
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the interplay between homogeneous and heterogeneous catalytic mechanisms in copper–iron nanoparticles working under chemically relevant tumour conditions†
Javier
Bonet-Aleta
 abc,
Miguel
Encinas-Gimenez
abc,
Esteban
Urriolabeitia
d,
Pilar
Martin-Duque‡
befg,
Jose L.
Hueso
*abcf and
Jesus
Santamaria
*abcf
abc,
Miguel
Encinas-Gimenez
abc,
Esteban
Urriolabeitia
d,
Pilar
Martin-Duque‡
befg,
Jose L.
Hueso
*abcf and
Jesus
Santamaria
*abcf
aInstitute of Nanoscience and Materials of Aragon (INMA), CSIC-Universidad de Zaragoza, Campus Río Ebro, Edificio I+D, C/Poeta Mariano Esquillor, s/n, 50018, Zaragoza, Spain. E-mail: jlhueso@unizar.es; jesus.santamaria@unizar.es
bNetworking Research Center in Biomaterials, Bioengineering and Nanomedicine (CIBER-BBN), Instituto de Salud Carlos III, 28029, Madrid, Spain
cDepartment of Chemical and Environmental Engineering, University of Zaragoza, Campus Rio Ebro, C/María de Luna, 3, 50018 Zaragoza, Spain
dInstituto de Síntesis Química y Catálisis Homogénea, ISQCH (CSIC-Universidad de Zaragoza), 50009 Zaragoza, Spain
eInstituto Aragonés de Ciencias de la Salud (IACS), Avenida San Juan Bosco, 13, 50009 Zaragoza, Spain
fInstituto de Investigación Sanitaria (IIS) Aragón, Avenida San Juan Bosco, 13, 50009 Zaragoza, Spain
gFundación Araid, Av. de Ranillas 1-D, 50018 Zaragoza, Spain
First published on 8th June 2022
Abstract
The present work sheds light on a generally overlooked issue in the emerging field of bio-orthogonal catalysis within tumour microenvironments (TMEs): the interplay between homogeneous and heterogeneous catalytic processes. In most cases, previous works dealing with nanoparticle-based catalysis in the TME focus on the effects obtained (e.g. tumour cell death) and attribute the results to heterogeneous processes alone. The specific mechanisms are rarely substantiated and, furthermore, the possibility of a significant contribution of homogeneous processes by leached species – and the complexes that they may form with biomolecules – is neither contemplated nor pursued. Herein, we have designed a bimetallic catalyst nanoparticle containing Cu and Fe species and we have been able to describe the whole picture in a more complex scenario where both homogeneous and heterogeneous processes are coupled and fostered under TME relevant chemical conditions. We investigate the preferential leaching of Cu ions in the presence of a TME overexpressed biomolecule such as glutathione (GSH). We demonstrate that these homogeneous processes initiated by the released by Cu–GSH interactions are in fact responsible for the greater part of the cell death effects found (GSH, a scavenger of reactive oxygen species, is depleted and highly active superoxide anions are generated in the same catalytic cycle). The remaining solid CuFe nanoparticle becomes an active catalyst to supply oxygen from oxygen reduced species, such as superoxide anions (by-product from GSH oxidation) and hydrogen peroxide, another species that is enriched in the TME. This activity is essential to sustain the homogeneous catalytic cycle in the oxygen-deprived tumour microenvironment. The combined heterogeneous–homogeneous mechanisms revealed themselves as highly efficient in selectively killing cancer cells, due to their higher GSH levels compared to healthy cell lines.
Introduction
Glutathione (GSH) is a key peptide in the regulation of intracellular reactive oxygen species (ROS) levels. Its role is of paramount importance in the tumour microenvironment (TME),1 where GSH is overexpressed to counteract the overproduction of oxidizing species such as H2O2 that may disrupt redox homeostasis leading to apoptosis.2 GSH counteracts the generation of ROS via enzymatic reaction with the glutathione peroxidase (GPx) enzyme.3 Consequently, GSH is quickly becoming the target of new cancer therapies.1 In addition, the high intratumoral GSH concentrations (up to mM levels4,5) may interfere with emerging cancer therapies (chemodynamic- (CDT), sonodynamic- (SDT), and photodynamic (PDT)) therapy that are ROS-dependent and become less effective in the presence of increased GSH levels.6Nanocatalytic cancer therapy is rapidly emerging as a novel alternative able to trigger selective catalytic reactions in cancer cells to induce their apoptosis.7,8 Transition metal nanocatalysts in particular are able to promote GSH depletion via oxidation mechanisms thereby interfering in the survival and protection mechanisms of cancer cells.7 The ideal scenario involves catalytic materials that can eliminate antioxidant molecules such as GSH, while simultaneously promoting the generation of ROS. It is obvious that a deeper understanding of the role of nanocatalysts in the TME is critical to enhance their efficient action. However, this still represents a formidable challenge: the catalytic mechanisms of the most promising nanoplatforms and their interplay with key biomolecules remains elusive due to the complexity of the interactions in the TME.
A fundamental aspect of the interaction between catalyst nanoparticles (NPs) and the TME that is often overlooked relates to the surface phenomena involved. In particular, leaching (i.e. metal ions lixiviated from the surface of the nanostructured catalysts into the surrounding fluid) is a phenomenon likely to have a strong influence on the final therapy outcome. Previous investigations have aimed at designing nanoplatforms with pH-triggered metal ion lixiviation given the mildly-acidic TME.9 The influence of acidity in metal lixiviation has been explored in several cancer-related works for Fe,10,11 Mn12–14 or Cu.15–20 It has also been shown that the complex chemical composition in biological environments includes molecular species such as amino acids, that may promote lixiviation especially in the case of Cu.21–23 However, the role that these species may play as catalysts and the interactions with heterogeneous processes fostered by the solid phase have not been investigated.
Some valuable insights can be derived from conventional, aqueous phase catalysis. Eremin et al.24 recently pictured an expanded vision of the nature of transition-metal-catalyzed reactions. These authors described the well-established scenarios of (i) molecular-based catalysis and (ii) nanoparticle-based catalysis (heterogeneous catalysis) and presented three additional intermediate scenarios given by (iii) lixiviation-driven catalysis; (iv) “cocktail” of catalysts derived from the nanoparticle (clusters, atoms, lixiviated ions) and (v) dynamic catalytic systems. The action of the catalyst nanoparticles used in medical applications is generally interpreted on the premises of purely heterogeneous mechanisms. Only in a few cases, (e.g. MnO2-based nanomaterials, where dissolution of the nano-oxide structure through reaction with GSH is followed by Fenton processes facilitated by the as-formed Mn2+ ions) an attempt has been made to describe processes closer to lixiviation-driven catalysis. It must be noticed that the nanoplatforms evaluated as catalysts for cancer therapy are usually composed by more than one metal, (e.g. Cu2MoS4,25 MnFe2O4,26 CuFe2O4,27 copper/manganese silicate,17 CuxFeySz (ref. 28) or SrCuSi4O10 (ref. 20)) and these may be affected to a different extent by lixiviation phenomena under the mildly-acidic, hypoxic and GSH-enriched conditions prevalent in the TME. In any case, issues such as the extent of the lixiviation process, the possibility of a preferential leaching of a specific transition metal or the influence of specific chemical species present in the TME remain mostly unexplored. At present, is not possible to state whether the therapeutic action of nanocatalysts in the TME is attributable to lixiviated ions, a purely heterogeneous reaction, or a combination of both phenomena.
Here, we have thoroughly evaluated the catalytic mechanisms behind the action of a copper–iron mixed oxide (CuFe) nanocatalyst under TME representative conditions. This is a bimetallic system, complex enough to illustrate the main phenomena taking place. Our results shed light on the interplay between heterogeneous and homogeneous processes occurring in the presence of GSH. We report for the first time how the presence of elevated levels of GSH induces a preferential lixiviation of Cu species, initiating a homogeneous catalytic cycle that efficiently oxidizes GSH into glutathione disulfide (GSSG), assisted by the in situ formation of Cu–GSH coordination complexes. Simultaneously, the progressively Fe-enriched NP gives rise to heterogeneous catalytic cycles using ROS generated in the homogeneous GSH oxidation cycle (Fig. 1) or the overexpressed H2O2 present in the tumoral media.29,30 The system chosen is of especial interest, considering the increasing relevance of Cu and its interaction with key processes in cancer development.31 Overall, the results presented in this work provide new insight on the dynamics of the chemical reactions inside the TME and valuable clues for the design of more efficient catalysts to operate in this environment.
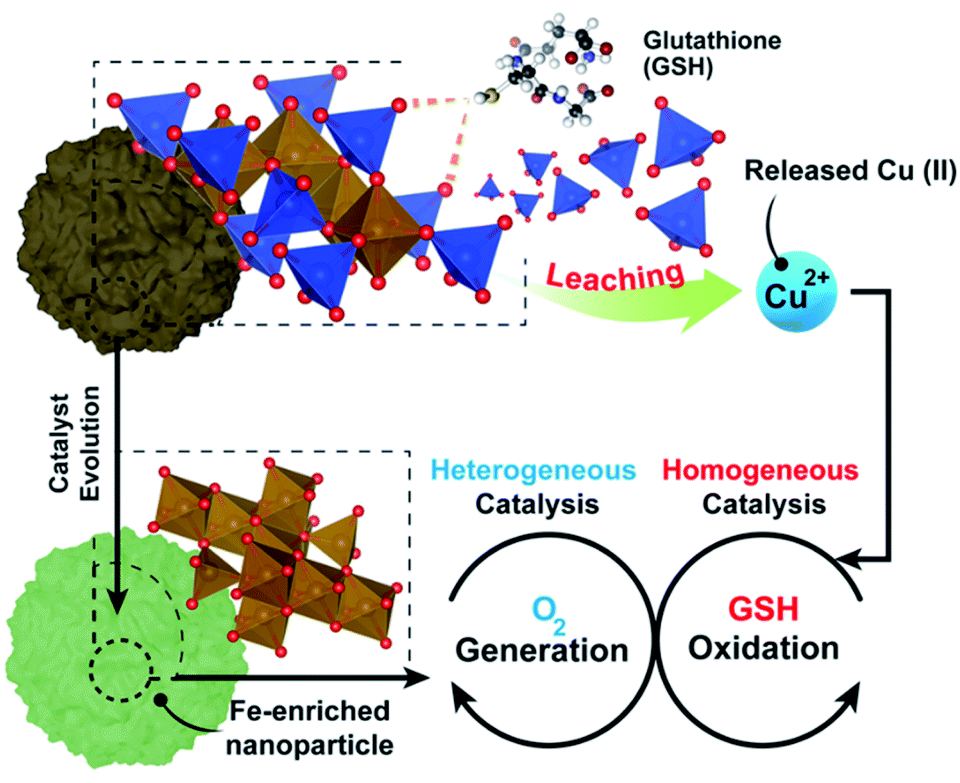 | ||
| Fig. 1 Simplified overview of the homogeneous–heterogeneous processes fostered by the CuFe nanocatalyst in the presence of GSH. After a GSH-triggered Cu release from the nanocatalyst, Cu2+ catalyzes the homogeneous oxidation of GSH into GSSG. Simultaneously, Fe(III) species present on the nanoparticle surface catalyze the conversion of H2O2 and ˙O2− species, considered as by-products from GSH oxidation into O2 necessary to sustain the GSH depletion homogeneous cycle. | ||
Results and discussion
Cation leaching in the presence of GSH
The influence of GSH on the release of ionic Cu and Fe from the CuFe2O4 nanocatalyst was quantified by microwave plasma-atomic emission spectroscopy (see Experimental section for details). A concentration of GSH of 5 mM was selected for the experiments in order to mimic the average intracellular GSH levels reported in the literature for cancer cells.32–35 The presence of GSH at a concentration of 5 mM changed considerably the leaching patterns. Thus, the release of Cu was strongly enhanced, with a cumulative release of ca. 70% of the initial Cu in the particle after 24 h (compared to about 20% in the absence of GSH). On the other hand, while Fe is not expected to be lixiviated significantly at this pH, we found that ca. 30% of the initial Fe content was released from the nanoparticle after 24 h (see Fig. 2a). We attribute this behaviour to the generation of high-energy vacancies after GSH-triggered Cu release that facilitates the transfer of Fe into the solution.36 The fact that Cu release is favored in the presence of GSH is not totally unexpected, as previous works had reported Cu lixiviation from CuO nanoparticles in the presence of amino acids or peptides.21,22 Interestingly, at lower pH values (pH of 5.80, close to the pH in a solid tumour medium) the extent of leaching for both Cu and Fe in the presence of GSH was reduced (Fig. 2b), although the percentage of Cu leached doubles that of iron. Considering the different ionic forms of GSH upon varying the pH (ref. 37) (Fig. 2c), this behaviour can be linked to the stronger nucleophilic character of –SH group from GSH species as pH increases.38 Compared to –SH, thiolate (–S−) form exhibits much stronger nucleophile behaviour and the processes where –S− is involved may occur even at pH values far below thiol pKa,39 which may explain the promotion of Cu leaching at higher pH.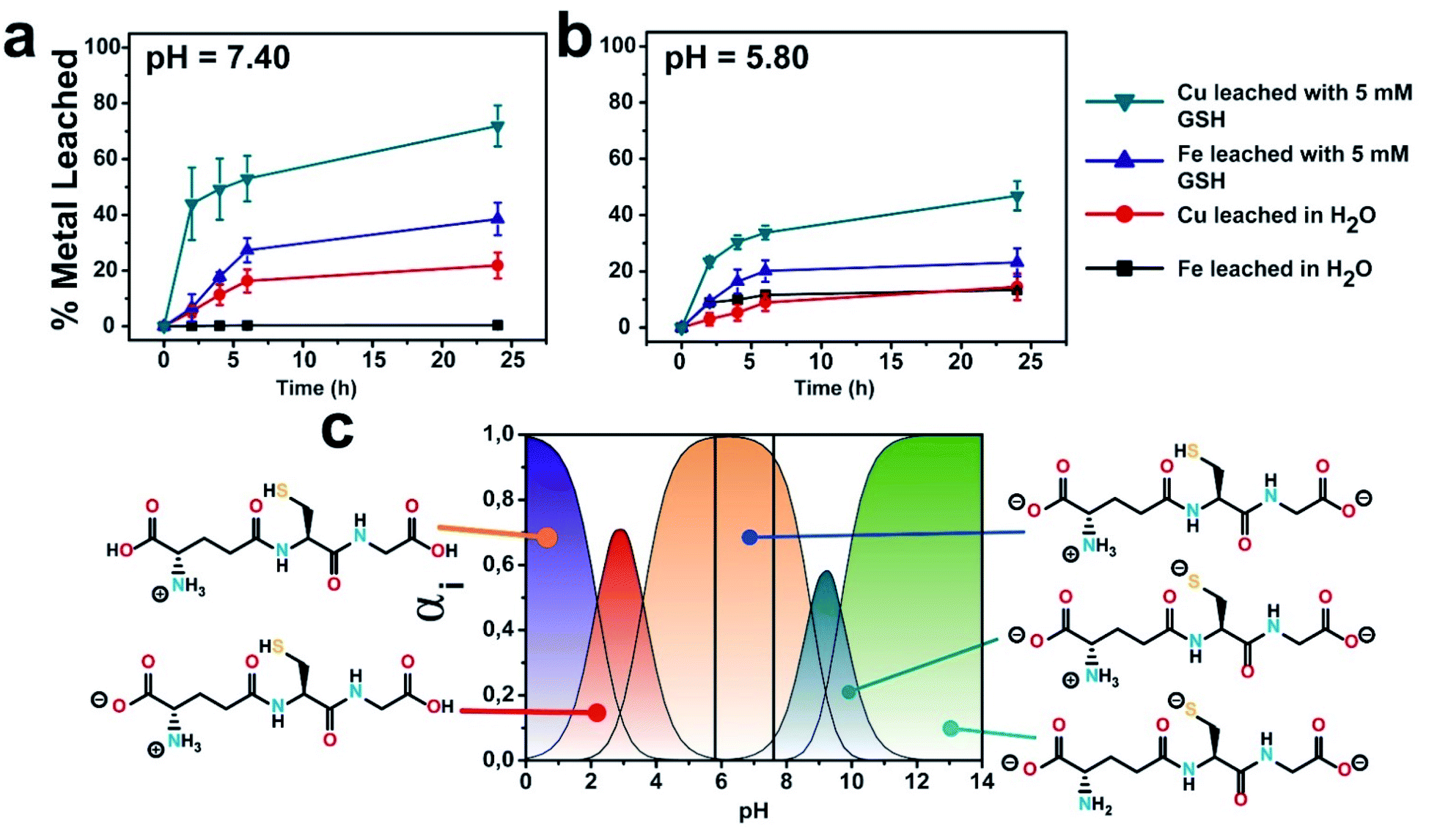 | ||
| Fig. 2 GSH effect on the evolution of copper and iron cations lixiviated at different pH media: (a) pH = 7.4; (b) pH = 5.80; (c) different GSH ionic species as a function of different pH values; vertical lines represent the pH of selected experimental conditions for a better identification of expected GSH species; GSH concentration was set to 5 mM. Speciation diagram was generated using pKa values obtained from.40 | ||
Homogeneous GSH oxidation by cations leached from CuFe nanoparticles
After the abrupt cation release observed in the presence of GSH (Fig. 2a and b) and its preferential action towards the lixiviation of Cu (roughly twice as much Cu is released, compared to Fe), it is reasonable to assume a catalytic scenario mainly composed by aqueous Cu2+ ions and GSH (5 mM). This is relevant because previous works have described a evolution of GSH into its oxidized form (GSSG) catalyzed by Cu2+.41 In this scenario, O2 could act as electron acceptor yielding reduced reactive oxygen species (˙O2− (ref. 42 and 43) and H2O2 (ref. 41)) and GSSG as products, respectively (see Fig. 3a). Encouraged by these perspectives, we explored the possibility of fostering homogeneous oxidation processes using the cations released from our CuFe2O4 nanocatalyst, while retaining the heterogeneous catalytic activity of the nanoparticles themselves.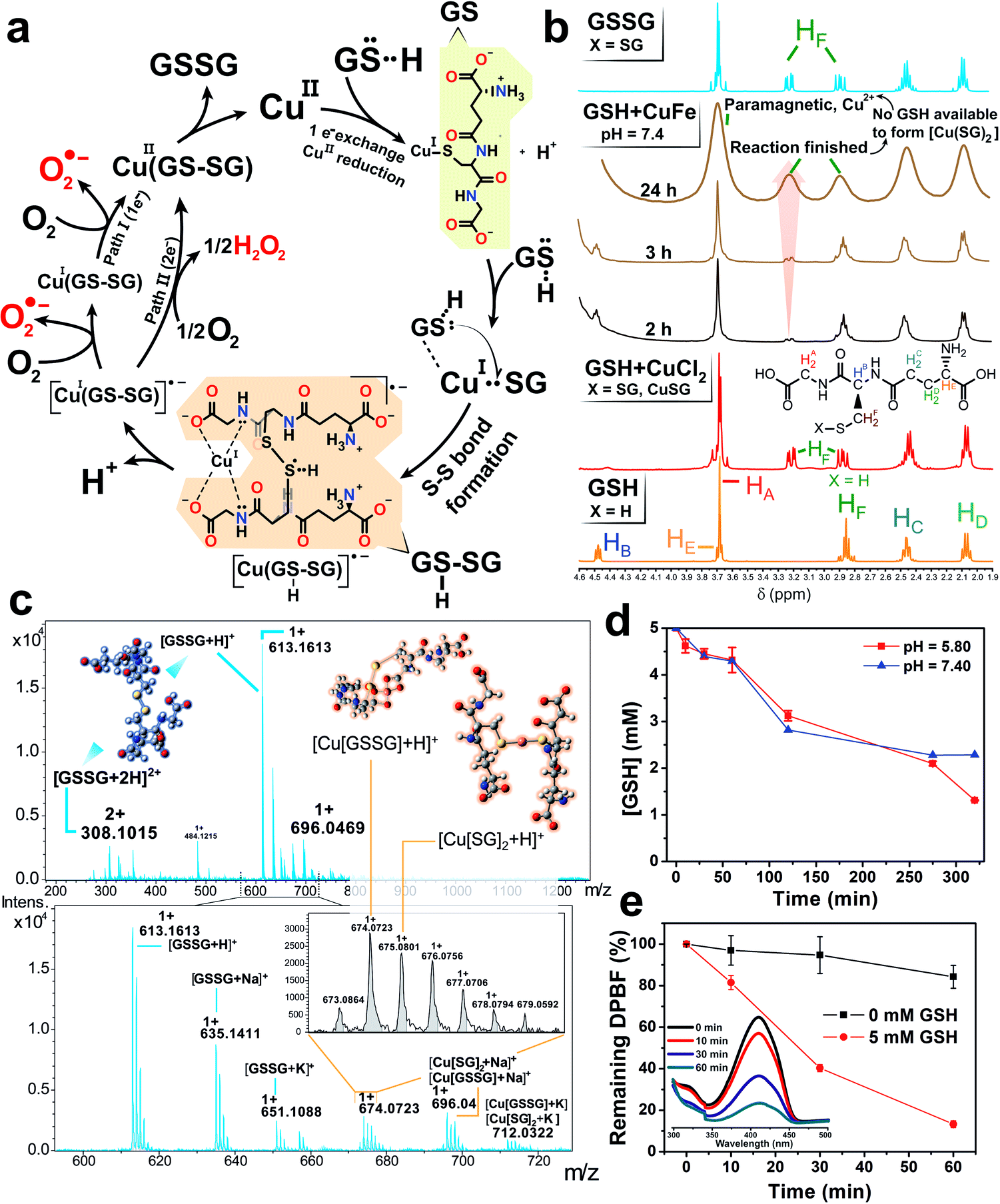 | ||
| Fig. 3 Homogeneous catalysis of ionic Cu and GSH with O2. (a) Proposed homogeneous catalytic cycle for the Cu-assisted GSH oxidation. The reoxidation of Cu(SG)2 to Cu(GSSG) involves a reaction between the Cu(GS–SG)˙− complex and O2, yielding superoxide radical species (˙O2−) as reaction by-product. The shaded areas correspond to the structure of GS and GS(H)SG, which are abbreviated for a better understanding; (b) 1H NMR spectra of GSH, GSSG, GSH + CuCl2 and GSH + CuFe at different reaction times (2, 3 and 24 h) with the corresponding proton assignments. 1H NRM signal at 3.22 ppm implies a chemical modification near –SH group of the native GSH molecule, either [Cu(SG)2]+ or GSSG formation. 1H NMR reaction spectra at 24 h is clearly altered due to paramagnetism induced by free Cu2+ in solution, since the reaction is complete and no GSH is available to coordinate Cu2+; (c) HRMS-ESI from control experiments with CuCl2 + GSH binary mixture in anaerobic conditions to quench the catalytic reaction. Two peaks at m/z = 613.16 and 635.14 corresponding to [GSSG − H]+ and [GSSG + Na]+ confirmed the generation of GSH oxidation product. The catalytic intermediate Cu(SG)2 is detected at m/z = 674 and 675, respectively; (d) evolution of GSH concentration in the presence of the CuFe nanocatalyst at pH 7.40 or 5.80 (adjusted with HCO3−), 37 °C, [GSH]0 = 5 mM; [CuFe] = 0.1 mg mL−1; (e) influence of GSH on the generation of anion superoxide species ˙O2− as side-product of the Cu-catalyzed GSH oxidation. The absorbance of DPBF at a wavelength of 411 nm was used as indirect probe; reaction conditions: pH = 7.40 (adjusted with HCO3−), [GSH]0 = 5 mM, [DPBF]0 = 0.1 mM, [CuFe] = 0.1 mg mL−1. | ||
In order to analyze the potential contribution of each lixiviated metal (Fe, Cu) in the homogeneous catalysis of GSH we performed a series of control experiments using chloride salt precursors (CuCl2 and FeCl3, respectively). We carried out 1H-nuclear magnetic resonance (1H-NMR) analysis of the mixture CuCl2 + GSH (Fig. 3b) to characterize the Cu(SG)2 complex.42–46 These assays were performed in the absence of O2 to prevent the total evolution of the reaction to products. The resulting spectra revealed a splitting of the β-CH2 protons of the Cys residue of GSH, appearing as an unresolved multiplet at 2.86 ppm into a well defined AB spin system at 3.22 and 2.88 ppm, due to the bonding of the S atom to the reduced Cu(I) center.43,47 Other signals remained almost unchanged, suggesting that only the S atom is involved in bonding of GSH to Cu(I). This is in accordance with hard soft acid base theory (HSAB), soft-basic thiol (–SH) groups from GSH tend to bond soft-acidic Cu(I) centers.48
Diffusion-ordered NMR spectroscopy (DOSY) experiments were also performed with GSH, GSSG and the CuCl2 + GSH binary mixture to determine the molecular size of each molecule and complex intermediates present in the solution based on their diffusion coefficients (D, m2 s−1). The diffusion coefficients from the control experiments with GSH and GSSG (Fig. ESI-1a and b†) were adjusted to 3.98 × 10−10 and 2.75 × 10−10 m2 s−1, respectively. Likewise, their corresponding hydrodynamic radii calculated through Stokes–Einstein equation were 0.6 and 0.9 nm. These values are in good agreement with intramolecular distances obtained in X-ray structures for GSH49–51 and GSSG.52,53 DOSY analysis of the CuCl2 + GSH binary mixture yielded a product with a D = 2.75 × 10−10 m2 s−1 (Fig. ESI-2a†). In the presence of Cu2+, the species formed are larger than GSH (Fig. ESI-1a†) but of a similar size in comparison with GSSG (Fig. ESI-1b†). The complex Cu(SG)2 is formed under these conditions,42–46 which exhibit a rather analogous coefficient D with respect to GSSG. Also, we were able to confirm the formation of the Cu complex through the homogeneous catalytic cycle of Fig. 3a using high resolution mass spectroscopy-electroSpray ionization (HRMS-ESI) (Fig. 3c), that allowed identification of peaks corresponding to [GSSG + H]+ (m/z = 613.1613) and [Cu(SG)2 + H]+ (m/z = 675.0801). Ngamchuea et al.41 studied the Cu(II)-mediated GSH catalytic oxidation and suggested a reaction pathway based on kinetic experiments where Cu(SG)2, the same species detected in our control experiments (Fig. 3c) also acted as reaction intermediate.
We also evaluated the potential contribution of Fe species to form complexes with GSH. In this case, control experiments with FeCl3 were carried out at pH = 3.60 to minimize the formation of iron hydroxide species which interfere with NMR measurements and the introduction of species that are not normally present at physiological pH. 1H-NMR and DOSY control experiments with FeCl3 confirmed the formation of [Fe–(SG)x] complexes (Fig. ESI-3a and b†), with proton chemical shifts at 3.04 and 2.76 ppm corresponding to assignments previously reported in the literature.54,55 In addition, HRMS-ESI analysis revealed the formation of [Fe2(SG)2 + H]+ (m/z = 725.0352) and [Fe(SG) + H]+ (m/z = 363.0173) complexes (Fig. ESI-4†).54
Once the formation of complexes with Cu and Fe cations were confirmed, analogous experiments with the CuFe nanocatalyst were subsequently carried out in the presence of GSH. The Cu lixiviated at pH = 7.4 interacted with the excess of GSH like in the control experiments. 1H-NMR and DOSY analysis of the solution (Fig. 3b and ESI-2b†), revealed both the presence of unreacted GSH (D = 4 × 10−10 m2 s−1) and the formation of species with D = 2.70 × 10−10 m2 s−1. Taking into account the control experiments with CuCl2, the formation of the Cu(SG)2 intermediates seemed also very likely in the presence of the CuFe catalyst. The widening on the spectra signals was attributed to different equilibria established between GSH and Cu(SG)2.45,56 The presence of very small amounts of paramagnetic [Fe–(SG)x] complexes cannot be discarded, and could also contribute to the widened signal. MS-ESI analysis of the solution at different reaction times (3–24 h) yielded a mixture of Cu–SG-derived fragments (Fig. ESI-5†) supporting the hypothesis of Cu-SG as reaction intermediate in the catalytic cycle.
GSH levels were monitored via ultra performance liquid chromatography-photo diode array (UPLC-PDA) and revealed an important decrease at pH = 7.40 and 5.80 due to the catalytic activity of the Cu lixiviated by GSH (Fig. 3d). Remarkably, the formation of H2O2 and ˙O2− were also detected in the presence of GSH (Fig. 3e) using 1,3-diphenylisobenzofuran (DPBF) as analytical probe (see also Experimental section and scheme of the reactions in Fig. ESI-6a†).57,58 The specific generation of ˙O2− was evaluated using dihydroethidium (DHE) (see Fig. ESI-6a†).58–61 An enhancement of 20% of the fluorescence signal of oxidized DHE in the presence of 5 mM of GSH was found (Fig. ESI-6b†) in comparison to the control experiment in the absence of catalyst. Considering this set of experimental results we concluded that the simultaneous formation of H2O2 and ˙O2− was taking place during the catalytic GSH oxidation. These findings are also in agreement with other reports available in the literature41,43,45,62 and allowed us to propose a homogeneous catalytic reaction taking place mainly between the Cu2+ cations released from the CuFe catalyst and GSH (reaction step displayed in Fig. 3a): (i) GSH is able to bind and reduce aqueous Cu2+ species into Cu+ through –SH group to form the CuI(SG) intermediate; (ii) a second GSH molecule is able to cleave the Cu–S bond to promote S–S formation through a radical process.39 This step is thermodynamically favoured since S–S bond energy is larger in comparison to Cu–S (429 vs. 285 kJ mol−1, respectively63). Following reported thiol oxidation kinetics,39 we propose the (iii) formation of the radical intermediate [CuI(GSSG)]˙−. O2 acting as electron acceptor withdraws one/two electron from this disulfide radical anion (Fig. ESI-7†) (iv) to yield the superoxide anion ˙O2− (path I, one electron) or H2O2 (path II, two electrons) that we have been able to detect together with CuI(GSSG). After this fast electron transfer, (v) the CuI center rapidly oxidizes into CuII in the presence of O2.45 (vi) The catalytic cycle is restored after GSSG is detached from the coordination sphere of CuII and replaced by a fresh GSH molecule. In the presence of a GSH excess, the CuII(GSSG) complex exchanges GSSG by GSH to restart the catalytic cycle.45 This process is also thermodynamically favoured, since the formation constant (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K′) of Cu(SG)2 is significantly higher (26.6)64 than Cu(GSSG) (3.63).65 The appearance of the Cu(SG)2 complex is also favored by pH values typically met in a tumour microenvironment (Fig. ESI-8 and ESI-9†). Although the lixiviation of Cu triggered by GSH proceeds at a slower pace at pH = 5.80 (see Fig. 2b), the percentage of Cu leached after 2 h of reaction reached ∼25%, enough to produce the oxidation of roughly half of the initial GSH at that time (Fig. 3d). Furthermore, lower pH values may also increase the reaction rate of Fenton and Fenton-like processes related to the Cu and Fe28,32,66 and enhance the GSH oxidation rates via ROS formation.
K′) of Cu(SG)2 is significantly higher (26.6)64 than Cu(GSSG) (3.63).65 The appearance of the Cu(SG)2 complex is also favored by pH values typically met in a tumour microenvironment (Fig. ESI-8 and ESI-9†). Although the lixiviation of Cu triggered by GSH proceeds at a slower pace at pH = 5.80 (see Fig. 2b), the percentage of Cu leached after 2 h of reaction reached ∼25%, enough to produce the oxidation of roughly half of the initial GSH at that time (Fig. 3d). Furthermore, lower pH values may also increase the reaction rate of Fenton and Fenton-like processes related to the Cu and Fe28,32,66 and enhance the GSH oxidation rates via ROS formation.
Similarly, to the control experiments carried out with FeCl3, the experiments between CuFe and GSH excess were carried out under acidic conditions. After 3 hours of reaction, 1H-NMR analysis (Fig. ESI-3a†) showed no meaningful fractions of GSSG/Fe(SG) complex formed. UPLC-PDA and MS-ESI analyses further corroborated the lower conversion of GSH in the presence of lixiviated Fe ions (Fig. ESI-10 and ESI-11,† respectively). While an increase of the [Fe(SG) + H]+ signal was found after 24 h of reaction (Fig. ESI-8a†), a significant concentration of GSH was still present (Fig. ESI-8b†). These GSH-oxidation results together with previous MP-AES results at neutral pH (Fig. 2a) further suggest a limited influence of Fe in the homogeneous conversion of GSH. Two factors are key to justify these phenomena: (i) the much larger standard reduction potential of Fe3+/2+ (E0 = +0.77 V compared to +0.153 V for Cu2+/+) implies a comparatively slower catalytic cycle since the regeneration of Fe3+ species to restart the cycle requires a higher energy demand, an energy penalty analogous to that observed in Fenton-like processes;67 (ii) the scarcity of labile Fe3+ available, both due to its slower leaching rate compared to Cu (Fig. 2a) and to the fact that at physiological pH released iron tend to rapidly form Fe(OH)3 species.68
Heterogeneous O2 generation of the Fe-enriched solid nanoparticles
Given the strong Cu release in comparison with the much less intense Fe lixiviation, we investigated the morphological and catalytic properties of the progressively Fe-enriched nanoparticles. After the interaction with GSH, most of Cu present in the NP is lixiviated into the aqueous media (see Fig. 2a). This was confirmed by high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) and energy dispersive X-ray spectroscopy (EDX) mapping analysis of the NPs before (Fig. 4a) and after (Fig. 4b) reaction, revealing a very significant Fe enrichment following the preferential leaching of Cu after its interaction with GSH. X-ray photoelectron spectroscopy (XPS) analysis of the CuFe nanocatalyst further confirmed the strong reduction of the atomic percentage of Cu at the surface in comparison with the original sample (0.28 atomic% Cu after exposure, compared to 7.80 before, a 28-fold decrease, see Tables ESI-1 and ESI-2†). This was further supported by the significant reduction of the Cu 2p3/2 photoemission contribution after incubation with GSH (Fig. 4c). | ||
| Fig. 4 Heterogeneous catalysis on the Fe-enriched nanoparticle surface: (a) and (b) STEM-EDX images before/after catalyst interaction with GSH. Prior to the GSH-triggered lixiviation of Cu, Fe and Cu co-localize within the crystalline network of the nanoparticle oxide. However, after GSH exposition, Cu starts to be released and its presence in the nanoparticle is strongly reduced; (c) X-ray photoemission spectra corresponding to the Cu 2p3/2 and Fe 2p regions before and after reaction of the CuFe nanocatalyst with GSH; as a consequence of leaching process, the intensity of Cu signal is close to noise; (d) TEM images of the CuFe nanocatalyst after 1 hour incubation with different GSH concentrations relevant at the intracellular and extracellular levels. Size analysis of individual nanoparticles reveals a certain reduction of size in the presence of larger GSH concentrations (5 mM); (e) scheme showing the transformation of the by-products generated via aerobic GSH oxidation into O2 in the presence of the solid Fe-enriched catalyst that enables the regeneration of O2 as electron acceptor to sustain the GSH oxidation cycle; (f) O2 generation capabilities of the Fe-enriched nanoparticles and of the supernatant containing leached Cu cations after the addition of H2O2; experimental conditions: pH = 7.40; [H2O2]0 = 1 mM, [CuFe] = 0.1 mg mL−1; addition of H2O2 is highlighted. | ||
In addition, the oxidation state of Cu was strongly affected by the process. The remaining Cu content was enriched in Cu+ as shown by the ratios of the contributions at 932.6 and 934.3 eV due to the reduction capability of GSH,69 compared to the original sample with a higher ratio of Cu(II) to Cu(I) states.70 In contrast, the Fe surface atomic percentage slightly increased (see Tables ESI-1 and ESI-2†). TEM analysis of the CuFe nanocatalyst after one hour of incubation with different GSH concentrations revealed that some NP size reduction takes place in the presence of average intracellular GSH concentrations (i.e. 5 mM) as a consequence of metal leaching (Fig. 4d). XRD analysis showed an important decrease of the intensity corresponding to the (400) diffraction peak71 which accounts for the crystalline plane that includes four tetrahedral Cu sites (Fig. ESI-12a and b†). This reduction is also consistent with the selective loss of copper sites upon the incubation with GSH (Fig. 2). In contrast, the Fe-enriched remaining solid nanoparticle matches well with the XRD patterns of Fe3O4 and CuFe2O4 (Fig. ESI-12a†).
Fe-based oxides have been reported as active catalysts to transform ROS species, such as ˙O2− (ref. 72 and 73) or H2O2 (ref. 7, 27 and 74) into H2O2 and O2, respectively. Specifically, both species (˙O2− and H2O2) are interesting in our system. ˙O2− is a reaction side-product resulting from the homogeneous catalytic cycle (Fig. 3b) and H2O2 is both an intratumoral overexpressed molecule75,76 and a reported by-product of Cu-catalyzed GSH oxidation in the homogeneous phase.41 Thus, the Fe-enriched fraction of the heterogeneous CuFe catalyst was not a mere spectator of the homogeneous catalytic cycle. On the contrary, it became an active agent in generating O2 from H2O2 under hypoxic conditions (i.e. low O2 concentration) that prevail in TME (Fig. 4e). This is clearly shown in Fig. 4f. After each H2O2 injection a clear increase in the concentration of dissolved O2 was observed in the presence of the Fe-enriched catalyst, acting as a catalase-mimicking surrogate. This experiment rendered the opposite result when carried out in the presence of the Cu leachate from the catalyst, where no O2 generation could be observed (Fig. 4f). The generated O2 allowed to complete and sustain the homogeneous GSH-oxidation cycle, which needs O2 as final electron acceptor.77 We also evaluated the potential capability of this Fe-enriched catalyst to transform ˙O2− into O2. KO2 was selected as superoxide source. In spite of the rapid self-dismutation of ˙O2− itself in the control experiments,78 the contribution of the catalyst could be clearly distinguished at longer reaction times (Fig. ESI-13†). Interestingly, while the Fe-enriched solid nanocatalyst was able to decompose H2O2 into O2 (Fig. 4f), it showed negligible activity towards ˙OH production via Fenton-like processes when tested with methylene blue (MB) (Fig. ESI-14†).
Interplay between homogeneous and heterogeneous catalysis in healthy and cancer cells with different GSH levels
To evaluate the biological action of the CuFe nanocatalyst, additional experiments were carried out against different cell lines. Different cancer cell lines were specifically selected for their intrinsic high GSH levels, while healthy cell lines (i.e. fibroblasts and mesenchymal cells) with lower GSH concentrations were used as control.79,80 Specifically, four different tumour cell lines were chosen to assess the cytotoxic effect of this nanocatalyst: U251-MG and U87-MG (both malignant glioblastoma cell lines), SKOV-3 (ovarian cancer cell line) and HeLa (cervical cancer cell line). The aim of the study was to compare the behaviour of cell lines against the action of our nanocatalyst, since we could expect variable effect depending on their specific GSH content (Fig. 5a). | ||
| Fig. 5 Cancer cell lines with overexpressed GSH levels are intensely affected by CuFe-triggered catalysis. (a) Left: scheme of catalyst evolution inside the cell, highlighting the role played by GSH; Right: intracellular GSH concentration levels detected for U251-MG, HeLa and hpMSC cell lines before and after 24 h of incubation with 25.0 μg mL−1 of CuFe nanocatalyst. Statistical analysis revealed significant differences for cancer cells with higher GSH levels (i.e. U251-MG and HeLa) in comparison to hpMSC, which likely enhance GSH-related pathways, affecting cell viability; (b) cell viability study of CuFe nanocatalyst with different cancer (SKOV-3, U251-MG, HeLa and U87) and non-tumoral (hpMSC and fibroblasts) cell lines. CC50 values stand for the cytotoxic concentration to kill 50% of cell populations after 48 h of incubation; [CuFe] concentration for each experiment was (1) 12.5 μg mL−1; (2) 25.0 μg mL−1; (3) 50.0 μg mL−1; (4) 100 μg mL−1 and (5) 200 μg mL−1; (c) confocal microscopy images corresponding to the U251-MG, HeLa and hpMSC cell lines before and after the incubation with CuFe NPs for 24 h using 25.0 μg mL−1 of nanocatalyst; (actin fibres are displayed in red after staining with phalloidin-Alexa 488, nuclei are shown in blue and stained with DAPI and CuFe appear in green colour, pointed by yellow arrows and seen by reflection). | ||
The results of the cytotoxicity studies after different incubation times with the CuFe nanocatalyst (Fig. 5b) revealed a clear effect: the viability of the four tumour cell lines was reduced, even at the lower concentrations of the catalyst and shorter incubation times (see Fig. 5b). In contrast, the non-tumoral cell lines remained viable in the presence of much higher concentrations of the catalyst. The 50% cytotoxic concentration (CC50) determined for the CuFe nanocatalyst was significantly lower in the cancer cell lines than in the healthy ones (Fig. 5b and Table ESI-6†). In addition, the monitoring and quantification of intracellular GSH levels in U251-MG and HeLa cell (cancerous) and hpMSC (healthy) cell lines helped to correlate with their respective viabilities in the absence and presence of the CuFe nanocatalyst (Fig. 5b). The corresponding GSH intracellular levels were quantified following a recently optimized protocol by Bonet-Aleta et al.32 (see Experimental section). Both cancer cell lines (i.e. U251-MG and HeLa) showed a significant decrease in GSH levels (40.6 and 20.6%, respectively) while GSH levels of the evaluated healthy cell line remain unaltered. This behaviour may be attributed to the differences between the intracellular GSH concentration levels of cancer and healthy cell lines, respectively.32,33 Catalytic conversion differences between U251-MG and HeLa may arise from distinct responses from different cells under a GSH-deprivation scenario. For instance, HeLa cell line treated with Cu salts at normal cysteine levels raise their intracellular GSH levels increasing its biosynthesis, which may be reflected as a reduced catalytic conversion.81,82 Representative optical (Fig. ESI-15†) and confocal microscopy (Fig. 5c) images of these cell lines treated with 25.0 μg mL−1 at 24 h confirmed the internalization of the catalyst within all cell lines to trigger the above described catalytic cycle.
Regarding the CuFe NPs internalization route, it has been widely reported that the internalization of both Cu or Fe-based nanomaterials mainly occurs through endocytosis23 (via clathrin-mediated pathway83). However surface coating also plays a key role, since for FexOy NPs coated with BSA and PEG, clathrin- or caveolin-mediated endocytosis were favoured, respectively.84 Moreover, the use of DMSA as coating agent has been also reported as a way to boost the uptake efficiency for Fe2O3 NPs.85
Up to now, the interaction between Cu and Fe based nanoparticles and GSH has been either interpreted from the perspective of a heterogeneous process taking place at the nanoparticle–liquid interface or directly neglected. We have detected both ˙O2− and H2O2 as the reaction by-products of Cu-catalyzed GSH oxidation, which is consistent with literature.41,45,62,86 This reaction is taking place homogeneously with leached Cu species, i.e. while GSH interacts with the surface to produce the release of Cu(I) species, the GSH oxidation itself would be a homogeneous process taking place in the bulk of the solution and not on the catalyst particle. However, both reaction by-products ˙O2− and H2O2 can interact with the remaining Fe-enriched nanoparticle which acts as an heterogeneous catalyst yielding O2, which in turn is necessary to sustain the homogeneous GSH oxidation.
Taking into account the above results, we propose the following reaction mechanism (see Fig. 6): (i) in a first step, nucleophilic thiol (–SH) groups from GSH promote the release of Cu species from the nanocatalyst crystalline network. GSH overexpressed in TME forms a coordination complex with released Cu, (Cu(SG)2). The formation of this complex effectively removes free Cu(I) species from the environment, shifting liquid phase equilibria and increasing the rate of Cu leaching; (ii) the Cu(SG)2 complex starts a homogeneous catalytic cycle where GSH is finally oxidized to GSSG by dissolved O2; this latter process entails a one/two electron transfer from [CuI(GSSG)]˙− to O2, promoting the generation of H2O2 and ˙O2− species which readily (iii) react with the FeIII sites remaining in the solid nanocatalyst through a Haber–Weiss reaction87,88 to regenerate O2 that feeds the homogeneously Cu-catalyzed reaction. Moreover, additional O2 is produced by the Fe-enriched catalytic NP using the intracellular H2O2 (Fig. 4f). Finally, (iv) GSSG is released from the coordination sphere of Cu2+, which becomes available to be reduced by GSH and re-start the catalytic cycle. The proposed mechanism constitutes a perfect example of how two catalytic processes, namely the homogeneous Cu-catalyzed GSH oxidation and the heterogeneously catalyzed processes of H2O2 decomposition and ˙O2 reaction are synergistically coupled to produce the efficient oxidation of a key tumour metabolite (GSH) allowing to circumvent O2-scarcity in the TME.
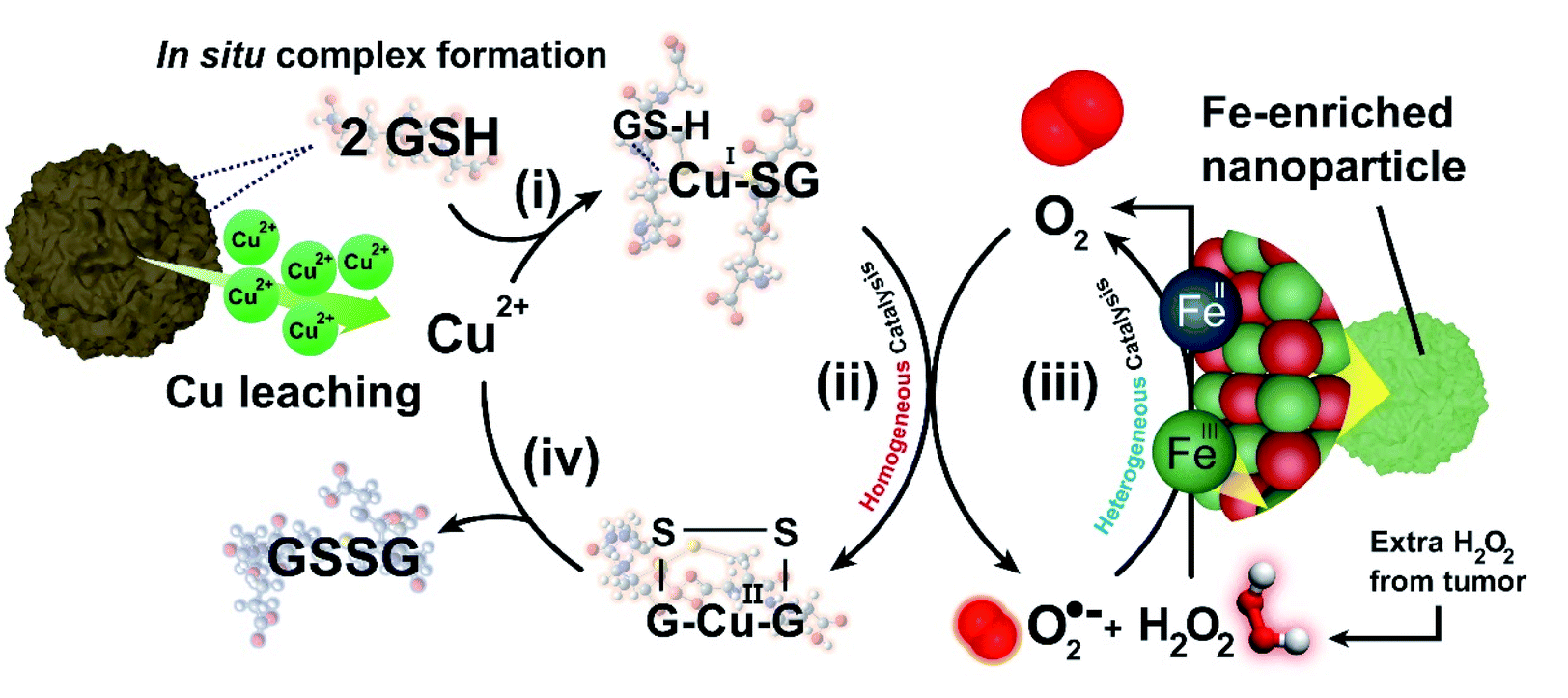 | ||
| Fig. 6 Complete homogeneous-heterogeneous interplay for the CuFe nanoparticles in the presence of GSH and O2: (i) in a first step, GSH triggers Cu-release from the spinel nanostructure (ii) excess GSH is able to form an organometallic complex with Cu through thiol (–SH) groups to stabilize CuI (iii) molecular O2 accepts electrons from Cu(SG)2 complex to yield O2 reduced species (H2O2 or ˙O2−, depending on the number of electrons transferred) and Cu(GSSG); (iv) in a heterogeneously coupled process, as-generated H2O2 and ˙O2− donates electrons to the remaining Fe-enriched surface of the solid heterogeneous nanocatalyst in a process that generates oxygen needed for step (iii). Moreover, intratumoural H2O2 decomposes on the Fe-enriched catalyst, contributing additional oxygen generation. | ||
Experimental
Chemicals
Iron(III) chloride hexahydrate (FeCl3·6H2O, 97%), copper(II) chloride dihydrate (CuCl2·2H2O, ≥99.0%), sodium acetate anhydrous (CH3COONa, 99.0%), bovine serum albumin (BSA), ethylene glycol (EG), dimercaptosuccinic acid (DMSA, 99.0%), glutathione, glutathione oxidized (GSSG, ≥98.0% HPLC), 5,5′-dithiobis(2-nitrobenzoic acid) (DNTB), hydrogen peroxide (H2O2, 33% v/v), sodium bicarbonate (NaHCO3, 99%), methylene blue (MB, ≥95.0), dihydroethidium (DHE, 99%), potassium dioxide (KO2, >99%), trichloroacetic acid (CCl3COOH, >99%) and tris(hydroxymethyl)aminomethane (TRIS, >99%) were purchased from Sigma Aldrich. Acetonitrile (ACN) was purchased from WVR (Avantor). UPLC grade water was obtained from a Milli-Q Advantage A10 System with resistivity of 18.2 mΩ (Merk Millipore, Germany). Phalloidin-Alexa 488 was acquired from Molecular Probes (A12379) and Fluoromount-G + DAPI from EMS (17984-24). Dulbecco's modified Eagle medium (DMEM) was purchased from Lonza (Ref. 12-614F). Dulbecco's phosphate buffered saline (DPBS 1×, Ref. 17-512F) and PBS (Ref. BE02-017F) were also purchased from Lonza.Characterization techniques
Transmission electron microscopy (TEM) was performed using a FEI TECNAI T20 microscope operated at 200 keV. High-resolution transmission electron microscopy (HRTEM) was performed using a FEI Titan (80–300 kV) microscope at an acceleration voltage of 300 kV. In both cases samples were prepared by drop casting 5 μL of the nanoparticle suspension on a holey carbon TEM grid. UV-vis spectra were obtained on a UV-vis double beam spectrophotometer Jasco V67. X-ray photoelectron spectroscopy (XPS) was performed with an Axis Supra spectrometer (Kratos Tech). The samples were mounted on a sample rod placed in the pretreatment chamber of the spectrometer and then evacuated at room temperature. The spectra were excited by a monochromatized Al Kα source at 1486.6 eV and subsequently run at 8 kV and 15 mA. A survey spectrum was acquired at 160 eV of pass energy, and for the individual peak regions, spectra were recorded with a pass energy of 20 eV. Analysis of the peaks was performed with the CasaXPS software using a weighted sum of Lorentzian and Gaussian component curves after Shirley background subtraction. The binding energies were referenced to the internal C 1s standard at 284.5 eV. X-ray diffraction (XRD) patterns were obtained in a PANalytical Empyrean equipment in Bragg–Brentano configuration using Cu Kα radiation and equipped with a PIXcel1D detector. 1H spectra (D2O) were recorded at 25 °C using a Bruker Avance 400 MHz NMR spectrometer with TMS as the internal standard and deuterated water as solvent in a 5 mm QNP probe. Nanoparticle tracking analysis was measured on Malvern Nanosight 300.Synthesis of CuFe–DMSA nanoparticles
CuFe2O4 nanoparticles were synthesized via a templated-growth method.32 Firstly, 250 mg of BSA were dissolved in 2.5 mL of Milli-Q water and consequently 12.5 mL of ethylene glycol were added. After that, 270.0 mg of FeCl3·6H2O, 85.0 mg of CuCl2 and 375.0 mg of CH3COONa were added into the reaction vessel. Reaction was kept stirring for 2 h at room temperature to ensure a correct binding of BSA–nucleophile groups to the metallic ions. Then, the reaction was transferred to a Teflon autoclave and the temperature was set to 180 °C for 24 h. Finally, the product was centrifuged at 12000 rpm for 20 min. 20 mL of a 25 mM solution of DMSA were poured to the reaction pellet and the dispersion was assisted with ultrasonication. 5 mL of 0.7 M NaOH solution were added to ensure the correct DMSA dissolution to decorate the nanocatalyst surface. The final product was purified by two centrifugation cycles at 12000 rpm for 20 min. The nanocatalyst was stored at 4 °C for further use. CuFe nanocatalysts were synthetized using bovine serum albumin (BSA) as template27,89 and ethylene glycol (EG) as a solvent to modulate particle size.90 The abundance of nucleophile functional groups in the BSA (–COO–, –NH2) can chelate the metal ions (Cu2+, Fe3+) and serve as starting point to growth the nanostructure. Although the distribution size of the nanoparticles is homogeneous, these nanoparticles suffer from aggregation in aqueous media, which may hinders cell internalization.91 As shown previously by Miguel-Sancho et al.,92 DMSA functionalization enhanced the dispersion and stability of the nanoparticles in aqueous media (Fig. ESI-16 and ESI-17†). The synthesis of these materials has been performed at the Platform of Production of Biomaterials and Nanoparticles of the NANBIOSIS ICTS, more specifically by the Nanoparticle Synthesis Unit of the CIBER in BioEngineering, Biomaterials & Nanomedicine (CIBER-BBN).
Metal leaching study by microwave plasma-atomic emission spectroscopy (MP-AES)
Each solution was prepared in an Eppendorf tube with CuFe–DMSA nanoparticle at a concentration of 0.1 mg mL−1 (total volume = 1 mL). The different solutions were introduced in an Eppendorf thermoshaker at 37 °C and constant stirring of 400 rpm. At every time point (2 h, 4 h, 6 h, 24 h), samples were centrifuged at 13300 rpm during 30′, and the supernatants were collected for further analysis. The nanoparticle pellet was resuspended with the corresponding solution, until the next analysis cycle, where the procedure was repeated. At the experiment endpoint, the supernatant samples were analyzed together with final nanoparticle pellet, to close mass balances and elucidate how much metal moved to the solution. All the samples were digested with HCl:HNO3 (3:1) mixture overnight. Cu and Fe concentrations were determined through the analysis with Agilent 4100 MP-AES.
1H-NMR-study of CuFe–GSH interaction
The interaction study of leached metals (Cu, Fe) with GSH was performed by the 1H-NMR and DOSY analysis of different reaction mixtures. For CuCl2–GSH experiments, the pH was fixed to 7.4 (buffer Na2HPO4/KH2PO4) and the molar ratio CuCl2:GSH was set to 1:4 ([GSH] = 20 mM, [CuCl2·2H2O] = 5 mM). For FeCl3–GSH experiments, the pH was not fixed (pH = 3.60) and the molar ratio FeCl3:GSH was set to 1:4 ([GSH] = 20 mM, [FeCl3·6H2O] = 4 mM). The reaction mixtures were analyzed after incubation for 5 minutes. For CuFe + GSH experiments, molar ratio CuFe:GSH was 1:44 ([GSH] = 20 mM, [CuFe2O4] = 0.5 mM). Reaction were incubated at pH = 7.4 (Na2HPO4/KH2PO4) and pH = 3.70 (free pH) for 3 and 24 h. All samples were filtered before analysis.
Mass spectroscopy analysis of the reaction
ESI (ESI+) mass spectra were recorded using an Esquire 3000 ion-trap mass spectrometer (Bruker Daltonic GmbH) equipped with a standard ESI/APCI source. Samples were introduced by direct infusion with a syringe pump. Nitrogen served both as the nebulizer gas and the dry gas. The HRMS mass spectra were recorded using an MicroToF Q, API-Q-ToF ESI with a mass range from 20 to 3000 m/z and mass resolution 15000 (FWHM). Samples prepared for 1H-NMR/DOSY experiments were analyzed by using this methodology.
Catalytic GSH depletion
Catalytic activity towards GSH oxidation of CuFe–DMSA nanoparticles was evaluated according to the following protocol. 5 mM of GSH were mixed with 0.1 mg mL−1 of CuFe–DMSA in a total volume of 2.5 mL. GSH concentration at different reaction times was measured by UPLC-PDA. 20 μL of reaction were mixed with 100 μL of 1 mM 5,5′-disulfanediylbis(2-nitrobenzoic acid) (DTNB) and 880 μL of 0.01 M 2-amino-2-hydroxymethyl-propane-1,3-diol (TRIS). Standards for the calibration curve were prepared following Table ESI-5.†UPLC-PDA equipment for GSH quantification
GSH analysis were performed on Waters ACQUITY system H-Class which consisted of a binary pump, an autosampler, a column thermostat and a photodiode array (PDA) detector. This system is coupled to a photo diode array (PDA) detector to monitor absorbance from derivatized GSH at 412 nm during analysis time. Data acquisition and processing were performed by using MASSLYNX software (Waters Corporation, USA). In order to analyze GSH from derivatized samples as describe below, chromatographic separation was performed using an ACQUITY UPLC BEH C18 column (130 Å, 1.7 μm 2.1 × 50 mm, from WATERS) at 40 °C under an isocratic flow of 0.3 mL min−1 containing 50% acetonitrile, 50% Milli-Q water.Generation of hydrogen peroxide H2O2, and superoxide radicals ˙O2−
1,3-Diphenylisobenzofuran (DPBF) was employed as a probe to measure the production of H2O2 and ˙O2− during homogeneous GSH oxidation. 20 μL of 10 mM DPBF solution (in ethanol) were added to 2.5 mL of a mixture ethanol:PBS(1×) 2:1. Catalyst and GSH concentration were 0.1 mg mL−1 and 5 mM, respectively. UV-vis analysis of remaining DPBF was performed after centrifuging the sample (100 μL of reaction + 400 μL mixture ethanol:PBS-1×) at 13000 rpm for 5′. Selective ˙O2− detection was carried on using dihydroethidium (DHE) as a probe.42,59–61 15 μL of concentrated DHE solution was added to a Na2HPO4/KH2PO4 buffer solution (pH = 7.4) to reach a final concentration of 100 μM. Catalyst and GSH concentration were 0.01 mg mL−1 and 5 mM, respectively. 500 μL were centrifuged, and 400 μL of the supernatant were mixed with 1000 μL PBS and measured on a LS 55 fluorescence spectrometer (PerkinElmer). λexc/λem = 480 nm/500–700 nm, slit-widths of 10 nm for both excitation and emission wavelengths.
Analysis of O2 consumption/generation
Molecular O2 was measured with a NeoFox oximeter equipped with FOSPOR-R probe. In order to analyze the O2-generation capabilities of the remaining nanoparticle, CuFe (0.1 mg mL−1) was incubated with 5 mM of GSH (pH = 7.40) for 3 h to induce selective Cu-release. The solution was centrifuged at 13300 rpm for 10′ to separate homogeneous and heterogeneous catalysts to analyze their individual catalytic activity towards O2 production using [H2O2]0 = 1 mM. To check O2 production using KO2, the heterogeneous catalyst was mixed with 100 μM KO2 at pH = 7.40 (adjusted with Na2HPO4/KH2PO4 buffer).
Cell culture conditions and study of CuFe cytotoxicity
In order to assess the effect that these nanoparticles produced on the chosen cell lines, a viability assay on 96 well plates was carried out. Briefly, cells were seeded at different densities depending on their type (3000 cells per well for tumoral cell lines, i.e. U251-MG, SKOV-1, HeLa, U87-MG and 4000 cells per well for healthy cell lines, i.e. hpMSC and fibroblasts), using Dulbecco's modified Eagle medium (DMEM), supplemented with 1% L-glutamine, 1% PSA (penicillin, streptomycin, amphotericin), and 10% fetal bovine serum (FBS); in the case of hpMSC, DMEM medium was also supplemented with fibroblast growth factor 2 (FGF-2) at 5 μg mL−1 24 h after the seeding, the wells were treated with CuFe nanoparticles, in such a way that the supernatant in each well was replaced with a suspension of CuFe nanoparticles in DMEM (DMEM + FGF-2 for hpMSC) at different concentrations, ranging from 200 to 12.5 μg mL−1. After 24, 48 and 72 h of incubation, the wells were washed with Dulbecco's phosphate buffered saline (DPBS), and then, a solution of blue cell viability reagent in DMEM (10% v/v) was used to assess the cytotoxic effect of these nanoparticles on the different cell lines. After 1 h of incubation with the blue cell viability reagent at 37 °C, the signal was measured with a BioTek plate reader, exciting at 528 nm and measuring the emission at 590 nm.Cellular analysis by confocal microscopy
Confocal microscopy assay was carried out to assess the capacity of internalization of CuFe NPs into 3 different cell lines: U251-MG, HeLa and hpMSC. Cells were seeded onto 12 mm Ø coverslips, which were deposited on a 24-well plate, at a density of 20000 cells per well, and incubated at 37 °C and 5% CO2. After 24 h, cells were treated with CuFe NPs dispersed in DMEM at a concentration of 25 μg mL−1 during 24 h (for negative control C-wells, DMEM was replaced with fresh media). After this time, cells were washed 3 times with DPBS, fixed with 4% paraformaldehyde, and then washed 3 more times with DPBS. In order to prepare the samples for confocal microscopy, cells were permeabilized with 0.1% saponine. After that, cytoplasmic actin fibres were stained with phalloidin-Alexa 488; (dilution 1:200), and samples were deposited onto a drop of Fluoromount-G + DAPI for nuclei staining. Nanoparticle aggregates could be observed due to the reflection of the incident light. In order to confirm the presence of the nanoparticles inside the cell, a Z-Stack assay of the whole cell, and its ulterior maximum orthogonal projection were performed. This assay was carried out in a confocal microscope (ZEISS LSM 880 Confocal Microscope), using a 63×/1.4 Oil DIC M27 objective.
Intracellular catalytic oxidation of GSH
In order to assess the catalytic effect of CuFe NPs and their correlation with intracellular GSH levels, U251-MG, HeLa and hpMSC cells were seeded in p100 dishes. Before they reached 90% of confluence, cells were treated with 25 μg mL−1 for 24 h (for C-dishes, DMEM was replaced with fresh media). Then, cells were washed with PBS–EDTA and trypsinized. Cells in suspension were counted with a Neubauer chamber and centrifuged at 500 g during 5 minutes, in order to obtain the cell pellet. Analysis of the intracellular GSH levels was performed as follows, based on a previously optimized protocol.32 Briefly, cells were first trypsinized, washed twice with PBS (500 g for 5 minutes) and collected. Cells were again centrifuged at 13300 rpm for 5 minutes. Supernatant was discarded and 400 μL of 12% CCl3COOH solution was added to cell pellet to precipitate proteins and avoid their interference during the analysis. Samples were sonicated and left 15 minutes at 4 °C. The resulting suspension was centrifuged for 13300 rpm for 5 minutes. The supernatant was isolated and subjected to a derivatization process: 50 μL of the sample were mixed with 930 μL of a 0.01 M TRIS solution and 20 μL of a 1 mg mL−1 DTNB solution. pH of each individual sample was adjusted at a value of 8.0 using 4–6 μL of a 3 M NaOH solution. Standards for the calibration curve were prepared following Table ESI-7.† Samples were analyzed by UPLC-PDA equipment following the analysis conditions presented before. For the calculation of intracellular GSH, an average volume of 1.2 × 10−11 L, 3.3 × 10−12 L and 2.6 × 10−12 L were selected for hpMSCs, U251-MG and HeLa cells, respectively.32,93
Statistical analysis
Results are expressed as mean ± standard deviation. Statistical analysis of biological experiments together with significance between means were evaluated by two-way analysis of variance (ANOVA) for multiple comparisons by Dunnett's multiple comparison test using GraphPad Software. Analysis of significance for intracellular catalytic oxidation of GSH was performed using t test. Statistically significant differences were expressed as follows: *p < 0.05, **p < 0.005, ***p < 0.0005 and ****p < 0.00005.Conclusions
Processes promoted by heterogeneous catalysts are not necessarily of a purely heterogeneous nature. Here we demonstrate that a combination of homogeneous and heterogeneous processes can originate from a copper–iron based nanocatalyst under conditions that are relevant in the tumour microenvironment. The main part of the contributing reactions take place in a homogeneous cycle catalyzed by released Cu species. The results shed light on the complexity of the processes taking place in developing fields such as nanocatalytic cancer therapy. As demonstrated in this work for a CuFe hybrid nanocatalyst, lixiviation mechanisms induced by specific biomolecules with a strong presence in the TME such as GSH may lead to new catalytic scenarios where homogeneous and heterogeneous processes are no longer unrelated events. This means that catalyst design becomes more complex, since it has to take into account the effect of environmental species on the stability of the catalyst, but also more powerful, since leaching processes can be engineered to yield synergistic catalytic actions. Under this scenario, the design of the catalyst will consider not only its ability to favour specific surface reactions, but also its role as a reservoir for the long-term release of active ions in response to stimuli from the chemical environment. This point of view will be key to develop novel nanoparticles capable of acting as successful bio-orthogonal catalysts.Data availability
All the data supporting the findings of this study are available within the article and its ESI† files and from the corresponding authors upon reasonable request.Author contributions
J. B.-A. and J. L. H. prepared and characterized the materials. J. B.-A., M. E., J. L. H. and E. U. performed the experiments, ran the characterization and analysed the data; E. U., P. M.-D., J. L. H. and J. S. designed, coordinated and supervised the research; J. B.-A., J. L. H. and J. S. wrote the paper with contribution and approval of all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Research Council (ERC Advanced Grant CADENCE number 742684) is gratefully acknowledged. The TEM measurements were conducted at the Laboratorio de Microscopias Avanzadas, Instituto de Nanociencia y Materiales de Aragon, Universidad de Zaragoza, Spain. The synthesis of materials has been performed by the Platform of Production of Biomaterials and Nanoparticles of the NANBIOSIS ICTS, more specifically by the Nanoparticle Synthesis Unit of the CIBER in BioEngineering, Biomaterials & Nanomedicine (CIBER-BBN). J. B.-A. acknowledges the Spanish Government for an FPU predoctoral contract. M. E. acknowledges the Diputacion General de Aragon (DGA) for a predoctoral contract. The authors thank Dr Javier Calzada and Dr Silvia Irusta for their help with UPLC and XPS data acquisition. E. P. U. thanks Gobierno de Aragón-FSE (Spain, grupo Aminoácidos y Péptidos, E19_20R) for funding. Finally, the authors thank the Scientific and Technical Services at CIBA (IACS).Notes and references
- Y. Ding, Y. Dai, M. Wu and L. Li, Chem. Eng. J., 2021, 426, 128880 CrossRef CAS.
- A. Acharya, I. Das, D. Chandhok and T. Saha, Oxid. Med. Cell. Longevity, 2010, 3, 23–34 CrossRef PubMed.
- D. L. Nelson, A. L. Lehninger and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, 2008 Search PubMed.
- G. K. Balendiran, R. Dabur and D. Fraser, Cell Biochem. Funct., 2004, 22, 343–352 CrossRef CAS PubMed.
- N. Traverso, R. Ricciarelli, M. Nitti, B. Marengo, A. L. Furfaro, M. A. Pronzato, U. M. Marinari and C. Domenicotti, Oxid. Med. Cell. Longevity, 2013, 2013, 972913 Search PubMed.
- L. Wang, M. Huo, Y. Chen and J. Shi, Adv. Healthcare Mater., 2018, 7, e1701156 CrossRef PubMed.
- S. Li, L. Shang, B. Xu, S. Wang, K. Gu, Q. Wu, Y. Sun, Q. Zhang, H. Yang, F. Zhang, L. Gu, T. Zhang and H. Liu, Angew. Chem., Int. Ed., 2019, 58, 12624–12631 CrossRef CAS PubMed.
- Q. Sun, Z. Wang, B. Liu, F. He, S. Gai, P. Yang, D. Yang, C. Li and J. Lin, Coord. Chem. Rev., 2022, 451, 214267 CrossRef CAS.
- X. Zhang, Y. Lin and R. J. Gillies, J. Nucl. Med., 2010, 51, 1167 CrossRef CAS PubMed.
- L. Yu, Y. Chen, M. Wu, X. Cai, H. Yao, L. Zhang, H. Chen and J. Shi, J. Am. Chem. Soc., 2016, 138, 9881–9894 CrossRef CAS PubMed.
- L. Yue, J. Wang, Z. Dai, Z. Hu, X. Chen, Y. Qi, X. Zheng and D. Yu, Bioconjugate Chem., 2017, 28, 400–409 CrossRef CAS PubMed.
- X. Jing, Y. Xu, D. Liu, Y. Wu, N. Zhou, D. Wang, K. Yan and L. Meng, Nanoscale, 2019, 11, 15508–15518 RSC.
- S. Wang, F. Li, R. Qiao, X. Hu, H. Liao, L. Chen, J. Wu, H. Wu, M. Zhao, J. Liu, R. Chen, X. Ma, D. Kim, J. Sun, T. P. Davis, C. Chen, J. Tian, T. Hyeon and D. Ling, ACS Nano, 2018, 12, 12380–12392 CrossRef CAS PubMed.
- J. Xu, W. Han, P. Yang, T. Jia, S. Dong, H. Bi, A. Gulzar, D. Yang, S. Gai, F. He, J. Lin and C. Li, Adv. Funct. Mater., 2018, 28, 1803804 CrossRef.
- F. Jiang, B. Ding, Y. Zhao, S. Liang, Z. Cheng, B. Xing, B. Teng, P. a. Ma and J. Lin, Sci. China Mater., 2020, 63, 1818–1830 CrossRef CAS.
- G. Guo, H. Zhang, H. Shen, C. Zhu, R. He, J. Tang, Y. Wang, X. Jiang, J. Wang, W. Bu and X. Zhang, ACS Nano, 2020, 14, 13391–13405 CrossRef CAS PubMed.
- J. Xu, R. Shi, G. Chen, S. Dong, P. Yang, Z. Zhang, N. Niu, S. Gai, F. He, Y. Fu and J. Lin, ACS Nano, 2020, 14, 9613–9625 CrossRef CAS PubMed.
- H. Wu, F. Chen, C. You, Y. Zhang, B. Sun and Q. Zhu, Small, 2020, 16, 2001805 CrossRef CAS PubMed.
- Y. Li, Z. Gao, F. Chen, C. You, H. Wu, K. Sun, P. An, K. Cheng, C. Sun, X. Zhu and B. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 30930–30935 CrossRef CAS PubMed.
- C. Yang, M. R. Younis, J. Zhang, J. Qu, J. Lin and P. Huang, Small, 2020, 16, 2001518 CrossRef CAS PubMed.
- C. Gunawan, W. Y. Teoh, C. P. Marquis and R. Amal, ACS Nano, 2011, 5, 7214–7225 CrossRef CAS PubMed.
- Z. Wang, A. von dem Bussche, P. K. Kabadi, A. B. Kane and R. H. Hurt, ACS Nano, 2013, 7, 8715–8727 CrossRef CAS PubMed.
- Z. Wang, N. Li, J. Zhao, J. C. White, P. Qu and B. Xing, Chem. Res. Toxicol., 2012, 25, 1512–1521 Search PubMed.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef CAS.
- M. Chang, M. Wang, M. Wang, M. Shu, B. Ding, C. Li, M. Pang, S. Cui, Z. Hou and J. Lin, Adv. Mater., 2019, 31, 1905271 CrossRef CAS PubMed.
- S.-Y. Yin, G. Song, Y. Yang, Y. Zhao, P. Wang, L.-M. Zhu, X. Yin and X.-B. Zhang, Adv. Funct. Mater., 2019, 29, 1901417 CrossRef.
- Y. Liu, W. Zhen, L. Jin, S. Zhang, G. Sun, T. Zhang, X. Xu, S. Song, Y. Wang, J. Liu and H. Zhang, ACS Nano, 2018, 12, 4886–4893 CrossRef CAS PubMed.
- Z. Wang, Y. Wang, H. Guo, N. Yu, Q. Ren, Q. Jiang, J. Xia, C. Peng, H. Zhang and Z. Chen, J. Colloid Interface Sci., 2021, 592, 116–126 CrossRef CAS.
- X. Wang, X. Zhong, Z. Liu and L. Cheng, Nano Today, 2020, 35, 100946 CrossRef CAS.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794–798 CAS.
- E. J. Ge, A. I. Bush, A. Casini, P. A. Cobine, J. R. Cross, G. M. DeNicola, Q. P. Dou, K. J. Franz, V. M. Gohil, S. Gupta, S. G. Kaler, S. Lutsenko, V. Mittal, M. J. Petris, R. Polishchuk, M. Ralle, M. L. Schilsky, N. K. Tonks, L. T. Vahdat, L. Van Aelst, D. Xi, P. Yuan, D. C. Brady and C. J. Chang, Nat. Rev. Cancer, 2021, 102–113 Search PubMed.
- J. Bonet-Aleta, M. Sancho-Albero, J. Calzada-Funes, S. Irusta, P. Martin-Duque, J. L. Hueso and J. Santamaria, J. Colloid Interface Sci., 2022, 617, 704–717 CrossRef CAS PubMed.
- A. L. Ortega, S. Mena and J. M. Estrela, Cancers, 2011, 3, 1285–1310 CrossRef CAS.
- N. S. Kosower and E. M. Kosower, Int. Rev. Cytol., 1978, 54, 109–160 CAS.
- S. C. Lu, Mol. Aspects Med., 2009, 30, 42–59 CrossRef CAS.
- J. Hedberg, E. Blomberg and I. Odnevall Wallinder, Environ. Sci. Technol., 2019, 53, 4030–4044 CrossRef CAS PubMed.
- A. K. Tummanapelli and S. Vasudevan, J. Phys. Chem. B, 2015, 119, 15353–15358 CrossRef CAS PubMed.
- S. Wall, J.-Y. Oh, A. Diers and A. Landar, Front. Physiol., 2012, 3, 1–9 Search PubMed.
- P. Nagy, Antioxid. Redox Signaling, 2013, 18, 1623–1641 CrossRef CAS PubMed.
- D. L. Rabenstein, J. Am. Chem. Soc., 1973, 95, 2797–2803 CrossRef CAS.
- K. Ngamchuea, C. Batchelor-McAuley and R. G. Compton, Chem.–Eur. J., 2016, 22, 15937–15944 CrossRef CAS PubMed.
- H. Speisky, M. Gómez, C. Carrasco-Pozo, E. Pastene, C. Lopez-Alarcón and C. Olea-Azar, Bioorg. Med. Chem., 2008, 16, 6568–6574 CrossRef CAS PubMed.
- H. Speisky, M. Gómez, F. Burgos-Bravo, C. López-Alarcón, C. Jullian, C. Olea-Azar and M. E. Aliaga, Bioorg. Med. Chem., 2009, 17, 1803–1810 CrossRef CAS PubMed.
- M. E. Aliaga, C. Carrasco-Pozo, C. López-Alarcón and H. Speisky, Transition Met. Chem., 2010, 35, 321–329 CrossRef CAS.
- M. E. Aliaga, C. López-Alarcón, R. Bridi and H. Speisky, J. Inorg. Biochem., 2016, 154, 78–88 CrossRef CAS PubMed.
- M. T. Morgan, L. A. H. Nguyen, H. L. Hancock and C. J. Fahrni, J. Biol. Chem., 2017, 292, 21558–21567 CrossRef CAS PubMed.
- M. R. Ciriolo, A. Desideri, M. Paci and G. Rotilio, J. Biol. Chem., 1990, 265, 11030–11034 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- S. A. Moggach, A. R. Lennie, C. A. Morrison, P. Richardson, F. A. Stefanowicz and J. E. Warren, CrystEngComm, 2010, 12, 2587–2595 RSC.
- C. Görbitz, Acta Chem. Scand., 1987, 41, 362–366 CrossRef.
- W. Wright, Acta Crystallogr., 1958, 11, 632–642 CrossRef CAS.
- C. Jelsch and C. Didierjean, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1538–1540 CrossRef.
- J. Kretzschmar, E. Brendler, J. Wagler and A.-C. Schmidt, J. Hazard. Mater., 2014, 280, 734–740 CrossRef CAS PubMed.
- Z. Gao, P. Carames-Mendez, D. Xia, C. M. Pask, P. C. McGowan, P. A. Bingham, A. Scrimshire, G. Tronci and P. D. Thornton, Dalton Trans., 2020, 49, 10574–10579 RSC.
- W. Qi, J. Li, C. Y. Chain, G. A. Pasquevich, A. F. Pasquevich and J. A. Cowan, J. Am. Chem. Soc., 2012, 134, 10745–10748 CrossRef CAS PubMed.
- A. Corazza, I. Harvey and P. J. Sadler, Eur. J. Biochem., 1996, 236, 697–705 CrossRef CAS PubMed.
- K. A. Jesse, S. W. Anferov, K. A. Collins, J. A. Valdez-Moreira, M. E. Czaikowski, A. S. Filatov and J. S. Anderson, J. Am. Chem. Soc., 2021, 143, 18121–18130 CrossRef CAS PubMed.
- K. Żamojć, M. Zdrowowicz, P. B. Rudnicki-Velasquez, K. Krzymiński, B. Zaborowski, P. Niedziałkowski, D. Jacewicz and L. Chmurzyński, Free Radical Res., 2017, 51, 38–46 CrossRef PubMed.
- J. Zielonka, S. Srinivasan, M. Hardy, O. Ouari, M. Lopez, J. Vasquez-Vivar, N. G. Avadhani and B. Kalyanaraman, Free Radical Biol. Med., 2008, 44, 835–846 CrossRef CAS PubMed.
- F. Gao, T. Shao, Y. Yu, Y. Xiong and L. Yang, Nat. Commun., 2021, 12, 745 CrossRef CAS PubMed.
- M. C. Ortega-Liebana, M. M. Encabo-Berzosa, A. Casanova, M. D. Pereboom, J. O. Alda, J. L. Hueso and J. Santamaria, Chem.–Eur. J., 2019, 25, 5539–5546 CrossRef CAS PubMed.
- X. Meng, D. Li, L. Chen, H. He, Q. Wang, C. Hong, J. He, X. Gao, Y. Yang, B. Jiang, G. Nie, X. Yan, L. Gao and K. Fan, ACS Nano, 2021, 15, 5735–5751 CrossRef CAS PubMed.
- J. A. Dean and N. A. Lange, Lange's Handbook of Chemistry, McGraw-Hill, 1992 Search PubMed.
- M. J. Walsh and B. A. Ahner, J. Inorg. Biochem., 2013, 128, 112–123 CrossRef CAS PubMed.
- V. G. Shtyrlin, Y. I. Zyavkina, V. S. Ilakin, R. R. Garipov and A. V. Zakharov, J. Inorg. Biochem., 2005, 99, 1335–1346 CrossRef CAS PubMed.
- T. Hu, L. Yan, Z. Wang, W. Shen, R. Liang, D. Yan and M. Wei, Chem. Sci., 2021, 12, 2594–2603 RSC.
- X. Shen, Z. Wang, X. Gao and Y. Zhao, ACS Catal., 2020, 10, 12657–12665 CrossRef CAS.
- R. Crichton, R. R. Crichton and J. R. Boelaert, Inorganic Biochemistry of Iron Metabolism: From Molecular Mechanisms to Clinical Consequences, Wiley, 2001 Search PubMed.
- B. Ma, S. Wang, F. Liu, S. Zhang, J. Duan, Z. Li, Y. Kong, Y. Sang, H. Liu, W. Bu and L. Li, J. Am. Chem. Soc., 2019, 141, 849–857 CrossRef CAS PubMed.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- Y. Liu, Z. Guo, F. Li, Y. Xiao, Y. Zhang, T. Bu, P. Jia, T. Zhe and L. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 31649–31660 CrossRef CAS PubMed.
- W. Wang, X. Jiang and K. Chen, Chem. Commun., 2012, 48, 7289–7291 RSC.
- S. Guo, Y. Han and L. Guo, Catal. Surv. Asia, 2020, 24, 70–85 CrossRef CAS.
- Y. Zhang, Y. Xu, D. Sun, Z. Meng, W. Ying, W. Gao, R. Hou, Y. Zheng, X. Cai, B. Hu and X. Lin, Chem. Eng. J., 2020, 390, 124521 CrossRef CAS.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794 CAS.
- C. Liu, D. Wang, S. Zhang, Y. Cheng, F. Yang, Y. Xing, T. Xu, H. Dong and X. Zhang, ACS Nano, 2019, 13, 4267–4277 CrossRef CAS PubMed.
- M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef PubMed.
- G. Wu, V. Berka, P. J. Derry, K. Mendoza, E. Kakadiaris, T. Roy, T. A. Kent, J. M. Tour and A.-L. Tsai, ACS Nano, 2019, 13, 11203–11213 CrossRef CAS PubMed.
- Y. Zhao, D. Wang, I. W. He, J. Liu, D. Jana, Y. Wu, X. Zhang, C. Qian, Y. Guo, X. Chen and A. K. Bindra, Angew. Chem., Int. Ed., 2021, 60, 26254–26259 CrossRef PubMed.
- P. Kuppusamy, H. Li, G. Ilangovan, A. J. Cardounel, J. L. Zweier, K. Yamada, M. C. Krishna and J. B. Mitchell, Cancer Res., 2002, 62, 307 CAS.
- M. Hultberg, A. Isaksson, A. Andersson and B. Hultberg, Chem.-Biol. Interact., 2007, 167, 56–62 CrossRef CAS PubMed.
- B. Hultberg, A. Andersson and A. Isaksson, Toxicology, 1997, 117, 89–97 CrossRef CAS PubMed.
- M. Arsianti, M. Lim, C. P. Marquis and R. Amal, Biomacromolecules, 2010, 11, 2521–2531 CrossRef CAS PubMed.
- B. Svitkova, V. Zavisova, V. Nemethova, M. Koneracka, M. Kretova, F. Razga, M. Ursinyova and A. Gabelova, Beilstein J. Nanotechnol., 2021, 12, 270–281 CrossRef CAS PubMed.
- Y. Xie, J. Jiang, Q. Tang, H. Zou, X. Zhao, H. Liu, D. Ma, C. Cai, Y. Zhou, X. Chen, J. Pu and P. Liu, Adv. Sci., 2020, 7, 1903323 CrossRef CAS PubMed.
- H. Speisky, M. Gómez, F. Burgos-Bravo, C. López-Alarcón, C. Jullian, C. Olea-Azar and M. E. Aliaga, Bioorg. Med. Chem., 2009, 17, 1803–1810 CrossRef CAS PubMed.
- J. Weiss and C. W. Humphrey, Nature, 1949, 163, 691 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- J. Zhang, X. Gao, D. Ma, S. He, B. Du, W. Yang, K. Xie, L. Xie and Y. Deng, Chem. Eng. J., 2021, 422, 130094 CrossRef CAS.
- C. Hai, S. Li, Y. Zhou, J. Zeng, X. Ren and X. Li, Journal of Asian Ceramic Societies, 2017, 5, 176–182 CrossRef.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- N. Miguel-Sancho, O. Bomatí-Miguel, G. Colom, J. P. Salvador, M. P. Marco and J. Santamaría, Chem. Mater., 2011, 23, 2795–2802 CrossRef CAS.
- L. Zhao, C. D. Kroenke, J. Song, D. Piwnica-Worms, J. J. H. Ackerman and J. J. Neil, NMR Biomed., 2008, 21, 159–164 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures and tables. See https://doi.org/10.1039/d2sc01379g |
| ‡ Current address: Surgery Department, Medical School, Zaragoza University, C/Domingo Miral s/n, 50009, Zaragoza, Spain. |
| This journal is © The Royal Society of Chemistry 2022 |