macrocyclic ligand Py[8]via four-fold Ag–N bonds. Another silver atom is clamped by two pyridyl nitrogen atoms at the bottom of the bowl. It should be emphasized that the metalated pyridyl diide is sandwiched by two parallel pyridyl rings of Py[8], leading to a fully eclipsed face-to-face π–π stacking with small dihedral angles (θI–II = 5.0° and θII–III = 4.4°) and displacement distances (DI–II = 0.738 Å and DII–III = 0.523 Å). This unusual eclipsed face-to-face π–π stacking is in sharp contrast to the common displaced π–π stacking between aromatic rings as a result of the requirement of mitigated π–π electron repulsion.15 Theoretical studies reveal that the pπ electrons of the central metalated pyridine largely participate in the C–Ag2 multi-centered bonding (Fig. S11†) and thus weaken the π–π repulsion with two bilateral pyridine rings belonging to the peripheral Py[8].
With the macrocycle-protected organosilver cluster intermediates in hand, we subsequently removed the protective Py[8] by protonation to initiate transformations of the diide–Ag4 clusters. After screening several acids with a wide range of pKa values (Fig. S12†), the strong trifluoromethanesulfonic acid CF3SO3H was selected because of the prompt generation of protonated Py[8] as a white precipitate (Fig. S13†) upon the addition of CF3SO3H to the acetone solution of MePyAg5. This protonation process was simultaneously accompanied by a gradual solution color change from light yellow to brown. Diffusion of diethyl ether into the brown solution deposited two types of brown and colorless crystals in a yield of 52% (Table S1†) and 13%, respectively.
X-ray crystallographic analysis of the MePyAg5-derived brown crystalline sample reveals that its structure includes a twisted cuboctahedral Ag13 core, which is peripherally wrapped by six 2-methylpyridyl diides via a μ4-C,C-η2,η2 mode and five coordinative triflate anions (Fig. 2a). There is one free CF3SO3− serving as an uncoordinated counter anion. This Ag13 nanocluster is different from the reported Ag13 cuboctahedron, each triangle face of which is capped by an Fe(CO)4 group to generate a [Ag13Fe8(CO)32]4− cluster.16 The electrospray ionization mass spectroscopy (ESI-MS) study on the solution of the brown crystals gives rise to two isotopically well-resolved peaks at m/z = 2847.7750 and 2865.7704, which can be assigned to the species {[Ag13(MePy–H)6](CF3SO3)6}+ and {[Ag13(MePy–H)6](CF3SO3)6·H2O}+, respectively (Fig. S14†). This result indicates that six 2-methylpyridyl diides around the cluster core are all protonated at the pyridyl nitrogen atoms. In the light of the charge balance requirement, we assume that the 13-membered silver cluster core shows twelve positive charges and has an unpaired electron, which is similar to the paramagnetic [Ag13Fe8(CO)32]4− cluster.16 This conjecture is supported by electron paramagnetic resonance (EPR) measurement (Fig. S15†)16 and X-ray photoelectron spectroscopy (XPS, (Fig. S16†)). The XPS spectra revealed two intensive peaks in the Ag 3d region at 374.9 and 368.9 eV corresponding to Ag 3d3/2 and Ag 3d5/2, respectively (Fig. S16b†). The peak could be deconvoluted into 375.5 eV (3d3/2, Ag0), 374.9 eV (3d3/2, Ag+), 369.5 eV (3d5/2, Ag0) and 368.9 eV (3d5/2, Ag+). The area ratios of Ag+/Ag0 for Ag 3d3/2 and Ag 3d5/2 are 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 10:1, basically consistent with the presence of two kinds of Ag+ and Ag0 in the crystal structure. Based on the Auger electron spectroscopy (AES) data under X-ray irradiation of 1486.6 eV, the Auger parameters of Ag+ and Ag0 are deduced to be 724.5 and 725.1 eV, which are in good agreement with those of Ag2O (Auger parameter: 724.5 eV)17 and Ag(0) (Auger parameter: 726.0 eV).18 Consequently, the molecular formula of the brown crystals is determined to be [Ag13(MePy–H)6](CF3SO3)6 (MePyAg13). The existence of low valence silver atoms in the [Ag13]12+ core of MePyAg13 implies that some Ag(I) ions were reduced during the construction of MePyAg13, which is supported by further X-ray crystallographic analysis of the colorless crystals (Fig. 1). The identification of the colorless crystalline product as protonated 2,2′-dimethyl-4,4′-bipyridine (Fig. S17 and S18†) clearly indicates the occurrence of a two-electron oxidative coupling between two C1–Ag2 units from two individual 2-methylpyridine–Ag4 motifs. This coupling process simultaneously generates low valence silver atoms. In view of the high similarity of pyridyl C2–Ag4 moieties between MePyAg5 and MePyAg13, we deduce that the 13-membered nanocluster core in MePyAg13 arises from the assembly of low valence silver atoms with remaining protonated 2-methylpyridine diide–Ag4 motifs in situ released from the Py[8] macrocycle of MePyAg5 (Fig. 1 and S19†).
:1 and 10:1, basically consistent with the presence of two kinds of Ag+ and Ag0 in the crystal structure. Based on the Auger electron spectroscopy (AES) data under X-ray irradiation of 1486.6 eV, the Auger parameters of Ag+ and Ag0 are deduced to be 724.5 and 725.1 eV, which are in good agreement with those of Ag2O (Auger parameter: 724.5 eV)17 and Ag(0) (Auger parameter: 726.0 eV).18 Consequently, the molecular formula of the brown crystals is determined to be [Ag13(MePy–H)6](CF3SO3)6 (MePyAg13). The existence of low valence silver atoms in the [Ag13]12+ core of MePyAg13 implies that some Ag(I) ions were reduced during the construction of MePyAg13, which is supported by further X-ray crystallographic analysis of the colorless crystals (Fig. 1). The identification of the colorless crystalline product as protonated 2,2′-dimethyl-4,4′-bipyridine (Fig. S17 and S18†) clearly indicates the occurrence of a two-electron oxidative coupling between two C1–Ag2 units from two individual 2-methylpyridine–Ag4 motifs. This coupling process simultaneously generates low valence silver atoms. In view of the high similarity of pyridyl C2–Ag4 moieties between MePyAg5 and MePyAg13, we deduce that the 13-membered nanocluster core in MePyAg13 arises from the assembly of low valence silver atoms with remaining protonated 2-methylpyridine diide–Ag4 motifs in situ released from the Py[8] macrocycle of MePyAg5 (Fig. 1 and S19†).
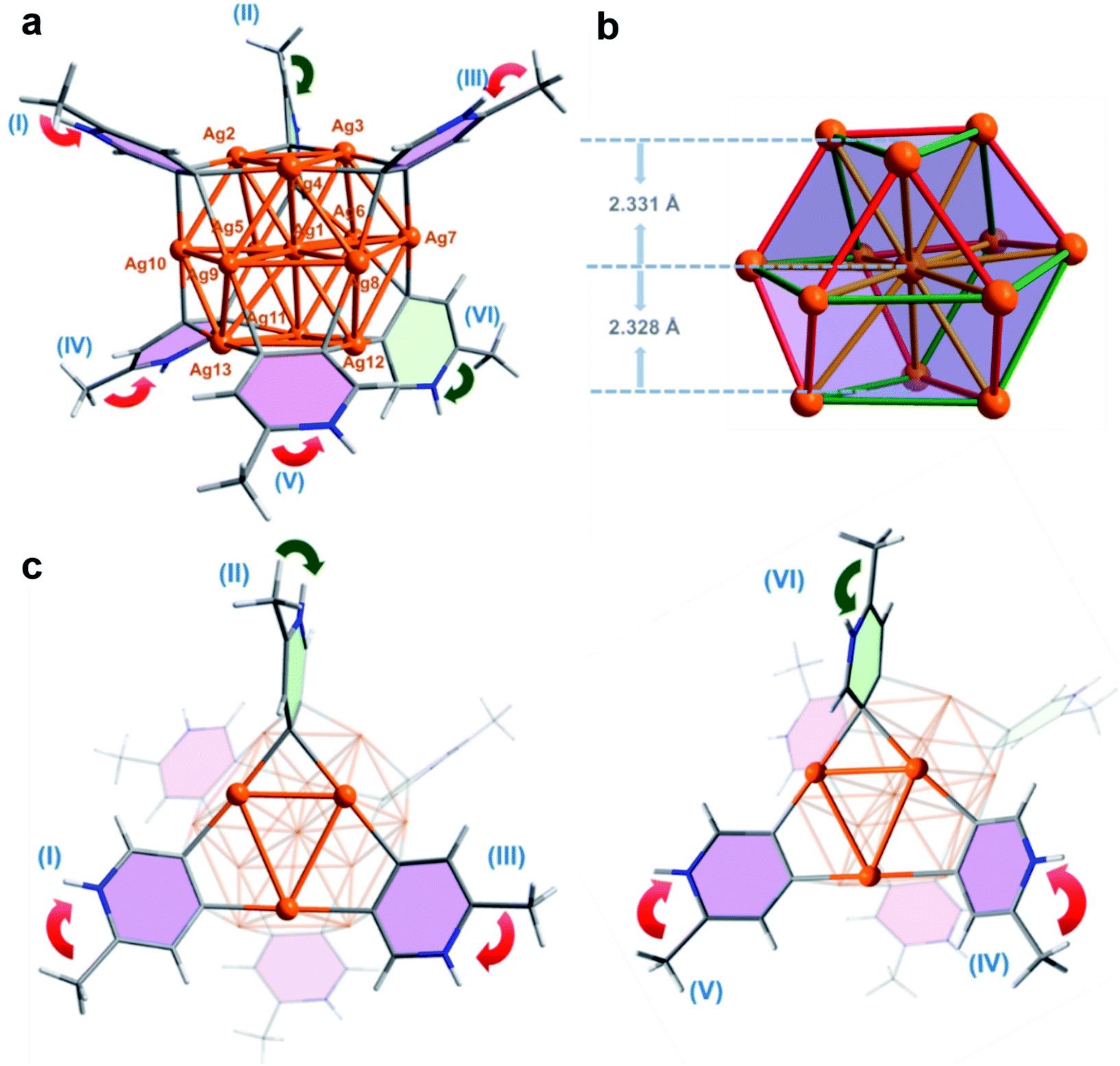 | ||
| Fig. 2 (a) Crystal structure of MePyAg13 containing equatorial (pink) and axial (light green) 2-methylpyridyl diide ligands. Peripheral CF3SO3− anions are omitted for clarity. (b) The Ag13 kernel in MePyAg13. Color coding: Ag, brown; C, gray; H, white; N, blue. (c) Arrangement of the orientated 2-methylpyridine rings attached on the Ag13 kernel at the upper and nether sides in MePyAg13. Selected bond lengths (Å) of MePyAg13 highlighted in yellow: Ag1–Ag2 2.885(2); Ag1–Ag3 2.897(2); Ag1–Ag4 3.025(2); Ag1–Ag5 2.947(2); Ag1–Ag6 3.167(2); Ag1–Ag7 2.986(2); Ag1–Ag8 2.930(2); Ag1–Ag9 3.062(2); Ag1–Ag10 2.915(2); Ag1–Ag11 2.929(2); Ag1–Ag12 2.863(2); Ag1–Ag13 3.038(2); red: Ag2–Ag3 2.780(2); Ag2–Ag10 2.825(3); Ag4–Ag9 2.716(3); Ag4–Ag8 2.758(2); Ag3–Ag7 2.763(2); Ag5–Ag6 2.718(2); Ag5–Ag11 2.736(2); Ag10–Ag13 2.716(2); Ag9–Ag13 2.737(3); Ag8–Ag12 2.817(2); Ag6–Ag7 2.701(2); Ag11–Ag12 2.796(2); green: Ag2–Ag4 3.218(2); Ag3–Ag4 3.256(2); Ag10–Ag9 3.089(2); Ag8–Ag7 3.253(2); Ag2–Ag5 3.281(2); Ag3–Ag6 3.078(2); Ag5–Ag10 3.223(2); Ag11–Ag13 3.024(2); Ag8–Ag9 3.022(2); Ag12–Ag13 3.322(2); Ag7–Ag12 3.196(2); Ag6–Ag11 3.164(2). | ||
The twisted cuboctahedral Ag13 core in MePyAg13 is divided into three layers of Ag3–Ag7–Ag3, and the distances between the central silver (Ag1) and the upper and lower Ag3 layers are 2.331 and 2.328 Å, respectively (Fig. 2b). The argentophilic distances between the central silver atom Ag1 and twelve surface silver ions are in the range of 2.863(2)–3.167(2) Å (yellow color in Fig. 2b), which are comparable with the Ag–Ag distances in [Ag13Fe8(CO)32]4− (2.923 Å). The other 24 surficial argentophilic edges among twelve peripheral silver atoms can be classified into two types. The short edges bridged by the same diide carbon atom (average distances: 2.755 Å, red color in Fig. 2b) and the long ones spanned by two diide carbon atoms of the same pyridine ring (average distances: 3.205 Å, green color in Fig. 2b) are comparable to those in the parent tetranuclear cluster MePyAg5 (2.703 Å and 3.192 Å, respectively). The maximum value of the argentophilic distance alternations between the long and short edges in MePyAg13 is 0.621 Å, which is significantly larger than that in [Ag13Fe8(CO)32]4−.16 In addition, six protonated 2-methylpyridine diide ligands each is enveloped by a rectangular plane of the Ag13 core through four-fold Ag–C bonds in the range of 2.12(2)–2.29(3) Å, which are slightly longer than the Ag–C bonds in MePyAg5. Notably, the protonated 2-methylpyridine diide ligands on both upper (I, II and III) and lower layers (IV, V and VI) exhibit different orientations (Fig. 2a). Therein, four diide ligands (I, III, IV and V in pink color) are equatorially bonded to the Ag13 core, while the other two (II and VI in light green color) are in an axial orientation. The biased spatial arrangement of the protonated 2-methylpyridine ligands can be described by the arrow vector from the methyl substituent toward the pyridyl nitrogen. As to the four equatorial 2-methylpyridine ligands, I and III on the upper layer are related by a C3-axis, while IV and V on the nether layer have a mirror symmetry (red arrows in Fig. 2c). On the other hand, the axial 2-methylpyridine ligand II is in an endo-form with the methyl group close to the silver nanocluster, while ligand VI shows an exo-form with the methyl group pointing away from the cluster core (green arrows in Fig. 2c). In this way, the twelve peripheral silver ions (from Ag2 to Ag13) of the Ag13 core are coordinated by two kinds of pyridyl carbon atoms (Cpara and Cmeta refer to the 4- and 5-position carbon atom on the pyridine ring, respectively) in three combinations of Cpara–Ag–Cpara, Cpara–Ag–Cmeta and Cmeta–Ag–Cmeta (Fig. S20†). The differentiated orientation of the total six 2-methylpyridyl diide ligands breaks the symmetry and thus imparts chirality to the whole cluster structure of MePyAg13, making it the first {M13}-type chiral metal nanocluster to our knowledge.19 Herein, both cluster enantiomers are present in the crystal structure of MePyAg13 (Fig. S21†).
We next carried out density functional theory (DFT) calculations to investigate the bonding and argentophilic interaction in MePyAg13. The Wiberg bond order20 based on the Löwdin orthogonalized basis was calculated and summarized in Table S2.† The sum of Ag–C bond orders for each CAg2 species is close to one (0.893–0.975) and the multi-center bond order (MCBO, Table S3†) of each CAg2 species ranges from 0.013 to 0.091, suggesting the formation of a 3c–2e bonding in each CAg2 species. In addition, the Ag–Ag Wiberg bond order of MePyAg13 is in the range of 0.189–0.384 as a result of significant argentophilic interaction between two silver atoms. Therein, the average Wiberg bond order of short Ag–Ag edges (red color in Fig. 2b) in a cuboctahedral Ag13 core is 0.359, while those of the long Ag–Ag edges (green color in Fig. 2b) and twelve central ones (yellow color in Fig. 2b) are 0.222 and 0.260, respectively. The independent gradient model based on Hirshfeld partition (IGMH)21 was further carried out via sign(λ2)ρ functions to examine intramolecular interactions in MePyAg13. From the IGMH map, the mazarine area between six 2-methylpyridyl diides and the Ag13 core (Fig. S22a†) indicates prominent electron density in these regions and covalent characteristics of the C–Ag bonds. Furthermore, there are blue areas in the isosurface between two adjacent silver atoms, suggesting the presence of apparent argentophilic interaction within the Ag13 core (Fig. S22b and c†).
The Atoms in Molecules (AIM) analysis22 also confirms strong argentophilic interaction in MePyAg13. As shown in Fig. S23,† the bond critical points exist in each pair of adjacent silver atoms. The negative bond degree parameter (BD = E(r)/ρ(r), E(r) and ρ(r) being the total electron energy density and the electron density value at the Ag–Ag (3, −1) critical point) and the ratio |V(r)|/G(r) > 1 (V(r) and G(r) being the pressures exerted on and by the electrons at the Ag–Ag (3, −1) critical point),23 indicate apparent argentophilic interactions in the Ag13 core (Table S4†). It is worth noting that some bond critical points lie between H atoms of 2-methylpyridyl diides and F or O atoms of CF3SO3−, suggesting that multiple hydrogen bonding is instrumental in the stabilization of MePyAg13.
Our two-step synthetic strategy is also applicable in the synthesis of n-PrPyAg13 and PhPyAg13 by the reaction of differently substituted n-PrPyAg5 or PhPyAg5 with CF3SO3H, respectively. Similarly, during the acidification of n-PrPyAg5 or PhPyAg5, the coupling product 2,2′-dipropyl-4,4′-bipyridine or 2,2′-diphenyl-4,4′-bipyridine was detected and successfully characterized by mass spectrometry (Fig. S24 and S25†) and single crystal XRD analysis (Fig. S26 and S27†). XRD crystallographic analysis, high-resolution ESI-MS and UV-vis absorption spectra reveal that n-PrPyAg13 (Fig. 3a, S28 and S29†) and PhPyAg13 (Fig. 3b and S30–S32†) are isostructural with MePyAg13 in the aspects of bond distances and angles. Nevertheless, the dihedral angles between the upper and lower Ag3 planes in n-PrPyAg13 and PhPyAg13 are 11.5° and 11.7°, respectively (Table S5†), a bit higher than that of MePyAg13 (9.7°). In addition, the mean deviation of the central Ag7 layer in n-PrPyAg13 and PhPyAg13 is 0.125 and 0.104 Å, respectively, also larger than that of MePyAg13 (0.094 Å). The more distorted Ag13 core in n-PrPyAg13 and PhPyAg13 should be ascribed to significant steric hindrance among peripheral n-propyl and phenyl substituents. Furthermore, in contrast to the exo and endo spatial orientations of two axial pyridyl diide ligands in n-PrPyAg13 and MePyAg13, PhPyAg13 has two axial 2-phenylpyridine ligands II and VI both in the exo-form with the phenyl group pointing away from the cluster core (green arrows in Fig. 3b). This structural difference underlines the oriented diversity of the pyridyl diide ligands around the Ag13 nanocluster core.
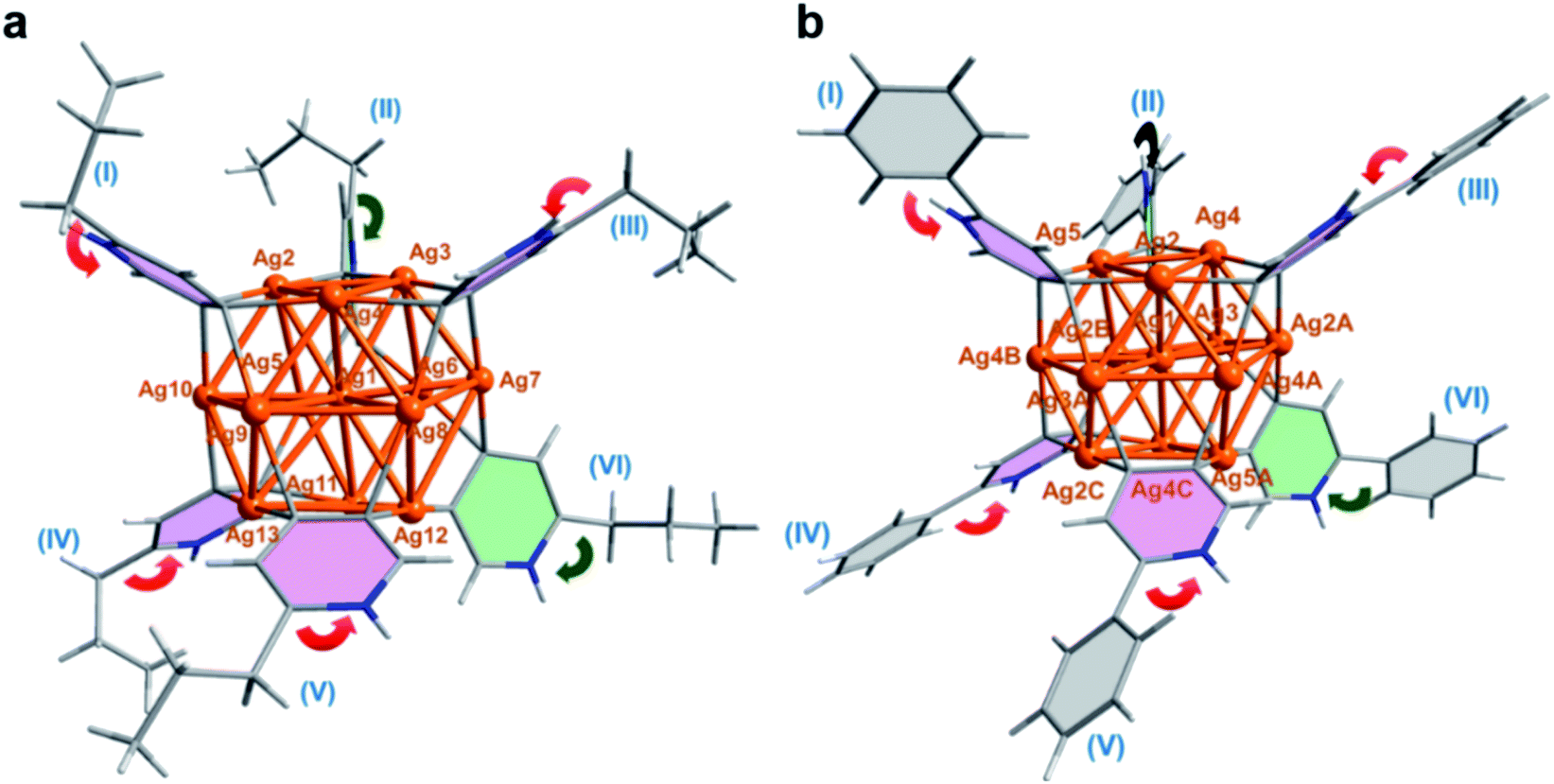 | ||
| Fig. 3 Crystal structures of the Ag13 nanoclusters in (a) n-PrPyAg13 and (b) PhPyAg13 with equatorial (pink) and axial (light green) pyridyl ligands. Peripheral CF3SO3− anions and solvent molecules are omitted for clarity. Color coding: Ag, brown; C, gray and blue gray; H, white; N, blue. | ||
The newly synthesized Ag13 nanoclusters retain their structures intact in solution based on the 1H-NMR and ESI-MS spectra. In the 1H NMR spectrum of MePyAg13, three sets of aromatic resonance peaks at 8.94, 8.31–8.19 and 7.98 ppm with a ratio of 1:10:1 were observed (Fig. S33†). This unusual integral ratio corresponding to the pyridyl protons in MePyAg13 may be due to different configurational isomers in solution or differentiated ligand orientations in MePyAg13. Diffusion ordered spectroscopy (DOSY) of MePyAg13 (Fig. S34†) shows only one diffusion coefficient of D = 7.35 × 10−10 m2 s−1 for the pyridyl proton peaks (Table S6†). Based on the Stokes–Einstein equation, the calculated diameter was 18.75 Å, which agrees well with the measured distances in the crystal structure of MePyAg13 (17.55 Å). Besides, variable temperature 1H NMR spectra (Fig. S35†) and UV-vis spectra (Fig. S36†) display negligible changes with the temperature increase from 298 to 353 K, excluding the possibility of other equilibrium cluster conformations in solution. Therefore, the aromatic integral ratio of 1:10:1 may correspond to 12 inequitable pyridyl protons of six diide ligands. In order to accurately assign the twelve pyridyl proton atoms, the NMR spectrum of MePyAg13 was simulated by the scaling method based on an optimized structure. The simulated results show that the proton signal at 8.94 ppm corresponds to the proton atom attaching on the carbon atom C6 (adjacent to the pyridyl nitrogen) in ligand VI, and the peak at 7.98 ppm is assigned to the C3 hydrogen atom (meta-position to the pyridyl nitrogen) of II (Fig. S37a†). In addition, the chemical shifts of the remaining ten protons are also inequivalent but very close, which are finely influenced by the ligand orientation and the C–Ag2 units (Fig. S37b†). The 1H NMR spectrum of n-PrPyAg13 (Fig. S38†) is very similar to that of MePyAg13 because of their completely consistent ligand orientation scenario. In contrast, the 1H NMR spectrum of PhPyAg13 shows four broad resonance peaks at 8.77, 8.53, 7.90 and 7.66 ppm with an integral ratio of 6:6:12:18 (Fig. S39†). The simulated 1H NMR spectrum of PhPyAg13 (Fig. S40a†) indicates that the resonances at 8.77 and 8.53 ppm are assigned to the protons on the meta and ortho positions of the pyridine rings in PhPyAg13, and the peaks at 7.90 and 7.66 ppm correspond to the protons on the phenyl rings (Fig. S40b†). The above experimental and theoretical NMR results suggest that distinct spatial arrangements of the pyridyl diide ligands lead to biased charge distribution on carbon atoms and thus account for inequivalent proton NMR peaks.
Inter- and intra-cluster coupling of diide ligands
To gain further insight into the formation process of Ag13 nanoclusters, we conducted a cross experiment between the same equivalent MePyAg5 and n-PrPyAg5 with CF3SO3H in acetone. The ESI-MS monitoring showed a normal distribution of various Ag13 nanoclusters containing a ligand combination of protonated MePy and n-PrPy in a ratio from 1:5 to 5:1 (Fig. 4a and Table S7†). In addition, the cross-coupling product 2-methyl-2′-propyl-4,4′-bipyridine was determined as the primary coupling species in ESI-MS spectra, wherein the minor homo-coupling products 2,2′-dimethyl-4,4′-bipyridine and 2,2′-dipropyl-4,4′-bipyridine were observed as well. This ESI-MS evidence suggests that the initial formation of the Ag13 nanoclusters mostly relies on random combinations of two types of resurgent pyridyl diide–Ag4 clusters as they are released from the protective macrocycles.
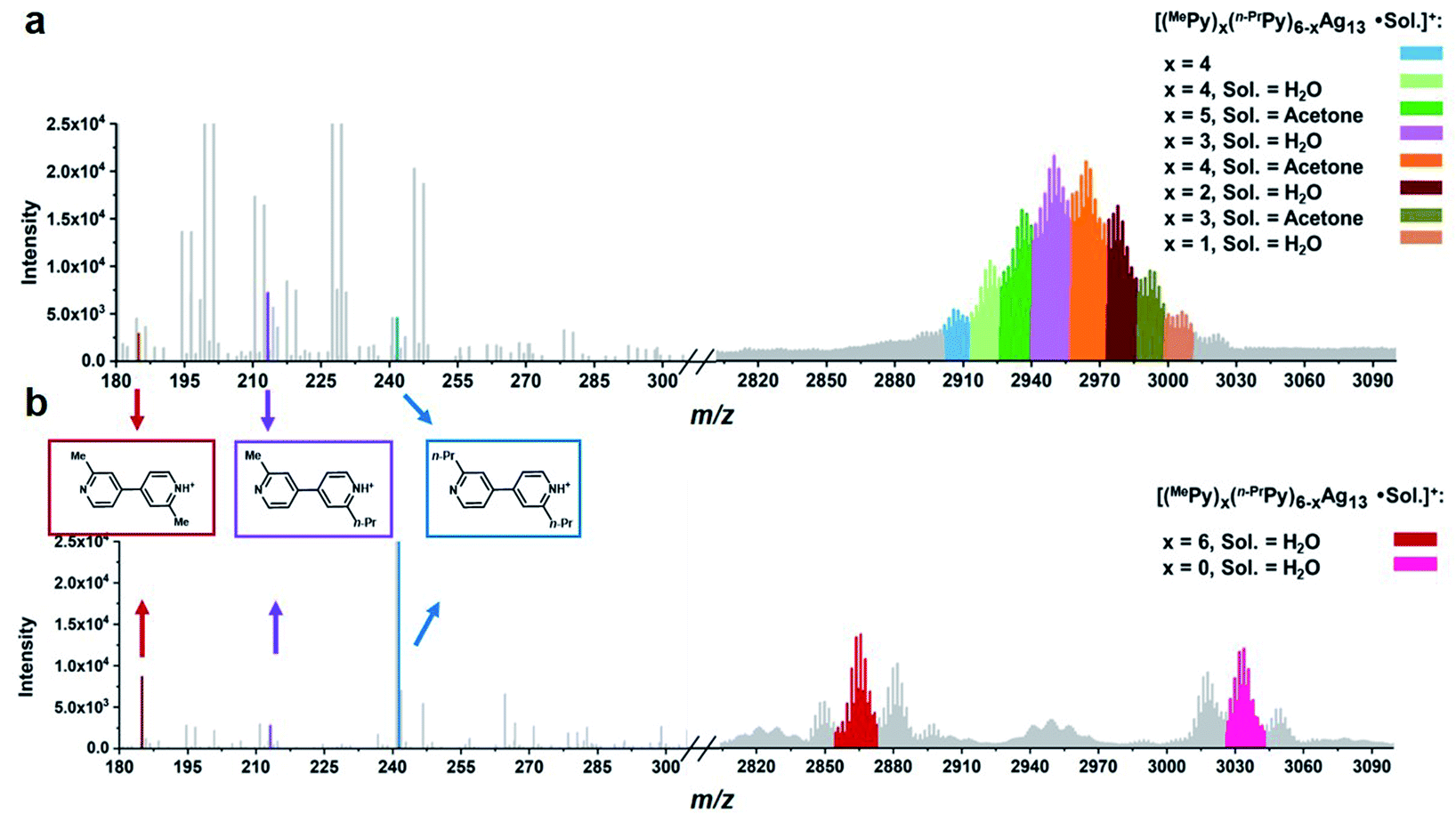 | ||
| Fig. 4 ESI mass spectra of the acidifying solution samples containing equivalent (a) MePyAg5 and n-PrPyAg5 in acetone, and (b) MePyAg13 and n-PrPyAg13 in acetone. | ||
We next studied the transformation of diide ligands on the surface of MePyAg13. The addition of CF3SO3H to the methanol solution of MePyAg13 also led to the coupling product 2,2′-dimethyl-4,4′-bipyridine in a high yield of 67%. In order to ensure the occurrence of intra-cluster coupling, the cross experiment by using equivalent MePyAg13 and n-PrPyAg13 was implemented. The homo-coupling compounds 2,2′-dimethyl-4,4′-bipyridine and 2,2′-dipropyl-4,4′-bipyridine were detected as major products in the ESI-MS monitoring (Fig. 4b), while only slight cross-coupling product 2-methyl-2′-propyl-4,4′-bipyridine was observed. In contrast to the prompt inter-cluster coupling among pyridyl diide–Ag4 clusters at the initial stage, the coupling reaction of diide ligands around a single Ag13 nanocluster at the second stage is relatively sluggish as indicated by the detection of unreactive Ag13 clusters even after four days (Fig. S41†). Meanwhile, spherical nanoparticles with the sizes in the range of 20–40 nm were identified by transmission electron microscopy (TEM) images (Fig. S42†), suggesting the formation of larger nanocluster species by the association and merging of Ag13 nanoclusters after the occurrence of intra-cluster oxidative coupling. Furthermore, infrared spectra of the solid originated from the Ag13–CF3SO3H mixture exhibit two stretching vibrations at 1540 cm−1 and 1443 cm−1 (Fig. S43†), which can be assigned to the characteristic absorption peaks of a pyridine ring. This result indicates that the larger Ag nanocluster species are still coordinated by pyridine ligands. Along with the prolongation of the reaction time, a black precipitate due to the further aggregation of large nanoparticles was observed.
Combining the above experimental results, we propose two phases of inter- and intra-cluster coupling accompanied by the structural evolution of silver cluster species from organic diide–Ag4 clusters through Ag13 nanoclusters to large nanoparticles. As shown in Fig. 5, the removal of protective Py[8] macrocycles by protonation makes the released organic diide–Ag4 clusters randomly combine together and undergo a two-electron oxidative coupling to generate low valence silver atoms. The remaining protonated diide–Ag4 clusters then assemble with the low valence silver atoms to produce the Ag13 nanoclusters. In the second phase, the aryl dicarbanion ligands on the surface of a Ag13 nanocluster experience protolysis at a [C–Ag2] position and the remaining [C–Ag2] site at the para position undergoes an intra-cluster oxidative coupling to generate homo-coupling bipyridine products and expedite the formation of large nanoparticle species. It is worth noting that the oxidative coupling of organic species around in situ generated metal subnano- or nano-clusters has been proposed in many metal-catalyzed reactions.1a,h However, there has been little knowledge about the rationality of such mechanism and structural details of related intermediates. The present transformation from isolated mononuclear metal complexes through low nuclearity clusters to nanoclusters and nanoparticles is reminiscent of the dynamic structural evolution of cluster species in those metal-catalyzed reactions, and also gives important insights into the coupling process of organic ligands at different reaction stages.
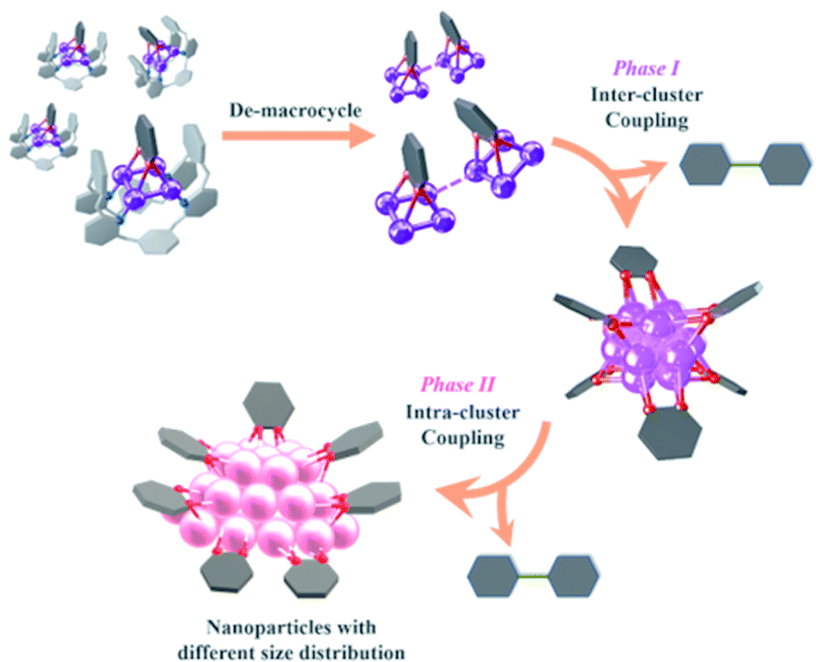 | ||
| Fig. 5 Structural evolution of organosilver species from organic diide–Ag4 clusters through nanoclusters to nanoparticles along with the occurrence of coupling reactions at different stages. | ||
Conclusion
In this work, a series of pyridyl vicinal dicarbanion bonded Ag13 nanoclusters with an open-shell electronic structure are synthesized and characterized for the first time. The newly developed two-step synthetic method involves the occurrence of an inter-cluster oxidative coupling reaction and the assembly of diide-centered Ag4 clusters with in situ generated low-valence metal atoms to generate organosilver nanoclusters. Moreover, the aryl dicarbanion ligands on the nanocluster surface further undergo an intra-cluster oxidative coupling to produce homo-coupling products and larger nanocluster species, providing a new perspective for the dynamic evolution of organometallic species in metal catalysis, from isolated metal atoms through low nuclearity organometallic clusters to nanoclusters and nanoparticles. As the in situ generated organometallic nanoclusters have been deemed to be active species in many metal-catalyzed reactions, we believe that this study not only reveals novel inter- and intra-cluster coupling steps in different stages, but also is reminiscent of the evolution of cluster species in metal-catalyzed reactions.Experimental
Materials and methods
All commercially available chemicals were used without further purification. The solvents used in this study were processed by standard procedures. 1H-NMR and COSY experiments were carried out on a JEOL ECX-400 MHz instrument. Mass spectra were obtained using a Thermo Scientific Exactive Orbitrap instrument and TIMS-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany). DOSY experiments were carried out on a Bruker Avance 600 MHz instrument using a 5 mm TXI H–C/N–D Z-GRD probe. 2D sequence for diffusion measurements were conducted using stimulated echo with 1 spoil gradient. UV-vis spectra were recorded on a Cary 7000 UV-vis-NIR spectrophotometer. EPR experiments were carried out using a JEOL JES-FA200 ESR spectrometer. Transmission electron microscopy (TEM) measurements were performed on a Hitachi H-7650 microscope. The XPS measurement was carried out in a Kratos ULTRA AXIS DLD ultrahigh vacuum photoelectron spectroscopy system. A monochromatic Al Kα X-ray (1486.6 eV) excitation source was used as an excitation source. The details of X-ray crystallographic measurements are summarized in the ESI.† Octamethylazacalix[8]pyridine (Py[8]) was synthesized according to the reported synthetic protocol by Pd-catalyzed fragment coupling reactions between a terminal dibrominated linear pentamer and a terminal diaminated linear trimer.24 Detailed synthesis procedures for aryl dicarbanion bonded Ag4 clusters and hexakis-aryl dicarbanion bonded Ag13 nanoclusters are summarized below.Synthesis of aryl dicarbanion bonded Ag4 clusters
:1) solution at room temperature. After stirring for five minutes, the propargylamine dissolved in 0.5 mL CH2Cl2/CH3OH (v/v 1:1) solution was added dropwise. Then acetone (23.2 mg, 0.4 mmol) was added to the system. After stirring for 10 hours under lucifugal conditions, a black precipitate was produced. After centrifugation, the supernatant was processed under vacuum to produce a yellow crude oily product. Yellow block crystals of complex MePyAg5 were isolated by diffusion of diethyl ether into a concentrated CH2Cl2/CH3OH (v/v 1:1) solution of crude product. Yield: 61% (23.2 mg). 1H NMR (400 MHz, acetone-d6): δ 8.51–8.23 (m, 2H), 8.08 (m, 4H), 7.67 (s, 2H), 7.61–7.37 (m, 10H), 6.91 (d, J = 7.3 Hz, 1H), 6.85 (d, J = 7.7 Hz, 3H), 6.15 (d, J = 8.2 Hz, 1H), 6.09 (d, J = 8.0 Hz, 3H), 3.87 (s, 12H), 3.14 (s, 12H), 2.42 (s, 3H). HR-MS (ESI) calcd for C56H53Ag5F6N17O6S ([MePyAg5–OTf−]+) 1777.8949, found 1777.9000. The crystal structure of the complex MePyAg5 is shown in Fig. 1.
Synthesis of hexakis-aryl dicarbanion bonded Ag13 nanoclusters
Data availability
The X-ray crystallographic coordinates for structures reported in this work have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition number CCDC-1996369 (MePyAg5), CCDC-2062353 (n-PrPyAg5), CCDC-2062351 (PhPyAg5), CCDC-1995296 (MePyAg13), CCDC-2062348 (n-PrPyAg13), CCDC-2062349 (PhPyAg13), CCDC-2175048 (protonated Py[8]), CCDC-1995303 (protonated 2,2′-dimethyl-4,4′-bipyridine), CCDC-2060712 (protonated 2,2′-dipropyl-4,4′-bipyridine). These data can be obtained free of charge from the Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/data_request/cif. For full characterization data including UV-vis spectra, High-resolution ESI-MS, EPR, NMR, XPS, IR, TEM, DFT calculations and experimental details, see the ESI.† Any further relevant data are available from the authors upon reasonable request.Author contributions
L. Z. conceived and supervised the project. The synthetic experiments and structural characterizations were carried out by C.-C. L. EPR measurements were performed by S. Z. R. J. and Y. X. provided assistance in ESI-MS measurements. C.-C. L., S. Z., J. T. and L. Z. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the National Natural Science Foundation of China (22025105, 21821001, 91956125 and 22074075) is gratefully acknowledged.References
- (a) D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2 CrossRef CAS; (b) A. M. Trzeciak and A. W. Augustyniak, Coord. Chem. Rev., 2019, 384, 1 CrossRef CAS; (c) L. Liu and A. Corma, Trends Chem., 2020, 2, 383 CrossRef CAS; (d) J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Dominguez, A. Leyva-Perez and A. Corma, Science, 2012, 338, 1452 CrossRef CAS PubMed; (e) M. Kolter and K. Koszinowski, Chem.–Eur. J., 2019, 25, 13376 CrossRef CAS PubMed; (f) J. Oliver-Messeguer, L. Liu, S. García-García, C. Canós-Giménez, I. Domínguez, R. Gavara, A. Doménech-Carbó, P. Concepción, A. Leyva-Pérez and A. Corma, J. Am. Chem. Soc., 2015, 137, 3894 CrossRef PubMed; (g) A. Corma, P. Concepción, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. López-Quintela, D. Buceta, E. Mendoza, G. Guilera and A. Mayoral, Nat. Chem., 2013, 5, 775 CrossRef CAS PubMed; (h) M. S. Szulmanowicz, A. Gniewek, W. Gil and A. M. Trzeciak, ChemCatChem, 2013, 5, 1152 CrossRef CAS.
- (a) A. V. Astakhov, O. V. Khazipov, A. Y. Chernenko, D. V. Pasyukov, A. S. Kashin, E. G. Gordeev, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2017, 36, 1981 CrossRef CAS; (b) S. Kim, F. Loose, M. J. Bezdek, X. Wang and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 17900 CrossRef CAS PubMed; (c) Y. Jin, C. Zhang, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chem. Soc. Rev., 2021, 50, 2297 RSC.
- (a) D. Pun, T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8213 CrossRef CAS PubMed; (b) B. L. Tran, J. L. Fulton, J. C. Linehan, J. A. Lercher and R. M. Bullock, ACS Catal., 2018, 8, 8441 CrossRef CAS; (c) B. L. Tran, J. L. Fulton, J. C. Linehan, M. Balasubramanian, J. A. Lercher and R. M. Bullock, ACS Catal., 2019, 9, 4106 CrossRef CAS; (d) A. Leyva-Pérez, J. Oliver-Meseguer, P. Rubio-Marqués and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 11554 CrossRef PubMed; (e) S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2014, 47, 816 CrossRef CAS PubMed; (f) R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346 CrossRef CAS PubMed; (g) J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084 CrossRef CAS PubMed; (h) I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208 CrossRef CAS PubMed.
- D. Canseco-Gonzalez, A. Gniewek, M. Szulmanowicz, H. Müller-Bunz, A. M. Trzeciak and M. Albrecht, Chem.–Eur. J., 2012, 18, 6055 CrossRef CAS PubMed.
- C. Li, S. Song, Y. Li, C. Xu, Q. Luo, Y. Guo and X. Wang, Nat. Commun., 2021, 12, 3813 CrossRef CAS PubMed.
- (a) T. Murahashi, Y. Hashimoto, K. Chiyoda, M. Fujimoto, T. Uemura, R. Inoue, S. Ogoshi and H. Kurosawa, J. Am. Chem. Soc., 2008, 130, 8586 CrossRef CAS PubMed; (b) S. Hu, T. Shima and Z. Hou, Nature, 2014, 512, 413 CrossRef CAS PubMed; (c) M. Teramoto, K. Iwata, H. Yamaura, K. Kurashima, K. Miyazawa, Y. Kurashige, K. Yamamoto and T. Murahashi, J. Am. Chem. Soc., 2018, 140, 12682 CrossRef CAS PubMed.
- (a) H. Someya, H. Yorimitsu and K. Oshima, Tetrahedron, 2010, 66, 5993 CrossRef CAS; (b) N. Takashi and H. Tamio, Chem. Lett., 2005, 34, 1152 CrossRef; (c) G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124 RSC.
- (a) Y. Kikukawa, Y. Kuroda, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 2434 CrossRef CAS PubMed; (b) A. K. Clarke, M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2016, 55, 13798 CrossRef CAS PubMed; (c) H. G. Ghalehshahi and R. Madsen, Chem.–Eur. J., 2017, 23, 11920 CrossRef CAS PubMed; (d) S. G. Sudrik, J. Sharma, V. B. Chavan, N. K. Chaki, H. R. Sonawane and K. P. Vijayamohanan, Org. Lett., 2006, 8, 1089 CrossRef CAS PubMed.
- (a) G. M. Whitesides, D. E. Bergbreiter and P. E. Kendall, J. Am. Chem. Soc., 1974, 96, 2806 CrossRef CAS; (b) B. K. Tate, A. J. Jordan, J. Bacsa and J. P. Sadighi, Organometallics, 2017, 36, 964 CrossRef CAS.
- (a) D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2006, 45, 745 CrossRef CAS PubMed; (b) T. A. Bender, M. Morimoto, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 1701 CrossRef CAS PubMed; (c) D. M. Kaphan, M. D. Levin, R. G. Bergman, K. N. Raymond and F. D. Toste, Science, 2015, 350, 1235 CrossRef CAS PubMed; (d) M. D. Levin, D. M. Kaphan, C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 9682 CrossRef CAS PubMed; (e) X. He, Y. Xue, C.-C. Li, Y. Wang, H. Jiang and L. Zhao, Chem. Sci., 2018, 9, 1481 RSC.
- (a) J. Preindl, K. Jouvin, D. Laurich, G. Seidel and A. Fürstner, Chem.–Eur. J., 2016, 22, 237 CrossRef CAS PubMed; (b) L. Jašíková, M. Anania, S. Hybelbauerová and J. Roithová, J. Am. Chem. Soc., 2015, 137, 13647 CrossRef PubMed; (c) D. Weber, M. A. Tarselli and M. R. Gagné, Angew. Chem., Int. Ed., 2009, 48, 5733 CrossRef CAS PubMed.
- G. Abbiati, A. Arcadi, G. Bianchi, S. Di Giuseppe, F. Marinelli and E. Rossi, J. Org. Chem., 2003, 68, 6959 CrossRef CAS PubMed.
- E.-X. Zhang, D.-X. Wang, Q.-Y. Zheng and M.-X. Wang, Org. Lett., 2008, 10, 2565 CrossRef CAS PubMed.
- (a) M. F. Ibad, A. Schulz and A. Villinger, Organometallics, 2015, 34, 3893 CrossRef CAS; (b) M. Håkansson, H. Eriksson and S. Jagner, Inorg. Chim. Acta, 2006, 359, 2519 CrossRef.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- (a) V. G. Albano, L. Grossi, G. Longoni, M. Monari, S. Mulley and A. Sironi, J. Am. Chem. Soc., 1992, 114, 5708 CrossRef CAS; (b) X. Liu, J. Chen, J. Yuan, Y. Li, J. Li, S. Zhou, C. Yao, L. Liao, S. Zhuang, Y. Zhao, H. Deng, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2018, 57, 11273 CrossRef CAS PubMed; (c) X.-J. Xi, J.-S. Yang, J.-Y. Wang, X.-Y. Dong and S.-Q. Zang, Nanoscale, 2018, 10, 21013 RSC.
- G. Schön, Acta Chem. Scand., 1973, 27, 2623 CrossRef.
- V. K. Kaushik, J. Electron Spectrosc. Relat. Phenom., 1991, 56, 273 CrossRef CAS.
- (a) K. K. Chakrahari, J.-H. Liao, S. Kahlal, Y.-C. Liu, M.-H. Chiang, J.-Y. Saillard and C. W. Liu, Angew. Chem., Int. Ed., 2016, 55, 14704 CrossRef CAS PubMed; (b) V. G. Albano, F. Calderoni, M. C. Iapalucci, G. Longoni, M. Monari and P. Zanello, J. Cluster Sci., 1995, 6, 107 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083 CrossRef CAS.
- Q. Chem and T. Lu, J. Comput. Chem., 2022, 43, 539 CrossRef PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, UK, 1990 Search PubMed.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
- H.-Y. Gong, X.-H. Zhang, D.-X. Wang, H.-W. Ma, Q.-Y. Zheng and M.-X. Wang, Chem.–Eur. J., 2006, 12, 9262 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1995296, 1995303, 1996369, 2060712, 2062348, 2062349, 2062351, 2062353 and 2175048. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00989g |
| This journal is © The Royal Society of Chemistry 2022 |