
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Generation of a transient base-stabilised arylalumylene for the facile deconstruction of aromatic molecules†
Debabrata
Dhara‡
ab,
Arumugam
Jayaraman‡
 ab,
Marcel
Härterich
ab,
Rian D.
Dewhurst
ab and
Holger
Braunschweig
*ab
ab,
Marcel
Härterich
ab,
Rian D.
Dewhurst
ab and
Holger
Braunschweig
*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 2nd May 2022
Abstract
While a stable base-free arylalumylene bearing a sterically encumbered terphenyl substituent has been reported previously, we herein report that our attempts to form a base-stabilised arylalumylene bearing a relatively small terphenyl substituent and an N-heterocyclic carbene base led instead to a “masked” dialumene (LRAl![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) AlRL), self-stabilised by one peripheral aromatic group. Intriguingly, examining the behavior of this species or its transient dialumene formed from reducing the diiodoarylalane in aromatic solvents under different conditions reveals that they both decouple into the desired base-stabilised arylalumylene. This transient acyclic, dicoordinate alumylene is highly reactive, deconstructing benzene and toluene to furnish dialuminium derivatives of pentalene, providing the first example of a neutral AlI compound able to deconstruct these less reactive arenes. Computational insights were also gained on the dialumene dissociation and on the mechanism of arene deconstruction by alumylene.
AlRL), self-stabilised by one peripheral aromatic group. Intriguingly, examining the behavior of this species or its transient dialumene formed from reducing the diiodoarylalane in aromatic solvents under different conditions reveals that they both decouple into the desired base-stabilised arylalumylene. This transient acyclic, dicoordinate alumylene is highly reactive, deconstructing benzene and toluene to furnish dialuminium derivatives of pentalene, providing the first example of a neutral AlI compound able to deconstruct these less reactive arenes. Computational insights were also gained on the dialumene dissociation and on the mechanism of arene deconstruction by alumylene.
Introduction
Aluminium and its compounds are most commonly found in their zerovalent (metallic Al, Al0) and +3 oxidation states (trivalent Al, AlIII). Thus it is not surprising that compounds of aluminium in its +1 and +2 oxidation states – generally rare and unstable species – have a marked tendency to disproportionate into Al0 (metal) and AlIII compounds.1 A number of AlI compounds ([:AlX], [:AlH], [Al2O]) have been realized by cryochemical methods.2 However, a major development in low-valent aluminium chemistry was the near-ambient temperature isolation of an AlII compound, R2Al-AlR2 (R = CH(SiMe3)2), the first Al–Al bonded molecular species, by Uhl in 1988.3 Several dialanes have since been isolated using a similar strategy.4 A further breakthrough in low-valent Al chemistry was the generation of transient dialumenes RAlAlR (R = m-terphenyl, aryl, or silyl groups), featuring AlI atoms, which were trapped as cycloaddition products with aromatic species.5 Only recently, Inoue and coworkers reported a stable, neutral, doubly NHC-stabilised dialumene (I), which contains silyl substituents (Scheme 1a).6a Shortly, the same group presented a doubly NHC-stabilised diaryldialumene, II.6b These two compounds have shown exciting abilities in small-molecule activation and catalysis.6 In 2021, Krämer and Cowley reported the third stable dialumene (III), which bears amino substituents with chelating phosphine bases.7 Interestingly, this species was demonstrated to exhibit a reversible dissociation behavior.
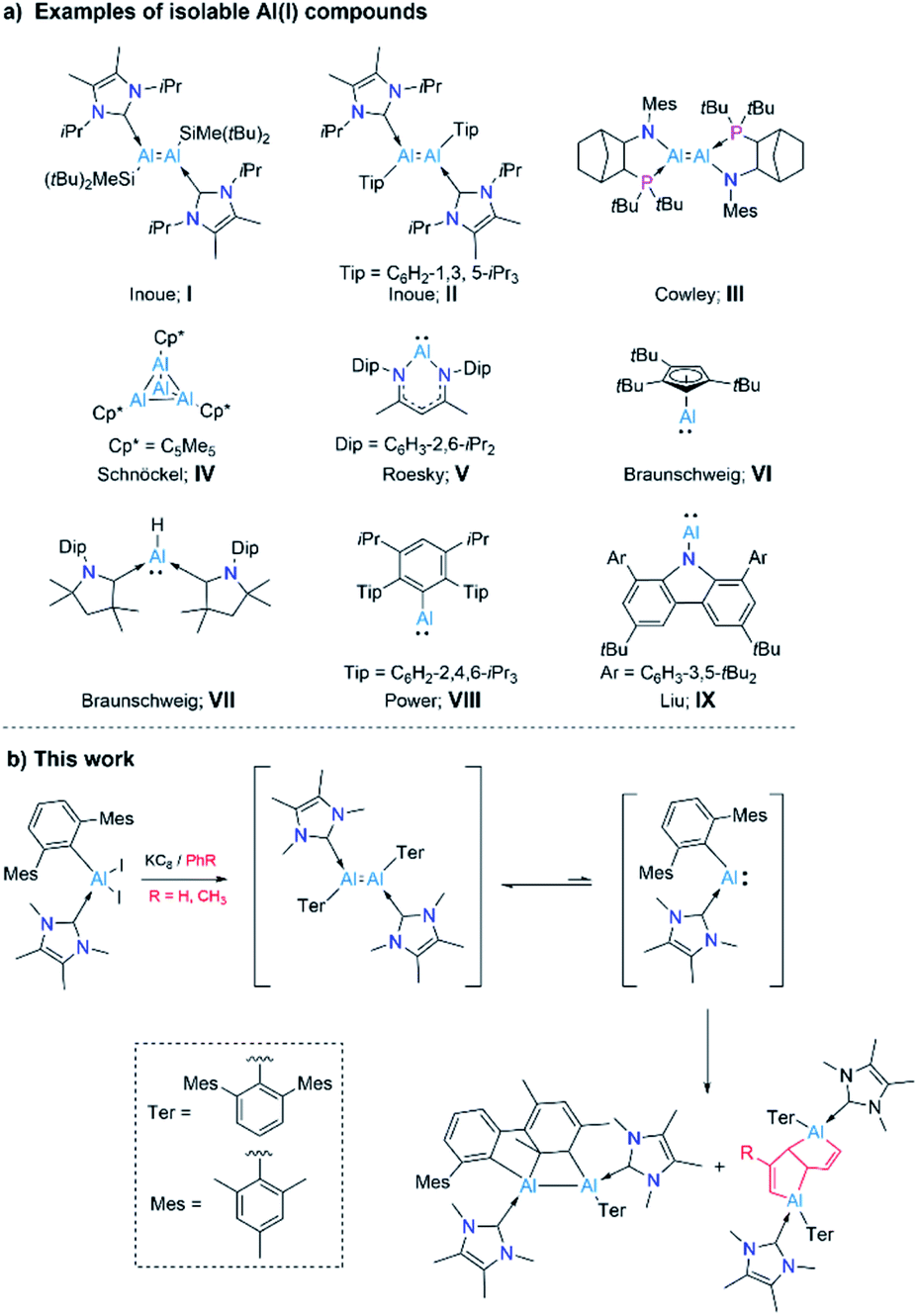 | ||
| Scheme 1 (a) Examples of isolable Al(I) species (I–IX); and (b) synthesis of an arrested dialumene and deconstruction of arenes by a masked alumylene (AlI) presented in this work. | ||
Monovalent aluminium species (AlI) have been known since 1991, with the isolation of the tetrameric AlI species  (Cp* = pentamethylcyclopentadienyl; IV, Scheme 1a),8 which was shown to dissociate into its monomer [AlCp*] upon heating.9 However, the isolation of a AlI compound with a β-diketiminato ligand by Roesky and coworkers (V, Scheme 1a) allowed access to a stable mono-AlI species.10 In 2018, our group reported the compound [(η5-Cp3t)AlI] (VI, Scheme 1a) (Cp3t = 1,3,5-tri-tert-butylcyclopentadienyl), a monomeric species that showed several interesting reactivity patterns.11 We also reported a parent alumylene (i.e., AlIH) species (VII, Scheme 1a) stabilised by two cyclic (alkyl)(amino)carbenes (CAACs) which showed 36% singlet diradical character (AlIII).12 While these discoveries provided mono-AlI species for further reaction, they both bear further stabilising groups, meaning that neither is monocoordinate. A stunning recent discovery by Power et al. presented a first stable, monomeric, base-free, and monocoordinate AlI species (VIII, Scheme 1a), bearing a sterically bulky terphenyl substituent.13 Shortly after this discovery, the groups of Liu and Hinz independently reported the synthesis of another base-free alumylene that carries a sterically hindered amino substituent (IX).14 We note that although various terminologies, including alanediyl,13 aluminylene,14a and aluminyl,7 have been used in the literature for AlI compounds of the type [:AlR(L)n] (n = 0–2), for simplicity and due to their analogy to carbenes and borylenes we use the terminology “alumylenes” for this type of compounds. In addition to these two types of AlI compounds, a third form of AlI species has also emerged relatively recently, namely aluminyl anions of the form [AlR2]−, which adopt diverse structures and show high Al-centered reactivity with electrophiles.15
(Cp* = pentamethylcyclopentadienyl; IV, Scheme 1a),8 which was shown to dissociate into its monomer [AlCp*] upon heating.9 However, the isolation of a AlI compound with a β-diketiminato ligand by Roesky and coworkers (V, Scheme 1a) allowed access to a stable mono-AlI species.10 In 2018, our group reported the compound [(η5-Cp3t)AlI] (VI, Scheme 1a) (Cp3t = 1,3,5-tri-tert-butylcyclopentadienyl), a monomeric species that showed several interesting reactivity patterns.11 We also reported a parent alumylene (i.e., AlIH) species (VII, Scheme 1a) stabilised by two cyclic (alkyl)(amino)carbenes (CAACs) which showed 36% singlet diradical character (AlIII).12 While these discoveries provided mono-AlI species for further reaction, they both bear further stabilising groups, meaning that neither is monocoordinate. A stunning recent discovery by Power et al. presented a first stable, monomeric, base-free, and monocoordinate AlI species (VIII, Scheme 1a), bearing a sterically bulky terphenyl substituent.13 Shortly after this discovery, the groups of Liu and Hinz independently reported the synthesis of another base-free alumylene that carries a sterically hindered amino substituent (IX).14 We note that although various terminologies, including alanediyl,13 aluminylene,14a and aluminyl,7 have been used in the literature for AlI compounds of the type [:AlR(L)n] (n = 0–2), for simplicity and due to their analogy to carbenes and borylenes we use the terminology “alumylenes” for this type of compounds. In addition to these two types of AlI compounds, a third form of AlI species has also emerged relatively recently, namely aluminyl anions of the form [AlR2]−, which adopt diverse structures and show high Al-centered reactivity with electrophiles.15
Despite these developments, examples of AlI species, such as monomeric alumylenes [:AlR], dialumenes ([RAlAlR]) and aluminyl anions ([AlR2]−) are still very scarce due to their highly reactive nature and lack of general synthetic strategies.15b,16 Our research interests within low-valent aluminium chemistry have been in preparing and probing the reactivity of monomeric AlI compounds. Given that a base-free arylalumylene has been isolated,13 while attempts to isolate an NHC-bound analogue led only to isolation of a diaryl dialumene,6b we have been interested in developing a method to prepare an NHC-coordinated arylalumylene. We reasoned that the reduction of an NHC-stabilised dihaloalane bearing a superbulky aryl group (Ar*), i.e. [(NHC)AlX2Ar*] (Ar* = 2,6-C6H3Mes2, Mes = 2,4,6-Me3C6H2, NHC = NHCMe4, X = I) might lead to a stable, monomeric alumylene of the form [(NHC)Ar*Al:] – effectively an acyclic version of Roesky's alumylene (V, Scheme 1a). This study has led instead to the discovery of highly unusual outcomes of reducing a dihaloalane: an intramolecular formal [2 + 2] cycloaddition product by dearomatization of an outlying aryl group (Mes) by the generated dialumene of the form ([(NHC)Ar*AlAlAr*(NHC)]), and bicyclic bis(alane) products arising from the deconstruction of aromatic solvent molecules by the anticipated transient alumylene of the form [(NHC)Ar*Al:] (Scheme 1b). Insights gained through a combination of experiments and computations hint that from the reduction reaction, the initial species formed is a dialumene of the form [(NHC)Ar*AlAlAr*(NHC)], a species responsible for the intramolecular cycloaddition product. An additional hint gained was that the diaryldialumene can unravel into its monomeric alumylene [(NHC)Ar*Al:], which engages in deconstructing the less reactive arenes benzene and toluene.
Results and discussion
We chose a bulky terphenyl moiety (2,6-C6H3Mes2, Mes = 2,4,6-Me3C6H2) and a small N-heterocyclic carbene (1,3,4,5-tetramethylimidazol-2-ylidene, NHCMe4) for our study. The bulky terphenyl-substituted, NHC-coordinated AlIII diiodide precursor 2 was synthesized by iodination of the corresponding hydride, 1, with an excess of methyliodide in toluene at room temperature (Scheme 2).17a | ||
| Scheme 2 Synthesis of compound 2. Mes = 2,4,6-Me3C6H2. | ||
The NHCMe4-substituted terphenylalane 1 was prepared by reacting TerLi18 with the corresponding carbene-stabilised alane.17a The synthetic routes to 1 and 2 are analogous to the procedures of Inoue.17 Formation of 1 and 2 were confirmed by 1H and 27Al{1H} NMR spectroscopy. The 1H NMR signal of the Al-bound hydrides of 1 appeared at high field 4.04 (br, s AlH2, FWHM: 257 Hz) ppm, while a 27Al{1H} NMR signal was found at low field 112.4 ppm compared to that of NHCMe4 AlH3 (1H: 4.45 (br, s AlH2, FWHM: 779 Hz) ppm; 27Al{1H}: 106.5 ppm). These values match well with those of similar reported compounds.6 The identities of 1 and 2 as NHC-stabilised terphenylalanes were further confirmed by their solid-state structures (Fig. 1).
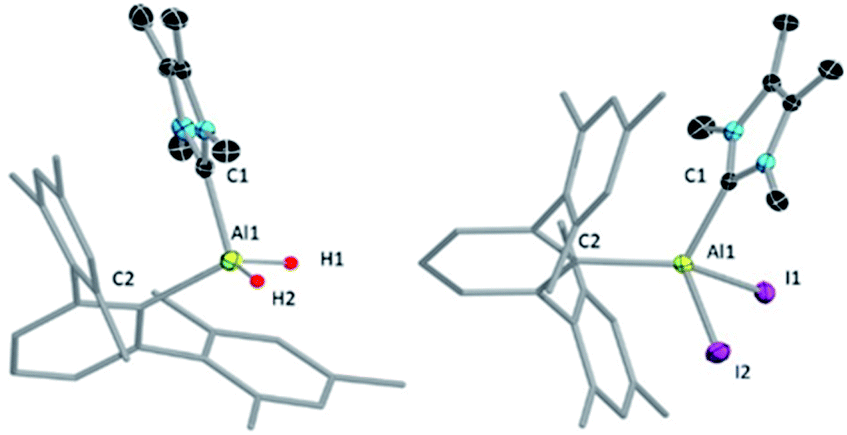 | ||
| Fig. 1 Molecular structures of 1 (left) and 2 (right) with ellipsoids at the 50% probability level. All hydrogen atoms except H1 and H2 are omitted for clarity in 1. All hydrogen atoms along with toluene (solvent of crystallization) are omitted for clarity in 2. The terphenyl substituent in 1 and 2 is depicted in wireframe style for simplicity. Selected bond lengths [Å] and bond angles [°] for 1: Al1–C1 2.040(8), Al1–C2 2.028(1); C1–Al1–C2 106.9(5); for 2: Al1–C1 2.033(6), Al1–C2 1.994(6); C1–Al1–C2 114.2(3). | ||
Reduction of 2 with four equivalents of KC8 at room temperature in benzene led to the compounds 3 and 4 (64% and 36% respectively, relative NMR conversion, Scheme 3). Separation of the compounds was achieved by taking advantage of their differing solubility. Repeated washing of the reaction mixture with hexanes allowed us to separate 4 (5% isolated yield, see ESI† for details). The remaining solid was dissolved in a minimum amount of benzene and allowed to evaporate slowly inside a glovebox to afford bright yellow crystals of 3 (20%, isolated yield). After extraction of 3 and 4, the remaining solution contained a mixture of the two compounds, which we could not separate further. Both 3 and 4 were characterized by multinuclear solution NMR spectroscopy and solid-state molecular structure determination. The 1H NMR signal for the vinylic proton of the activated mesityl group in 3 (attached to C5, Scheme 3) appeared at 5.14 ppm, while the signal for the allylic proton (attached to C1) appeared much more upfield (0.7 ppm) than those of conventional allylic protons (ca. 1.8 ppm). This is likely due to aluminium attachment to the C1 center, making the proton attached to C1 more hydridic. The 13C{1H} NMR spectroscopic signal of the vinylic carbon C5 was found at 116 ppm (as confirmed by HSQC). The 1H NMR spectroscopic signal for the C1,4 vinylic protons of 4 was found at 5.01 ppm (d, 3J = 12 Hz), in the usual range for signals of vinylic protons but slightly upfield compared to that of 3. The signal for the C2,5 vinylic protons was found significantly further downfield (7.32 ppm), which is in line with the 1H resonances of analogous protons in a similar compound reported by Crimmin.19 Solid samples of 4 were poorly soluble while also being unstable in benzene, partly explaining its poor isolated yield. Partial decomposition of 4 was noted after ca. 4 h in the presence of benzene, as indicated by 1H NMR spectroscopy.
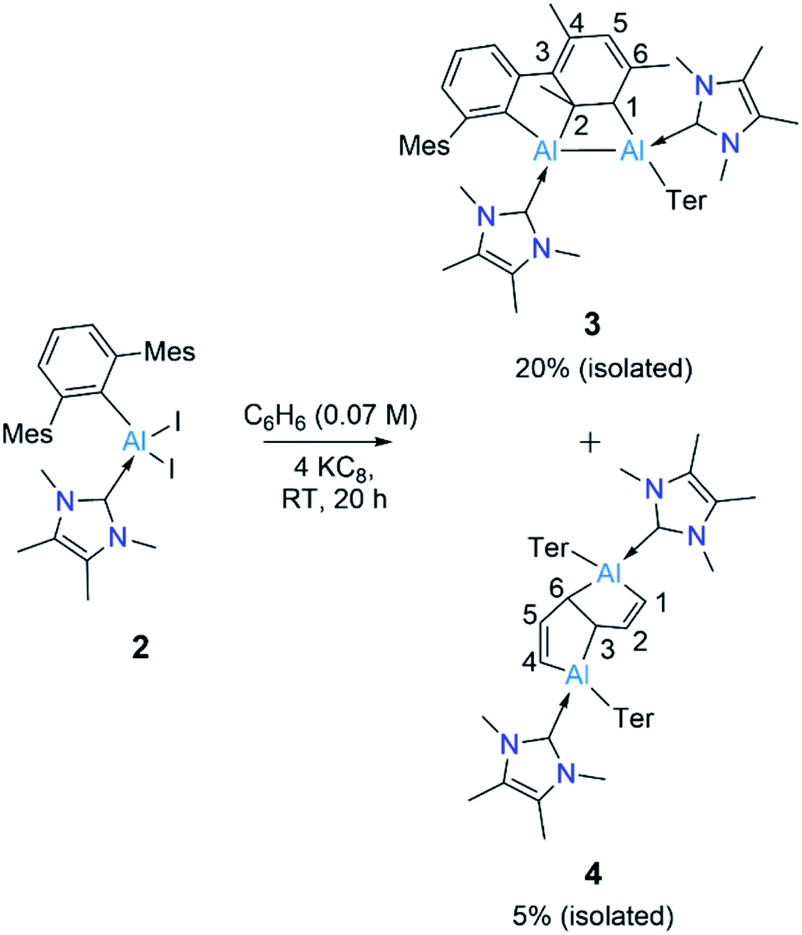 | ||
| Scheme 3 Synthesis of 3 and 4. Ter = 2,6-C6H3Mes2, Mes = 2,4,6-Me3C6H2. | ||
Despite the instability of the latter, X-ray quality single-crystals of 3 and 4 were obtained from saturated benzene solutions. The solid-state structure of 3 (Fig. 2) showed that one of the mesitylene rings loses its planarity (and thus also its aromaticity), forming an Al2C2 four-membered ring. The Al–Al distance in 3 is 2.6091(5) Å, significantly longer than those of the carbene-stabilised dialumene II (AlAl: 2.4039(8) Å)6b and the 1,2-dialuminacyclobutene [R2Al2(CSiMe3)2] (R = 2,6-C6H3Dip2, Dip = 2,6-C6H3iPr2) Al–Al: 2.4946(9) Å,20 slightly shorter than a carbene-stabilised dialuminacyclobutane, [(NHC)(tBu2SiMe)AlCH2CH2Al(NHC)(tBu2SiMe)] (NHC = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) Al–Al 2.6503(10) Å (ref. 6a) but similar to a range of other Al–Al single-bond distances (2.5 to 2.95 Å).16 The four-membered Al2C2 ring deviates from planarity with an Al1–Al2–C2–C1 dihedral angle of 23.51(6)°, which matches well with previously reported Al2C2 rings.20
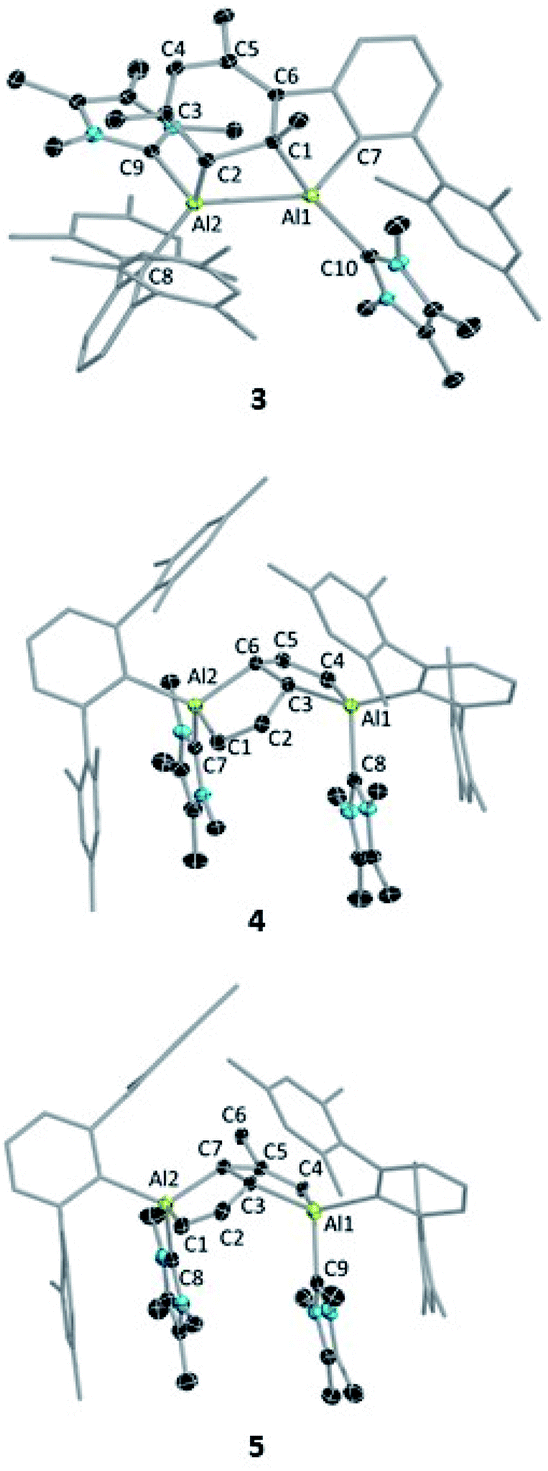 | ||
| Fig. 2 Molecular structures of 3, 4 and 5 with ellipsoids at the 50% probability level. All hydrogen atoms and two benzene molecules in 3 (solvent of crystallization), and one benzene molecule in 5 are omitted for clarity. The terphenyl substituents in all structures are depicted in wireframe style for simplicity. Selected bond lengths [Å] and bond angles [°]: for 3: Al1–Al2 2.6091(5), C3–C4 1.3520(13), C6–C5 1.3662(12), C1–C2 1.5629(12), Al1–C10 2.0828(9), Al2–C9 2.0753(10), Al1–C7 2.0062(9), Al2–C8 2.0605(9), Al2–C2 2.0849(9), Al1–C1 2.0485(9); C7–Al1–C1 91.15(4), Al1–Al2–C2 74.56(3), Al2–Al1–C1 73.82(3), Al1–C1–C2 104.64(5), Al2–C2–C1 100.93(5); for 4: Al1–C3 = Al2–C6 2.0277(12), C3–C2 = C6–C5 1.4862(17), C1–C2 = C4–C5 1.3503(18), Al2–C1 = Al1–C4 1.9591(13), Al1–C8 = Al2–C7 2.0770(13); C6–Al2–C1 = C3–Al1–C4 92.08(5), C2–C3–Al1 = Al2–C6–C5 114.75(9), C2–C3–C6–C5 130.7 (1); for 5: Al1–C3 2.089(15), Al2–C7 1.982(19), C3–C2 1.48(3), C7–C5 1.53(2), C2–C1 1.345(14), C5–C4 1.45(2), Al1–C4 1.910(15), C3–C7 1.576(15); Al1–C3–C2 115.5(11), Al2–C7–C5 126.8(12), C1–Al2–C7 91.2(6), C3–Al1–C4 92.0(5), C2–C3–C7–C5 148.1(1). | ||
The structure of 4 (Fig. 2) exhibits two fused five-membered rings resembling a dihydropentalene structure, connected in an endo fashion (torsion angle Al1–C3–C6–Al2: 118.36(7)°; C2–C3–C6–C5: 130.7(1)°). Each ring of the dihydropentalene structure contains one tetrahedral AlIII center, while the carbon backbone is clearly derived from the activation of the benzene solvent. A similar dearomatized product was observed in the reaction of biphenylene with Roesky's AlI complex V, reported by Crimmin et al.,19 however, this AlI complex does not undergo a similar activation of benzene or toluene.
The ring-opening of benzene derivatives is rare and is generally considered to be very difficult. An early work demonstrating the ring-opening of benzene by a main-group compound was reported by Meller in 1988 on the reduction of a dihaloaminoborane in a benzene/dimethoxyethane solvent mixture, which led to the formation of a diboron derivative of pentalene.21 In 2019, the groups of Goicoechea and Aldridge presented the reversible insertion of a reactive aluminium anion into benzene at 80 °C, the product of which was trapped using Me2SnCl2.22 In our case, to confirm that the solvent was the source of the backbone of the product 4, we repeated the reaction in toluene at room temperature. The 1H NMR spectrum of the reaction mixture showed the formation of 3 (ca. 68%) along with two new peaks, a doublet of doublets and a singlet in the vinyl region, indicating the formation of 5 (ca. 28%, Scheme 4). Compound 5 is more soluble than 3, allowing its isolation by hexane washing and crystallization (3% isolated yield). The remaining yellow solid was dried, providing 3 in 40% isolated yield. Repeating the reaction with a short reaction time (ca. 10 h) and starting at −78 °C led to greater selectivity for 3, allowing its isolation in 70% yield. The 1H NMR spectrum of 5 exhibited two characteristic signals for the vinyl protons attached to C1 and C4 at 5.01 ppm (dd, 4J = 2 Hz, 3J = 12 Hz) and 4.83 ppm (s), respectively. The signal for the proton attached to C2 was found further downfield at 7.34 ppm as doublet-of-doublets (3J = 3 Hz (coupling with bridging proton, confirmed by COSY), 3J = 12 Hz (coupling with vinylic proton, confirmed by COSY)). Like 4, 5 has a similar endo geometry but unlike 4 it has no center of inversion as observed from its solid-state structure (Fig. 2). The two localized C–C double bonds significantly differ in length: the length of C2–C1 (1.345(14) Å) is closer to that of the C2–C1 bond distance of 4, whereas the C5–C4 distance in 5 (1.45(2) Å) is longer than typical C–C double bonds. The structures of 4 and 5 indicate that they arise from the deconstruction of benzene and toluene, respectively, by a low-valent aluminium species. To further support this, and to provide 3 more selectively, we performed the reduction of 2 in hexane (Scheme 4). As expected, this reaction resulted in 3 almost exclusively according to 1H NMR of the crude reaction mixture and afforded it in 90% isolated yield.
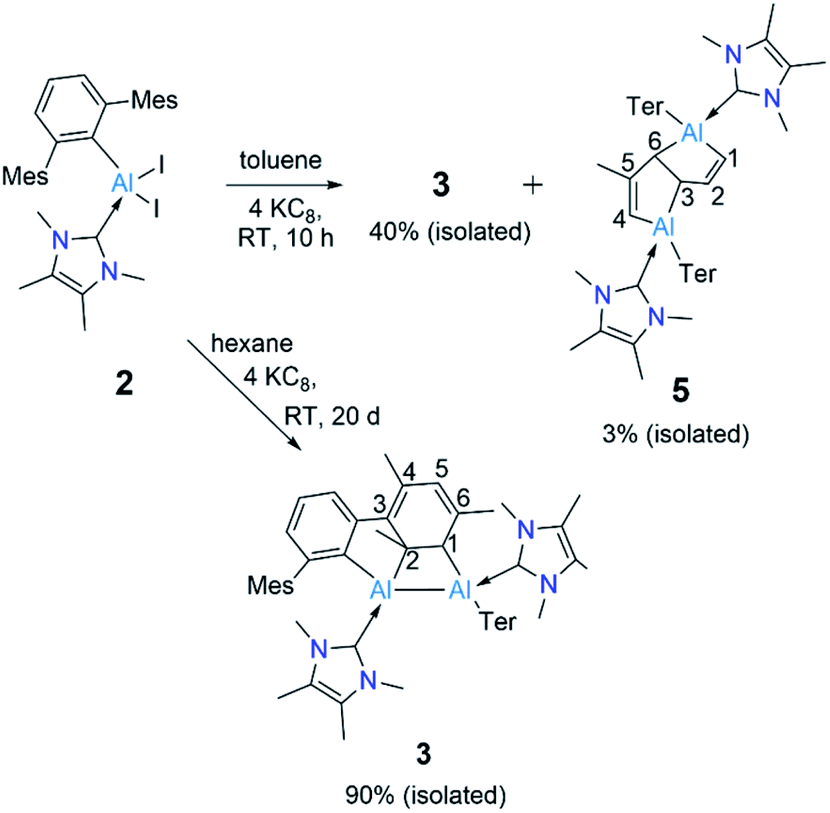 | ||
| Scheme 4 Reduction of 2 in toluene and hexanes to obtain 3 and 5. (Ter = 2,6-C6H3Mes2, Mes = 2,4,6-Me3C6H2.) | ||
Compound 3 can be thought of as a pseudo-dialumene internally stabilised by [2 + 2] cycloaddition between an AlAl bond and the π system of a peripheral mesitylene ring. Earlier studies by Power5a and Tokitoh5b independently disclosed the synthesis of bicyclic masked base-free dialumenes formed by [4 + 2] cycloaddition of toluene or benzene with the AlAl bond of intermediate dialumenes. It is worth mentioning that while [2 + 2] cycloaddition is symmetry forbidden, it can occur with bent double bonds between heavier main group elements,23 which show “charge-shift” bonding having substantial amounts of second-order Jahn–Teller effects. Doubly base-coordinated compounds with bent AlAl bonds are also an exception for the symmetry forbidden [2 + 2] cycloaddition reactions, as several examples of addition reactions have recently been reported with substrates such as alkynes, alkenes and CO2.6 However, thus far, there is no precedence demonstrating an addition reaction with arenes. Compound 3 serves as the first example in this regard, though the addition is an intramolecular one.
We presume that dialumene 6 is the initial species formed from the two-fold reduction of 2 before it transforms to 3. We suggest this because dialumene II was the main product of reducing an NHC-coordinated diiodoalane supported by the relatively small aryl group 2,4,6-tri-iso-propylphenyl (Tipp).6b To ascertain if dialumene 6 is the initially formed species, attempts were made to halt the reduction at the formation of the diiodoalane intermediate [(NHC)(I)Ar*Al–AlAr*(I)(NHC)] by employing substoichiometric amounts of KC8 (1.5 equiv) at room temperature. Calculations performed at the PBE1PBE-D3BJ/Def2TZVP/SMD(benzene)//PBE1PBE-D3BJ/Def2SVP level of theory on this reaction hinted that diiododialane formation from the homocoupling of the early NHC-stabilised iodo(aryl)aluminyl radical [(NHC)(I)Ar*Al]˙ is highly likely, as the coupled species is markedly stable (ΔG = −25.2 kcal mol−1). Unfortunately, experiments invariably formed 3 and the starting material in a ratio ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, contrasting the results of reducing a base-free diiodoarylalane, wherein the diiododialane is the primary product.5a This may have resulted from reducing the in situ generated diiododialane to dialumene 6 more rapidly than the starting material 2.
:3, contrasting the results of reducing a base-free diiodoarylalane, wherein the diiododialane is the primary product.5a This may have resulted from reducing the in situ generated diiododialane to dialumene 6 more rapidly than the starting material 2.
Since compound 3 can be viewed as a masked dialumene, we anticipated that simple heating might allow the generation of a dialumene. However, no decomposition or changes were observed during a variable-temperature NMR experiment on a benzene solution of 3 from 25 °C up to 100 °C. Prolonged heating at 100 °C (24 h) led to the formation of 8, the product of a formal intermolecular C–H activation (Scheme 5), along with a small batch of crystals of 4. The reduction of 2 in benzene at 100 °C also provided a similar outcome, with 8 as the major product. The 1H NMR spectrum of 8 showed a distinctive, broad signal (FWHM = 129 Hz) at 4.41 ppm corresponding to the Al-bound hydride, which is somewhat downfield of that of aluminium hydride 1 but in the range of similar carbene-stabilised dialumene hydrides.6 A broad signal (FWHM = 75 Hz) at 1.68 ppm was also observed corresponding to the newly formed methylene group. Interestingly, in the solid-state structure of 8 (Fig. 3), the Al–Al bond (Al1–Al2 2.6228(5) Å) was found to be significantly longer than that of 3 (Al–Al: 2.6091(5) Å (Fig. 3)).
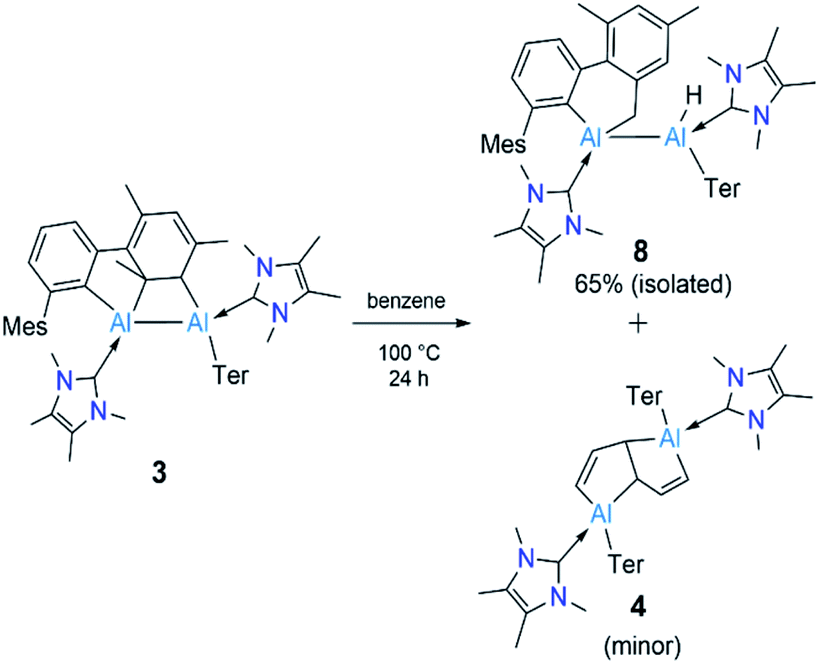 | ||
| Scheme 5 Thermolysis of 3 to form 8 and 4. Ter = 2,6-C6H3Mes2, Mes = 2,4,6-Me3C6H2. | ||
 | ||
| Fig. 3 Molecular structure of 8 with ellipsoids at the 50% probability level. All but one hydrogen atom and a single hexane molecule (solvent of crystallization) are omitted for clarity. The terphenyl substituent and residues of the C–H activated terphenyl are depicted in wireframe style for simplicity. Selected bond lengths [Å] and bond angles [°]: Al1–Al2 2.6228 (5), Al1–C4 2.0331(14), Al1–C3 2.0866(14), Al1–C8 2.0449(14), Al2–C1 2.0638(14), Al2–C2 2.07840(15); Al1–C4–C5 105.69(9), C8–Al1–C4 90.90(6), C3–Al1–Al2 113.22(4), C3–Al1–C8 107.19(6), C2–Al2–C1 105.12(6), C2–Al2–Al1–C3 173.03(7), C8–Al1–Al2–C1 178.57(7). | ||
The outcome of this experiment suggests that upon heating, 3 first reverts to 6, which then transforms to 8 (Fig. 4). In other words, 3 is a kinetic product and 8 is a thermodynamic product from 6. The thermodynamics computed for 3, 6 and 8 are in line with our experimental observation that 3 is significantly more stable than 6 (ΔG = −13.0 kcal mol−1), while 8 is substantially more stable than 3 (ΔG = −24.4 kcal mol−1) and 6 (ΔG = −37.4 kcal mol−1). It can be assumed that 3 forms via an intramolecular [2 + 2] cycloaddition in 6. For 8, however, given that the added methylene and hydride functionalities in 8 are trans-oriented, a straightforward C–H activation across the AlAl bond in 6 may not be viable as it can only lead to a cis-isomer of 8. Conversion of this cis-isomer to the obtained trans-isomer via rotation of the Al–Al single bond may not be feasible due to sterics. Thus, we suggest 8 forms via an intramolecular C–H activation in alumylene 7 that was generated from the dissociation of dialumene 6, leading to a hydridoalane intermediate (9), followed by a formal insertion of another 7 into the Al–H bond in 9 (Fig. 4). Only a minor amount of the pentalene product 4 obtained from the same thermal reaction signals that the dialumene 6 generated from the reduction reaction somehow reacts with benzene before self-stabilising to form 3. Even for this reaction, we propose the involvement of alumylene 7via dissociation of 6 (Fig. 4).
 | ||
| Fig. 4 Plausible route to the C–H activation product 8 and the benzene-deconstructed product 4 from masked dialumene 3. Ter = 2,6-C6H3Mes2, Mes = 2,4,6-Me3C6H2. Free energy differences (ΔG), in kcal mol−1, computed among all species are given in parentheses. | ||
Compelling evidence for the dissociation of compounds with heavier main group multiple bonds under mild conditions has been known for nearly five decades.24 However, only very recently, a report by Krämer and Cowley demonstrated the reversible dissociation of a doubly base-stabilised dialumene, III, for which the dissociation energy (ΔGdiss) was calculated to be small (7.1 kcal mol−1).7 The ΔGdiss computed for dialumene 6 is 17.3 kcal mol−1, which is lower than the ΔGdiss of dialumene II (29.8 kcal mol−1). This suggests 6 can easily dissociate into 7 at room temperature.§ Experimental evidence for the dissociation of 6 comprises: (i) a low yield of 5 and a high yield of 3 when the reduction was carried out at low temperatures in toluene (vide supra). The low yield of 5 can be primarily attributed to suppressing the dissociation of 6 into 7 at low temperatures; and (ii) conducting the reduction in a much more dilute benzene solution (0.02 M), for which the 1H NMR spectrum showed the formation of a greater proportion of 4 (65%) than 3 (35%).¶ Under these dilute conditions, alumylene 7, which is dissociated from 6 can persist long enough to react with benzene to form the deconstruction product 4. From comparing the outcomes of the reduction of 2 at two different temperatures and that of the elevated temperature reaction of 3, it becomes evident that the room-temperature reduction reaction is the optimal condition to obtain arene deconstruction products in good yields.
Given that a stable base-free arylalumylene has been isolated recently, and this does not seem to react with benzene,13 the fact that our base-stabilised aryl alumylene 7 is transient and tends to react with benzene and toluene suggests the presence of a crucial disparity in the frontier orbitals between base-stabilised aryl alumylenes and their respective base-free versions. The computed structure of 7 and its frontier orbitals are shown in Fig. 5. Analogous data for the corresponding base-free alumylene is provided in the ESI (Fig. S34†). The frontier orbitals of 7 display that the HOMO and LUMO are both located on the aluminium center and that the HOMO–LUMO gap (HLG) is 3.232 eV (Fig. 5), which is a significantly decreased energy gap than that found in its base free alumylene (HLG = 3.818 eV, Fig. S34†). Importantly, NHC coordination raises the HOMO energy. This is in accord with the proposal by Liu25 that occupation of the LUMO of free alumylenes is a prerequisite to altering the HOMO energy and enhancing their reactivity.
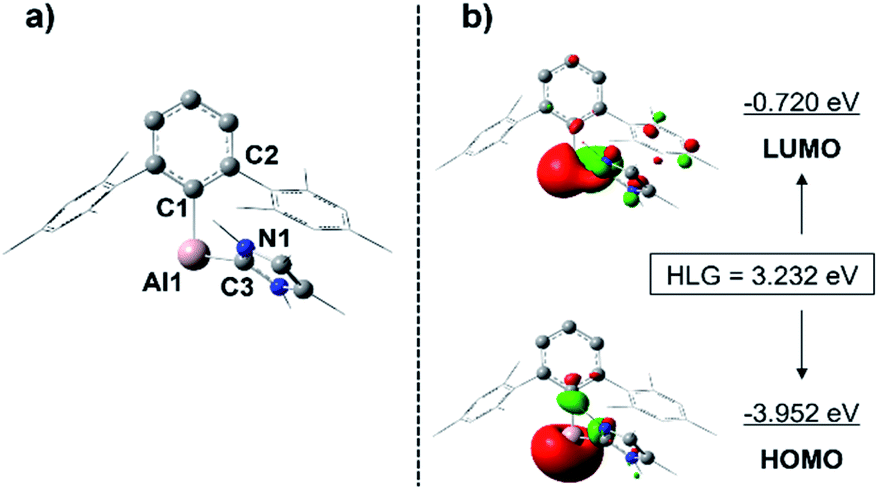 | ||
| Fig. 5 Computed structure (a) and frontier orbitals (±0.05 isovalue) (b) of alumylene 7. HLG: HOMO–LUMO gap. Hydrogens are omitted for clarity. Mesityl groups of the terphenyl substituent and the methyl groups of the NHC are depicted in wireframe style for simplicity. Selected bond lengths [Å] and bond angles [°]: Al1–C1 2.072, Al1–C3 2.128, C1–Al1–C3 96.4, C2–C1–Al1–C3 69.2, N1–C3–Al1–C1 61.6. | ||
For the pentalene product 4 formed from alumylene 7 and benzene, two mechanistic pathways, as shown in Fig. 6, have been conceived and explored computationally. We utilized a truncated terphenyl group for these calculations where the mesityl groups were replaced with 2,6-dimethylphenyl groups and NHCMe4 was replaced by NHCMe2 (commonly known as IMe).|| Note that the identification numbers of the computational model compounds are derived from their experimental counterparts, with the addition of a prime symbol. The first pathway entails a (1 + 4) cycloaddition between alumylene and benzene, followed by a (1 + 2) cycloaddition by the second alumylene and subsequent transformation (black profile). This pathway follows the mechanistic steps proposed by Crimmin et al. to deconstruct biphenylene by Roesky's alumylene V.19 The second pathway involves an initial formal insertion of alumylene into the aromatic C–C bond of benzene, followed by a second alumylene addition and subsequent transformation (blue profile). The insertion route of the latter pathway was assumed from the report of Aldridge et al., which demonstrates the reversible insertion of an anionic AlI compound into the benzene ring.22 The mechanistic study results reveal that both pathways are feasible under the employed reaction conditions and that they may both be (almost equally) competing to form the pentalene product 4.
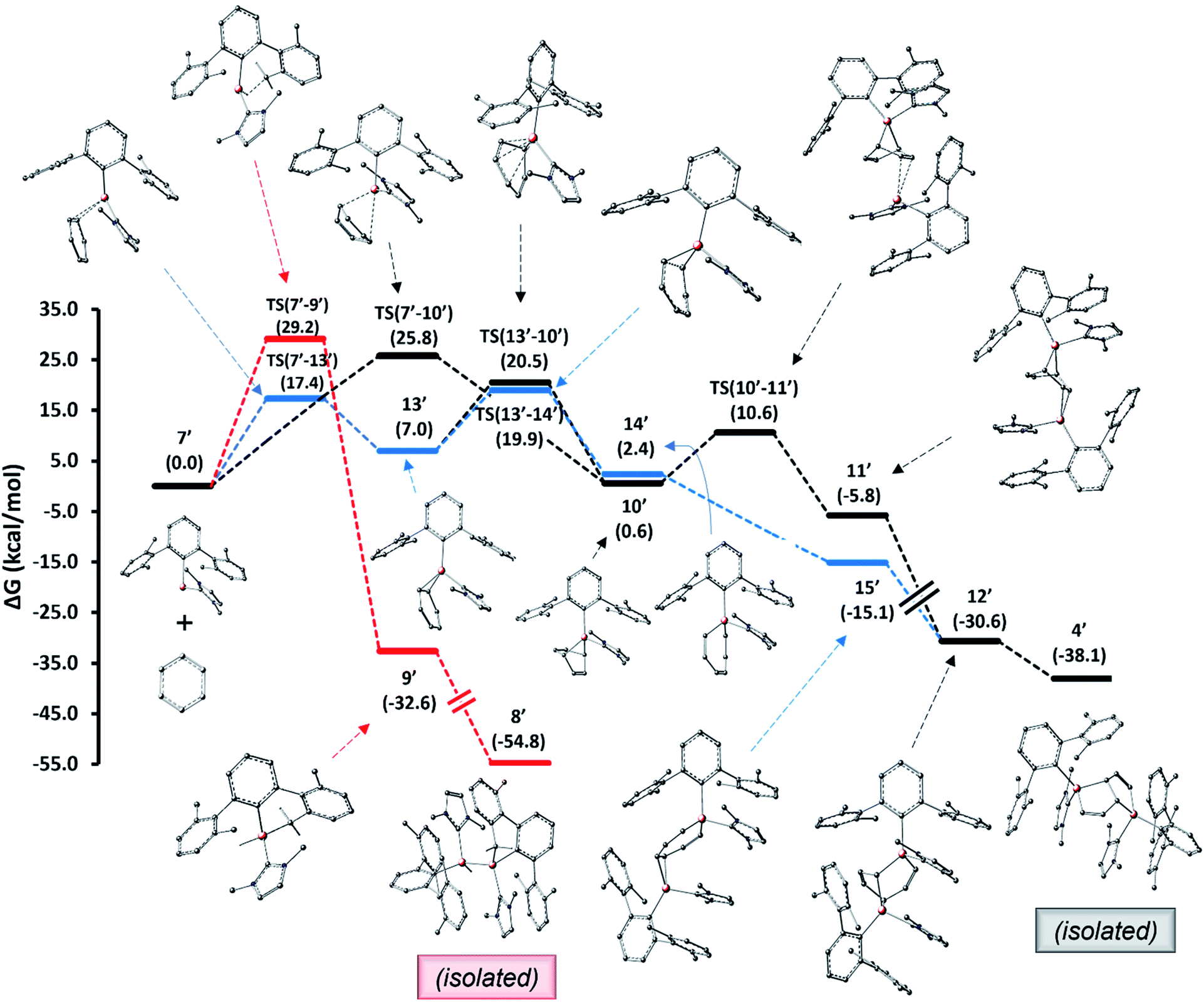 | ||
| Fig. 6 Mechanisms computed for the formation of benzene deconstruction product 4 (black and blue profiles) and the C–H activation product 8 (red profile) from alumylene 7. Free energies in parentheses are in kcal mol−1. | ||
The computed pathways show that the initial process involved in the reaction of alumylene 7′ with benzene is a [1 + 2] cycloaddition, leading to the alumirane intermediate 13′. Intriguingly, this intermediate presents similar kinetics and thermodynamics in the next stage to form the bicyclic intermediate 10′ and the insertion intermediate 14′. This paves the way for divergent pathways for the arene deconstruction reaction. Forming 10′ from 7′ and benzene directly via the [1 + 4] cycloaddition transition state TS(7′–10′), as proposed by Crimmin for the biphenylene deconstruction,19 can be less regarded, as this route holds a relatively large activation barrier. As intermediates 10′ and 14′ lie only slightly uphill from their starting materials, 7′ and benzene, and hold easily attainable, reversible activation barriers, we propose they form reversibly from their starting materials. In contrast, the C–H activation intermediate 9′ (red profile), which we presumed to form at high temperatures from 7′ and to be responsible for the formation of 8′, while having a larger barrier than predicted for forming 10′ and 14′, lies significantly lower in energy from its starting material 7′; thus, the reversibility of this step is less likely. These disparities explain why 8 was the major product when the reduction was carried out at high temperatures and when the benzene solution of 3 was heated.
Conclusions
We report that reducing an NHC-stabilised diiodoalane bearing a sterically encumbered aryl substituent, 2,6-dimesitylphenyl, in different hydrocarbon solvents leads to different outcomes. In hexanes, a highly unusual self-stabilised dialumene species was the product, instead of the anticipated acyclic, dicoordinate alumylene (AlI). In benzene or toluene, the primary product was a dialuminadihydropentalene arising from C–C bond activation and deconstruction of the respective arene solvent. Subsequent probing through experiments and DFT calculations showed that the self-stabilised dialumene and its dialumene precursor dissociate to form the initially-sought alumylene before reacting with benzene and toluene. This reactivity of the transient alumylene suggests that it is more reactive than the previously reported di- and monocoordinate AlI species of Roesky and Power, respectively. We envisage that careful tuning of the aryl and base groups could enable isolation of a similar base-stabilised alumylene and allow the full exploitation of the fascinating reactivity of this family of compounds.Data availability
Full experimental and computational details are provided as part of the ESI.†Author contributions
H. B. supervised the project. D. D. carried out the synthetic work. D. D. and M. H. carried out the X-ray crystallographic analyses. A. J. carried out the computational studies. D. D., R. D. D. and A. J. prepared the manuscript. D. D. and A. J. prepared the ESI. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft is gratefully acknowledged. D. D. and A. J. thank the Alexander von Humboldt Foundation for postdoctoral fellowships. A. J. also thanks the Natural Sciences and Engineering Research Council (Canada) for a postdoctoral fellowship. We thank Mr Christoph Mahler for helping with mass spectrometry and Ms Laura Wolz for the VT-NMR data collection.Notes and references
- S. Aldridge and A. J. Downs, The Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities, Wiley, Chichester, 2011 Search PubMed.
- (a) C. Dohmeier, D. Loos and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1996, 35, 129 CrossRef CAS; (b) G. V. Chertihin and L. Andrews, J. Phys. Chem., 1993, 97, 10295 CrossRef CAS.
- W. Uhl, Z. Naturforsch.,B:J.Chem.Sci., 1988, 43, 1113 CrossRef CAS.
- (a) A. Purath, C. Dohmeier, A. Ecker, H. Schnöckel, K. Amelunxen, T. Passler and N. Wiberg, Organometallics, 1998, 17, 1894 CrossRef CAS; (b) A. Purath and H. Schnöckel, J. Organomet. Chem., 1999, 579, 373 CrossRef CAS; (c) M. Schiefer, N. D. Reddy, H. W. Roesky and D. Vidovic, Organometallics, 2003, 22, 3637 CrossRef CAS; (d) C. Schnitter, H. W. Roesky, C. Röpken, R. Herbst-Irmer, H.-G. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 1998, 37, 1952 CrossRef CAS; (e) S. J. Bonyhady, D. Collis, G. Frenking, N. Holzmann, C. Jones and A. Stasch, Nat. Chem., 2010, 2, 865 CrossRef CAS PubMed.
- (a) R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 10784 CrossRef CAS PubMed; (b) T. Agou, K. Nagata and N. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10818 CrossRef CAS PubMed.
- (a) P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384 CrossRef CAS PubMed; (b) C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Chem. Sci., 2020, 11, 4817 RSC; (c) C. Weetman, P. Bag, T. Szilvási, C. Jandl and S. Inoue, Angew. Chem., Int. Ed., 2019, 58, 10961 CrossRef CAS PubMed.
- R. L. Falconer, K. M. Byrne, G. S. Nichol, T. Krämer and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 24702 CrossRef CAS PubMed.
- C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1991, 30, 564 CrossRef.
- S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke and A. Kuhn, Angew. Chem., Int. Ed. Engl., 1993, 32, 1729 CrossRef.
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274 CrossRef CAS PubMed.
- (a) A. Hofmann, T. Tröster, T. Kupfer and H. Braunschweig, Chem. Sci., 2019, 10, 3421 RSC; (b) M. Fischer, S. Nees, T. Kupfer, J. T. Goettel, H. Braunschweig and C. Hering-Junghans, J. Am. Chem. Soc., 2021, 143, 4106 CrossRef CAS PubMed; (c) S. Nees, F. Fantuzzi, T. Wellnitz, M. Fischer, J. E. Siewert, J. T. Goettel, A. Hofmann, M. Härterich, H. Braunschweig and C. Hering-Junghans, Angew. Chem., Int. Ed., 2021, 60, 24318 CrossRef CAS PubMed.
- S. K. Mellerup, Y. Cui, F. Fantuzzi, P. Schmid, J. T. Goettel, G. Bélanger-Chabot, M. Arrowsmith, I. Krummenacher, Q. Ye, V. Engel, B. Engels and H. Braunschweig, J. Am. Chem. Soc., 2019, 141, 16954 CrossRef CAS PubMed.
- J. D. Queen, A. Lehmann, J. C. Fettinger, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2020, 142, 20554 CrossRef CAS PubMed.
- (a) X. Zhang and L. L. Liu, Angew. Chem., Int. Ed., 2021, 60, 27062 CrossRef CAS PubMed; (b) A. Hinz and M. P. Müller, Chem. Commun., 2021, 57, 12532 RSC.
- (a) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92 CrossRef CAS PubMed; (b) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702 CrossRef CAS PubMed.
- (a) P. Bag, C. Weetman and S. Inoue, Angew. Chem., Int. Ed., 2018, 57, 14394 CrossRef CAS PubMed; (b) K. Hobson, C. J. Carmalt and C. Bakewell, Chem. Sci., 2020, 11, 6942 RSC.
- (a) See the ESI† for details;; (b) H. Xu, C. Weetman, F. Hanusch and S. Inoue, Chem.–Eur. J., 2021, e202104042 Search PubMed.
- D. Dhara, M. Mandal, A. Maiti, C. B. Yildiz, P. Kalita, N. Chrysochos, C. Schulzke, V. Chandrasekhar and A. Jana, Dalton Trans., 2016, 45, 19290 RSC.
- R. Y. Kong and M. R. Crimmin, Angew. Chem., Int. Ed., 2021, 60, 2619 CrossRef CAS PubMed.
- C. Cui, X. Li, C. Wang, J. Zhang, J. Cheng and X. Zhu, Angew. Chem., Int. Ed., 2006, 45, 2245 CrossRef CAS PubMed.
- A. Meller, D. Bromm, W. Maringgele, D. Böhler and G. Elter, J. Organomet. Chem., 1988, 347, 11 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000 CrossRef CAS PubMed.
- (a) P. P. Power, Organometallics, 2020, 39, 4127 CrossRef CAS; (b) T. L. Allen, W. H. Fink and P. P. Power, J. Chem. Soc., Dalton Trans., 2000, 407 RSC; (c) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877 CrossRef CAS PubMed; (d) P. P. Power, Acc. Chem. Res., 2011, 44, 627 CrossRef CAS PubMed; (e) C. A. Caputo, Z. Zhu, Z. D. Brown, J. C. Fettinger and P. P. Power, Chem. Commun., 2011, 47, 7506 RSC; (f) C. A. Caputo, J. Koivistoinen, J. Moilanen, J. N. Boynton, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2013, 135, 1952 CrossRef CAS PubMed; (g) T. Sasamori, T. Sugahara, T. Agou, K. Sugamata, J. D. Guo, S. Nagase and N. Tokitoh, Chem. Sci., 2015, 6, 5526 RSC; (h) G. Trinquier and J. P. Malrieu, J. Am. Chem. Soc., 1987, 109, 5303 CrossRef CAS.
- See, for example: P. J. Davidson and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1973, 317a RSC.
- X. Zhang and L. L. Liu, Angew. Chem., Int. Ed., 2022, 61, e202116658 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR and UV-vis spectra, X-ray crystallographic and computational details. CCDC 2107311, 2107312, 2107317, 2107329, 2107337, 2107370. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01436j |
| ‡ These authors contributed equally. |
| § The ground state for alumylene 7 is singlet (ΔGsinglet–triplet = 17.0 kcal mol−1). |
| ¶ Note that the proportion mentioned is only a relative ratio as per 1H NMR spectroscopic data, and it is not scaled against any external standard. |
| || A few mechanistic steps computed on both pathways using the full experimental system displayed similar energetics to the truncated model, suggesting that the model used can reliably represent the pathways followed by the experimental system. |
| This journal is © The Royal Society of Chemistry 2022 |