
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceParallel imaging of coagulation pathway proteases activated protein C, thrombin, and factor Xa in human plasma†
Sylwia
Modrzycka
a,
Sonia
Kołt
a,
Stéphanie G. I.
Polderdijk
b,
Ty E.
Adams
b,
Stanisław
Potoczek
c,
James A.
Huntington
b,
Paulina
Kasperkiewicz
 a and
Marcin
Drąg
*a
a and
Marcin
Drąg
*a
aDepartment of Chemical Biology and Bioimaging, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: marcin.drag@pwr.edu.pl; sylwia.modrzycka@pwr.edu.pl; sonia.kolt133@gmail.com; paulina.kasperkiewicz@pwr.edu.pl
bDepartment of Haematology, Cambridge Institute for Medical Research, University of Cambridge, The Keith Peters Building, Hills Road, Cambridge, CB2 0XY, UK. E-mail: stephanie.polderdijk@cantab.net; tea22@cam.ac.uk; jah52@cam.ac.uk
cDepartment of Haematology, Blood Neoplasms, and Bone Marrow Transplantation, Wrocław Medical University, Pasteura 1, 50-367 Wrocław, Poland. E-mail: stanislaw.potoczek@umw.edu.pl
First published on 27th April 2022
Abstract
Activated protein C (APC), thrombin, and factor (f) Xa are vitamin K-dependent serine proteases that are key factors in blood coagulation. Moreover, they play important roles in inflammation, apoptosis, fibrosis, angiogenesis, and viral infections. Abnormal activity of these coagulation factors has been related to multiple conditions, such as bleeding and thrombosis, Alzheimer's disease, sepsis, multiple sclerosis, and COVID-19. The individual activities of APC, thrombin, and fXa in coagulation and in various diseases are difficult to establish since these proteases are related and have similar substrate preferences. Therefore, the development of selective chemical tools that enable imaging and discrimination between coagulation factors in biological samples may provide better insight into their roles in various conditions and potentially aid in the establishment of novel diagnostic tests. In our study, we used a large collection of unnatural amino acids, and this enabled us to extensively explore the binding pockets of the enzymes' active sites. Based on the specificity profiles obtained, we designed highly selective substrates, inhibitors, and fluorescent activity-based probes (ABPs) that were used for fast, direct, and simultaneous detection of APC, thrombin, and fXa in human plasma.
Introduction
Signaling pathways are exceptional mechanisms that maintain homeostasis, execute many vital processes, and fight pathological conditions. Their activities are based on the synergistic occurrence of receptor–ligand interactions and the peptide bond hydrolysis performed by proteases. Proteolytic signaling plays the leading role in the blood coagulation pathway, which is a cascade of protease activation events that results in the formation of thrombin, an effector enzyme. The cascade can be induced by two different stimuli – exposure of blood to tissue factor on the subendothelium (extrinsic pathway)1 or through contact with certain surfaces (intrinsic pathway)2 – before merging into the common pathway and generation of thrombin.3 The coagulation cascade involves several reactions that convert inactive zymogens into active serine proteases, the key participants in this process. In this pathway, there are several zymogens, namely, factors (f) II, VII, IX, X, XI, XII, and protein C.4 Once activated, the zymogens are tagged with the suffix “a” (some exceptions include activated protein C (APC), fII and fIIa, which are generally referred to as prothrombin and thrombin, respectively). One of the most extensively studied reactions in the process of blood clotting is the proteolytic activation of prothrombin to active thrombin, and this is performed by prothrombinase, which is the complexation of fXa with fVa.5 Thrombin activates platelets by cleaving protease-activated receptors, PAR1 and PAR4;6 by cleaving fibrinogen to fibrin,7 the two major components of a hemostatic clot; and by stimulating its own formation through proteolytic activation of fV, fVIII and fXI.8 On the other hand, when excess thrombin is generated, it shuts down the coagulation cascade by binding to the endothelial cell receptor thrombomodulin and converting PC to APC, which proteolytically inactivates the intrinsic Xase complex (fIXa–fVIIIa) and the prothrombinase complex.9 All proteases involved in this intricate cascade of events play a part in maintaining the balance between coagulation and blood circulation. However, the balance seems to be mostly dependent on the activity of APC, thrombin, and fXa from the common pathway. Disruption of the naturally occurring levels of these proteases in plasma may lead to severe disorders. Abnormal activities of these coagulation factors have been associated with conditions such as hemophilia,10 thrombosis,11–13 disseminated intravascular coagulation,14 Alzheimer's disease,15,16 sepsis,17 stroke,18,19 cancer,20 multiple sclerosis,21 and COVID-19.22,23Despite major progress in understanding the coagulation system, the individual roles of APC, thrombin, and fXa in various conditions have not yet been fully established, nor are there any sufficiently sensitive and selective methods to detect their individual levels in biological samples, such as blood plasma. Therefore, the development of selective chemical tools to enable coagulation factors in human plasma to be detected and discriminated would aid in unveiling their individual roles in various states of diseases. The most convenient approach is to determine the specificity profiles for enzyme substrates and use these profiles as a basis for designing individual peptide substrates, inhibitors, and ABPs that are tagged with detectable moieties. Since APC, thrombin, and fXa are related vitamin K-dependent serine proteases with considerable structural homology and similar substrate preferences,24–27 it is difficult to distinguish between them using only natural amino acids. To overcome this limitation, we utilized a defined P1 library and the Hybrid Combinatorial Substrate Library (HyCoSuL). Both libraries contained a large pool of unnatural amino acids, which allowed for the chemical space in the P4–P1 positions to be more extensively explored. The library screening results enabled us to design and synthesize active and selective APC, thrombin, and fXa substrates with the ability to distinguish these proteases from other coagulation factors. The most selective substrates were then converted into inhibitors and ABPs with Cy3/Cy5/Cy7 fluorophores. Finally, we demonstrated that our fluorescent activity-based probes could selectively label APC, thrombin, and fXa using mixtures of purified enzymes and human plasma.
Results
Substrate specificity of APC, thrombin, and fXa at the P1 position
To develop selective chemical tools to study coagulation proteases, we established the substrate specificity profiles for APC, thrombin, and fXa using a set of natural and unnatural amino acids. To determine the specificity of P1, we tested the activity of these enzymes against a tailored library of fluorogenic substrates with a constant P4–P2 sequence and various amino acid residues at the P1 position.28 The general structure of the library was Ac–Ala–Arg–Leu–P1–ACC, in which ACC was 7-amino-4-carbamoylmethylcoumarin, Ac was an acetyl group, and P1 was an individual amino acid. A comparison of the substrate specificity of APC, thrombin, and fXa at the P1 position revealed that all three proteases exhibited substrate preferences that were overlapping (Fig. 1). We found that they interacted exclusively with positively charged L-Arg (100%), and no other amino acid residue fit in that subsite, which was consistent with previous studies that found arginine at the P1 position in all APC, thrombin, and fXa physiological substrates.26,29–31 | ||
| Fig. 1 Substrate specificity profiles for APC, thrombin, and fXa at the P1 position presented as heatmaps. The defined library was treated with individual enzymes, and the substrate hydrolysis rate was measured as an increase in fluorescence over time (RFU s−1). The substrate cleavage was monitored for 30 min (λex = 355 nm, λem = 460 nm). The substrate specificity profiles were obtained by setting the highest RFU s−1 value as 100% and adjusting the other results accordingly. The average relative activity was determined as a percentage of the best-recognized amino acid (n = 2, where n represents the number of independent experiments). | ||
Substrate specificity for the APC, thrombin, and fXa at the P4–P2 positions
Next, we utilized the Hybrid Combinatorial Substrate Library (HyCoSuL) approach that was developed by our group32 to determine the binding pocket preferences at the P4–P2 positions. HyCoSuL extends far beyond the classic Positional Scanning Substrate Combinatorial Library (PS-SCL) and can provide enhanced selectivity for individual coagulation factors. Since all three enzymes preferentially accommodated arginine in the S1 pocket, we employed the previously described combinatorial library with arginine fixed at the P1 position.28,33,34 This library consisted of three tetrapeptide sublibraries (Ac–P4–Mix–Mix–Arg–ACC, Ac–Mix–P3–Mix–Arg–ACC, Ac–Mix–Mix–P2–Arg–ACC) and contained a natural and a large pool of unnatural amino acids at the investigated position (P4, P3 or P2) and an equimolar mixture of natural amino acids (Mix) at the remaining positions. The activity of enzymes toward the libraries was tested, and the data resulted in a highly detailed picture of the active site preferences of APC, thrombin, and fXa (Fig. 2).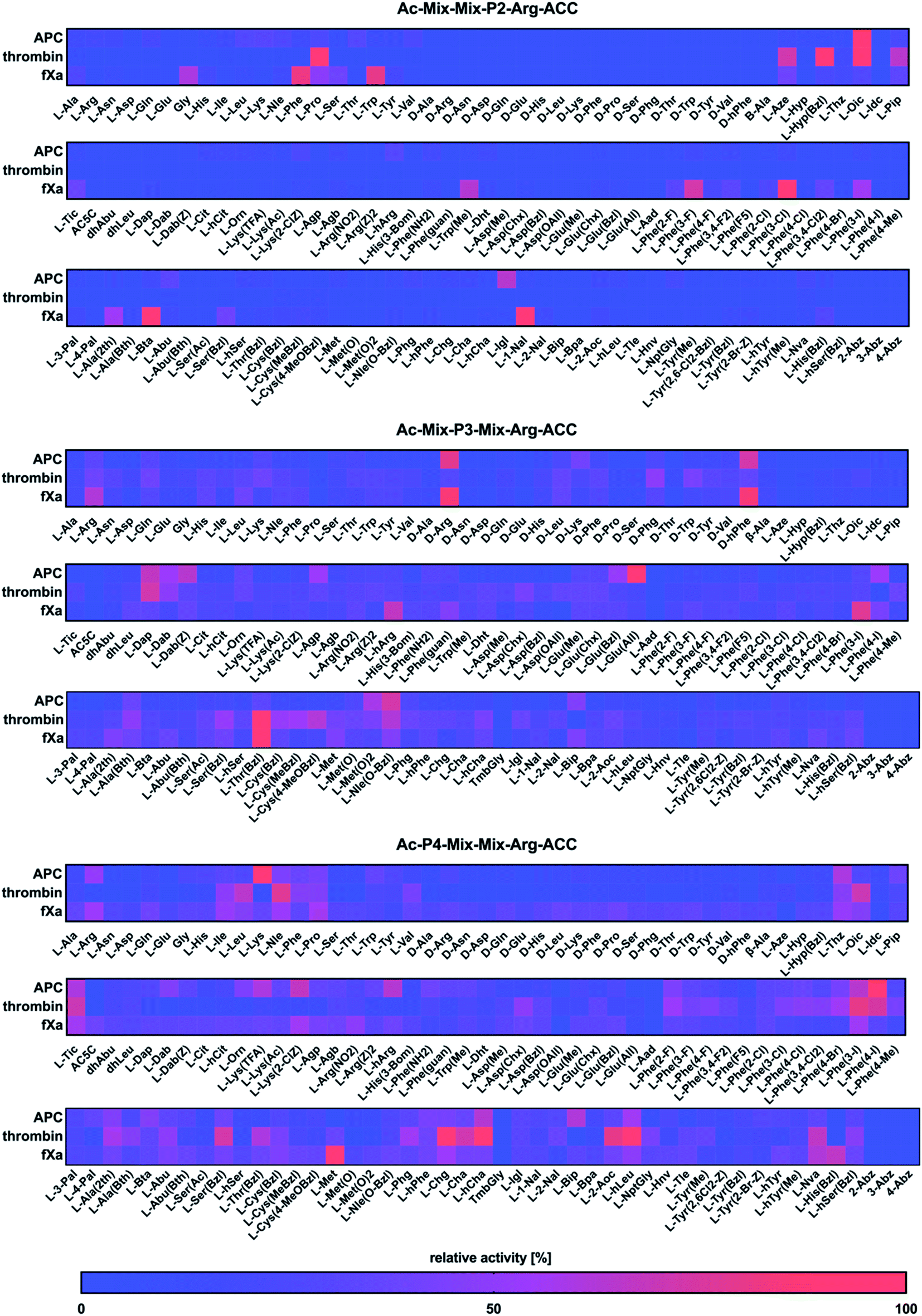 | ||
| Fig. 2 Substrate specificity profiles for APC, thrombin, and fXa at the P4–P2 positions, which are presented as heatmaps. The HyCoSuL library was treated with individual enzymes, and the substrate hydrolysis rate was measured as an increase in fluorescence over time (RFU s−1). The release of ACC was monitored for 30 min (λex = 355 nm, λem = 460 nm). The substrate specificity profiles were established by setting the highest RFU s−1 value as 100% and adjusting the other results accordingly. The average relative activity is presented as a percentage of the best-recognized amino acid (n = 2, where n represents the number of independent experiments). | ||
P2 position analysis
The APC, thrombin, and fXa S2 pockets exhibited a very high level of specificity, and each pocket accommodated only a few natural and unnatural residues. The relative activity of other amino acids did not exceed 30%. Negatively charged (L-Asp, L-Glu, L-Aad (L-alpha-homoglutamic acid 6-t-butyl ester)) and aminobenzoic acid residues (2-Abz, 3-Abz, 4-Abz) were completely ignored by the investigated proteases. Moreover, all tested enzymes exhibited no activity toward D-amino acids, indicating that their S2 pocket is stereochemically specific. In the case of APC from the pool of natural amino acids, the S2 pocket accommodated many structurally different residues, including polar, nonpolar, and basic amino acids, and the best residue was L-Lys (30%). These findings are consistent with the data obtained by the Huntington group.35,36 However, the most preferred amino acid was the unnatural proline derivative L-octahydroindole-2-carboxylic acid (L-Oic, 100%). L-2-Indanylglycine (L-Igl, 64%), which is bulky and hydrophobic, was also well tolerated at this position, and more importantly, it allowed us to distinguish APC from both thrombin and fXa. The specificity profile for the thrombin substrate at the P2 position was narrower than that of APC and fXa, since its S2 pocket, which is formed by Tyr60A, Trp60D, His57 and Leu99, is hydrophobic and restricted.37–39 Among the natural amino acids, there was a noticeable preference for L-Pro (93%), which confirmed previous findings.26,29,40,41 We observed a similar regularity in the set of unnatural amino acids, since, compared to other structures, proline derivatives were more potently hydrolyzed. Additionally, L-azetidine-2-carboxylic acid (L-Aze, 72%) and L-piperidine-2-carboxylic acid (L-Pip, 65%) were recognized approximately three times better by thrombin than by APC and fXa. We found that the fXa S2 pocket was capable of binding various natural amino acids with a preference for hydrophobic L-Phe (82%), L-Trp (80%) and the nonpolar Gly (60%) without a side chain. This finding is in agreement with the previous data reported by Gosalia et al.42 What distinguished the specificity of fXa from the other investigated proteases was the high activity toward bulky unnatural amino acids such as 3-benzothienyl-L-alanine (L-Bta, 100%) and 3-(1-naphthyl)-L-alanine (L-1-Nal, 94%) as well as phenylalanine analogs with halide substituted in meta position L-Phe(3-F) (73%), L-Phe(3-Cl) (100%), L-Phe(3-I) (55%).P3 position analysis
Compared to the S2 pocket, the S3 pocket appeared to be more tolerant to structural diversity. In the case of natural amino acids, all three proteases displayed similar substrate preferences. Therefore, extending the classic PS-SCL library with a large pool of unnatural amino acids allowed the chemical space of the S3 subsite to be more extensively explored. We observed that the amino acids with D-stereochemistry were well tolerated at this position, but proline and its unnatural derivatives demonstrated little to no activity. The APC S3 pocket preferentially accommodated the basic residues of L-Arg (32%) and L-Lys (28%). Interestingly, their D-enantiomers were recognized even better (D-Arg, 79%, D-Lys, 41%), which is in agreement with previous results.43 Additionally, a significant increase in the relative activity was also observed in the case of the L-Arg and L-Lys derivatives with shorter side chains and the same chemical character, such as L-Agp (54%) and L-Dap (67%). Bulky amino acid residues with a benzyl group, namely, benzyloxycarbonyl-L-2,4-diaminobutyric acid (L-Dab(Z), 64%), L-glutamic-acid-gamma-benzyl ester (L-Glu(Bzl), 52%), and 6-benzyloxy-L-norleucine (L-Nle(O-Bzl), 73%), were both well recognized and selectively recognized at this position. Thrombin revealed a broad preference for natural amino acids, mainly long and basic L-Arg (39%) and L-Lys (39%). The hydrophobic unnatural residues equipped with a benzyl group were fairly well tolerated at this position. Among them, two cysteine derivatives, L-Cys(MeBzl) (54%) and L-Cys(4-MeOBzl) (62%), were found to be actively and selectively recognized only by thrombin. These observations demonstrated that the APC and thrombin S3 pockets have dual properties. They are built from negatively charged residues, since they accept arginine and lysine, but they can also accommodate large, hydrophobic side chains. For the fXa from the pool of natural amino acids, the best-recognized amino acid was L-Arg (61%). The fXa S3 subsite also accommodated D-amino acids with the highest activity toward D-Arg (86%) and D-hPhe (85%). On the other hand, these structures were also well recognized by APC and therefore could not be used for the further synthesis of selective substrates. Surprisingly, we found that compared to its shorter analogs (L-Agb, 23%, L-Agp, 20%), the arginine derivative elongated with one methylene group (L-hArg, 67%) was more active. This preference distinguished fXa from APC and thrombin.P4 position analysis
In the case of APC, the most preferred natural amino acid residues were long and basic L-Arg (51%) and L-Lys (100%). Negatively charged and D-enantiomers were mostly rejected at this position. From the set of unnatural structures, lysine and arginine derivatives L-Lys(TFA) (60%), Lys(2-ClZ) (67%), and L-hArg (63%) fit well in this subsite. We observed that L-Phe(4-I) (89%), a phenylalanine analog with an iodine substitution in the para position, was also recognized well. Unlike APC, thrombin did not recognize arginine and lysine residues but was capable of binding branched aliphatic amino acids, such as L-Ile (49%), L-Leu (65%), and L-Val (42%). These findings are consistent with the data obtained by the Craik26 and Hellman groups.44 Unnatural phenylalanine derivatives were also tolerated fairly well; however, they were not selective because APC also recognized them. Our data demonstrated that side chain elongation enhanced substrate processing, as in the case of 2-amino-octanoic acid (L-2-Aoc, 80%), L-homoleucine (L-hLeu, 98%), and L-homocyclohexylalanine (L-hCha, 94%). The analysis of the P4 position demonstrated that fXa exhibited almost no stereospecificity in the S4 pocket since it recognized natural amino acid residues and their D-enantiomers at very similar levels. The most preferred amino acid was L-Met (100%); however, more importantly, D-Pro (39%) and L-His(Bzl) (66%) were exclusively recognized only by fXa and therefore could be further applied in the design of specific tools for this enzyme.Design and kinetic analysis of the selective substrates
To design selective substrates for all investigated proteases, we combined the results obtained from the P1 library screening with the HyCoSuL approach and selected the amino acid residues that were preferred by only one enzyme. We then synthesized several promising tetrapeptides with various P4–P2 regions and performed an initial screening (Fig. 3). For the most promising substrates, we performed a detailed kinetic analysis (kcat, KM, kcat/KM) (Table 1).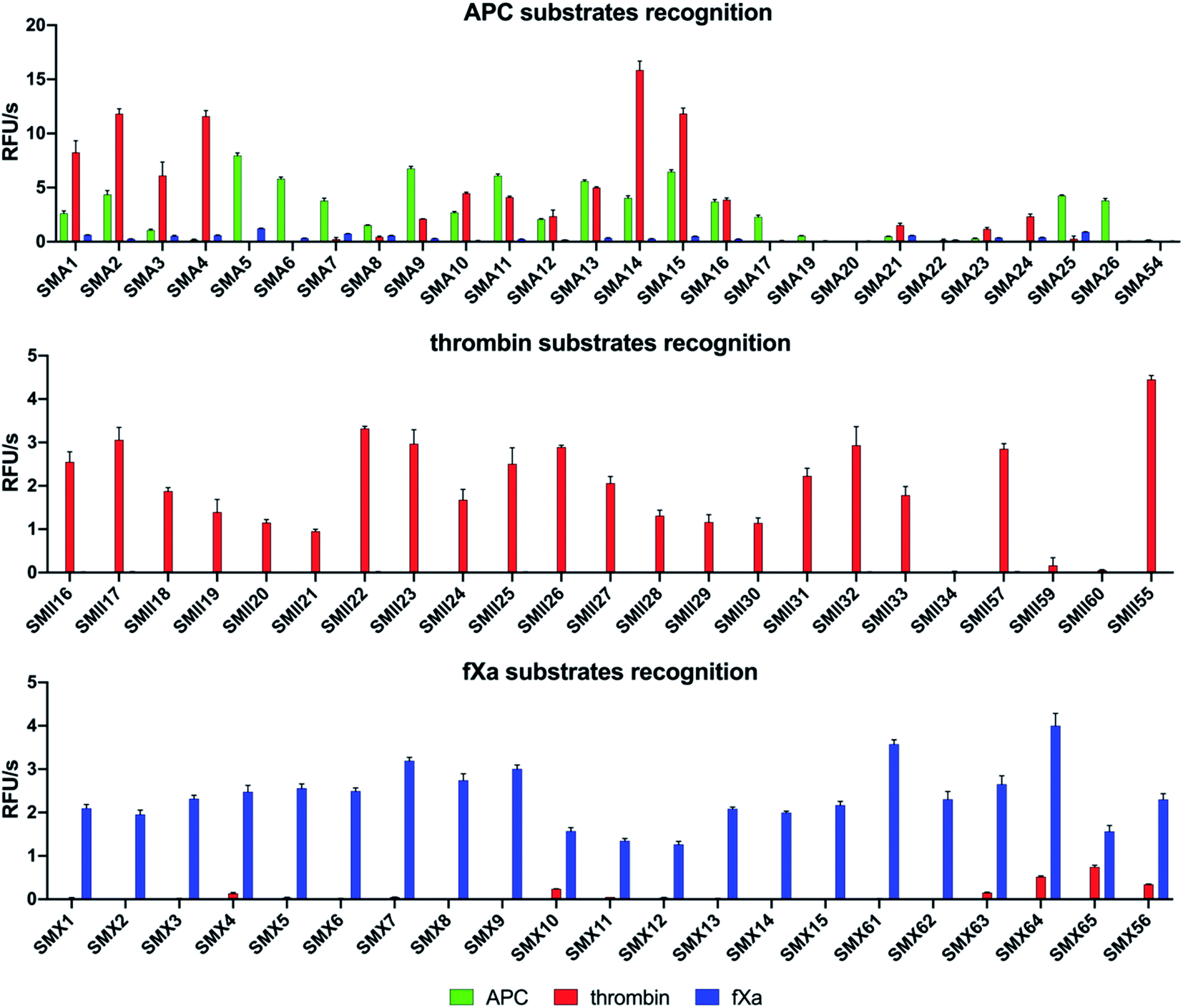 | ||
| Fig. 3 Results of the substrate screening with APC, thrombin, and fXa. The rate of substrate hydrolysis as relative fluorescence units per second (RFU s−1) was measured for 30 min (λex = 355 nm, λem = 460 nm). The data are presented as the mean values ± s.d.; n = 3, where n represents the number of independent experiments. The substrate sequences are presented in the ESI section (Tables S2–S4†). | ||
| Substrate | k cat/KM [M−1 s−1] | |||
|---|---|---|---|---|
| APC | Thrombin | fXa | ||
| SMA5 | Ac–Lys–Dab(Z)–Igl–Arg–ACC | 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 2000 000 ± 2000 |
1800 ± 80 | 4000 ± 150 |
| SMA6 | Ac–Lys–Agp–Igl–Arg–ACC | 40000 ± 1600 |
180 ± 7 | 1300 ± 90 |
| SMA17 | Ac–Lys–Dab(Z)–Lys–Arg–ACC | 10000 ± 600 |
130 ± 8 | 190 ± 6 |
| SMA26 | Ac–Lys–βhLys–Igl–Arg–ACC | 15500 ± 800 |
60 ± 2 | 380 ± 13 |
| SMII22 | Ac–hCha–Cys(MeBzl)–Pip–Arg–ACC | 2560 ± 180 | 8660000 ± 260000 |
53400 ± 3900 |
| SMII32 | Ac–hLeu–Cys(4-MeOBzl)–Pip–Arg–ACC | 1840 ± 70 | 7660000 ± 230000 |
64000 ± 5800 |
| SMII55 | Ac–Nle–Lys–Pro–Arg–ACC | 3160 ± 80 | 6680000 ± 460000 |
55500 ± 500 |
| SMX8 | Ac–Gln–hArg–Bta–Arg–ACC | 520 ± 10 | 800 ± 80 | 182000 ± 13000 |
| SMX9 | Ac–DPro–hArg–Bta–Arg–ACC | 560 ± 20 | 750 ± 50 | 217000 ± 15000 |
| SMX15 | Ac–DPro–hArg–1-Nal–Arg–ACC | 590 ± 10 | 400 ± 20 | 143000 ± 6500 |
| SMX61 | Ac–Met–hArg–1-Nal–Arg–ACC | 720 ± 40 | 1450 ± 70 | 221000 ± 12000 |
APC substrate design and validation
We designed and synthesized twenty-five APC tetrapeptide fluorogenic substrates (named SMA1–SMA26) that contained the most selective natural and unnatural amino acid residues and one substrate with only natural amino acids in its sequence (SMA54). Our initial screening showed that the sequences with L-Igl (SMA5–SMA8), L-Val (SMA9–SMA12), and L-Oic (SMA13–SMA16) at P2 were more efficiently cleaved by APC than those with L-Lys(2-ClZ) (SMA1–SMA4), L-Lys (SMA17–SMA20) and L-Cha (SMA21–SMA24). This is in agreement with our previous HyCoSuL screening results (Fig. 2). P2 was also crucial in terms of discriminating APC from thrombin and fXa since L-Igl and L-Lys were the most selective amino acids for APC and were poorly recognized by the other coagulation factors. Moreover, we discovered that exchanging the long L-Nle(O-Bzl) (SMA4) at P3 for the shorter L-Dab(Z) (SMA1), L-Agp (SMA2) and L-Glu(Bzl) (SMA3) resulted in significant increases in activity (13-, 22- and 5.5-fold, respectively). Compared to the best natural substrate (SMA54), the best substrate that contained unnatural amino acids (SMA5) was cleaved approximately 50-fold more efficiently, suggesting that the use of the HyCoSuL approach allowed us to obtain active and selective chemical tools for APC. To minimize potential cross reactivity, we performed a detailed kinetic analysis of the four APC-selective substrates against the other proteases. The catalytic efficiency was determined and revealed that the most potent substrate, SMA5 (Ac–Lys–Dab(Z)–Igl–Arg–ACC, kcat/KM = 71000 ± 2000 M−1 s−1), was also recognized by thrombin (kcat/KM = 1800 ± 80 M−1 s−1) and fXa (kcat/KM = 4000 ± 150 M−1 s−1); therefore, this structure was excluded from further consideration. However, we were able to find several selective substrates, among which SMA17 (Ac–Lys–Dab(Z)–Lys–Arg–ACC, kcat/KM = 10000 ± 600 M−1 s−1) displayed a very high kcat/KM selectivity ratio and almost nondetectable hydrolysis when tested with other coagulation factors.
Thrombin substrate design and kinetic evaluation
For thrombin, we synthesized twenty-three optimal fluorogenic substrates (SMII16–SMII55) with natural and unnatural amino acid residues. We noticed that the exchange of only one amino acid at the P2 position, short L-Pro (SMII25), L-Aze (SMII28) or L-Pip (SMII31) for longer L-Hyp(Bzl) (SMII34), caused a significant decrease in activity by over 99%, strongly suggesting that this pocket is small and has a critical role in substrate interactions, and this result was consistent with the previous results for thrombin.41 However, the preference of P4 for bulky and branched L-hCha (SMII22), L-hLeu (SMII23), and L-Chg (SMII57), which was greater than that for the long L-2-Aoc (SMII24) obtained in our initial substrate screening, matched the specificity profiling for HyCoSuL (Fig. 2). To determine the leading candidates for inhibitors and activity-based probes, we performed a detailed kinetic analysis of the promising substrates (SMII22, SMII32, SMII55) with all investigated proteases. Our data revealed that the most potent thrombin substrate, SMII22 (Ac–hCha–Cys(MeBzl)–Pip–Arg–ACC, kcat/KM = 8660000 ± 260000 M−1 s−1), was weakly hydrolyzed by APC and fXa, and 3383- and 162-fold selectivity values were observed for thrombin, respectively. The mostly natural substrate SMII55 (Ac–Nle–Lys–Pro–Arg–ACC) displayed the lowest kcat/KM selectivity ratio among all tested substrates; thus, it was eliminated from further consideration.
fXa substrate design and validation
Based on the library screening results, we designed and synthesized twenty-one fXa individual fluorogenic substrates (SMX1–SMX56). After an initial screening, we found that among the fXa substrates, those containing L-Bta at P2 (SMX7–SMX9) exhibited the highest activity, and a 1.5-fold reduction in hydrolysis was observed with L-Phe at P2 (SMX10–SMX12). We also noticed that replacing D-Pro (SMX15) at P4 with L-Arg(NO2) (SMX13) or L-Gln (SMX14) caused only a slight decrease in the overall activity. However, exchanging D-Pro at P4 (SMX15, kcat/KM = 143000 ± 6500 M−1 s−1) for L-Met (SMX61, kcat/KM = 221000 ± 12000 M−1 s−1) increased the kcat/KM value by 55%. These findings mirror the HyCoSuL screening data described above. Moreover, P2 and P4 have an important role in distinguishing fXa from thrombin, since the most selective substrate, SMX15 (Ac–DPro–hArg–1-Nal–Arg–ACC), was 358 times more hydrolyzed by fXa than by thrombin. Based on that, SMX15 was a leading candidate for the conversion to inhibitor and ABP, and it was selected for use in biological samples.
Development of biotinylated ABPs
To date, the in situ study of coagulation factors relies on antibody-related techniques, which poorly discriminate between active and inactive (e.g., zymogen, inhibited protease) forms. Since the presence of active protein defines its biological function, we used what was learned above to design chemical tools for the detection of the active forms of APC, thrombin, and fXa. We converted the most selective substrates into first-generation biotin-labeled ABPs. Such tools allow us to track the active enzymes in biological samples via SDS–PAGE followed by protein transfer. Additionally, these tools can be utilized to isolate a protein of interest from other proteins by applying affinity purification/enrichment on appropriate beads.45,46 Our probes were equipped with a biotin tag that was separated from the peptide sequence by a 6-aminohexanoic acid linker, which was applied to reduce the steric hindrance of the compounds. As a reactive warhead, we selected a diphenyl phosphonate, an electrophile known to covalently react with the active site of serine proteases.47 Using a mixed solid- and solution-phase approach, we synthesized three biotin-labeled ABPs that contained the most selective APC sequences as follows: P-SMA61 (biotin–6-Ahx–Lys–Agp–Igl–ArgP(OPh)2), P-SMA171 (biotin–6-Ahx–Lys–Dab(Z)–Lys–ArgP(OPh)2) and P-SMA261 (biotin–6-Ahx–Lys–βhLys–Igl–ArgP(OPh)2). For thrombin, we chose two potent and selective substrates as scaffolds for biotinylated probes as follows: P-SMII221 (biotin–6-Ahx–hCha–Cys(MeBzl)–Pip–ArgP(OPh)2) and P-SMII321 (biotin–6-Ahx–hLeu–Cys(4-MeOBzl)–Pip–ArgP(OPh)2). For fXa, we synthesized three first-generation ABPs: P-SMX91 (biotin–6-Ahx–DPro–hArg–Bta–ArgP(OPh)2), P-SMX151 (biotin–6-Ahx–DPro–hArg–1-Nal–ArgP(OPh)2), and P-SMX611 (biotin–6-Ahx–Met–hArg–1-Nal–ArgP(OPh)2) (Fig. 4A and B).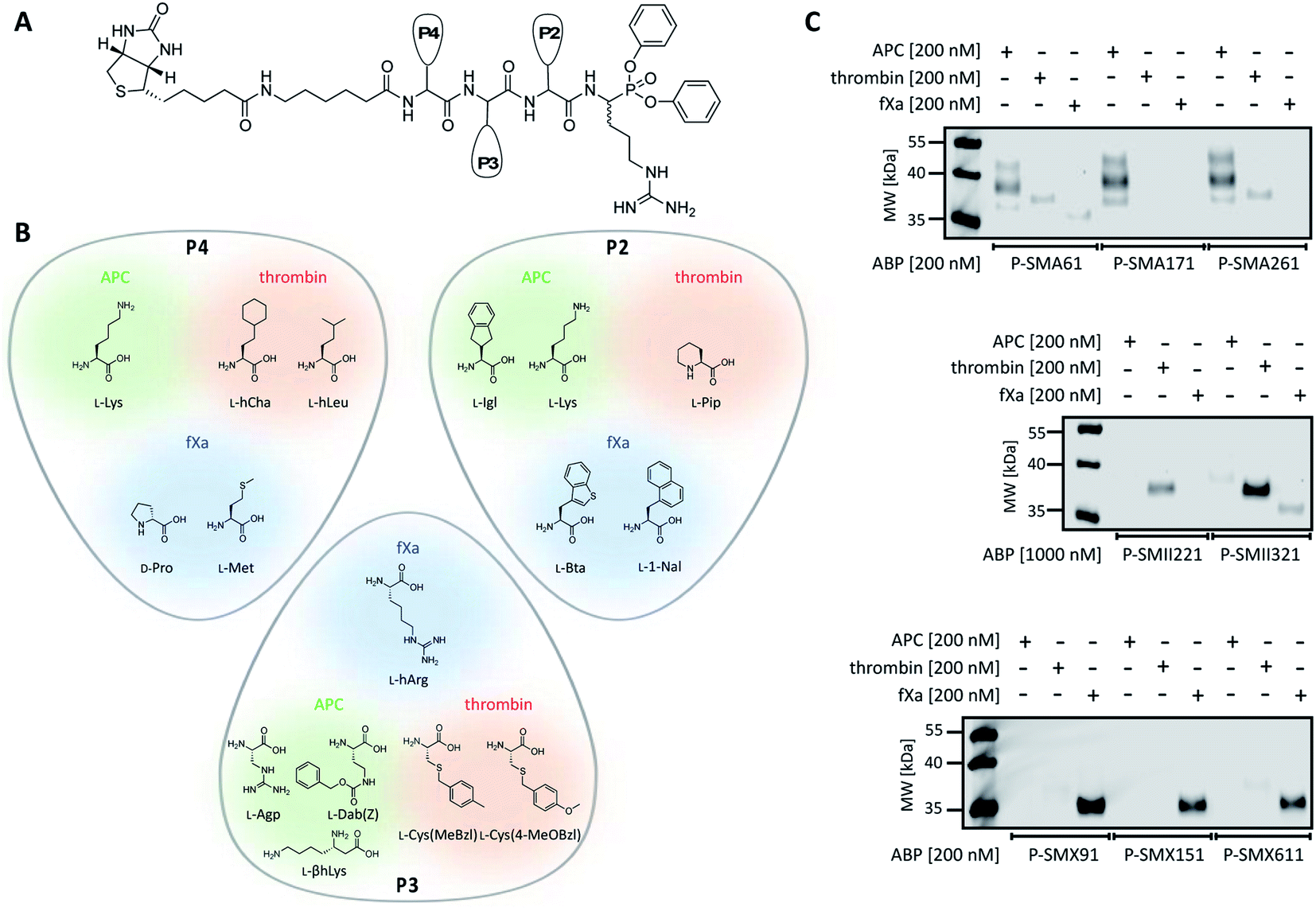 | ||
| Fig. 4 APC, thrombin, and fXa biotinylated ABPs. (A) A general structure of a biotinylated ABP for APC, thrombin, and fXa. (B) Amino acid residues selected for the synthesis of biotinylated ABPs based on the substrate kinetics evaluation. (C) Labeling of purified coagulation factors (APC, thrombin, fXa) using eight biotinylated ABPs. The enzymes (200 nM) were incubated separately with each probe (the probe:enzyme ratio was 1 or 5, depending on the ABP used) for 30 min at 37 °C. The samples were then subjected to SDS–PAGE analysis, transferred to a membrane, incubated with fluorescent streptavidin Alexa Fluor 647 conjugate, and detected at 658 nm using an Azure Biosystems Sapphire Biomolecular Imager. The results are representative of at least 3 replicates. | ||
Detection of APC, thrombin, and fXa using biotinylated ABPs
Since we introduced two main modifications in the substrate structure (replacing ACC with a diphenyl phosphonate and changing the N-terminal acetyl to a biotin tag), we had to verify whether the selectivity of our probes was maintained. To confirm the utility of biotinylated ABPs for membrane detection, we performed in vitro labeling using purified coagulation factors (APC, thrombin, fXa) (Fig. 4C and S2†). All probes were incubated separately with each active protease (the probe:enzyme ratio was 1 or 5, depending on the ABP used) for 30 min. Then, SDS–PAGE analysis was performed, the proteins were transferred to nitrocellulose membranes and visualization was performed with streptavidin conjugated with fluorophore. First, we tested the efficiency of P-SMA61, P-SMA171, and P-SMA261 toward purified APC. As a selectivity control, we used other relevant serine proteases from the coagulation pathway, namely, thrombin and fXa. This analysis revealed that all three probes were capable of APC labeling; however, P-SMA61 displayed cross-reactivity with thrombin and fXa and P-SMA261 labeled thrombin. We observed that P-SMA171 exhibited both APC potency and selectivity, and because our prior goal was to find a highly selective peptide sequence, we chose P-SMA171 for further biological studies. Next, we analyzed the binding activity of P-SMII221 and P-SMII321, which are the first-generation probes we designed for thrombin. After reaction with both ABPs, the western blot showed a signal from a protein between 35 and 40 kDa, corresponding to the size of thrombin. It appeared that compared to P-SMII321, P-SMII221 was less potent but more selective toward thrombin, so it was chosen for the further detection of thrombin in biological samples. All three fXa ABPs were potent and selective. However, careful examination revealed that P-SMX91 and P-SMX611 slightly labeled thrombin. Modifying the P-SMX611 structure by replacing L-Met at P4 with D-Pro allowed us to obtain P-SMX151. This change reduced the off-target thrombin labeling while retaining the potency of this probe.
Design of selective fluorescent ABPs and inhibitors
Since biotinylated ABPs are not suitable for the simultaneous and direct detection of protease activity, we exchanged the biotin tag for fluorescent cyanine derivatives (Cy3, Cy5, Cy7), which have high quantum yields and allow labeled proteins to be detected at low concentrations.48 Using previously obtained APC-, thrombin-, and fXa-selective sequences, we synthesized three second-generation ABPs with the general structure of Cy5–6-Ahx–Lys–Dab(Z)–Lys–ArgP(OPh)2 (P-SMA172) for APC, Cy7–6-Ahx–hCha–Cys(MeBzl)–Pip–ArgP(OPh)2 (P-SMII222) for thrombin, and Cy3–6-Ahx–DPro–hArg–1-Nal–ArgP(OPh)2 (P-SMX152) for fXa (Fig. 5A). Since our second-generation ABPs are equipped with a diphenyl phosphonate as a reactive group, they also exhibit the properties of a covalent inhibitor. Therefore, in the next step, we replaced the fluorescent tag with the N-terminal acetyl group and synthesized a set of simple peptide-based inhibitors, namely, I-SMA17 (Ac–Lys–Dab(Z)–Lys–ArgP(OPh)2), I-SMII22 (Ac–hCha–Cys(MeBzl)–Pip–ArgP(OPh)2) and I-SMX15 (Ac–DPro–hArg–1-Nal–ArgP(OPh)2), for the investigation of APC, thrombin, and fXa, respectively (Fig. 5A).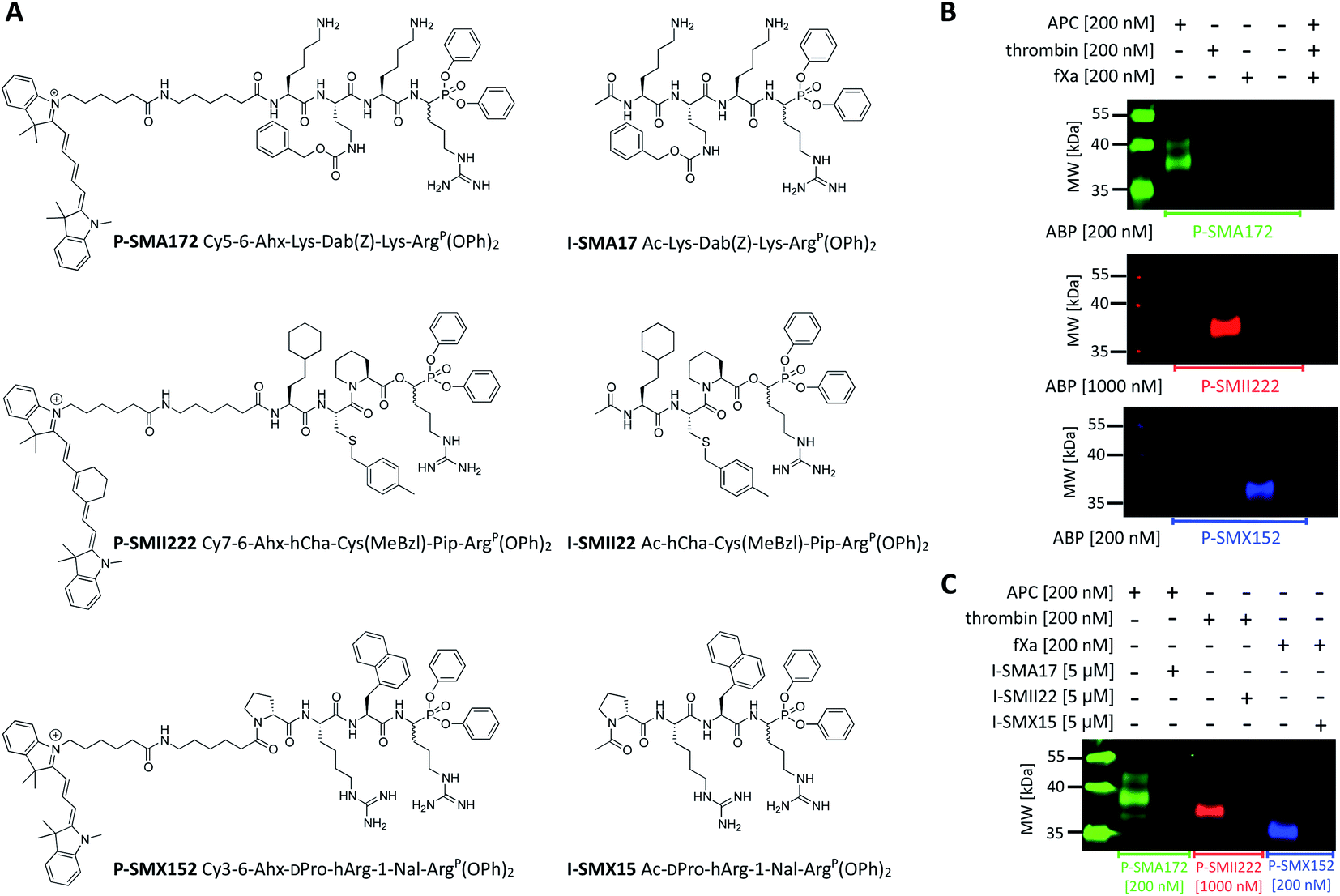 | ||
| Fig. 5 APC, thrombin, and fXa fluorescent ABPs and inhibitors. (A) Structures of fluorescent ABPs and inhibitors dedicated to APC, thrombin, and fXa. (B) Labeling of purified coagulation factors (APC, thrombin, fXa) using fluorescent ABPs. The enzymes (200 nM) were incubated separately with each probe (the probe:enzyme ratio was 1 or 5, depending on the ABP used) for 30 min at 37 °C. The samples were then subjected to SDS–PAGE analysis, transferred to a membrane, and imaged with lasers of 520 nm for Cy3, 658 nm for Cy5, and 784 nm for Cy7 using an Azure Biosystems Sapphire Biomolecular Imager. The results are representative of at least 3 replicates. (C) Simultaneous APC, thrombin, and fXa detection and inhibition. Each enzyme (200 nM) was incubated with the appropriate inhibitor (the final inhibitor concentration was 5 μM) for 60 min prior to probe addition (the probe:enzyme ratio was 1 or 5, depending on the ABP used; 30 min). The samples were subjected to electrophoresis and membrane transfer. Visualization was performed with lasers of 520 nm for Cy3, 658 nm for Cy5, and 784 nm for Cy7 using an Azure Biosystems Sapphire Biomolecular Imager. The results are representative of at least 3 replicates. | ||
Labeling the coagulation factor by fluorescent ABPs
To evaluate the selectivity of our fluorescent probes, we performed in vitro labeling and SDS–PAGE analysis under identical conditions as in the case of biotinylated ABPs (the probe:enzyme ratio was 1 or 5, depending on the ABP used, 30 min) (Fig. 5B and S3†). As controls, we used probes and enzymes alone. After protein transfer to nitrocellulose membranes, the samples were analyzed using different wavelengths. We observed three clear fluorescent signals that were generated by labeling each protease with its specific probe and did not observe any additional bands that originated from the potential cross-reactivity of the probes. Therefore, we concluded that our second-generation ABPs were potent and highly selective and could be employed simultaneously in biological samples that contained APC, thrombin, and fXa. In the next experiment, we preinhibited APC with I-SMA17, thrombin with I-SMII22, and fXa with I-SMX15 (5 μM of each inhibitor, 60 min incubation) prior to the probe addition (the probe:enzyme ratio was 1 or 5, depending on the ABP used, 30 min) (Fig. 5C and S4†). As a control, we analyzed proteases that were treated only with suitable ABPs. Western blot analysis revealed that all three inhibitors prevented probe binding and blocked the investigated enzymes; therefore, we concluded that our ABPs bound within the active site, confirming the utility of these chemical tools for the study of coagulation factors.
Kinetic evaluation of the fluorescent ABPs and inhibitors
To obtain better insight into the kinetics of the inhibitors and ABPs, we measured the apparent second-order rate constants for inhibition (kobs(app)/I) under pseudo first-order conditions and then calculated substrate-independent kobs/I values (Table 2). The kinetic analysis revealed that I-SMA17 and P-SMA172 exhibited very good inhibitory efficiencies toward APC, with kobs/I values equal to 1980 ± 20 M−1 s−1 and 2250 ± 170 M−1 s−1, respectively. As predicted, we observed no irreversible inhibition of thrombin and fXa, reflecting the in vitro labeling results (Fig. 5B). The comparison of kobs/I values demonstrated that thrombin inhibition was 348- and 2087-fold more rapid with I-SMII22 (kobs/I = 208700 ± 13800 M−1 s−1) than with APC (kobs/I = 600 ± 30 M−1 s−1) and fXa (kobs/I = 100 ± 0.1 M−1 s−1), respectively. Changing the N-terminus from acetyl (I-SMII22) to the large cyanine derivative Cy7 (P-SMII222) dramatically reduced the kobs/I value to 3500 ± 300 M−1 s−1. However, P-SMII222 retained its selectivity, as indicated by western blot and kinetic analyses (Fig. 5B, Table 2). These results support the use of P-SMII222 for thrombin detection in biological samples. Next, we observed that I-SMX15 and P-SMX152 bound to fXa with similar inhibition rate constants, which were kobs/I = 1200 ± 100 M−1 s−1 and kobs/I = 1800 ± 100 M−1 s−1, respectively. Importantly, cross-reactivity of P-SMX152 with thrombin was observed, while the inhibition of APC was 75-fold less efficient, indicating a high selectivity toward fXa.
| Compound | k obs/I [M−1 s−1] | |||
|---|---|---|---|---|
| APC | Thrombin | fXa | ||
| a (NI) not determined due to low enzyme inhibition. | ||||
| P-SMA172 | Cy5–6-Ahx–Lys–Dab(Z)–Lys–ArgP(OPh)2 | 2250 ± 170 | NI | NI |
| I-SMA17 | Ac–Lys–Dab(Z)–Lys–ArgP(OPh)2 | 1980 ± 20 | NI | NI |
| P-SMII222 | Cy7–6-Ahx–hCha–Cys(MeBzl)–Pip–ArgP(OPh)2 | 292 ± 4 | 3500 ± 300 | 375 ± 36 |
| I-SMII22 | Ac–hCha–Cys(MeBzl)–Pip–ArgP(OPh)2 | 600 ± 30 | 209000 ± 13800 |
100 ± 0,1 |
| P-SMX152 | Cy3–6-Ahx–DPro–hArg–1-Nal–ArgP(OPh)2 | 24 ± 1 | NI | 1800 ± 100 |
| I-SMX15 | Ac–DPro–hArg–1-Nal–ArgP(OPh)2 | 90 ± 6 | 90 ± 1 | 1200 ± 100 |
Detection of APC, thrombin, and fXa in human plasma
Our initial goal was to develop chemical tools that can not only visualize and distinguish between purified coagulation factors but can also detect them in complex biological samples. Previous in vitro labeling and kinetic analysis indicated that P-SMA172, P-SMII222 and P-SMX152 were potent and selective for the detection of APC, thrombin, and fXa, respectively. Thus, to test the utility of our probes in a biological sample, we selected human plasma, in which all coagulation factors are present25,49 (Fig. 6A, S5–S7†). We characterized the labeling by incubating EDTA-chelated plasma with three fluorescent ABPs (separately) at concentrations ranging from 1 to 20 μM. To test the off-target reactivity of each probe, we doubled the incubation time to one hour. Next, we subjected the samples to SDS–PAGE, followed by western blotting with anti-PC, anti-fII, and anti-fX. The results demonstrated that all three investigated enzymes (APC, thrombin, and fXa) can be detected in plasma with our selective ABPs, as confirmed by containing with appropriate antibodies. Even at the lowest probe concentration used (1 μM), P-SMA172 bound to APC in plasma and maintained its selectivity up to 10 μM. Using a 20 μM probe, we detected only minor additional labeling between 25 and 35 kDa. After incubation of P-SMII222 with plasma, we observed a strong band that corresponded to thrombin. Moreover, selectivity was maintained with thrombin labeling even at a very high probe concentration (20 μM), which confirmed that P-SMII222 can be easily employed for thrombin detection in plasma. Furthermore, we noticed that P-SMX152 primarily labeled fXa, but at higher concentrations (10 μM and 20 μM), it displayed some cross-reactivity with an unknown protein, suggesting that the probe concentration is crucial in achieving selectivity in plasma. Therefore, to visualize all investigated enzymes simultaneously, we determined the optimal probe concentration required for selective detection of APC, thrombin, and fXa (5 μM of each: P-SMA172, P-SMII222, and P-SMX152). We treated the plasma with the fluorescent probe cocktail for one hour and obtained three clear bands that represented each of the three coagulation factors (Fig. 6C and S8†).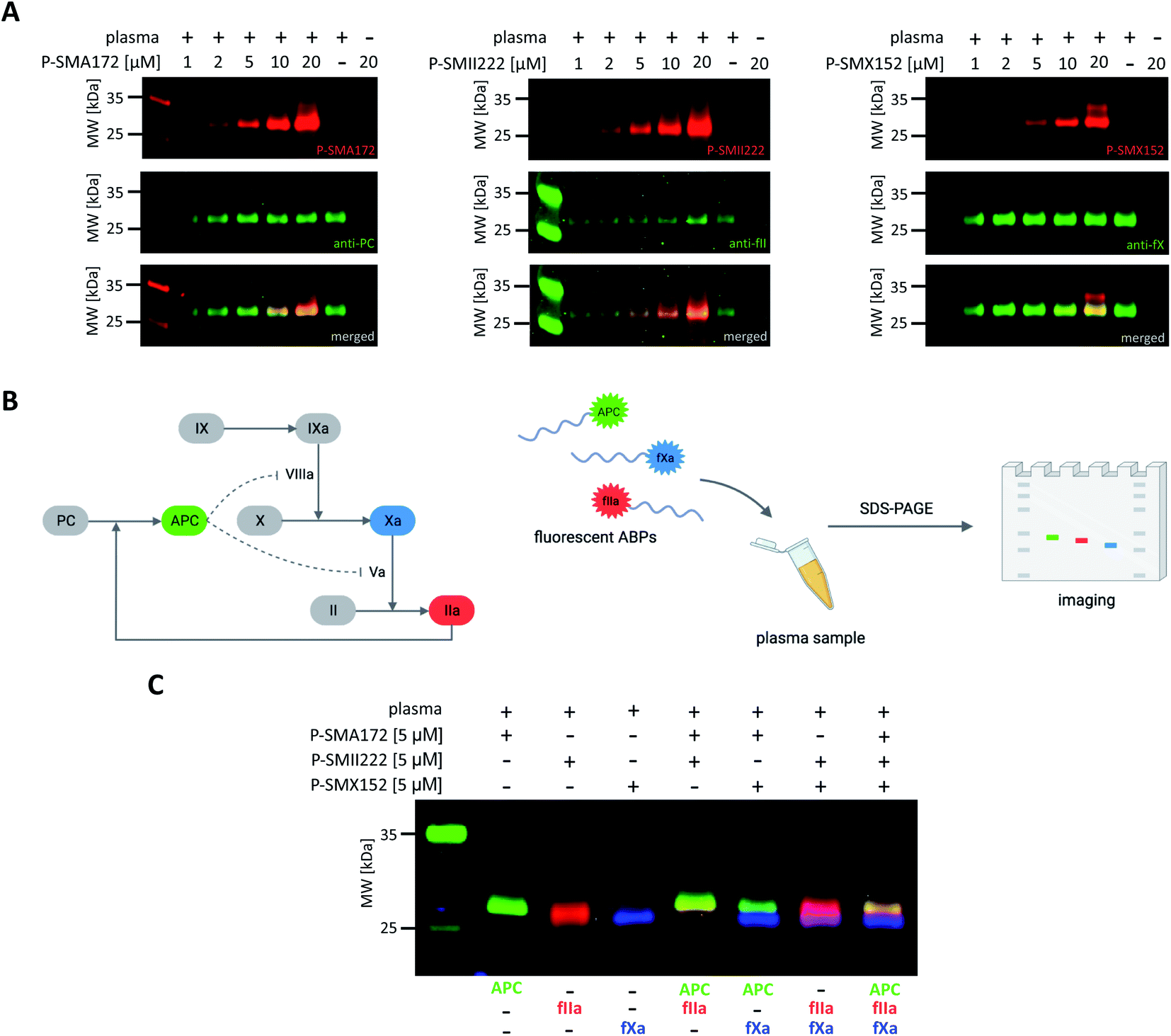 | ||
| Fig. 6 Coagulation factor (APC, thrombin, fXa) labeling in human plasma. (A) Probe concentration optimization assay. Human plasma was incubated with each fluorescent ABP separately at various probe concentrations ranging from 1 to 20 μM for 60 min at 37 °C. The samples were then subjected to SDS–PAGE analysis, transferred to a membrane, immunostained with the appropriate antibody, and imaged using an Azure Biosystems Sapphire Biomolecular Imager as follows: for APC at 658 nm (for Cy5 detection) and 488 nm (for antibody detection), for thrombin at 784 nm (for Cy7 detection) and 658 nm (for antibody detection), for fXa at 520 nm (for Cy3 detection) and 658 nm (for antibody detection). The results are representative of at least 3 replicates. (B) Graphical scheme of the methodology used for simultaneous coagulation factor detection in human plasma. (C) Simultaneous coagulation factor labeling in human plasma. Human plasma was incubated with 5 μM of each fluorescently labeled ABP and subjected to SDS–PAGE analysis. Direct in-gel analysis was performed with lasers of 520 nm for Cy3, 658 nm for Cy5, and 784 nm for Cy7 using an Azure Biosystems Sapphire Biomolecular Imager. The results are representative of at least 3 replicates. | ||
Discussion
APC, thrombin, and fXa are related vitamin K-dependent serine proteases that maintain the balance between coagulation and blood circulation.24–26 Beyond coagulation, they possess multiple other physiological functions. For example, APC exerts anti-inflammatory and cytoprotective activity, which includes endothelial barrier protection, modulation of gene expression and antiapoptotic activity.18,50 Thrombin is involved in inflammation and plays an important role in fibrosis, angiogenesis, and viral infections.22 Therefore, downregulation and upregulation of APC, thrombin, and fXa activity may lead to life-threatening and debilitating diseases such as hemophilia,10,50 thrombosis11,12 and disseminated intravascular coagulation.14 Lower circulation levels of APC were also found in patients with thrombosis and postinfection ischemic stroke.13,18 Additionally, thrombin activity contributes to stroke pathology and is considered to be a natural target for its prevention.19 Innate immunity and the coagulation cascade are intricately linked to cooperatively respond to infection and injury. Dysregulation of this crosstalk and hypercoagulation has been associated with numerous disorders. The serum concentration of APC is markedly reduced and correlated with morbidity and mortality in sepsis.51 Increased thrombin generation and activity have been reported in the pathogenesis of multiple sclerosis21 and Alzheimer's disease.15,16 The activity of thrombin has also been linked to cancer progression.20 Additionally, recent studies have indicated that thrombin and fXa inhibitors can be used in treating COVID-19 patients.22,23 The many roles of APC, thrombin, and fXa in both physiological and pathophysiological processes reinforce the importance of monitoring their activity in biological samples as pharmacodynamic and diagnostic/prognostic markers. Therefore, the goal of our study was to develop potent and selective fluorescent activity-based probes for the simple and straightforward labeling of APC, thrombin, and fXa in biological samples.Since the early 1980s, the substrate specify profiles for APC, thrombin, and fXa have been determined based on the recognition motif in physiological substrates26,29–31 and using various techniques, including chromogenic peptide substrates,44,52 PS-SCL,29 fluorescence-quenched substrates53–55 or phage display.44 These studies reported that all three proteases interact almost exclusively with positively charged arginine at the P1 position, making the design of selective peptide sequences for APC, thrombin, and fXa challenging. However, the previously obtained substrate specificity profiles were based only on natural amino acids, which significantly limits the development of selective chemical tools that are capable of discriminating between APC, thrombin, and fXa. To address this problem, in the present work, we applied a defined (P1) library and HyCoSuL (P4–P2) technology32 for an in-depth profiling of APC, thrombin, and fXa substrate preferences. The main advantage of this approach was the use of a very large collection of unnatural amino acids, which enabled us to extensively explore the binding pockets of the enzymes' active sites.
Our findings on the substrate preferences of APC, thrombin, and fXa at the P1 position are consistent with previous studies. All three enzymes are preferentially cleaved after basic arginine, suggesting that the S1 pocket is wide, deep, and highly conserved. This is also in line with the previously obtained crystal structures of all three enzymes,27,56 in which the S1 pocket is considered hydrophobic, and the Asp189 residue therein serves as the recognition site for the basic side chain. Therefore, extending the classic PS-SCL library with a large pool of unnatural amino acids at the P4–P2 positions was necessary to achieve selective sequences. Our HyCoSuL screening showed that in the APC S2 pocket, L-Lys was the best recognized natural amino acid, which confirmed previous findings;35,36 however, we noticed a significant increase in APC activity toward L-Oic, which is an unnatural proline derivative with a flexible cyclohexane ring. Interestingly, bulky and hydrophobic L-Igl was also well tolerated at this position, suggesting the presence of an additional cavity near the APC S2 subsite that, compared to the natural amino acids, was capable of accommodating more complex structures. Previous studies reported that thrombin prefers L-Pro at the P2 position, which is also characteristic of other serine proteases, such as granzyme A,57 cathepsin G,58 proteinase 3,58 and neutrophil elastase;58 therefore, to achieve selectivity and reduce cross-reactivity, we also used two proline derivatives (L-Aze and L-Pip) to construct thrombin substrates. fXa, on the other hand, could be distinguished from other coagulation factors using bulky unnatural amino acids such as L-Bta and L-1-Nal. In the case of the S3 pocket, APC, thrombin, and fXa displayed similar substrate preferences regarding natural amino acids. We distinguished APC from thrombin and fXa by incorporating unnatural L-Dab(Z) at the P3 position. For thrombin, we chose cysteine derivatives with L-Cys(MeBzl) as the best hit. Furthermore, the preference for an arginine derivative elongated with one methylene group L-hArg discriminated fXa from APC and thrombin. At the APC P4 position, the most preferred amino acid residues were L-Arg and L-Lys and their derivatives, with L-Lys exhibited a superior selectivity. We observed a similar result as that found by the Craik26 and Hellman groups44 in the preference of thrombin for branched aliphatic amino acids, such as L-Ile, L-Leu and L-Val; however, it was the unnatural elongated L-hCha residue that allowed us to obtain the desired selectivity. For fXa in the P4 position, the most preferred amino acid was L-Met; however, D-Pro was the residue that was exclusively recognized only by fXa.
The specificity maps obtained clearly demonstrate that the selectivity lies in the P4–P2 region; therefore, in the next step, we selected the best hits from the HyCoSuL profiling and synthesized a selection of fluorogenic tetrapeptide substrates for each coagulation factor. Using unnatural amino acids, we obtained SMA5 (Ac–Lys–Dab(Z)–Igl–Arg–ACC), which is currently the best APC substrate reported. Since our goal was to create the most selective substrate possible, we also found SMA17 (Ac–Lys–Dab(Z)–Lys–Arg–ACC), which displayed a very high kcat/KM selectivity ratio and almost nondetectable hydrolysis when tested with thrombin and fXa. Several fluorogenic tetrapeptide substrates for thrombin have been already described;26,42 however, they all are based on natural amino acids and therefore lack selectivity and cannot be used for monitoring the activity of thrombin in a complex system. In contrast, our most potent thrombin substrate, SMII22 (Ac–hCha–Cys(MeBzl)–Pip–Arg–ACC), was weakly hydrolyzed by APC and fXa and exhibited 3383- and 162-fold selectivity values for thrombin, respectively. The majority of fXa substrates described in the literature are nonselective natural fluorogenic tetrapeptide substrates42 or internally quenched fluorescent (IQF) substrates59 that do not bind within the enzyme active site and therefore do not allow the discrimination of enzymes via western blotting. Compared to IQF substrates, our substrate SMX15 (Ac–DPro–hArg–1-Nal–Arg–ACC) was less sensitive; however, it was 358 times more hydrolyzed by fXa than by thrombin and could be converted into inhibitor and activity-based probes, whereas IQFs could not.
Our HyCoSuL-derived fluorescent ABPs enable the simultaneous detection of the investigated proteases in complex biological samples. They are equipped with a diphenyl phosphonate as a reactive group, which gives them the properties of a covalent inhibitor and enables the investigated protease to undergo chemical knockdown. More importantly, covalent and fluorescent ABPs can also be used for the direct in-gel visualization of coagulation factors, which gives them an advantage over the enzyme-activatable probes obtained thus far.19,60–62 We synthesized the following second-generation ABPs: P-SMA172 (Cy5–6-Ahx–Lys–Dab(Z)–Lys–ArgP(OPh)2) for APC, P-SMII222 (Cy7–6-Ahx–hCha–Cys(MeBzl)–Pip–ArgP(OPh)2) for thrombin, and P-SMX152 (Cy3–6-Ahx–DPro–hArg–1-Nal–ArgP(OPh)2) for fXa and tested their utility using kinetic assays and simple SDS–PAGE analysis. We confirmed that our second-generation ABPs were selective. The P-SMA172 and P-SMX152 selectivity for APC and fXa, respectively, was especially important since thrombin is generally a more active protease.27 Additionally, we applied the same recognition motif as that in the second-generation ABPs and synthesized a set of novel, selective inhibitors (I-SMA17 (Ac–Lys–Dab(Z)–Lys–ArgP(OPh)2) for APC, I-SMII22 (Ac–hCha–Cys(MeBzl)–Pip–ArgP(OPh)2) for thrombin, and I-SMX15 (Ac–DPro–hArg–1-Nal–ArgP(OPh)2) for fXa) for further study. In the last stage of this research, we selected human plasma, a complex mixture of proteases, to test the selectivity of our second-generation ABPs in a more biological setting. We incubated our fluorescent probes in the plasma sample and observed three clear species, which were confirmed to be APC, thrombin, and fXa by western blotting.
In summary, in this work, we determined the substrate specificity of APC, thrombin, and fXa and developed a set of selective and potent tools for the investigation of these proteases. We designed and obtained substrates, inhibitors, and fluorescent activity-based probes for fast, direct, and simultaneous detection of APC, thrombin, and fXa in human plasma. Our probes can be used in the future to visualize and compare the levels of APC, thrombin, and fXa in physiological and pathophysiological samples. Additionally, since our ABPs exhibit the properties of covalent inhibitors, they can also be applied for the pharmacological knockdown of enzymes, allowing their role in coagulation and in various diseases to be studied. The ability of our fluorescent ABPs to selectively detect APC, thrombin, and fXa in biological samples is of great importance since these proteases serve as diagnostic and prognostic markers for multiple disorders. In the future, our ABPs may be used as diagnostic tools and facilitate the choice of appropriate therapy for several disorders, such as bleeding, thrombosis, stoke, sepsis, and many others.
Materials and methods
Reagents
All chemical reagents for substrate, inhibitor and ABP synthesis were purchased from commercial suppliers and used without further purification. The Rink amide RA resin (particle size 200–300 mesh, loading 0.74 mmol g−1, for ACC-labeled substrates), 2-chlorotrityl chloride resin (particle size 100–200 mesh, loading 1.60 mmol g−1, for peptides further converted into inhibitors and ABPs), Fmoc–6-Ahx–OH, biotin, diisopropylcarbodiimide (DICI, peptide grade), 1-[bis(dimethylamino)-methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU, peptide grade), piperidine (PIP, peptide grade), O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU, peptide grade), and trifluoroacetic acid (TFA, purity 99%) were all obtained from Iris Biotech GmbH (Marktredwitz, Germany). Fmoc-protected amino acids (purity > 98%) were purchased from various suppliers: Iris Biotech GmbH, Creosalus (Louisville, KY, USA), Bachem (Torrance, CA, USA), and P3 BioSystems (Louisville, KY, USA). P2O5 (phosphorus pentoxide, purity 98%) was purchased from Avantor (Gliwice, Poland). N-Hydroxybenzotriazole (HOBt, monohydrate, purity > 98%) was purchased from Creosalus. 2,4,6-Trimethylpyridine (2,4,6-collidine, peptide grade), 2,2,2-trifluoroethanol (TFE), and triisopropylsilane (TIPS, purity 99%) were all purchased from Sigma-Aldrich (Poznan, Poland). N,N-Diisopropylethylamine (DIPEA, peptide grade) was purchased from VWR International (Gdansk, Poland). The following solvents were purchased from Avantor: dichloromethane (DCM, pure for analysis), N,N′-dimethylformamide (DMF, peptide grade), diethyl ether (Et2O, pure for analysis), methanol (MeOH, pure for analysis), acetonitrile (ACN, HPLC grade), and AcOH (acetic acid, purity 99%). Fluorescent tags (Cy3-NHS, Cy5-NHS and Cy7-NHS) were purchased from Lumiprobe GmbH (Hannover, Germany). Anti-PC antibody (chicken, polyclonal, ABIN597333) was purchased from Antibodies-online GmbH (Aachen, Germany). Anti-fII (sheep, polyclonal, PAHFII-S) and anti-fX (sheep, polyclonal, PAHFX-S) antibodies were obtained from Hematologic Technologies Inc. (Essex Junction, VT, USA).All individual substrates, inhibitors and ABPs were purified by HPLC on a Waters M600 solvent delivery module with a Waters M2489 detector system using a semipreparative Wide Pore C8 Discovery column (Waters sp z o.o., Warszawa, Poland). The solvent composition was as follows: phase A (water: 0.1% TFA) and phase B (acetonitrile: 0.1% TFA). The purity of each compound was confirmed with an analytical HPLC system using a Discovery Bio Wide Pore C8 analytical column. The solvent composition was as follows: phase A (water: 0.1% TFA) and phase B (acetonitrile: 0.1% TFA); gradient, from 95% A to 5% A over a period of 15 min. The purity of all compounds was ≥95%. The molecular weight of each compound was confirmed by high-resolution mass spectrometry using a WATERS LCT Premier XE with electrospray ionization (ESI) and a time-of-flight (TOF) module.
Library syntheses
Detailed protocols for the synthesis of the defined library Ac–Ala–Arg–Leu–P1–ACC and the combinatorial library with Arg at the P1 position are provided elsewhere.28,32,33 The synthesis of the fluorogenic leaving group ACC (7-amino-4-carbamoylmethylcoumarin) was carried out according to an efficient large-scale method described by Maly et al.63Enzyme preparation
Proteins were purified from fresh frozen plasma. Factor X and prothrombin were purified by a modified method of Bajzar and colleagues.64 Protein C was purified by a modified method of Kisiel.65 Factor X was activated with the factor X activator from Russell's viper venom as described.66 Thrombin was prepared by activation of prothrombin with Echis carinatus snake venom as described.67 Protein C was activated using thrombin–thrombomodulin to activate protein C (APC) as previously described.68 Protein purity was confirmed using SDS–polyacrylamide gel electrophoresis (SDS–PAGE) according to the methods described by Laemmli.69Kinetic assays
All kinetic experiments were performed using a spectrofluorometer (Molecular Devices SpectraMax Gemini XPS) on 96-well plates (Corning) with the following parameters: excitation/emission wavelength, 355/460 nm (cutoff, 455 nm), varying concentrations of substrates, inhibitors, ABPs and enzymes. The assay buffer contained 20 mM Tris-base, 150 mM NaCl, and 5 mM CaCl2, pH 7.4. The buffer was prepared at room temperature, and the enzyme kinetic studies were performed at 37 °C. All enzymes were preincubated in assay buffer for 15 min at 37 °C before being added to the wells. Each assay was repeated at least twice, and the data represent the average of these repetitions. The obtained results were analyzed using SoftMax (Molecular Devices), GraphPad Prism, and Microsoft Excel software.Defined library screening
To determine the substrate specificity of APC, thrombin, and fXa at the P1 position, we used the defined library with the general structure of Ac–Ala–Arg–Leu–P1–ACC containing 133 individual fluorogenic substrates.28 The assay conditions were as follows: 1 μL of each library substrate was placed in one well, and 99 μL of preincubated enzyme (15 min, 37 °C) was added. The final library concentration was 200 μM, and the enzyme concentration was 75 nM for APC, 9 nM for thrombin, and 21 nM for fXa. The substrate cleavage was monitored for 30 min (λex = 355 nm, λem = 460 nm), but only the linear portion of each progress curve was used to determine the substrate hydrolysis rate (RFU s−1, relative fluorescence unit per second). Substrate specificity profiles were established by setting the highest RFU s−1 value as 100% and adjusting other results accordingly.HyCoSuL screening
The APC, thrombin, and fXa substrate specificity profiles at the S4–S2 subsites were determined using the HyCoSuL P1-Arg library, which comprised over 100 natural and unnatural amino acids in each position of three sublibraries (P4, P3 and P2).32,33 Standard experimental conditions for P4, P3, and P2 sublibrary screening were as follows: 1 μL of substrate and 99 μL of preincubated enzyme (15 min, 37 °C), with a final substrate mixture concentration of 100 μM and final enzyme concentrations of 40 nM for APC, 1 nM for thrombin, and 5 nM for fXa. The release of ACC was measured for 30 min (λex = 355 nm, λem = 460 nm), and the linear part of each progress curve was used to determine the substrate hydrolysis rate. The results from screening analysis were based on the obtained RFU s−1 values for each sublibrary with the best recognized amino acid at each position set to 100% and other amino acids normalized accordingly.Individual substrate synthesis
The designed substrates were synthesized on Rink amide RA resin (loading 0.74 mmol g−1) using the standard solid phase peptide synthesis (SPPS) method,63 in which the Fmoc-protected amino acids and ACC as a reporter group were employed. In the first step, 7.5 g of resin was placed in a glass peptide synthesis vessel and swollen in DCM for 30 min. The N-terminal Fmoc-protecting group deprotection was performed using 20% piperidine in DMF (5, 5, and 25 min) and confirmed with a ninhydrin test. Next, the resin was washed with DMF (six times). Fmoc–ACC–OH (2.5 equiv., 6.12 g) was attached to Rink amide RA resin using HOBt (2.5 equiv., 2.08 g) and DICI (2.5 equiv., 1.80 mL) as coupling reagents. After 24 h, the mixture was filtered, the resin was washed with DMF (three times), and the reaction was repeated with 1.5 equiv. of all of the above reagents to improve the yield of Fmoc–ACC–OH coupling. After Fmoc-group removal, the resin was washed with DMF (six times), and the P1 amino acid Fmoc–Arg(Pbf)–OH (3 equiv., 10.80 g) with HATU (3 equiv., 6.33 g) and 2,4,6-collidine (3 equiv., 2.20 mL) in DMF was poured onto H2N–ACC–resin and gently agitated for 24 h. Next, the mixture was filtered, the resin was washed with DMF (three times), and the reaction was repeated with 1.5 equiv. of all of the above reagents to improve the yield of Fmoc–Arg(Pbf)–OH coupling. Then, the Fmoc-protecting group was removed in the same manner as described above, and deprotection was confirmed with a ninhydrin test. The resin was washed with DMF (six times), DCM (three times), and MeOH (three times) and dried over P2O5. Dried resin was divided into 74 portions (100 mg each) and placed in a multiwell peptide synthesis vessel. Next, a solution of the appropriate Fmoc–P2–OH (2.5 equiv.), HOBt (2.5 equiv., 28 mg), and DICI (2.5 equiv., 24 μL) in DMF was added to the H2N–P1–ACC–resin and gently agitated for 2.5 h. After Fmoc-protecting group removal, the P3 and P4 amino acids were attached to the H2N–P2–P1–ACC–resin with HOBt (2.5 equiv., 28 mg) and DICI (2.5 equiv., 24 μL) as coupling reagents in coupling/deprotection cycles. A ninhydrin test was performed after every amino acid coupling and Fmoc-group removal. The free amino group of the P4 residue was acetylated using 5 equiv. of AcOH, 5 equiv. of HBTU, and 5 equiv. of DIPEA in DMF. After 1 h, the solution was filtered, and the resin was washed with DMF (six times), DCM (three times), and MeOH (three times) and dried over P2O5. Finally, the substrates were cleaved from the resin with a mixture of TFA/TIPS/H2O (%, v/v/v, 95:2.5:2.5), precipitated in Et2O, purified by HPLC, and lyophilized. The purity of the ACC-labeled substrates was confirmed by analytical HPLC and analyzed using HRMS. Each substrate was dissolved in peptide grade DMSO to a final concentration of 10 mM and stored at −80 °C until use.
Substrate screening
All individual substrates were screened for their selectivity against the following coagulation factors: APC, thrombin, and fXa. The assay conditions were as follows: substrates with an initial concentration of 10 mM were diluted 100 times in DMSO, and 1 μL of each substrate dilution was placed in separate wells on the plate, followed by the addition of 99 μL of preincubated enzyme (15 min, 37 °C) in assay buffer. The final substrate concentration was 1 μM, and the enzyme concentration was selected individually for each set of substrates to obtain a robust fluorescence signal. The final enzyme concentration was in the range of 50–90 nM for APC, 0.15–35 nM for thrombin, and 7–100 nM for fXa, depending on the substrate used. Substrate hydrolysis was measured for 30 min (λex = 355 nm, λem = 460 nm), and the linear part of each progress curve was used to determine the substrate hydrolysis rate (RFU s−1). The obtained RFU s−1 were then adjusted to represent the value corresponding to the same activity of the enzymes.Substrate kinetics
For the selected ACC-labeled substrates, the kinetic parameters (kcat, KM, kcat/KM) were determined by measuring the increase in fluorescence over time. Each substrate was serially diluted until the eighth well to obtain substrate concentrations ranging from 4.68 nM to 741 μM, depending on the substrate used. Next, each enzyme was preincubated (15 min, 37 °C) in assay buffer to final enzyme concentrations in the range of 2.19–100 nM for APC, 0.07–346 nM for thrombin, and 2.80–224 nM for fXa and was added to the wells containing eight different substrate concentrations. The release of ACC was measured for 30 min (λex = 355 nm, λem = 460 nm). The linear part of each progression curve was used to calculate the kinetic parameters with Michaelis–Menten nonlinear regression using GraphPad Prism and Microsoft Excel software.ABPs and inhibitor synthesis
The peptide sequences of the best substrates were utilized to design irreversible ABPs and inhibitors. In the first step, 100 mg of 2-chlorotrityl resin (for each ABP) was placed in the peptide synthesis vessel and swollen in anhydrous DCM for 30 min, and then, the resin was washed once with DCM. Next, Fmoc–P2–OH (2.5 equiv.) was dissolved in anhydrous DCM, preactivated with DIPEA (3 equiv., 84 μL), added to the resin under an argon atmosphere, and stirred gently for 12 h. After that, the mixture was filtered, the resin was washed with DCM (three times), and the remaining active sites on the 2-chlorotrityl resin were deactivated with DCM/MeOH/DIPEA (%, v/v/v, 80:15:5) solution for 1 h. Next, the mixture was filtered, the resin was washed with DMF (six times), and N-terminal Fmoc-protecting group deprotection was performed using 20% piperidine in DMF (5, 5, and 25 min). After Fmoc-protecting group removal, the P3, P4 amino acids, and Fmoc–Ahx–OH were attached to the H2N–P2–resin with HOBt (2.5 equiv., 60 mg) and DICI (2.5 equiv., 52 μL) as coupling reagents in coupling/deprotection cycles. The biotin tag was coupled to H2N–Ahx–P4–P3–P2–resin using HBTU (2.5 equiv., 152 mg) and DIPEA (2.5 equiv., 70 μL) as coupling reagents in a DMF/DMSO mixture (%, v/v, 50:50). After 3 h, the resin was washed with DMF (six times), DCM (three times), and MeOH (three times) and dried over P2O5. The crude peptide was cleaved from the resin with a mixture of DCM/TFE/AcOH (%, v/v/v, 80:10:10). The solution was filtered, concentrated, and lyophilized. Next, the reactive warhead diphenyl phosphonate (Cbz–Arg(Boc)2P(OPh)2) was synthesized according to a previously described methodology.70 The Cbz-protecting group was removed using hydrogen and palladium on carbon. The obtained H2N–Arg(Boc)2P(OPh)2 (1 equiv.) was coupled with a peptide sequence biotin–6-Ahx–P4–P3–P2–COOH (1.2 equiv.) with HATU (1.2 equiv.) and 2,4,6-collidine (4 equiv.). The reaction was monitored with analytical HPLC, and after 2 h, the product was extracted to ethyl acetate with 5% NaHCO3, 5% citric acid, and brine. The organic phase was collected, dried over MgSO4, and evaporated. Finally, the side chain amino acid protecting groups were removed with a mixture of TFA/DCM/TIPS (%, v/v/v, 80:15:5). After 30 min, solvents were removed with argon flow, and the obtained product (biotin–6-Ahx–P4–P3–P2–ArgP(OPh)2) was dissolved in peptide grade DMSO, purified on HPLC, and lyophilized. The ABP purity was confirmed by analytical HPLC and analyzed using HRMS. ABP was then dissolved in peptide grade DMSO to a final concentration of 10 mM and stored at −80 °C until use.
To obtain the inhibitor, the amino group of H2N–P4–P3–P2–resin was acetylated using 5 equiv. of AcOH, 5 equiv. of HBTU, and 5 equiv. of DIPEA in DMF. After 1 h, the solution was filtered, and the resin was washed with DMF (six times), DCM (three times), and MeOH (three times) and dried over P2O5. The remaining steps were analogous to the synthesis of the biotin-labeled ABPs.
In the case of attaching the fluorescently labeled probes to the amine group of H2N–Ahx–P4–P3–P2–resin, the Cy3-NHS, Cy5-NHS or Cy7-NHS fluorophore was coupled. The amount of 2-chlorotrityl resin was reduced to 50 mg. Cy3-NHS (0.3 equiv., 15 mg), Cy5-NHS (0.3 equiv., 15 mg) or Cy7-NHS (0.3 equiv., 18 mg) ester was dissolved in DMF, preactivated with DIPEA (1.2 equiv., 17 μL), and stirred with the resin overnight. Next, the resin was washed with DMF (six times), DCM (three times), and MeOH (three times) and dried over P2O5. The remaining steps were analogous to the synthesis of the biotin-labeled ABPs.
Determination of the inhibition kinetics (kobs/I) for the fluorescently labeled ABPs and inhibitors
The second-order rate constants for inhibition (kobs/I) toward APC, thrombin, and fXa were measured under pseudo first-order conditions. Each ABP or inhibitor was serially diluted until the seventh well to obtain concentrations ranging from 308 nM to 100 μM, depending on the ABP/inhibitor used. The assay conditions were as follows: 20 μL of selected substrate (154 μM SMA5 for APC, 100 μM SMA4 for thrombin, and 50 μM SMII18 for fXa) was added to the wells containing 20 μL of seven different ABP/inhibitor concentrations. Next, 60 μL of APC, thrombin, or fXa (at a concentration of 10 nM) preincubated at 37 °C was added, and the fluorescence increase over time was measured (λex = 355 nm, λem = 460 nm) for 30 min. The kobs/I values were calculated using GraphPad Prism and Microsoft Excel software as described previously.71Coagulation factor labeling by ABPs based on SDS–PAGE analysis
In the first assay, to determine biotin-labeled ABPs selectivity, three purified coagulation factors (APC, thrombin, fXa) with a constant concentration of 200 nM were incubated separately with each probe (the probe:enzyme ratio was 1 or 5, depending on the ABP used) in assay buffer (20 mM Tris-base, 150 mM NaCl, 5 mM CaCl2, pH 7.4) for 30 min at 37 °C. Each enzyme was incubated with each probe in a volume of 40 μL, followed by reduction with 20 μL of 3× SDS/DTT for 5 min at 95 °C. The first well was loaded with 0.5 μL of the protein marker PageRuler Prestained Protein Ladder (Thermo Scientific), and then 10 μL of each sample was run onto a 12% (w/v) 15-well gel. SDS–PAGE separation was performed at 200 V for 39 min, followed by transfer to a nitrocellulose membrane (0.2 μm, Bio-Rad) at 10 V for 60 min. The membrane was blocked with 2.5% BSA in TBS–T (Tris-buffered saline with 0.1% (v/v) Tween 20) for 60 min at room temperature. Next, the membrane was incubated with fluorescent streptavidin Alexa Fluor 647 conjugate (dilution 1:10000 in TBS–T with 1% BSA) for 60 min. The biotin-labeled ABPs were detected at 658 nm using an Azure Biosystems Sapphire Biomolecular Imager and Azure Spot Analysis Software.
For preparing the fluorescently-labeled probe samples, electrophoresis and membrane transfer were performed in the same manner as described above. After the membrane was blocked with 2.5% BSA in TBS–T (60 min), the labeled proteins were directly imaged with lasers of 520 nm for Cy3, 658 nm for Cy5, and 784 nm for Cy7 using an Azure Biosystems Sapphire Biomolecular Imager and Azure Spot Analysis Software.
When testing the inhibitors, 200 nM of each enzyme was incubated with the appropriate inhibitor (final inhibitor concentration 5 μM) for 60 min, a prior to probe addition (the probe:enzyme ratio was 1 or 5, depending on the ABP used, 30 min). The total sample volume was 40 μL, to which 20 μL of 3× SDS/DTT was added and boiled for 5 min at 95 °C. Electrophoresis, membrane transfer and visualization were performed in the same manner as in the case of fluorescently labeled probes.
Coagulation factor labeling by ABPs in human plasma
Human plasma was isolated from whole blood (which was collected in tubes that contained anticoagulant EDTA) using Polymorphprep (Fisher Scientific) and Ficoll Paque Plus (Sigma-Aldrich). Then, 4 mL of Polymorphprep and 1 mL of Ficoll Paque Plus were added to a 15 mL falcon tube. Five milliliters of whole blood was layered into the tube and centrifuged at 450g for 35 min at room temperature. The obtained plasma was then carefully removed from the cell pellet using a Pasteur pipette and stored at −80 °C until use. Before labeling, human plasma was diluted 7.5 times in assay buffer (20 mM Tris-base, 150 mM NaCl, 5 mM CaCl2, pH 7.4).In the probe concentration optimization assay, human plasma was incubated with each fluorescently labeled probe separately at varying probe concentrations ranging from 1 to 20 μM. P-SMA172, P-SMII222, and P-SMX152 were used for APC, thrombin, and fXa detection, respectively. Incubation was carried out in assay buffer (20 mM Tris-base, 150 mM NaCl, 5 mM CaCl2, pH 7.4) for 60 min at 37 °C. The plasma was incubated with the probe in a total volume of 40 μL (20 μL of plasma and 20 μL of probe), followed by reduction with 20 μL of 3× SDS/DTT for 5 min at 95 °C. The first well was loaded with 0.5 μL of the protein marker PageRuler Prestained Protein Ladder (Thermo Scientific), and then 5 μL of each sample was run onto a 12% (w/v) 15-well gel. SDS–PAGE separation was performed at 200 V for 39 min, followed by transfer to a nitrocellulose membrane (0.2 μm, Bio-Rad) at 10 V for 60 min. The membrane was blocked with 2.5% BSA in TBS–T (Tris-buffered saline with 0.1% (v/v) Tween 20) for 60 min at room temperature. For APC detection, the membrane was treated with chicken anti-human polyclonal PC antibody (Antibodies-online GmbH, ABIN597333, 1:1000) for 7 h, followed by incubation with Alexa Fluor 488 goat anti-chicken secondary antibody (Life Technologies, A11039, 2:10000) for 1 h (both at room temperature). For the thrombin detection, the membrane was treated with sheep anti-human polyclonal fII antibody (Hematologic Technologies Inc., PAHFII-S, 1:1000) for 7 h, followed by incubation with Alexa Fluor 680 donkey anti-sheep secondary antibody (Life Technologies, A21102, 2:10000) for 1 h (both at room temperature). For fXa detection, the membrane was treated with sheep anti-human polyclonal fX antibody (Hematologic Technologies Inc., PAHFX-S, 1:1000) for 7 h, followed by incubation with Alexa Fluor 680 donkey anti-sheep secondary antibody (Life Technologies, A21102, 2:10000) for 1 h (both at room temperature). The membranes were then scanned using an Azure Biosystems Sapphire Biomolecular Imager and Azure Spot Analysis Software as follows: for APC at 658 nm (for Cy5 detection) and 488 nm (for antibody detection), for thrombin at 784 nm (for Cy7 detection) and 658 nm (for antibody detection), for fXa at 520 nm (for Cy3 detection) and 658 nm (for antibody detection).
For the simultaneous coagulation factor labeling, human plasma was incubated with 5 μM of each fluorescently labeled probe: P-SMA172, P-SMII222, and P-SMX152. Incubation was carried out in the same assay buffer as that used above (60 min, 37 °C). Plasma was incubated with the probe in a total volume of 40 μL (20 μL of plasma and 20 μL of probe mixture), followed by reduction with 20 μL of 3× SDS/DTT for 5 min at 95 °C. The first well was loaded with 2 μL of the protein marker PageRuler Prestained Protein Ladder (Thermo Scientific), and then 5 μL of each sample was run onto a 10% MES (w/v) 15-well gel. SDS–PAGE separation was performed at 100 V for 140 min. The gel was then directly scanned at 520 nm for Cy3, 658 nm for Cy5, and 784 nm for Cy7 detection using an Azure Biosystems Sapphire Biomolecular Imager and Azure Spot Analysis Software.
Safety statement
No unexpected or unusually high safety hazards were encountered.Author contributions
S. M., P. K., J. A. H., and M. D. designed the research. S. M. and S. K. performed the experiments. S. M., S. K., P. K., and M. D. analyzed the results. T. E. A., S. G. I. P., and J. A. H. contributed enzymes. S. P. contributed blood. S. M. wrote the paper. All authors critically revised the paper.Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.Conflicts of interest
Wrocław University of Science and Technology has filed a patent application covering the compounds Cy5–6-Ahx–Lys–Dab(Z)–Lys–ArgP(OPh)2, Ac–Lys–Dab(Z)–Lys–ArgP(OPh)2, Cy7–6-Ahx–hCha–Cys(MeBzl)–Pip–ArgP(OPh)2, Ac–hCha–Cys(MeBzl)–Pip–ArgP(OPh)2, Cy3–6-Ahx–DPro–hArg–1-Nal–ArgP(OPh)2, Ac–DPro–hArg–1-Nal–ArgP(OPh)2 with S. M., P. K., and M. D. as inventors.Acknowledgements
The Drąg laboratory is supported by the ‘TEAM/2017-4/32’ project, which is conducted within the TEAM program of the Foundation for Polish Science cofinanced by the European Union under the European Regional Development Fund.References
- N. Mackman, R. E. Tilley and N. S. Key, Arterioscler., Thromb., Vasc. Biol., 2007, 27, 1687–1693 CrossRef CAS.
- S. P. Grover and N. Mackman, Arterioscler., Thromb., Vasc. Biol., 2019, 39, 331–338 CrossRef CAS.
- S. Palta, R. Saroa and A. Palta, Indian J. Anaesth., 2014, 58, 515–523 CrossRef CAS.
- R. Chaudhry, S. M. Usama and H. M. Babiker, in StatPearls, Treasure Island (FL), 2021 Search PubMed.
- S. Krishnaswamy, J. Thromb. Haemostasis, 2013, 11(suppl. 1), 265–276 CrossRef PubMed.
- A. Arachiche, M. M. Mumaw, M. de la Fuente and M. T. Nieman, J. Biol. Chem., 2013, 288, 32553–32562 CrossRef CAS.
- M. W. Mosesson, J. Thromb. Haemostasis, 2005, 3, 1894–1904 CrossRef CAS.
- K. G. Mann, Chest, 2003, 124, 4S–10S CrossRef CAS.
- J. H. Griffin, Nature, 1995, 378, 337–338 CrossRef CAS.
- C. Negrier, M. Shima and M. Hoffman, Blood Rev., 2019, 38, 100582 CrossRef CAS PubMed.
- F. R. Rosendaal, Lancet, 1999, 353, 1167–1173 CrossRef CAS.
- G. X. Shen, Front. Biosci., 2006, 11, 113–120 CrossRef CAS PubMed.
- J. H. Griffin, B. Zlokovic and J. A. Fernandez, Semin. Hematol., 2002, 39, 197–205 CrossRef CAS PubMed.
- M. Levi and H. Ten Cate, N. Engl. J. Med., 1999, 341, 586–592 CrossRef CAS PubMed.
- H. Akiyama, K. Ikeda, H. Kondo and P. L. McGeer, Neurosci. Lett., 1992, 146, 152–154 CrossRef CAS.
- J. Iannucci, W. Renehan and P. Grammas, Front. Neurosci., 2020, 14, 762 CrossRef PubMed.
- J. Simmons and J. F. Pittet, Curr. Opin. Anaesthesiol., 2015, 28, 227–236 CrossRef CAS PubMed.
- J. H. Griffin, J. A. Fernandez, L. O. Mosnier, D. Liu, T. Cheng, H. Guo and B. V. Zlokovic, Blood Cells, Mol., Dis., 2006, 36, 211–216 CrossRef CAS.
- B. Chen, B. Friedman, M. A. Whitney, J. A. Winkle, I. F. Lei, E. S. Olson, Q. Cheng, B. Pereira, L. Zhao, R. Y. Tsien and P. D. Lyden, J. Neurosci., 2012, 32, 7622–7631 CrossRef CAS PubMed.
- R. Cantrell and J. S. Palumbo, Thromb. Res., 2020, 191(suppl. 1), S117–S122 CrossRef CAS.
- K. R. Jordan, I. Parra-Izquierdo, A. Gruber, J. J. Shatzel, P. Pham, L. S. Sherman, O. J. T. McCarty and N. G. Verbout, Metab. Brain Dis., 2021, 36, 407–420 CrossRef CAS.
- K. F. Aliter and R. A. Al-Horani, Cardiovasc. Drugs Ther., 2021, 35, 195–203 CrossRef CAS.
- R. A. Al-Horani, Am. J. Cardiovasc. Drugs, 2020, 20, 525–533 CrossRef CAS.
- G. Ferland, Nutr. Rev., 1998, 56, 223–230 CrossRef CAS.
- J. W. Suttie, FASEB J., 1993, 7, 445–452 CrossRef CAS PubMed.
- J. L. Harris, B. J. Backes, F. Leonetti, S. Mahrus, J. A. Ellman and C. S. Craik, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7754–7759 CrossRef CAS.
- T. Mather, V. Oganessyan, P. Hof, R. Huber, S. Foundling, C. Esmon and W. Bode, EMBO J., 1996, 15, 6822–6831 CrossRef CAS.
- W. Rut, L. Zhang, P. Kasperkiewicz, M. Poreba, R. Hilgenfeld and M. Drag, Antiviral Res., 2017, 139, 88–94 CrossRef CAS PubMed.
- B. J. Backes, J. L. Harris, F. Leonetti, C. S. Craik and J. A. Ellman, Nat. Biotechnol., 2000, 18, 187–193 CrossRef CAS PubMed.
- T. L. Yang, J. Cui, A. Rehumtulla, A. Yang, M. Moussalli, R. J. Kaufman and D. Ginsburg, Blood, 1998, 91, 4593–4599 CrossRef CAS.
- K. Kamata, H. Kawamoto, T. Honma, T. Iwama and S. H. Kim, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6630–6635 CrossRef CAS.
- M. Poreba, G. S. Salvesen and M. Drag, Nat. Protoc., 2017, 12, 2189–2214 CrossRef CAS.
- P. Kasperkiewicz, M. Poreba, S. J. Snipas, S. J. Lin, D. Kirchhofer, G. S. Salvesen and M. Drag, PLoS One, 2015, 10, e0132818 CrossRef.
- W. Rut, K. Groborz, L. Zhang, S. Modrzycka, M. Poreba, R. Hilgenfeld and M. Drag, Antiviral Res., 2020, 175, 104731 CrossRef CAS.
- S. G. I. Polderdijk and J. A. Huntington, Sci. Rep., 2018, 8, 8793 CrossRef PubMed.
- S. G. Polderdijk, T. E. Adams, L. Ivanciu, R. M. Camire, T. P. Baglin and J. A. Huntington, Blood, 2017, 129, 105–113 CrossRef CAS.
- W. Bode, I. Mayr, U. Baumann, R. Huber, S. R. Stone and J. Hofsteenge, EMBO J., 1989, 8, 3467–3475 CrossRef CAS.
- M. Karle, W. Knecht and Y. Xue, Bioorg. Med. Chem. Lett., 2012, 22, 4839–4843 CrossRef CAS PubMed.
- A. Hoffmann-Roder, E. Schweizer, J. Egger, P. Seiler, U. Obst-Sander, B. Wagner, M. Kansy, D. W. Banner and F. Diederich, ChemMedChem, 2006, 1, 1205–1215 CrossRef.
- S. Kawabata, T. Miura, T. Morita, H. Kato, K. Fujikawa, S. Iwanaga, K. Takada, T. Kimura and S. Sakakibara, Eur. J. Biochem., 1988, 172, 17–25 CrossRef CAS PubMed.
- H. M. Petrassi, J. A. Williams, J. Li, C. Tumanut, J. Ek, T. Nakai, B. Masick, B. J. Backes and J. L. Harris, Bioorg. Med. Chem. Lett., 2005, 15, 3162–3166 CrossRef CAS PubMed.
- D. N. Gosalia, C. M. Salisbury, D. J. Maly, J. A. Ellman and S. L. Diamond, Proteomics, 2005, 5, 1292–1298 CrossRef CAS PubMed.
- S. Butenas, V. Drungilaite and K. G. Mann, Anal. Biochem., 1995, 225, 231–241 CrossRef CAS PubMed.
- M. Gallwitz, M. Enoksson, M. Thorpe and L. Hellman, PLoS One, 2012, 7, e31756 CrossRef CAS PubMed.
- L. E. Sanman and M. Bogyo, Annu. Rev. Biochem., 2014, 83, 249–273 CrossRef CAS PubMed.
- M. H. Wright and S. A. Sieber, Nat. Prod. Rep., 2016, 33, 681–708 RSC.
- J. C. Powers, J. L. Asgian, O. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS PubMed.
- J. R. Carreon, K. M. Stewart, K. P. Mahon Jr, S. Shin and S. O. Kelley, Bioorg. Med. Chem. Lett., 2007, 17, 5182–5185 CrossRef CAS.
- A. Hunfeld, M. Etscheid, H. Konig, R. Seitz and J. Dodt, FEBS Lett., 1999, 456, 290–294 CrossRef CAS PubMed.
- S. G. I. Polderdijk, T. P. Baglin and J. A. Huntington, Curr. Opin. Hematol., 2017, 24, 446–452 CrossRef CAS PubMed.
- H. J. Mann, M. A. Short and D. E. Schlichting, Am. J. Health-Syst. Pharm., 2009, 66, 1089–1096 CrossRef CAS PubMed.
- R. Lottenberg, J. A. Hall, M. Blinder, E. P. Binder and C. M. Jackson, Biochim. Biophys. Acta, 1983, 742, 539–557 CrossRef CAS.
- B. F. Le Bonniec, T. Myles, T. Johnson, C. G. Knight, C. Tapparelli and S. R. Stone, Biochemistry, 1996, 35, 7114–7122 CrossRef CAS PubMed.
- P. E. Marque, R. Spuntarelli, L. Juliano, M. Aiach and B. F. Le Bonniec, J. Biol. Chem., 2000, 275, 809–816 CrossRef CAS PubMed.
- E. P. Bianchini, V. B. Louvain, P. E. Marque, M. A. Juliano, L. Juliano and B. F. Le Bonniec, J. Biol. Chem., 2002, 277, 20527–20534 CrossRef CAS.
- H. Nar, Trends Pharmacol. Sci., 2012, 33, 279–288 CrossRef CAS PubMed.
- S. Kolt, T. Janiszewski, D. Kaiserman, S. Modrzycka, S. J. Snipas, G. Salvesen, G. M. Dra, P. I. Bird and P. Kasperkiewicz, J. Med. Chem., 2020, 63, 3359–3369 CrossRef CAS PubMed.
- P. Kasperkiewicz, Y. Altman, M. D'Angelo, G. S. Salvesen and M. Drag, J. Am. Chem. Soc., 2017, 139, 10115–10125 CrossRef CAS PubMed.
- S. A. Andrade, E. M. Santomauro-Vaz, A. R. Lopes, A. M. Chudzinski-Tavassi, M. A. Juliano, W. R. Terra, M. U. Sampaio, C. A. Sampaio and M. L. Oliva, Biol. Chem., 2003, 384, 489–492 CAS.
- M. J. Page, A. L. Lourenco, T. David, A. M. LeBeau, F. Cattaruzza, H. C. Castro, H. F. VanBrocklin, S. R. Coughlin and C. S. Craik, Nat. Commun., 2015, 6, 8448 CrossRef CAS.
- X. Jin, X. Liu, X. H. Zhu, H. Li, W. Li, Y. Huang and S. Z. Yao, Anal. Methods, 2016, 8, 3691–3697 RSC.
- F. A. Jaffer, C. H. Tung, R. E. Gerszten and R. Weissleder, Arterioscler., Thromb., Vasc. Biol., 2002, 22, 1929–1935 CrossRef CAS PubMed.
- D. J. Maly, F. Leonetti, B. J. Backes, D. S. Dauber, J. L. Harris, C. S. Craik and J. A. Ellman, J. Org. Chem., 2002, 67, 910–915 CrossRef CAS PubMed.
- L. Bajzar and M. Nesheim, J. Biol. Chem., 1993, 268, 8608–8616 CrossRef CAS.
- W. Kisiel, J. Clin. Invest., 1979, 64, 761–769 CrossRef CAS PubMed.
- J. Jesty and Y. Nemerson, Methods Enzymol., 1976, 45, 95–107 CAS.
- E. Briet, C. M. Noyes, H. R. Roberts and M. J. Griffith, Thromb. Res., 1982, 27, 591–600 CrossRef CAS PubMed.
- N. L. Esmon, W. G. Owen and C. T. Esmon, J. Biol. Chem., 1982, 257, 859–864 CrossRef CAS.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS PubMed.
- A. Sivaraman, D. G. Kim, D. Bhattarai, M. Kim, H. Y. Lee, S. Lim, J. Kong, J. I. Goo, S. Shim, S. Lee, Y. G. Suh, Y. Choi, S. Kim and K. Lee, J. Med. Chem., 2020, 63, 5139–5158 CrossRef CAS PubMed.
- P. Kasperkiewicz, M. Poreba, S. J. Snipas, H. Parker, C. C. Winterbourn, G. S. Salvesen and M. Drag, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2518–2523 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Graphical scheme of the methodology used for the design of selective chemical tools; purity and MS analysis for substrates, inhibitors, and activity-based probes; full-size blots. See https://doi.org/10.1039/d2sc01108e |
| This journal is © The Royal Society of Chemistry 2022 |