
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDipole-mediated exciton management strategy enabled by reticular chemistry†
Ruomeng
Wan
 a,
Dong-Gwang
Ha
a,
Jin-Hu
Dou
a,
Woo Seok
Lee
b,
Tianyang
Chen
a,
Julius J.
Oppenheim
a,
Jian
Li
c,
William A.
Tisdale
*b and
Mircea
Dincă
*a
a,
Dong-Gwang
Ha
a,
Jin-Hu
Dou
a,
Woo Seok
Lee
b,
Tianyang
Chen
a,
Julius J.
Oppenheim
a,
Jian
Li
c,
William A.
Tisdale
*b and
Mircea
Dincă
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge 02139, Massachusetts, USA
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge 02139, Massachusetts, USA
cDepartment of Materials and Environmental Chemistry, Stockholm University, Stockholm, Sweden
First published on 24th August 2022
Abstract
Selectively blocking undesirable exciton transfer pathways is crucial for utilizing exciton conversion processes that involve participation of multiple chromophores. This is particularly challenging for solid-state systems, where the chromophores are fixed in close proximity. For instance, the low efficiency of solid-state triplet–triplet upconversion calls for inhibiting the parasitic singlet back-transfer without blocking the flow of triplet excitons. Here, we present a reticular chemistry strategy that inhibits the resonance energy transfer of singlet excitons. Within a pillared layer metal–organic framework (MOF), pyrene-based singlet donors are situated perpendicular to porphyrin-based acceptors. High resolution transmission electron microscopy and electron diffraction enable direct visualization of the structural relationship between donor and acceptor (D–A) chromophores within the MOF. Time-resolved photoluminescence measurements reveal that the structural and symmetry features of the MOF reduce the donor-to-acceptor singlet transfer efficiency to less than 36% compared to around 96% in the control sample, where the relative orientation of the donor and acceptor chromophores cannot be controlled.
Introduction
Controlling exciton transfer processes, either by inhibiting them, or by making them faster, is critical for a range of technologies that involve light interacting with matter. Whereas increasing the efficiency of exciton transfer has received considerable attention, developing deliberate means to inhibit exciton transport is less studied. Indeed, the latter challenge can be dismissed as almost trivial, because exciton transport pathways can always be shut down by increasing the donor–acceptor (D–A) separation in a two-chromophore pair. This trivial solution, however, is not an option when the overall efficiency of a photophysical process depends on multiple, antagonistic exciton transfer processes that require chromophores to be in close proximity (Fig. 1). A classic example involves triplet upconversion, which exhibits much lower efficiency in the solid-state1 compared to solution2 or mixed-phase3 systems primarily because back-transfer4 of the high energy singlet from the annihilator to the sensitizer becomes more pronounced when the chromophores occupy fixed positions in sufficient proximity to allow triplet transfer. This dilemma is also highlighted in the exciton management of white organic light emitting devices,5,6 as well as in emerging excitonic processes that consist of a mixture of Förster and Dexter energy transfer pathways.7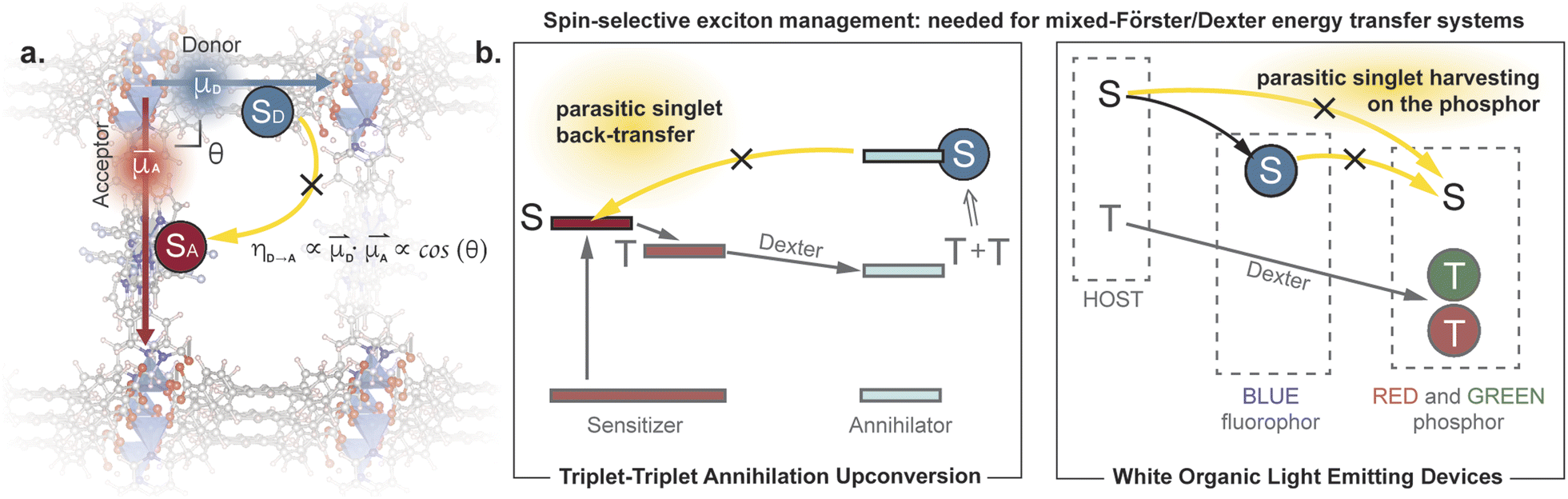 | ||
| Fig. 1 (a) Conceptual illustration of managing parasitic singlet transfer by perpendicular arrangement of transition dipoles. (b) Examples of systems that suffer from parasitic singlet transfer, yet meanwhile need to maintain efficient triplet transfer between the same moieties.4–6 | ||
One strategy for inhibiting communication between chromophores despite their close physical distance is to change how the donor “sees” the acceptor. When donors and acceptors are close (i.e. < 10 nm), singlet transfer occurs through Förster Resonance Energy Transfer (FRET).8 The efficiency of this nonradiative process is proportional to the inner product of the donor and acceptor transition dipole moments. Because this is a vectorial product, the FRET efficiency can theoretically be reduced to zero when the two transition dipole moments are exactly perpendicular to each other (Fig. 1). In other words, one can, in principle, eliminate singlet back-transfer if the acceptor is placed in the “blind spot” of the donor. Importantly, this perpendicular arrangement does not block the Dexter transfer9 of triplet excitons, and opens up the possibility for spin-selective control of exciton transport.
Although conceptually simple, this design is difficult to implement in practice because of entropic fluctuations. For instance, a complex comprising an anthracene donor and a porphyrin acceptor was reported to still exhibit highly efficient singlet transfer of over 90% despite the perpendicular arrangement of the D and A chromophores.10 The relaxation of the FRET selection rule in this complex was assigned to flexible binding in the D–A pair, which deviates the D–A angle away from 90°.10,11 Here, we demonstrate that placing perpendicular D–A pairs in a rigid solid framework greatly minimizes entropic fluctuations and leads to singlet transfer efficiency below 36%.
Owing to their rigid structures that place organic ligands at well-defined distances and angles from each other, metal–organic frameworks (MOFs) have recently gained traction as platforms for controlling excitons.12–23 One MOF topology that allows perpendicular arrangement of donor and acceptor moieties at close distance is a square lattice defined by dinuclear metal paddlewheel tetracarboxylates with rectangular ligands, pillared by a second type of ligand, typically linear bipyridyl molecules (Fig. 2).24 The presence of two distinct structural motifs—“linkers” and “pillars”— allows the localization of donors and acceptors at desired angles in a rigid scaffold.
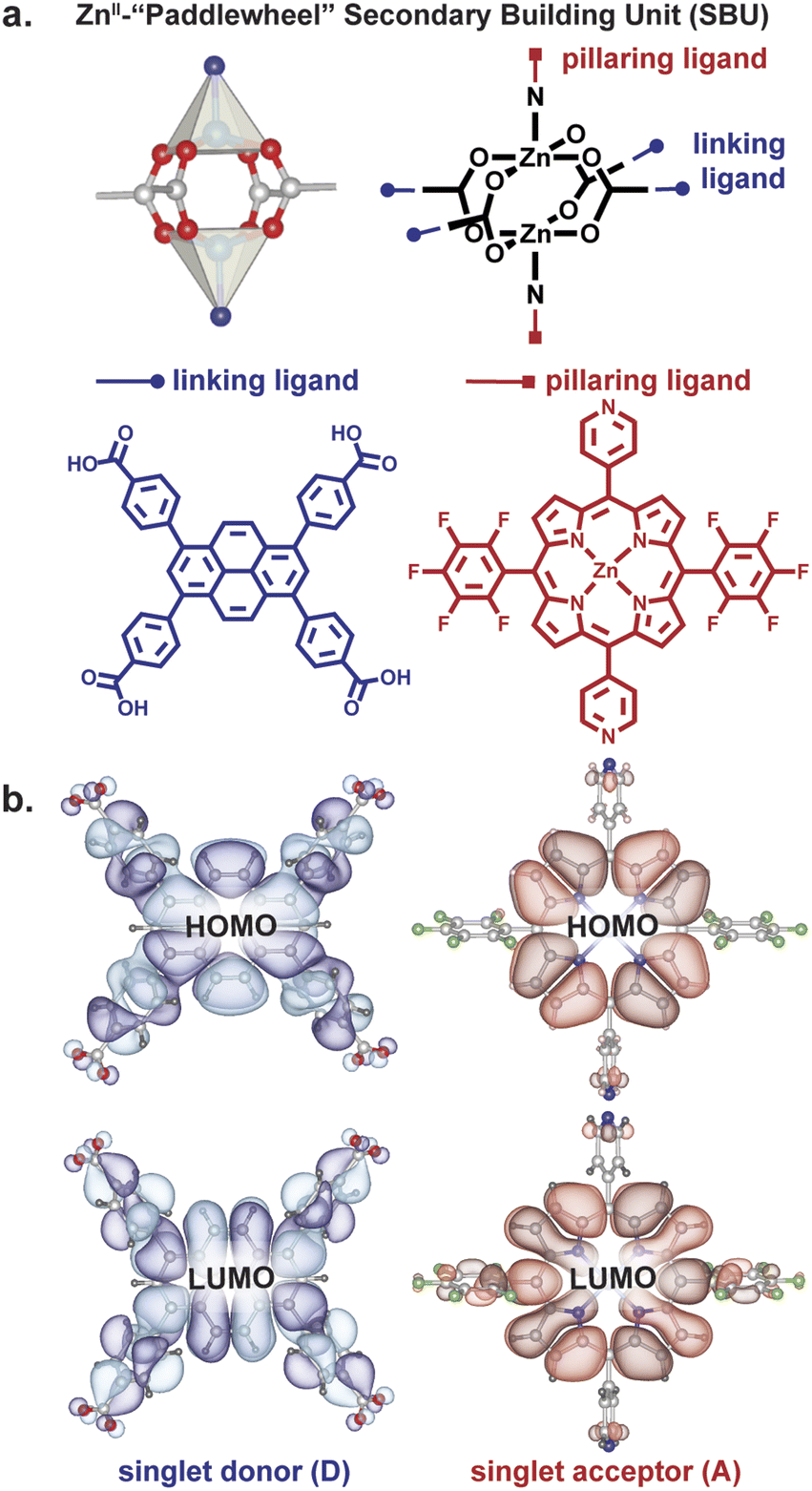 | ||
| Fig. 2 (a) Building blocks of the paddlewheel pillared PyP-MOF. Rectangular 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetrabenzoic acid forms two-dimensional grids, which are connected by porphyrin-based pillaring ligands into a three-dimensional network. (b) Frontier orbitals calculated for the donor and acceptor moieties. | ||
Results and discussion
Structural characterization and perpendicular D–A moieties
We employed a pyrene-based singlet donor (4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetrabenzoic acid, H4TBAPy) and a porphyrin-based singlet acceptor ([5,15-dipyridyl-10,20-bis (pentafluorophenyl)-porphyrinato]-zincII, H2P)24,25 as the linker and pillar ligands, respectively. Pyrenes and porphyrins are well known annihilators and sensitizers for triplet upconversion.26,27 We targeted closed-shell ZnII paddlewheel secondary building units (SBUs) to avoid interference of metal-based electronic transitions with the photophysical processes within the ligands.19 Treating a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of H4TBAPy:H2P in N,N-dimethylformamide (DMF) with Zn(NO3)2·6H2O and heating to 100 °C for 48 h produced red crystals of Zn2(Zn-P)(TBAPy) (PyP-MOF; Zn-P = porphyrin metalated with Zn2+). Pawley refinement of a powder X-ray diffraction (PXRD) pattern of these crystals obtained with synchrotron radiation (Fig. 3a) revealed a monoclinic centered unit cell with parameters a = 22.80(9) Å, b = 21.86(6) Å, c = 21.82(0) Å, β = 95.69(9)°. These unit cell parameters were confirmed by electron diffraction analysis (Fig. 3b–e), which also revealed strict perpendicularity between the pyrene donor and porphyrin acceptor. High resolution transmission electron microscopy (HR-TEM) provided real-space visualization of the D–A geometric relationship with nearly atomic precision (Fig. 4), corroborating the PXRD and electron diffraction studies.
:1 mixture of H4TBAPy:H2P in N,N-dimethylformamide (DMF) with Zn(NO3)2·6H2O and heating to 100 °C for 48 h produced red crystals of Zn2(Zn-P)(TBAPy) (PyP-MOF; Zn-P = porphyrin metalated with Zn2+). Pawley refinement of a powder X-ray diffraction (PXRD) pattern of these crystals obtained with synchrotron radiation (Fig. 3a) revealed a monoclinic centered unit cell with parameters a = 22.80(9) Å, b = 21.86(6) Å, c = 21.82(0) Å, β = 95.69(9)°. These unit cell parameters were confirmed by electron diffraction analysis (Fig. 3b–e), which also revealed strict perpendicularity between the pyrene donor and porphyrin acceptor. High resolution transmission electron microscopy (HR-TEM) provided real-space visualization of the D–A geometric relationship with nearly atomic precision (Fig. 4), corroborating the PXRD and electron diffraction studies.
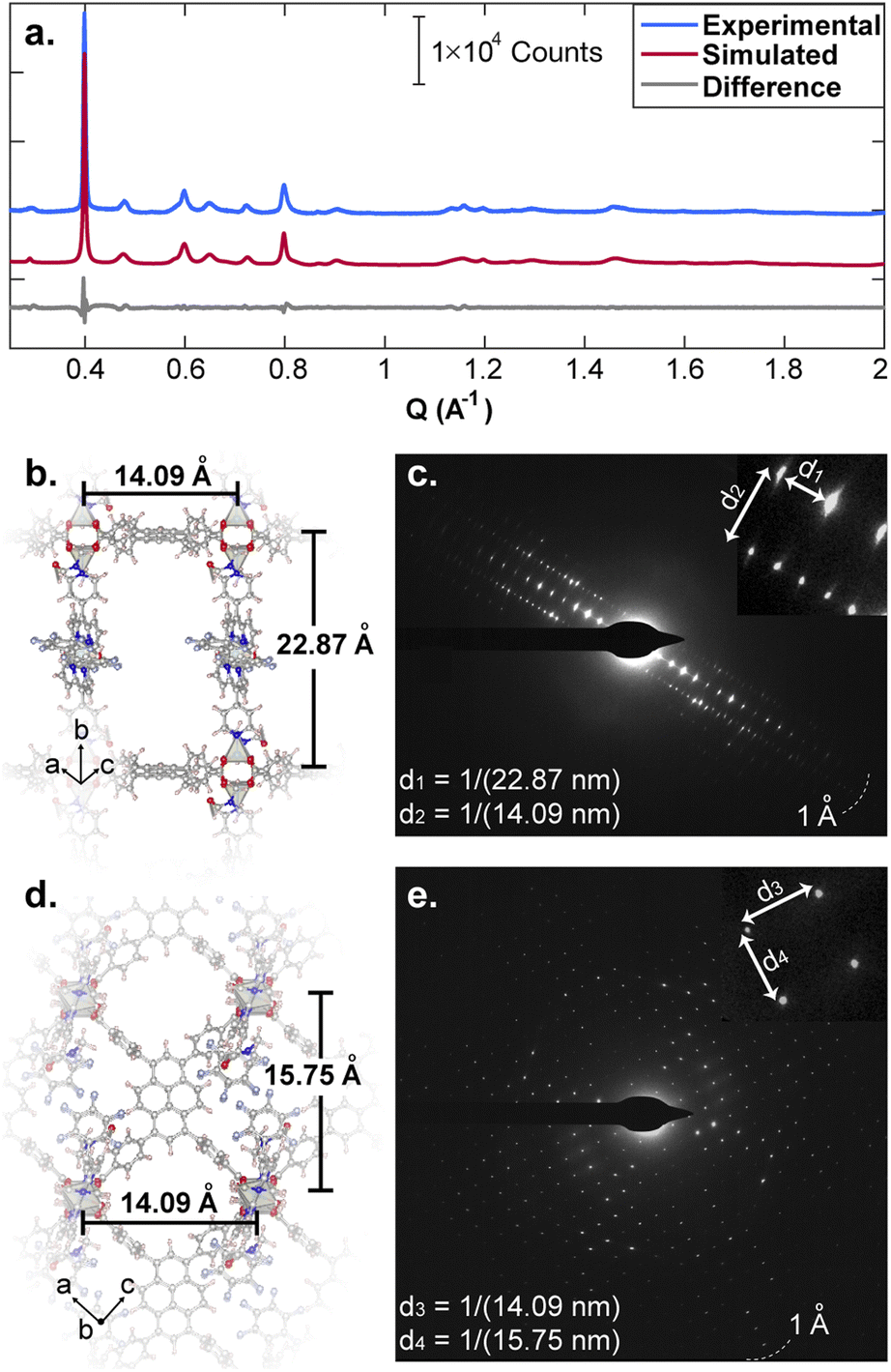 | ||
| Fig. 3 (a) Experimental (blue) and Pawley-refined simulated (red) PXRD patterns for PyP-MOF. The grey trace corresponds to the difference between the experimental and the simulated data. (b–e) Electron diffraction with the beam perpendicular to the crystallographic b axis, which coincides with the pillaring direction (b and c), and parallel to the b axis (d and e). | ||
 | ||
| Fig. 4 HR-TEM data revealing real-space perpendicular relationship between the D and A moieties. The insert shows the fast Fourier transform (FFT) of the real space micrographs. | ||
Relevantly, the effective transition dipole moments of both the pyrene linker and the porphyrin pillars are coplanar with their π systems (ESI Section 9†).28–30 Given the structural details discussed above, the donor and acceptor transition dipole moments themselves are thus strictly perpendicular within PyP-MOF.
Singlet transfer dynamics and the role of sample preparation
We tested whether the structural D–A arrangement in PyP-MOF indeed decreases the efficiency of singlet transfer (η![[D with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/char_0044_20d1.gif) A) by tracing the lifetime of the donor singlet excited state (S1D) in the presence (τDA) or absence (τD) of an acceptor:
A) by tracing the lifetime of the donor singlet excited state (S1D) in the presence (τDA) or absence (τD) of an acceptor: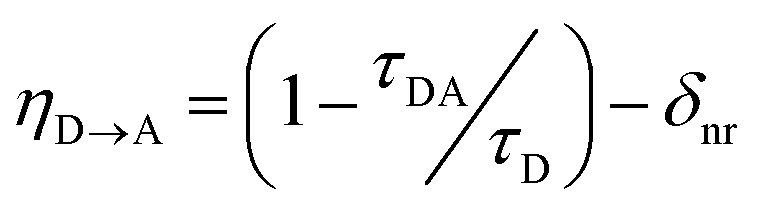 | (1) |
The decay of S1D of pure H4TBAPy diluted in polystyrene can be approximated by a single exponential function to give a lifetime τD of 1.83 ns (Fig. 5a). Importantly, both the energy level (∼2.72 eV) and the lifetime for this sample are in line with those reported for dilute solutions of H4TBAPy,31 confirming that incorporation in polystyrene eliminates the aggregation induced effects (i.e. excimer formation and quenching) common in solid-state samples. No significant variation of τD is observed when the H4TBAPy concentration is increased from 1 wt% to either 2 wt% or 4 wt% in polystyrene. However, upon further doubling the concentration to 8 wt%, a broad red-shifted feature grows in at 2.18 eV–2.48 eV, with a long lifetime (∼4 ns) characteristic of excimer emission (Fig. S8†).
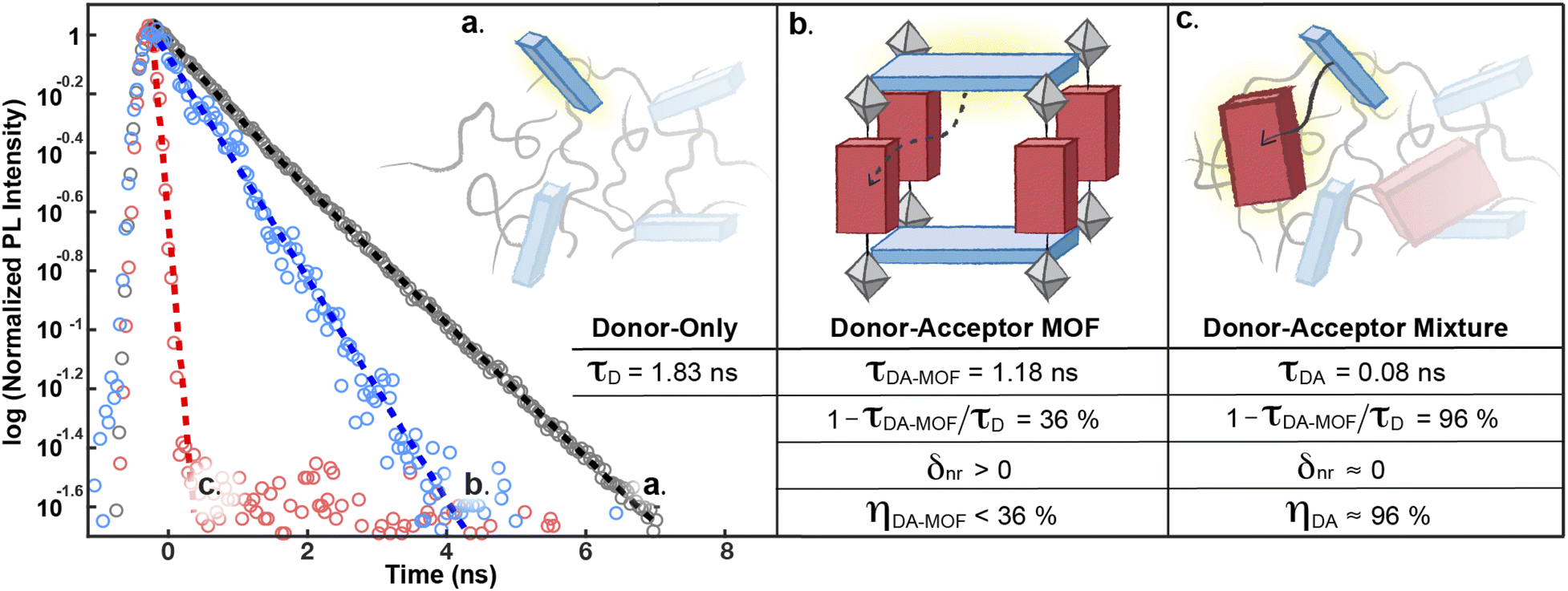 | ||
| Fig. 5 Comparison of the decay dynamics of the S1D state in: (a) the H4TBAPy donor, (b) the donor–acceptor PyP-MOF, and (c) the donor–acceptor mixture (control). All samples are measured as solid thin films diluted in polystyrene and spin-coated on quartz substrates (ESI Section 7†). The corresponding energy transfer efficiencies (η) are derived according to eqn (1). | ||
Particle aggregation effects can affect the photophysical properties of MOF samples and must be accounted for when interpreting the data as intrinsic to the MOF itself. Indeed, time-resolved PL measurements show that undiluted PyP-MOF samples exhibit broad red-shifted emission between 500 nm and 575 nm (2.16 eV–2.48 eV) compared to PyP-MOF samples diluted with polystyrene, which emit at ∼460 nm (2.70 eV) (Fig. 6b).
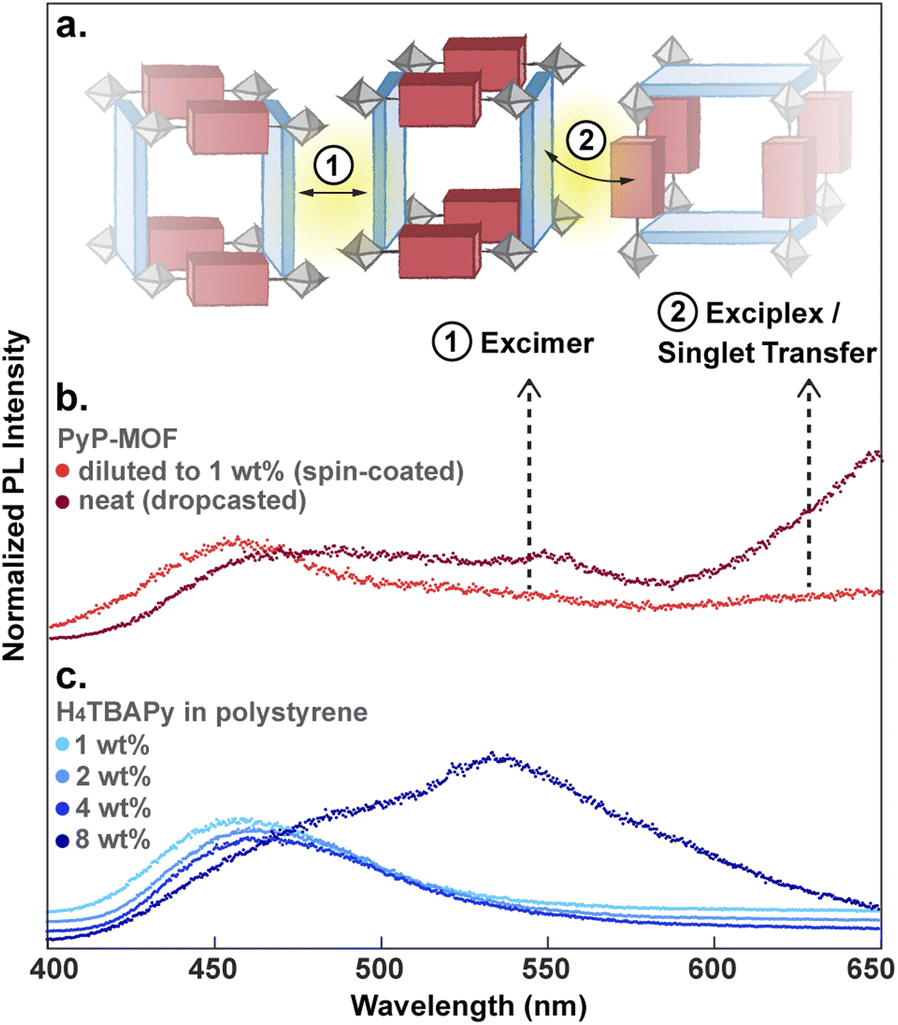 | ||
| Fig. 6 (a) Schematic illustration of the observed inter-crystallite D–D excimer formation and potential inter-crystallite D–A exciplex formation as well as D to A singlet energy transfer. (b) Comparison of the integrated PL (0–20 ns) of PyP-MOF sample diluted in polystyrene matrix (1 wt%) and that of neat PyP-MOF sample from direct drop-casting. (c) Concentration-dependent integrated PL of H4TBAPy diluted in polystyrene to 1 wt%, 2 wt%, 4 wt% and 8 wt%. | ||
This redshift matches that of the excimer formed in concentrated H4TBAPy sample (8 wt%, Fig. 6c). Importantly, because the distance between TBAPy moieties in PyP-MOF is fixed at around 20 Å by the porphyrin linkers, intra-framework excimer formation is not possible in this material. Instead, we attribute the observed red-shifted emission in concentrated PyP-MOF samples to excimer formation between pyrene moieties on neighboring MOF crystallites. The observation of inter-particle D–D excimer formation highlights the importance of diluting the MOF samples: if D–D inter-particle excimer formation is possible, so would be inter-particle D–A interactions (Fig. 6a). Such interactions are not bound by the geometric restrictions intrinsic to the MOF structure because the orientation of two neighboring particles is random. PL data from undiluted MOF samples would then convolute both intra-particle D–A interactions, intrinsic to the MOF, and inter-particle D–A interactions.
Indeed, PL data for polystyrene-diluted PyP-MOF samples integrated over 20 ns reveal no significant redshift, confirming effective suppression of excimer formation between MOF particles. In other words, we can reasonably assume that the measured τDA for the diluted MOF sample, ∼1.18 ns (Fig. 5b), is intrinsic to the MOF structure and is not influenced by inter-particle interactions. Combining this value with τD (1.83 ns) yields (1 − τDA/τD) = 36%. Previous studies in literature have identified that incorporation of H4TBAPy ligand into MOF framework can introduce additional non-radiative pathways,32 which is consistent with our PL quantum yield (QY) measurements (ESI Section 8.2†). Thus for PyP-MOF, δnr>0, suggesting that the singlet energy transfer efficiency ηD→A is lower than 36%.
Control system for bridging structural and photophysical study
This efficiency is best compared with ηD→A of a control D–A system comprising the same pyrene and porphyrin chromophores, dispersed in a polystyrene matrix in the same molar ratio as in PyP-MOF (1:1), but free from the geometric constraints imposed by the rigid MOF. This control system is also designed to ensure that all additional factors that could influence the electric and dielectric environment experienced by the H4TBAPy fluorophore are kept the same as those of the donor-only sample (except for the introduction of an acceptor), in order to avoid significant changes in other nonradiative recombination pathways. Therefore, the observation of an increased S1D decay rate in this control sample can reasonably be attributed to singlet transfer from free donor to free acceptor, which are oriented randomly in the control sample. As illustrated in Fig. 5c, τDA= 0.08 ns (and δnr ≈ 0) yields an energy transfer efficiency of ≈96%, suggesting that H4TBAPy and H2P possess sufficient spectral overlap for efficient energy transfer. The energetic accessibility of the D → A transfer pathway for this donor–acceptor pair is further supported by (i) a significant concentration-dependence of the donor QY in donor–acceptor DMF solution mixtures (ESI Table S3†); and (ii) the presence of spectral overlap directly observable from comparing the emission and absorption energy levels (ESI Fig. S6†). FRET efficiency is determined by three factors: the spectral overlap, the dipole orientation, and the distance between D and A. Since both close proximity and proper spectral overlap are satisfied for the D–A pair in PyP-MOF, we can conclude that the markedly reduced FRET efficiency in the framework is due to the orientation control imposed by the MOF, and not by distance or by energetic mismatches.
These results and our conclusion above are substantiated by recent observations related to triplet dynamics in a MOF with analogous topology.21 As discussed in the introduction, the perpendicular arrangement of D and A chromophores only affects excited states with certain spin values. In this recent report, it was shown that a paddlewheel pillared MOF analogous to ours enables efficient transfer of triplet excitons. Our results show that the same perpendicular arrangement of D and A chromophores also suppresses singlet transfer, suggesting spin-selective control of energy transfer.
Conclusions
The foregoing results demonstrate the utility of the reticular chemistry toolbox in exciton management, here by obstructing parasitic FRET pathways. By incorporating D and A moieties into a rigid framework with translational symmetry, the angular relationship between the D and A transition dipoles can be tuned, which in turn controls Förster energy transfer. In contrast to discrete coordination complexes, extended frameworks feature higher structural rigidity that allows stricter angular control and minimization of entropic fluctuations. These geometric concepts lead to a dramatic reduction in the efficiency of singlet transfer, from around 96% in control samples of D and A chromophores with random orientation, to less than 36% in the MOF. These results highlight the promise of reticular chemistry to tackle the challenge of parasitic FRET faced in fields such as triplet upconversion4 and white light emitting devices.5,6 In light of the growing varieties of excitonic processes that involve complex combinations of Förster and Dexter pathways,7 we envision an increasing need for enabling spin-dependent exciton management strategies.Data availability
All data supporting the findings of this study—PyP–MOF preparation, synchrotron PXRD, HR-TEM, electron diffraction, time-resolved PL, diffuse reflectance UV-vis, quantum yield characterization and TD-DFT calculation are available in the article and the ESI.†Author contributions
R. W., D.-G. H., W. A. T. and M. D. conceived the project. R. W. synthesized materials and prepared samples. R. W. and D.-G. H. carried out time-resolved PL measurement and analysis. R. W. and J.-H. D. performed HR-TEM and electron diffraction characterization. W. S. L. and R. W. collected and interpreted PL quantum yield data. T. C., J. L. and R. W. analyzed structrual data from synchrotron PXRD. J. J. O. performed and interpreted TD-DFT calculations. W. A. T. and M. D. supervised the synthesis and characterization of the materials. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Science Foundation (NSF DMR-2105495). W. A. T. was supported by the Camille & Henry Dreyfus Foundation (TC-17-005). HR-TEM and electron diffraction data were collected at the Automated Cryogenic Electron Microscopy Facility in MIT.nano on a Talos Arctica microscope, which was a gift from the Arnold and Mabel Beckman Foundation. We thank Dr Edward Brignole for assistance with HR-TEM and electron diffraction data collection. We also thank Prof. Marc Baldo for sharing access to the streak camera (HAMAMATSU C10627) set-up.References
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef CAS.
- T. Ogawa, N. Yanai, A. Monguzzi and N. Kimizuka, Sci. Rep., 2015, 5, 10882 CrossRef CAS PubMed.
- J. Perego, J. Pedrini, C. X. Bezuidenhout, P. E. Sozzani, F. Meinardi, S. Bracco, A. Comotti and A. Monguzzi, Adv. Mater., 2019, 31, 1903309 CrossRef CAS PubMed.
- T.-A. Lin, C. F. Perkinson and M. A. Baldo, Adv. Mater., 2020, 32, 1908175 CrossRef CAS PubMed.
- Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908 CrossRef CAS PubMed.
- G. Schwartz, S. Reineke, T. C. Rosenow, K. Walzer and K. Leo, Adv. Funct. Mater., 2009, 19, 1319 CrossRef CAS.
- E. W. Evans, A. J. Gillett, Q. Gu, J. Ding, Z. Chen, T. J. H. Hele, R. H. Friend and F. Li, 2021, arXiv:2109 13522.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- V. Gray, K. Börjesson, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, J. Phys. Chem. C, 2016, 120, 19018–19026 CrossRef CAS.
- V. Gray, B. Küçüköz, F. Edhborg, M. Abrahamsson, K. Moth-Poulsen and B. Albinsson, Phys. Chem. Chem. Phys., 2018, 20, 7549–7558 RSC.
- M. C. So, G. P. Wiederrecht, J. E. Mondloch, J. T. Hupp and O. K. Farha, Chem. Commun., 2015, 51, 3501–3510 RSC.
- Q. Zhang, C. Zhang, L. Cao, Z. Wang, B. An, Z. Lin, R. Hang, Z. Zhang, C. Wang and W. Lin, J. Am. Chem. Soc., 2016, 138, 5308–5315 CrossRef CAS PubMed.
- E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Chem. Soc. Rev., 2018, 47, 4710–4728 RSC.
- J. Yu, X. Li and P. Deria, ACS Sustainable Chem. Eng., 2019, 7, 1841–1854 CrossRef CAS.
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858–15861 CrossRef CAS PubMed.
- H.-J. Son, S. Jin, S. Patwardhan, S. J. Wezenber, N. C. Jeong, M. So, C. E. Wilmer, A. A. Sarjeant, G. C. Schatz, R. Q. Snurr, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 862–869 CrossRef CAS PubMed.
- S. Goswami, M. Chen, M. R. Wasielewski, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2018, 10, 34409–34417 CrossRef CAS PubMed.
- J. Cornelio, T.-Y. Zhou, A. Alkas and S. G. Telfer, J. Am. Chem. Soc., 2018, 140, 15470–15476 CrossRef CAS PubMed.
- M. Adams, M. Kozlowska, N. Baroni, M. Oldenburg, R. Ma, D. Busko, A. Turshatov, G. Emandi, M. O. Senge, R. Haldar, C. Wöll, G. U. Nienhaus, B. S. Richards and I. A. Howard, ACS Appl. Mater. Interfaces, 2019, 11, 15688–15697 CrossRef CAS PubMed.
- I. Roy, S. Goswami, R. M. Young, I. Schlesinger, M. R. Mian, A. E. Enciso, X. Zhang, J. E. Hornick, O. K. Farha, M. Wasielewski, J. T. Hupp and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 5053–5059 CrossRef CAS PubMed.
- A. D. Sample, J. Guan, J. Hu, T. Reese, C. R. Cherqui, J.-E. Park, F. Freire-Fernández, R. D. Schaller, G. C. Schatz and T. W. Odom, Nano Lett., 2021, 21, 7775–7780 CrossRef CAS PubMed.
- S. M. Shaikh, S. Ilic, B. J. Gibbons, X. Yang, E. Jakubikova and A. M. Morris, J. Phys. Chem. C, 2021, 125, 22998–23010 CrossRef CAS.
- A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 4204–4205 CrossRef CAS PubMed.
- T. C. Wang, N. A. Vermeulen, I. S. Kim, A. B. F. Martinson, J. F. Stoddart, J. T. Hupp and O. K. Farha, Nat. Protoc., 2016, 11, 149–162 CrossRef CAS PubMed.
- W. Zhao and F. N. Castellano, J. Phys. Chem. A, 2006, 110, 11440–11445 CrossRef CAS PubMed.
- A. Harriman, G. Porter and M.-C. Richoux, J. Chem. Soc., Faraday trans. II, 1981, 77, 833–844 RSC.
- M. K. Roos, S. Reiter and R. de Vivie-Riedle, Chem. Phys., 2018, 515, 586–595 CrossRef CAS.
- S. Matile, N. Berova, K. Nakanishi, J. Fleischhauer and R. W. Woody, J. Am. Chem. Soc., 1996, 118, 5198–5206 CrossRef CAS.
- G. Pescitelli, S. Gabriel, Y. Wang, J. Fleischhauer, R. W. Woody and N. Berova, J. Am. Chem. Soc., 2003, 125, 7613–7628 CrossRef CAS PubMed.
- P. Deria, J. Yu, T. Smith and R. P. Balaraman, J. Am. Chem. Soc., 2017, 139, 5973–5983 CrossRef CAS PubMed.
- J. Yu, J.-H. Park, A. Van Wyk, G. Rumbles and P. Deria, J. Am. Chem. Soc., 2018, 140, 10488–10496 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01127a |
| This journal is © The Royal Society of Chemistry 2022 |