
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into electrochemiluminescence dynamics by synchronizing real-time electrical, luminescence, and mass spectrometric measurements†
Xuemeng
Zhang
a,
Weifeng
Lu
a,
Cheng
Ma
 ac,
Tao
Wang
a,
Jun-Jie
Zhu
a,
Richard N.
Zare
b and
Qianhao
Min
*a
ac,
Tao
Wang
a,
Jun-Jie
Zhu
a,
Richard N.
Zare
b and
Qianhao
Min
*a
aState Key Laboratory of Analytical Chemistry for Life Science, Chemistry and Biomedicine Innovation Center, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: minqianhao@nju.edu.cn
bDepartment of Chemistry, Stanford University, Stanford, California 94305, USA
cSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, China
First published on 5th May 2022
Abstract
Electrochemiluminescence (ECL) comprises a sophisticated cascade of reactions. Despite advances in mechanistic studies by electrochemistry and spectroscopy, a lack of access to dynamic molecular information renders many plausible ECL pathways unclear or unproven. Here we describe the construction of a real-time ECL mass spectrometry (MS) platform (RT-Triplex) for synchronization of dynamic electrical, luminescent, and mass spectrometric outputs during ECL events. This platform allows immediate and continuous sampling of newly born species at the Pt wire electrode of a capillary electrochemical (EC) microreactor into MS, enabling characterization of short-lived intermediates and the multi-step EC processes. Two ECL pathways of luminol are validated by observing the key intermediates α-hydroxy hydroperoxide and diazaquinone and unraveling their correlation with applied voltage and ECL emission. Moreover, a “catalytic ECL route” of boron dipyrromethene (BODIPY) involving homogeneous oxidation of tri-n-propylamine with the BODIPY radical cation is proposed and verified.
Introduction
Electrochemiluminescence (also known as electrogenerated chemiluminescence, ECL) is an electrochemically triggered photoemission process involving electron transfer reactions on the surface of an electrode.1,2 Despite its extensive applications in immunoassay,3 diagnostics,4 and cell imaging,5,6 the understanding of mechanisms for ECL reactions remains incomplete.7–11 To date, many electrochemical and spectroscopic methods12 such as cyclic voltammetry (CV),10,13 differential pulse voltammetry (DPV),14 scanning electrochemical microscopy (SECM),15 electron spin resonance spectroscopy (ESR),7 and ECL microscopy9 have been developed for capturing the intermediates and unveiling the mechanisms of ECL systems. However, these methodologies mainly depend on the utilization of electrical and spectral parameters to reflect the transient state for deducing plausible pathways, and lack the ability to acquire directly molecular information during the entire ECL process. Additionally, ECL events always implicate the existence of multiple reactants with time-varying concentrations, which cannot be simultaneously distinguished and traced by electrochemical or spectroscopic approaches in a multiplexed manner.Mass spectrometry (MS) is capable of providing direct compositional and structural information on multiple target analytes at the molecular level with high sensitivity and specificity.16,17 Thus, the successful marriage of electrochemistry (EC) and MS may offer opportunities for gaining greater insight into electrochemical reactions, including ECL.18–20 Among the endeavors devoted to connecting EC with MS, differential electrochemical mass spectrometry (DEMS) and EC-electrospray ionization mass spectrometry (ESI-MS) are the classic strategies for the detection of gaseous and liquid species, respectively.20–23 However, the distance between the electrode–electrolyte interface (EEI) and the MS inlet brings about a loss of information on fleeting intermediates, and the applied high voltage for ionization may cause extra electrochemical changes.23,24 To overcome these barriers, EC reactors featuring a “waterwheel-shaped” electrode,19,25 hybrid ultramicroelectrode18 and bipolar ultramicroelectrode26 were developed and coupled with MS for rapid capture of transient species newly generated from the electrode. Furthermore, by treating ECL as a unique EC process, researchers have also identified several short-lived radicals and intermediates (typically TPrA˙+, TPrA˙, and Ru(bpy)3+) previously speculated to participate in ECL pathways by ultrafast in situ MS characterization.26–28 Despite excellent temporal resolution for a MS snapshot of a chemical transformation in ECL, dynamic correlation between chemical change and luminescence with respect to applied voltage at the EEI was usually neglected. Unlike common EC reactions, ECL involves more complicated reaction networks in terms of electron transfer, chemical transformation, and energy level transition,29,30 underlining the necessity and significance of simultaneously tracking redox current, labile electrogenerated species, and luminescence behavior in ECL mechanism studies. Unfortunately, aiming at the capture of reactive intermediates at the EEI, the existing EC-MS setups had to minimize the electrode dimension or shorten the transport distance to the gas phase,18,19,25,26,31 thus, physically constraining the simultaneous recording of electrical and especially luminescent signals. An ideal monitoring platform for dissecting ECL processes should be intended to observe electrogenerated species while tracing their dynamics correlated with electrochemical and luminescence variation, but hitherto, such three-channel synchronous tracking in ECL remains challenging.
Herein, a real-time ECL mass spectrometry platform (RT-Triplex) was designed and constructed for the interpretation of ECL reaction mechanisms by using a capillary EC microreactor to combine the photomultiplier tube (PMT) with a Venturi easy ambient sonic-spray ionization MS (V-EASI-MS) (Fig. 1), enabling real-time synchronous acquisition of electrical, luminescent, and mass spectrometric signals. In addition to the identification of electrochemical short-lived (half-life of sub-millisecond) intermediates and radicals, multi-step electrochemical redox processes implicating multi-electron transfer could also be molecularly tracked and distinguished with high potential and temporal resolution. More importantly, thanks to the orchestration of electricity-luminescence-mass three channels, the intermediate-regulated ECL pathways of luminol speculated by electrochemical or spectroscopic techniques were definitely validated at the molecular level by this method, and a “catalytic ECL route” of boron dipyrromethene (BODIPY) involving homogeneous oxidation of the co-reactant tri-n-propylamine (TPrA) with BODIPY radical cation (BODIPY˙+) in aprotic solvent was revealed and confirmed. Moreover, dynamic inspection of multiple participants in ECL including luminescent reagents, co-reactants and by-products could also be implemented owing to the multiplexing capability of MS, thus affording opportunities for globally delineating the essence of EC/ECL.
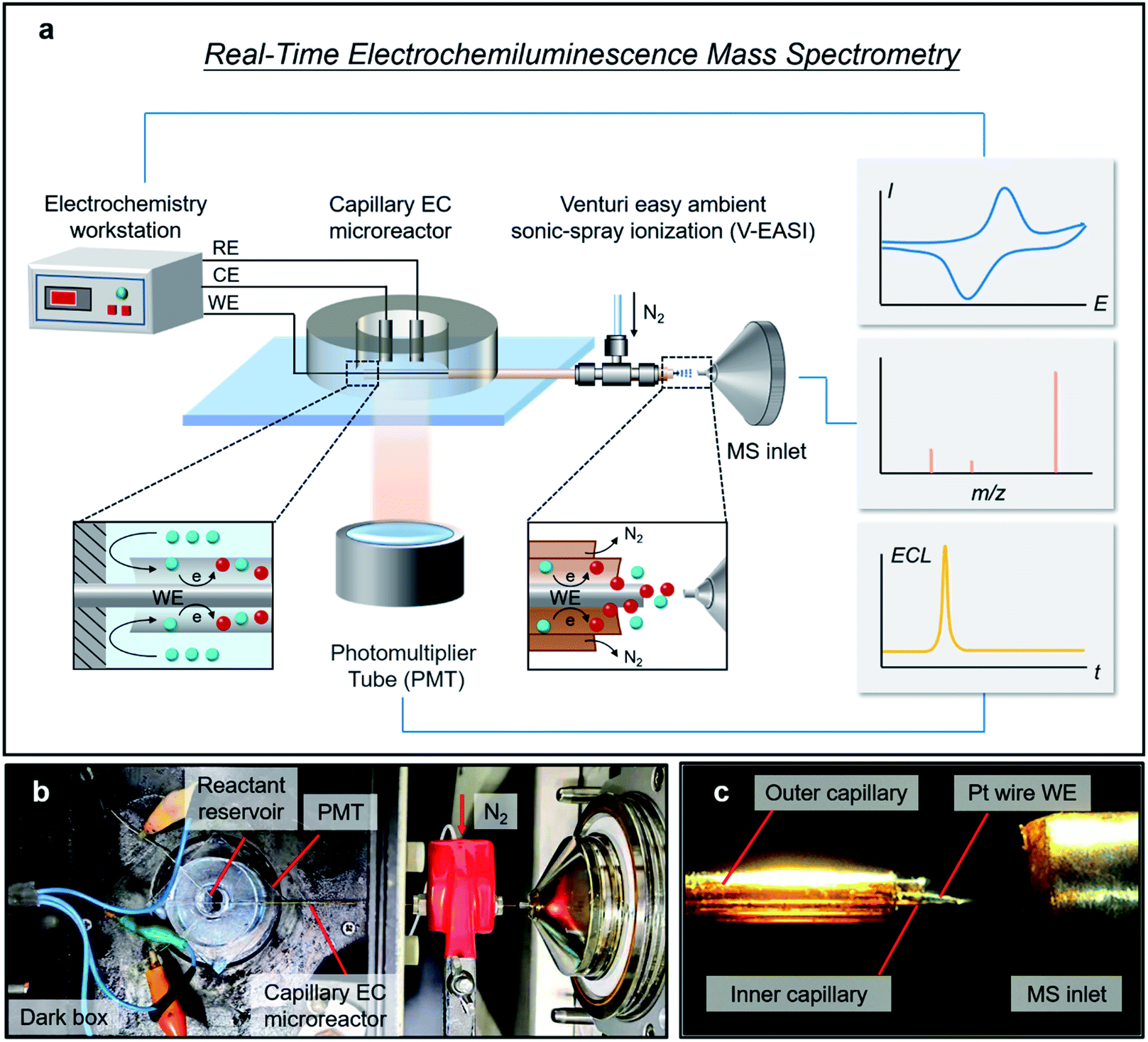 | ||
| Fig. 1 Design and configuration of the RT-Triplex setup. (a) Schematic of the RT-Triplex setup for electricity-luminescence-mass synchronization. Insets show the sideview schematic diagrams of the EC reaction occurring at the two ends of the capillary. (b) Photograph of the capillary EC microreactor interfaced with a V-EASI-MS. (c) Photograph showing the spray generated at the end of the capillary EC microreactor during sampling. The high-speed N2 stream in the outer capillary caused Venturi self-pumping of the sample solution in the inner capillary, and further nebulization at the tail end of the Pt wire WE. | ||
Results and discussion
Design and configuration of RT-Triplex
As illustrated in Fig. 1a, a capillary EC microreactor was fabricated by inserting a Pt wire (WE) through a fused silica capillary and extending it to the MS inlet at one end. To continuously supply reactant solution, a polydimethylsiloxane (PDMS) reservoir was fabricated and coupled to the other end of the capillary, while a Pt wire counter electrode (CE) and an Ag wire quasi-reference electrode (QRE) were threaded into the PDMS reservoir to construct a three-electrode system (Fig. S1†). To avoid extra in-source EC reaction, V-EASI with no need for high voltage was used to generate sonic spray (Fig. 1b and c). In this microreactor, the newborn species at the EEI were confined in the interspace between the Pt wire WE and capillary and were promptly delivered into the MS, ensuring regeneration of the EEI and consecutive readout of molecular information at the EEI (Fig. 1a, insets). The polyimide coating of the capillary part inside the reservoir was burned off to make ECL on the Pt wire WE accessible to the PMT placed below (Fig. S1†). Once potential was applied to trigger the ECL reaction, chemical variations at the EEI were tracked in real time by MS, while electrical signals and ECL emission were simultaneously recorded using the electrochemical workstation and the PMT, respectively. The configuration of the capillary EC microreactor and the RT-Triplex platform are described in the ESI.†Characterization of ultrafast and multi-step EC reactions by using RT-Triplex
To evaluate the capability of RT-Triplex in the transfer and identification of transient species, the well-studied electrooxidation of N,N-dimethylaniline (DMA) was chosen as a model reaction (Fig. 2a).25 After applying an oxidization potential of 1.5 V to the WE, a peak at m/z 121.0886 attributed to DMA˙+ was resolved in the mass spectrum (Fig. 2b). This ion signal correlated well with the applied potential pulse (Fig. 2c), indicating that this radical cation originated from the occurrence of electrochemical reactions (Fig. S2†). Successful capture of DMA˙+ indicated that our device is able to isolate fleeting intermediates (half-life of sub-millisecond) generated in the ultrafast EC reaction,25,32 which should be attributed to the close proximity of the WE terminal to the MS inlet and the rapid mobility (flow rate of 152 μL min−1 and linear velocity of 0.108 m s−1) of newly created species driven by the Venturi effect (Fig. S3†). Due to the dimerization of DMA, the ion signals of the protonated N,N,N′,N′-tetramethylbenzidine (TMB) and TMB˙+ were also observed at m/z 241.1699 and m/z 240.1621 when the potential was applied (Fig. 2b and S4†). | ||
Fig. 2 Real-time monitoring of the EEI during electrooxidation of DMA and electroreduction of flunitrazepam. The proposed mechanism for (a) electrooxidation of DMA and (d) electroreduction of flunitrazepam. (b) Positive-ion-mode mass spectrum of 94 μM DMA in acetonitrile (ACN) containing 1 mM lithium triflate (LiOTf) with a potential of 1.5 V applied to the Pt wire WE. (c) The EIC of m/z 121.0885 as a function of the applied potential. (e) Positive-ion-mode mass spectra of 25 μM flunitrazepam in MeOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) containing 1% formic acid with a potential of 0 V (top panel) and −1.5 V (bottom panel) applied to the Pt wire WE. Insets show the zoomed-in spectra of m/z 302.1295 and 286.1348 differentiated from an isotopic peak of other species (labeled with an asterisk). (f) The measured current, and (g) ion signals of m/z 314.0935, 298.0985, 300.1141, 284.1192, 302.1295, and 286.1348 versus potential during the CV scanning from 0 V to −1.5 V at a scan rate of 20 mV s−1. The left panel shows the zoomed-in EICs in the time range of 0–1.2 min. The voltages applied to the WE in this work were all versus the Ag wire QRE unless otherwise specified. :1, v/v) containing 1% formic acid with a potential of 0 V (top panel) and −1.5 V (bottom panel) applied to the Pt wire WE. Insets show the zoomed-in spectra of m/z 302.1295 and 286.1348 differentiated from an isotopic peak of other species (labeled with an asterisk). (f) The measured current, and (g) ion signals of m/z 314.0935, 298.0985, 300.1141, 284.1192, 302.1295, and 286.1348 versus potential during the CV scanning from 0 V to −1.5 V at a scan rate of 20 mV s−1. The left panel shows the zoomed-in EICs in the time range of 0–1.2 min. The voltages applied to the WE in this work were all versus the Ag wire QRE unless otherwise specified. | ||
Different from most EC-MS approaches solely focusing on capturing unproven intermediates, the presented RT-Triplex also aims to deconstruct multi-step electrochemical processes by following the molecular information at the EEI in each step. The electroreduction of flunitrazepam experiences a step-by-step reduction process from the nitro group to nitroso, N-hydroxyl, and eventually amino groups (Fig. 2d).33,34 Repeated potential sweeps between 0 V and −1.5 V were applied to the WE while recording the current and ion signals. As shown in Fig. 2e, upon lowering the potential to −1.5 V, all the intermediates F2–F5 (m/z 298.0985, 300.1141, 284.1192, and 302.1295) and final product F6 (m/z 286.1348) depicted in Fig. 2d were unambiguously observed. Compared with conventional CV measurements which monitor multi-step electrochemical processes using I–E curves (Fig. 2f, S5 and S28†),35,36 our method can offer a more sensitive and specific means to distinguish the onset potential of every step by recording the emergence of the product ion signal from zero background. As illustrated in Fig. 2g, the six-plex ion signal output gave exact onset potential (vs. Ag wire QRE) of the five intermediates and products (F2: −0.38 V; F3: −0.38 V; F4: −0.42 V; F5: −0.72 V; and F6: −0.76 V). In this context, the RT-Triplex platform possesses an excellent ability to resolve multi-step EC reactions within the potential range by characteristic ion signals, laying a technical foundation for correlating luminescence with crucial intermediates in ECL pathways.
Real-time monitoring and mechanistic study of the ECL reaction of luminol
Having demonstrated the capability of identifying short-lived intermediates and resolving multi-step EC systems in real time, we turned to exploring ECL processes by using RT-Triplex. Luminol is one of the most classic ECL systems, and its mechanism has been extensively studied by using electrochemical approaches and also referring to the understanding of chemiluminescence.7,10,37–41 However, few reports provided direct molecular evidence of the proposed intermediates or validated their roles in ECL processes at the molecular level. As compared to the tendency to form dimers in an acidic medium (Fig. S6†),42 the widely accepted ECL mechanism of luminol in aqueous alkaline solution is summarized in Fig. 3a.10,39,43 Deprotonated luminol (LH−) first undergoes one-electron, one-proton oxidation to form a luminol radical (L˙−), followed by an ultrafast step of oxidation to diazaquinone (L). Further oxidation of L in the presence of OH−, O2, or H2O2 leads to the generation of an excited 3-aminophthalate anion (AP−*), which then emits light on relaxation to complete the ECL process. The key intermediate α-hydroxy hydroperoxide or luminol endoperoxide (both denoted as L−OOH) was hypothesized to experience N2 elimination and then excitation activated by peroxide bond breaking (specifically for the endoperoxide intermediate)41,44 to create AP−*, thus playing an essential role in the progress of this reaction.10,41,43,45,46 | ||
| Fig. 3 Mechanistic study of the ECL reaction of luminol by using the RT-Triplex. (a) Proposed mechanisms for the ECL reaction of luminol at pH ≥ 7 and electrochemical dimerization of luminol at pH ≤ 7. The isomers of the dimer and oxidized dimer of luminol are shown in the ESI.† The structure of L−OOH is drawn according to one of the isomers, α-hydroxy hydroperoxide. (b) Negative-ion-mode mass spectra of 1 mM luminol in ACN/H2O (1:1, v/v) containing 10 mM ammonium acetate with a potential of 0 V and 1.5 V applied to the Pt wire WE at pH 7 and 11. (c) The applied potential and measured current, (d) ECL–time curve, and (e) EICs of dehydrated AP− (AP−–H2O, m/z 162.0195), deprotonated L (m/z 174.0308), dehydrated L−OOH (L−OOH–H2O, m/z 190.0255), L−OH (m/z 192.0255), L−OOH (m/z 208.0358), the deprotonated dimer (m/z 351.0847), and the oxidized dimer (m/z 349.0689) along with the potential switched between 0 V and 1.5 V. (f) Radar chart of the ion signal intensity ratio of AP−–H2O (m/z 162.0195), deprotonated L (m/z 174.0308), L−OOH–H2O (m/z 190.0255), L−OH (m/z 192.0255), L−OOH (m/z 208.0358), the deprotonated dimer (m/z 351.0847), and the oxidized dimer (m/z 349.0689) to the corresponding deprotonated luminol (m/z 176.0469) correlated with ECL emission at different pH values. The ratio and ECL intensity were the average of the values obtained during three continuous 1.5 V potential pulses in the potential step method. (g) The measured current, (h) ECL intensity and (i) MS signals of characteristic ions versus potential during the CV scanning from 0 V to 1.5 V at a scan rate of 10 mV s−1 at pH 7 and 11. | ||
After excluding the influence of cathode luminescence of luminol (Fig. S7†),47 we next systematically investigated the luminol ECL process by concurrent acquisition of electrical, luminescent, and mass spectrometric signals. In the potential step experiments (Fig. 3b), the deprotonated luminol was observed at m/z 176.0469 in negative ionization mode at an initial potential of 0 V (Fig. 3b and S8†). Upon switching the WE potential to 1.5 V, ion signals at m/z 162.0195, 174.0308, 351.0847, and 349.0689 appeared and were assigned to dehydrated AP− (AP−–H2O), deprotonated L, dimer, and oxidized dimer, as determined using RT-Triplex in the neutral medium (pH = 7) (Fig. 3b), accompanied by a fairly weak ECL (Fig. 3d). By comparison, reactions in an alkaline solution (pH = 11) gave rise to additional peaks at m/z 190.0255, 192.0412, and 208.0358 corresponding to dehydrated L−OOH (L−OOH–H2O), L−OH, and L−OOH in the mass spectrum, with ECL emission markedly intensified in the meantime (Fig. 3d). The above intermediates and products were identified by the comparison with their theoretical masses and MS/MS spectra (Table S3 and Fig. S9–S14†). Of note, the ion signal increases of these newly formed species (dehydrated L−OOH, L−OH, and L−OOH) were all synchronized with the elevated ECL intensity in the alkaline medium, thus proving the pivotal role of L−OOH in the ECL behavior (Fig. 3e). Although AP−–H2O and L were also found at pH 7, the ECL cascade reaction was greatly suppressed due to the less formed LH− and AP−* caused by OH− deficiency (Fig. 3e). Another striking difference between acidic and basic conditions lies in the dimerization of luminol that prefers to occur at low pH but slows down as pH increases, as evidenced by the substantial MS signals of the dimer and oxidized dimer at pH ≤ 7 (Fig. 3e and S15–S17†). The radar chart further depicts the variation of luminol-derived species and ECL intensity in the media of different pH (Fig. 3f). The ECL intensity and ion signals of m/z 190.0255 and 192.0412 simultaneously peaked at pH 11, reflecting that the formation of L−OOH in alkaline solvent highly contributes to the luminol ECL emission. In contrast, considerable amounts of luminol molecules are apt to dimerize in neutral or acidic solutions and hinder the ECL pathways, consistent with the previous finding that high pH is necessary for luminol ECL.39,43,48
To further unscramble the ECL behavior in details of what active species are generated to elicit the ECL process at what potential, CV measurement between 0 V and 1.5 V was carried out while recording the potential-dependent MS signal and ECL intensity by RT-Triplex (Fig. 3g). Similar to observations in the potential step method, the neutral environment brought about negligible ECL emission and almost no ion signal of L−OOH and its derivatives (Fig. 3h and i). On the contrary, in the pH 11 medium those L−OOH-related characteristic ions became detectable when the potential climbed to 0.96 V (the potential was calibrated to be 0.94 V vs. the saturated calomel electrode (SCE) using the ferrocene/ferrocenium couple). Because the well-known electrochemical conversion of OH− to O2 occurs at around 1.0 V vs. SCE,39,49,50 the above results thereby verified the pathway P1 that OH−-derived oxygen takes charge of transforming the intermediates LH˙ and L˙− to L−OOH. However, the generation of L−OOH was inconsistent on the timeline with ECL emission, which started to increase earlier at a potential of 0.30 V, implying that another route governs the ECL other than the L−OOH-mediated pathway P1. We also noticed that only the intermediate L among the ions of interest exhibits an onset potential at around 0.30 V. By ruling out the influence of dissolved oxygen (Fig. S18†), we deduced that the direct electrochemical oxidation of L to AP−* with the assistance of OH− (P2) may account for the ECL emission between 0.30 V and 0.96 V. In this way, concrete molecular information for the two previously speculated pathways of luminol ECL can be presented by this electricity-luminescence-mass triplex monitoring method.
In addition, electrochemical impedance spectroscopy characterization of the proposed RT-Triplex was also performed in the solvent of the luminol ECL system, ACN/H2O (1:1, v/v) containing 10 mM ammonium acetate. As shown in Fig. S19 and S20,† the extension of the Pt wire WE toward the MS inlet and relatively low concentration of supporting electrolyte to some extent contributed to ohmic resistance. However, as reflected by the instantly responsive mass spectrometric signals, these factors had little effect on the capability of the RT-Triplex platform to capture and monitor electrochemically generated species at the EEI in the ECL system.
Mechanistic insights into the EC and ECL processes of BODIPY
Encouraged by the success in verifying the anodic ECL mechanism of luminol, we next turned to exploring other under-researched ECL systems by using RT-Triplex. BODIPY dyes are a class of fluorescent compounds with unique spectroscopic and electrochemical properties.51,52 Bard et al. have described the EC and ECL properties of BODIPY with various substitution patterns by CV,53,54 but the details on chemical transformation at the EEI remain elusive. In line with Bard's conclusion, the radical cation BODIPY˙+ was unambiguously captured by this RT-Triplex platform with ion signals at m/z 318.2081 and 262.1441 respectively for electrochemical oxidation of BODIPY into B1 and B2 (Fig. 4a, b, d, e and S21†). Interestingly, extra peaks at m/z 317.2004 and 261.1362 were also observed with a voltage-dependent signal response identical to BODIPY˙+ (Fig. 4b, c, e and f). These species were speculated to be the dehydrogenated products B1b and B2b that contain a double bond in one of the alkyl substituents (Fig. 4a, d and S22†). Since radical ions with unsubstituted positions tend to undergo dimerization,55 the dimerized B2 was also readily tracked by our method in real time (Fig. S23†). MS/MS spectra of the radical cations and other participants in the electrooxidation of BODIPY are all summarized in Fig. S24.†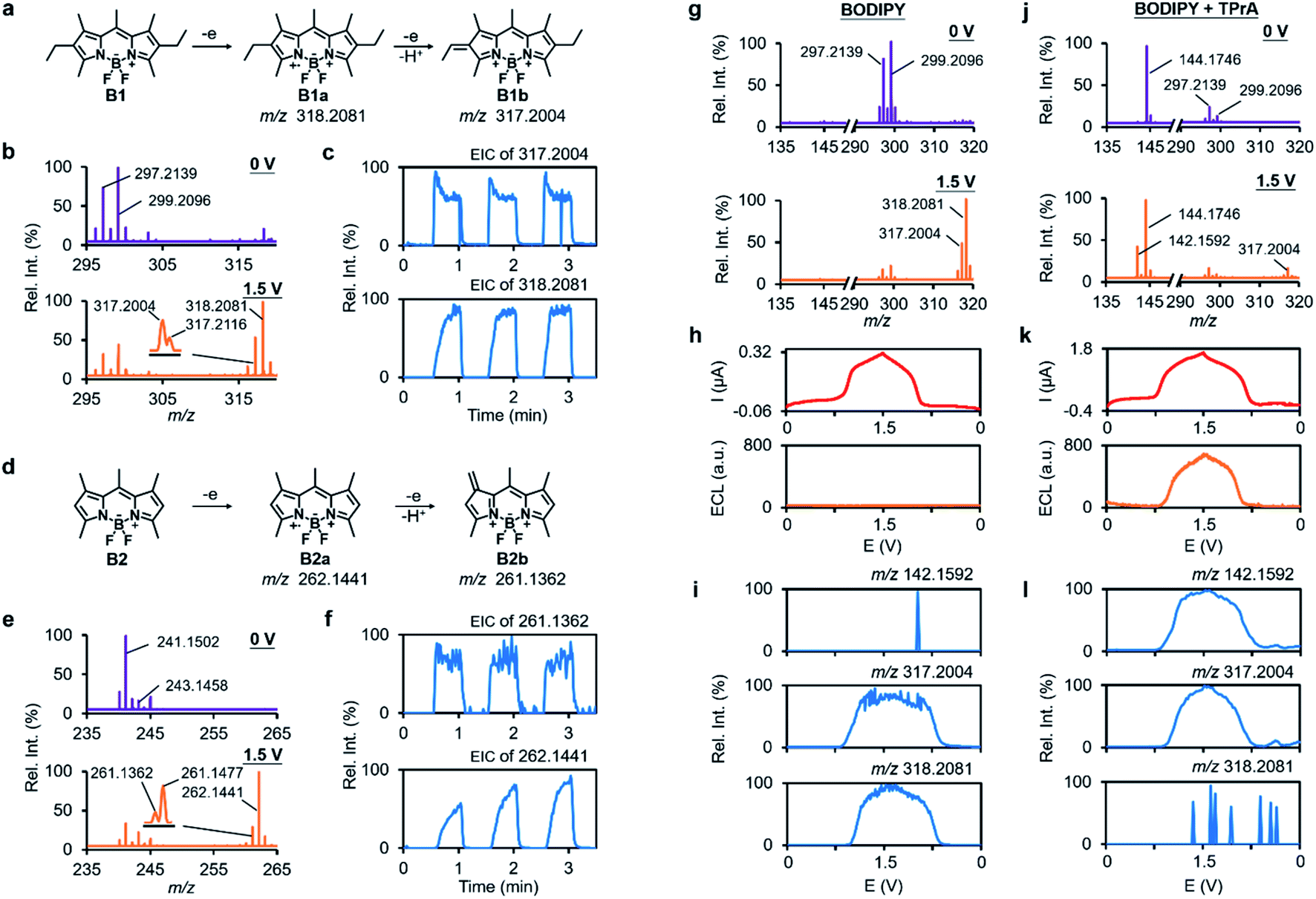 | ||
| Fig. 4 Dissection of EC and ECL processes of BODIPY by using the RT-Triplex. Proposed pathways for electrooxidation of (a) BODIPY B1 and (d) BODIPY B2. Positive-ion-mode mass spectra of 250 μM (b) B1 and (e) B2 in ACN containing 1 mM LiOTf with 0 V and 1.5 V applied to the Pt wire WE. Insets show the zoomed-in spectra. The EICs of (c) m/z 317.2004 and 318.2081 for B1, and (f) m/z 261.1362 and 262.1441 for B2 as the potential was switched between 0 V and 1.5 V. Positive-ion-mode mass spectra of 250 μM BODIPY B1 in the (g) absence or (j) presence of 25 μM TPrA with 0 V and 1.5 V applied to the Pt wire WE. (h) and (k) The measured current and ECL intensity and (i) and (l) ion signals of m/z 144.1746, 317.2004 and 318.2081 versus potential in the B1 system during the CV scanning from 0 V to 1.5 V at a scan rate of 20 mV s−1 in the (h) and (i) absence or (k) and (l) presence of TPrA. | ||
Bard and other researchers pioneered the study on the ECL properties of BODIPY in terms of annihilation and co-reactant routes,53,56 and here we intend to deepen the understanding of the tri-n-propylamine (TPrA)-involved ECL mechanism of BODIPY by our method. According to the previous knowledge of the BODIPY/TPrA system, both BODIPY and TPrA are supposed to be oxidized on the WE to generate BODIPY˙+ and TPrA˙+, among which TPrA˙+ becomes highly reductive TPrA˙ via deprotonation and then reacts with BODIPY˙+ to produce the excited BODIPY (BODIPY*) (Fig. 5a, black route).11,57 As detected in the luminescence channel during potential sweep, the introduction of TPrA led to apparent ECL emission in comparison with the system merely containing B1 (Fig. 4h and k). Real-time MS interrogation also showed that an extra peak at m/z 142.1592 assigned to [Pr2N = CHEt]+ from electrooxidation of TPrA popped up when the anodic potential was ramped to 0.76 V (Fig. 4g and j), which complied with a dynamic curve similar to that of luminescence (onset potential measured at 0.79 V, Fig. 4k and l). Along with the emerging [Pr2N = CHEt]+, the ion signal of BODIPY˙+ showed a fast attenuation (Fig. 4j, l and S25†). The same experimental results were observed in the B2 system (Fig. S26†). All this molecular evidence indicates that BODIPY˙+ is the vital intermediate to react with the co-reactant TPrA in transition to BODIPY* for ECL emission.
The next question is how TPrA participates in the chemistry that creates BODIPY* for luminescence. To clarify this issue, we modified our device by inserting another capillary into the reservoir near the end of the capillary microreactor for feeding the co-reactant TPrA freely (Fig. 5b), by which the electrochemical oxidation of BODIPY and further reaction with TPrA could be spatiotemporally separated. As shown in Fig. 5c and d, rapid infusion of TPrA (10 μL, 1 mM) only gave rise to its own protonated signal in the B1 system without voltage applied, while no significant ECL enhancement was detected. By contrast, feeding TPrA immediately after switching off the voltage applied to the WE (1.5 V) resulted in a distinct ECL spike (Fig. 5f), meaning that the BODIPY˙+-involved ECL reaction took place within this short time window.
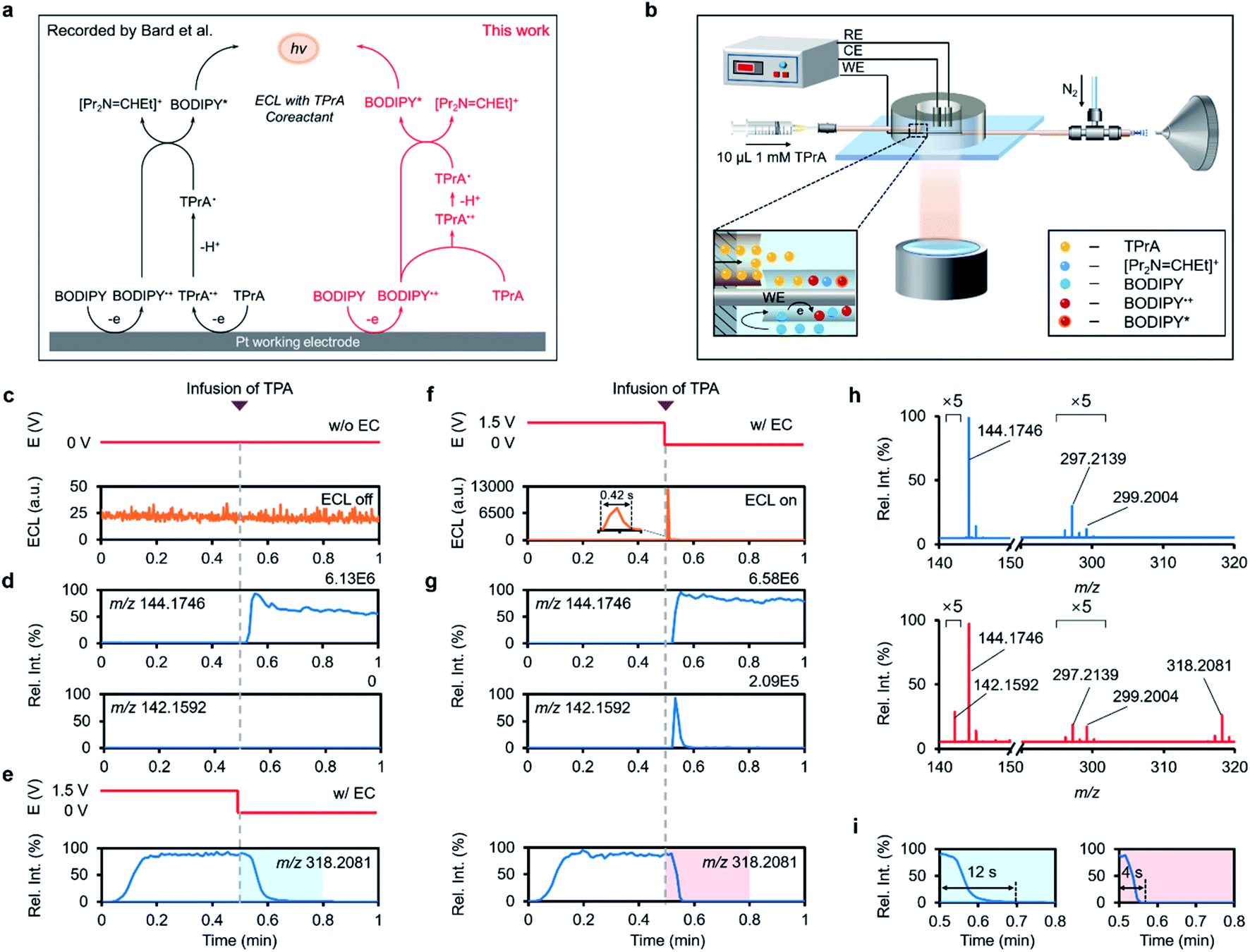 | ||
| Fig. 5 “Catalytic route” of the ECL reaction between BODIPY and TPrA. (a) The mechanism for the ECL reaction of the BODIPY/TPrA system proposed by previous studies (black pathway) and this work (red pathway). (b) Schematic of a modified RT-Triplex setup that allows for post-EC introduction of the co-reactant TPrA. (c) and (f) The applied potential, ECL–time curve and (d) and (g) EICs of characteristic ions before and after infusion of 10 μL 1 mM TPrA into ACN solution containing 250 μM BODIPY B1 and 1 mM LiOTf (c) and (d) while applying no voltage, or (f) and (g) immediately after switching the voltage from 1.5 V to 0 V. (e) The applied potential and EIC of m/z 318.2081 before and after infusion of blank solution after switching off the WE voltage. (h) Positive-ion-mode mass spectra of BODIPY B1 in ACN after injection of TPrA while applying no voltage (top), or immediately after switching off WE voltage (bottom). (i) Zoomed-in EICs of m/z 318.2081 after infusion of blank solution and TPrA immediately after switching off WE voltage at 0.5 min. | ||
To deliberate this interesting phenomenon, we turned to the channel for monitoring the dynamics of BODIPY˙+ around the inflection point of voltage and found that there was a carry-over effect of BODIPY˙+ lasting for 12 s in the absence of TPrA (Fig. 5e and i). This should be attributed to the high stability of fully substituted BODIPY˙+ and its adsorption on the electrode surface during anodization.56,58 By comparison, TPrA injection largely reduced the tailing of BODIPY˙+ to 4 s, along with a sharp peak in the EIC of m/z 142.1592 (Fig. 5g and i). The sudden increase in [Pr2N = CHEt]+ at the EEI was again verified in the mass spectra (Fig. 5h). As a control, neither ECL nor [Pr2N = CHEt]+ was generated in the system that excludes BODIPY from the electrolyte (Fig. S27 and S28†). Conclusively, the above discovered consumption of BODIPY˙+ in parallel with a surge of [Pr2N = CHEt]+ and a flash of light emission in this time frame indicated that the luminescence exclusively originated from the physical mixing of BODIPY˙+ and TPrA. These results mean that the BODIPY˙+-dominated ECL reaction could still occur even if TPrA was not pre-oxidized by anodization, motivating us to consider the likelihood that the stable BODIPY˙+ could solely oxidize TPrA to TPrA˙+ for relaying the ECL process.
With further evidence from the CVs of BODIPY and TPrA (Fig. S29†), an ECL “catalytic route” for the BODIPY/TPrA system is proposed in Fig. 5a (red pathway). In this pathway, the BODIPY˙+ newly produced on the WE diffused into bulk solution to initiate homogenous oxidation of TPrA. The resulting TPrA˙+ further loses a proton to transform into strong reducing TPrA˙, which subsequently converges with the remaining BODIPY˙+ to produce BODIPY* for light emission. Despite the similar “catalytic route” presented for other inorganic metal complex luminophores (e.g. Ru(II), Pt(II), and Ir(III) complexes) through the cycle of metal valence change,7,59,60 this mechanism for the BODIPY/TPrA ECL reaction appears not to have been previously recognized, and we have substantiated it with electrical, luminescence, and mass spectrometric triplex measurements. In this pathway, BODIPY both acts as a catalyst to oxidize the co-reactant and also as a luminophore to emit light, which affords the opportunity to develop an ECL cell imaging strategy based on the intracellular bio-co-reactants and electrochemically generated BODIPY˙+.5,61
Conclusions
In summary, we have developed an integrated mass spectrometric platform, RT-Triplex, to gain comprehensive insights into EC/ECL reactions by real-time acquisition and synchronization of electrical, luminescent, and mass spectrometric signals. We demonstrated the excellent ability of RT-Triplex in the capture of fleeting radicals at the EEI and potential-resolved molecular characterization of multi-step EC processes. Based on global interrogation of multiple intermediates and real-time monitoring of the time-varying ECL process in triple signal channels, two ECL pathways of luminol are validated by tracking key intermediates along with luminescence. More importantly, a “catalytic route” for the ECL reaction between BODIPY and TPrA is proposed and established by thoroughly unfolding the dynamic molecular information on all reactants, intermediates, and products. Taken together, our work creates a multi-dimensional operando analytical tool for in-depth understanding of ECL processes by precisely correlating molecular variation with luminescence events. We envision that this real-time hyphenated mass spectrometry technique, which we call RT-Triplex, also holds great promise in revealing other dynamic interfacial processes featuring electron transfer, chemical transformation, and light adsorption/emission (e.g. electrocatalysis,62 photoelectrocatalysis,63 synthetic photoelectrochemistry,64etc.) for catalyst screening and mechanistic investigations.Author contributions
Q. M. and J.-J. Z. conceived and directed the project. Q. M. and X. Z. designed and built the RT-Triplex. X. Z., W. L., and T. W. performed the experiment and gathered experimental data. X. Z. analyzed and interpreted the experimental data. X. Z. and Q. M. co-wrote the paper. Q. M., R. N. Z., and C. M. corrected the manuscript, finalized the manuscript draft, and guided the revision. All authors contributed to the discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (21974062 and 92053102), the Fundamental Research Funds for the Central Universities (020514380255), and the program B for Outstanding Ph.D. candidate of Nanjing University (202102B015). C. M. is also grateful to the National Natural Science Foundation of China (21904063) and the Natural Science Foundation of Jiangsu Province (BK20190279). We also sincerely thank Prof. Lingling Li from Nanjing Medical University for helpful discussion and suggestions.Notes and references
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS PubMed.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS PubMed.
- Y. Wang, G. Zhao, H. Chi, S. Yang, Q. Niu, D. Wu, W. Cao, T. Li, H. Ma and Q. Wei, J. Am. Chem. Soc., 2021, 143, 504–512 CrossRef CAS PubMed.
- W. Guo, H. Ding, C. Gu, Y. Liu, X. Jiang, B. Su and Y. Shao, J. Am. Chem. Soc., 2018, 140, 15904–15915 CrossRef CAS PubMed.
- C. Ma, S. Wu, Y. Zhou, H.-F. Wei, J. Zhang, Z. Chen, J.-J. Zhu, Y. Lin and W. Zhu, Angew. Chem., Int. Ed., 2021, 60, 4907–4914 CrossRef CAS PubMed.
- H. Ding, P. Zhou, W. Fu, L. Ding, W. Guo and B. Su, Angew. Chem., Int. Ed., 2021, 60, 11769–11773 CrossRef CAS PubMed.
- W. Miao, J.-P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 CrossRef CAS PubMed.
- T. Kai, M. Zhou, S. Johnson, H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2018, 140, 16178–16183 CrossRef CAS PubMed.
- A. Zanut, A. Fiorani, S. Canola, T. Saito, N. Ziebart, S. Rapino, S. Rebeccani, A. Barbon, T. Irie, H.-P. Josel, F. Negri, M. Marcaccio, M. Windfuhr, K. Imai, G. Valenti and F. Paolucci, Nat. Commun., 2020, 11, 2668 CrossRef CAS PubMed.
- Irkham, R. R. Rais, T. A. Ivandini, A. Fiorani and Y. Einaga, Anal. Chem., 2021, 93, 2336–2341 CrossRef CAS PubMed.
- M. Hesari, J. S. Lu, S. Wang and Z. Ding, Chem. Commun., 2015, 51, 1081–1084 RSC.
- Y. Wang, W. Guo, Q. Yang and B. Su, J. Am. Chem. Soc., 2020, 142, 1222–1226 CrossRef CAS PubMed.
- S. Chen, H. Ma, J. W. Padelford, W. Qinchen, W. Yu, S. Wang, M. Zhu and G. Wang, J. Am. Chem. Soc., 2019, 141, 9603–9609 CrossRef CAS PubMed.
- M. Hesari, K. N. Swanick, J. S. Lu, R. Whyte, S. Wang and Z. Ding, J. Am. Chem. Soc., 2015, 137, 11266–11269 CrossRef CAS PubMed.
- J. Rodríguez-López, M. Shen, A. B. Nepomnyashchii and A. J. Bard, J. Am. Chem. Soc., 2012, 134, 9240–9250 CrossRef PubMed.
- W. Wen, S. Yu, C. Zhou, H. Ma, Z. Zhou, C. Cao, J. Yang, M. Xu, F. Qi, G. Zhang and Y. Pan, Angew. Chem., Int. Ed., 2020, 59, 4873–4878 CrossRef CAS PubMed.
- S. A. Miller and V. Bandarian, J. Am. Chem. Soc., 2019, 141, 11019–11026 CrossRef CAS PubMed.
- C. Gu, X. Nie, J. Jiang, Z. Chen, Y. Dong, X. Zhang, J. Liu, Z. Yu, Z. Zhu, J. Liu, X. Liu and Y. Shao, J. Am. Chem. Soc., 2019, 141, 13212–13221 CrossRef CAS PubMed.
- T. A. Brown, H. Chen and R. N. Zare, J. Am. Chem. Soc., 2015, 137, 7274–7277 CrossRef CAS PubMed.
- B. Hasa, M. Jouny, B. H. Ko, B. Xu and F. Jiao, Angew. Chem., Int. Ed., 2020, 60, 3277–3282 CrossRef PubMed.
- T. Herl and F.-M. Matysik, Anal. Chem., 2020, 92, 6374–6381 CrossRef CAS PubMed.
- S. Moller, S. Barwe, J. Masa, D. Wintrich, S. Seisel, H. Baltruschat and W. Schuhmann, Angew. Chem., Int. Ed., 2019, 59, 1585–1589 CrossRef PubMed.
- S. Tang, H. Cheng and X. Yan, Angew. Chem., Int. Ed., 2020, 59, 209–214 CrossRef CAS PubMed.
- A. T. Blades, M. G. Ikonomou and P. Kebarle, Anal. Chem., 1991, 63, 2109–2114 CrossRef CAS.
- T. A. Brown, H. Chen and R. N. Zare, Angew. Chem., Int. Ed., 2015, 54, 11183–11185 CrossRef CAS PubMed.
- J. Hu, N. Zhang, P.-K. Zhang, Y. Chen, X.-H. Xia, H.-Y. Chen and J.-J. Xu, Angew. Chem., Int. Ed., 2020, 59, 18244–18248 CrossRef CAS PubMed.
- J. Liu, K. Yu, H. Zhang, J. He, J. Jiang and H. Luo, Chem. Sci., 2021, 12, 9494–9499 RSC.
- R. Qiu, X. Zhang, H. Luo and Y. Shao, Chem. Sci., 2016, 7, 6684–6688 RSC.
- Z. Liu, W. Qi and G. Xu, Chem. Soc. Rev., 2015, 44, 3117–3142 RSC.
- W. Guo, P. Zhou, L. Sun, H. Ding and B. Su, Angew. Chem., Int. Ed., 2021, 60, 2089–2093 CrossRef CAS PubMed.
- J. Hu, T. Wang, W.-J. Zhang, H. Hao, Q. Yu, H. Gao, N. Zhang, Y. Chen, X.-H. Xia, H.-Y. Chen and J.-J. Xu, Angew. Chem., Int. Ed., 2021, 60, 18494–18498 CrossRef CAS PubMed.
- F. Cao, J. Kim and A. J. Bard, J. Am. Chem. Soc., 2014, 136, 18163–18169 CrossRef CAS PubMed.
- P. Liu, I. T. Lanekoff, J. Laskin, H. D. Dewald and H. Chen, Anal. Chem., 2012, 84, 5737–5743 CrossRef CAS PubMed.
- M. Lu, C. Wolff, W. Cui and H. Chen, Anal. Bioanal. Chem., 2012, 403, 355–365 CrossRef CAS PubMed.
- K. Liu, S. Tang, T. Wu, S. Wang, M. Zou, H. Cong and A. Lei, Nat. Commun., 2019, 10, 639 CrossRef PubMed.
- C. Sandford, L. R. Fries, T. E. Ball, S. D. Minteer and M. S. Sigman, J. Am. Chem. Soc., 2019, 141, 18877–18889 CrossRef CAS PubMed.
- E. H. White, O. Zafiriou, H. H. Kagi and J. H. M. Hill, J. Am. Chem. Soc., 1964, 86, 940–941 CrossRef CAS.
- G. Merenyi and J. S. Lind, J. Am. Chem. Soc., 1980, 102, 5830–5835 CrossRef CAS.
- H. Cui, G.-Z. Zou and X.-Q. Lin, Anal. Chem., 2003, 75, 324–331 CrossRef CAS PubMed.
- C. M. Wang and H. Cui, Luminescence, 2007, 22, 35–45 CrossRef CAS PubMed.
- A. Giussani, P. Farahani, D. Martinez-Munoz, M. Lundberg, R. Lindh and D. Roca-Sanjuan, Chem.–Eur. J., 2019, 25, 5202–5213 CrossRef CAS PubMed.
- J. N'Diaye and K. Lian, J. Electroanal. Chem., 2019, 839, 90–95 CrossRef.
- H. Cui, Y. Xu and Z.-F. Zhang, Anal. Chem., 2004, 76, 4002–4010 CrossRef CAS PubMed.
- J. Michl, Photochem. Photobiol., 1977, 25, 141–154 CrossRef CAS.
- A. L. Rose and T. D. Waite, Anal. Chem., 2001, 73, 5909–5920 CrossRef CAS PubMed.
- J. Lind, G. Merenyi and T. E. Eriksen, J. Am. Chem. Soc., 1983, 105, 7655–7661 CrossRef CAS.
- G. Zhang, H. Chai, M. Tian, S. Zhu, L. Qu and X. Zhang, Anal. Chem., 2020, 92, 7354–7362 CrossRef CAS PubMed.
- J. Shu, Z. Han, T. Zheng, D. Du, G. Zou and H. Cui, Anal. Chem., 2017, 89, 12636–12640 CrossRef CAS PubMed.
- X. Shi, S. Back, T. M. Gill, S. Siahrostami and X. Zheng, Chem, 2021, 7, 38–63 CAS.
- X. Shi, S. Siahrostami, G.-L. Li, Y. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. Zheng and J. K. Nørskov, Nat. Commun., 2017, 8, 701 CrossRef PubMed.
- C. S. Wijesooriya, J. A. Peterson, P. Shrestha, E. J. Gehrmann, A. H. Winter and E. A. Smith, Angew. Chem., Int. Ed., 2018, 57, 12685–12689 CrossRef CAS PubMed.
- N. Kaplaneris, J. Son, L. Mendive-Tapia, A. Kopp, N. D. Barth, I. Maksso, M. Vendrell and L. Ackermann, Nat. Commun., 2021, 12, 3389 CrossRef CAS PubMed.
- A. B. Nepomnyashchii, M. Broring, J. Ahrens and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 8633–8645 CrossRef CAS PubMed.
- J. Suk, K. M. Omer, T. Bura, R. Ziessel and A. J. Bard, J. Phys. Chem. C, 2011, 115, 15361–15368 CrossRef CAS.
- A. B. Nepomnyashchii, M. Broring, J. Ahrens and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 19498–19504 CrossRef CAS PubMed.
- A. B. Nepomnyashchii and A. J. Bard, Acc. Chem. Res., 2012, 45, 1844–1853 CrossRef CAS PubMed.
- H. Qi, J. J. Teesdale, R. C. Pupillo, J. Rosenthal and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 13558–13566 CrossRef CAS PubMed.
- J. Fangmeyer, A. Behrens, B. Gleede, S. R. Waldvogel and U. Karst, Angew. Chem., Int. Ed., 2020, 59, 20428–20433 CrossRef CAS PubMed.
- L. Chen, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, L. C. Henderson, T. U. Connell, Y. H. Nai, R. Alexander, S. Carrara, C. F. Hogan, P. S. Donnelly and P. S. Francis, Chem. Sci., 2019, 10, 8654–8667 RSC.
- Z. Chen, K. M.-C. Wong, E. C.-H. Kwok, N. Zhu, Y. Zu and V. W.-W. Yam, Inorg. Chem., 2011, 50, 2125–2132 CrossRef CAS PubMed.
- S. Voci, B. Goudeau, G. Valenti, A. Lesch, M. Jovic, S. Rapino, F. Paolucci, S. Arbault and N. Sojic, J. Am. Chem. Soc., 2018, 140, 14753–14760 CrossRef CAS PubMed.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- X. Chang, T. Wang, P. Yang, G. Zhang and J. Gong, Adv. Mater., 2019, 31, 1804710 CrossRef PubMed.
- J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01317g |
| This journal is © The Royal Society of Chemistry 2022 |