
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective construction of cis-hydroindole scaffolds via an asymmetric inverse-electron-demand Diels–Alder reaction: application to the formal total synthesis of (+)-minovincine†
Fangqing
Zhang
ab,
Bing-Tao
Ren
b,
Yuqiao
Zhou
 c,
Yangbin
Liu
*b and
Xiaoming
Feng
*bc
c,
Yangbin
Liu
*b and
Xiaoming
Feng
*bc
aState Key Laboratory of Chemical Oncogenomics, Guangdong Provincial Key Laboratory of Chemical Genomics, Peking University Shenzhen Graduate School, Shenzhen, Guangdong 518055, China
bShenzhen Bay Laboratory, Shenzhen, 518055, China. E-mail: liuyb@szbl.ac.cn
cKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, 610064, China. E-mail: xmfeng@scu.edu.cn
First published on 15th April 2022
Abstract
cis-Hydroindole scaffolds widely exist in a large number of natural products, pharmaceuticals, and organocatalysts. Therefore, the development of efficient and enantioselective methods for the construction of cis-hydroindoles is of great interest and importance. Herein, a novel approach for the enantioselective synthesis of cis-hydroindole scaffolds has been realized through a chiral N,N′-dioxide/Mg(OTf)2 complex catalyzed asymmetric inverse-electron-demand Diels–Alder (IEDDA) reaction of 2-pyrones and cyclic enamines. A series of substituted cis-hydroindole derivatives bearing multiple contiguous stereocenters and functional groups were obtained in good to excellent yields and enantioselectivities (up to 99% yield, and 95% ee) under mild reaction conditions. Moreover, the enantioselective formal total synthesis of (+)-minovincine was concisely furnished with high efficiency and stereoselectivity to demonstrate the synthetic potential of this method.
Introduction
Chiral cis-hydroindole is a privileged scaffold present in numerous biologically active natural products1–8 such as minovincine, kopsinine, vindolinine, and aeruginosin 298-A, pharmaceutical products9 such as the antihypertensive drug perindopril, and proline analogue organocatalysts10,11 (Scheme 1a). Complex molecular architectures and fascinating biological properties have long motivated the development of synthetic methods towards enantioselective construction of chiral cis-hydroindoles.12–15 In this context, most strategies for the stereoselective construction of cis-hydroindoles are primarily based on using optically active starting materials.12,13a,b,13d In contrast, catalytic asymmetric reactions that rely on the use of readily accessible prochiral substrates to achieve enantioenriched cis-hydroindoles are still relatively rare. Mechanistically, these synthetic tactics are largely carried out by asymmetric (aza-)Michael additions.13c,14,15 Therefore, the development of a general and novel strategy for concise and efficient manipulation of densely functionalized cis-hydroindole derivatives with multiple stereocenters remains a significant challenge.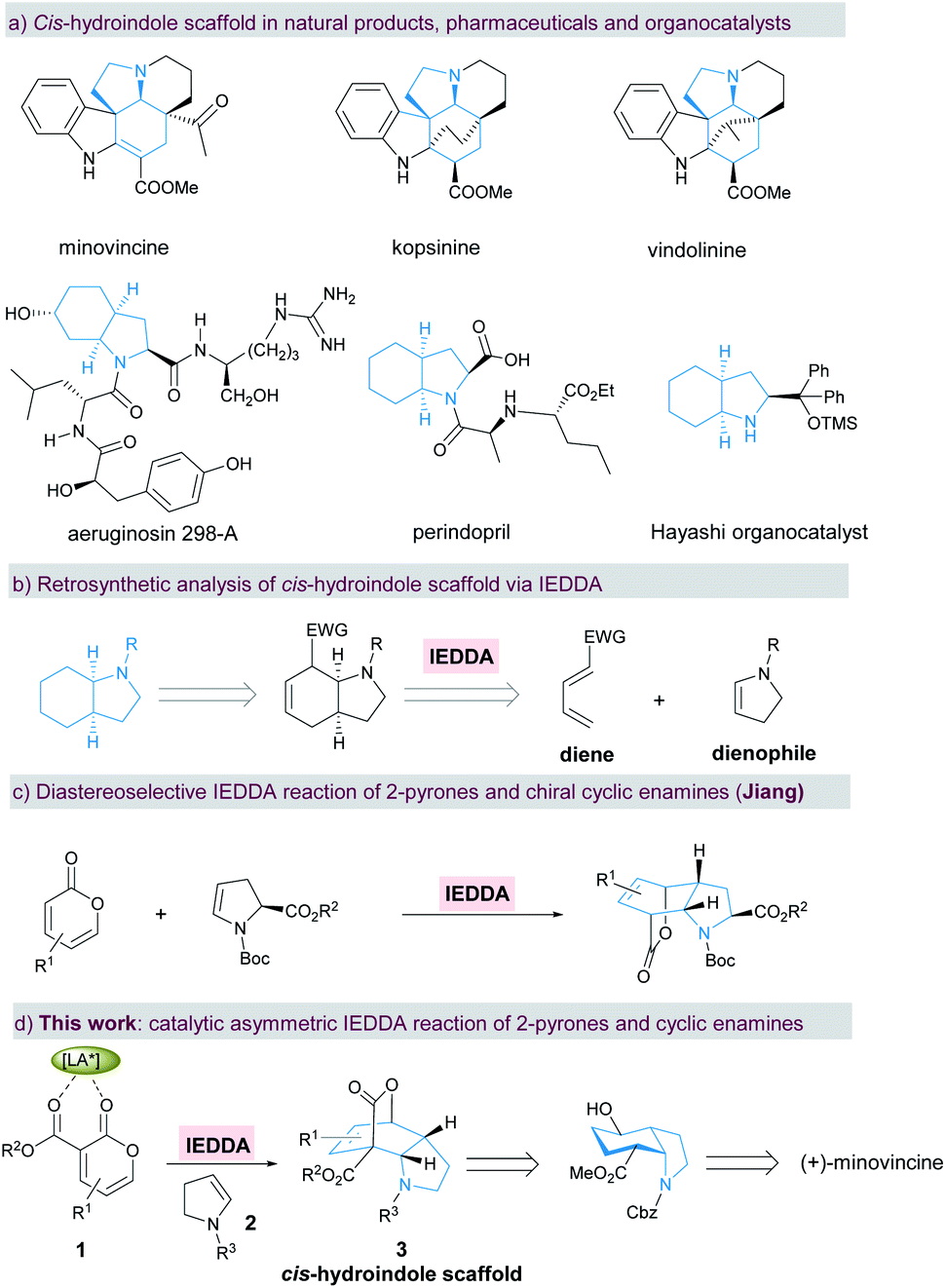 | ||
| Scheme 1 Enantioselective synthesis of the cis-hydroindole scaffold. | ||
As one of the most important and fundamental reactions in organic chemistry, the Diels–Alder reaction between a conjugated diene and dienophile is widely applied to construct a six-membered carbo/hetero-cyclic ring.16,17 By retrosynthetic analysis of cis-hydroindole, this chiral motif can be readily assembled from an electron-deficient diene and electron-rich cyclic enamine18via an enantioselective inverse-electron-demand Diels–Alder (IEDDA) reaction (Scheme 1b). Simultaneously, multiple stereocenters and dense functionalities can also be conveniently introduced into the resulting cis-hydroindole scaffolds in a single-step, which could be used for further functional group transformations and natural product synthesis. Due to the hemi-aromatic and adjustable electronic properties, electron-deficient 2-pyrone has become a favored diene component in the IEDDA reaction with a wide range of applications in aromatic compounds and complex natural product synthesis.19,20 Particularly, the Cai group demonstrated the enantioselective IEDDA reaction of 3-carboalkoxyl-2-pyrone with electron-rich dienophiles, such as 2,2-dimethyl-1,3-dioxole,20g silyl cyclohexadienol20i and 1-naphthyl acetylenes,20j affording the products in high yield, excellent ee, and high dr. In spite of the above achievements, catalytic asymmetric synthesis of cis-hydroindoles via the enantioselective IEDDA reaction of 2-pyrones with cyclic enamines is still in its infancy so far. Recently, Jiang and co-workers disclosed an elegant diastereoselective IEDDA reaction of electron-deficient 2-pyrones with chiral cyclic enamines, affording the bridged cis-hydroindole derivatives in high yield with a moderate exo/endo ratio (Scheme 1c).21 Herein, we describe our efforts towards an enantioselective IEDDA reaction catalyzed by the chiral N,N′-dioxide/Mg(OTf)2 complex22 using 3-carboalkoxyl-2-pyrone 1 and cyclic enamine 2 as the reaction partners (Scheme 1d). This reaction provided a facile and rapid route to access the bridged cis-hydroindole motif bearing four contiguous stereocenters with excellent levels of diastereo- and enantioselectivity. Furthermore, a formal total synthesis of bioactive (+)-minovincine alkaloid was furnished concisely and enantioselectively by subsequent transformation of the enantiomerically enriched products.
Results and discussion
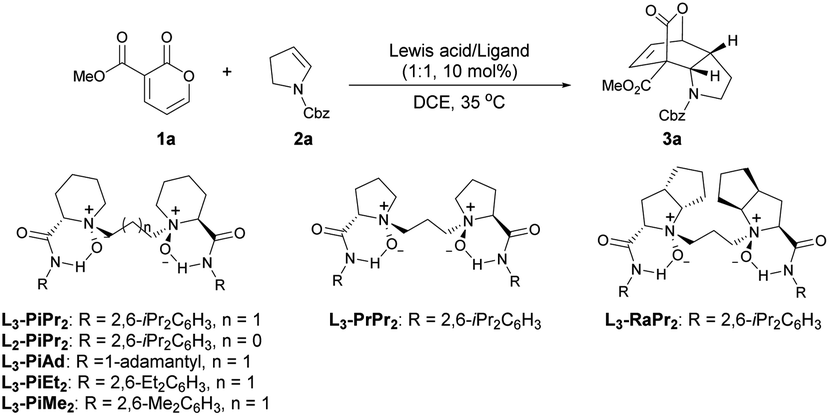
Our studies commenced by using 3-carbomethoxy-2-pyrone 1a and cyclic enamine 2a as model substrates to optimize the reaction conditions (Table 1). First of all, different metal salts coordinated with the N,N′-dioxide ligand L3-PiPr2 were evaluated in DCE at 35 °C (entries 1–4). The results showed that Sc(OTf)3 and In(OTf)3 only led to trace yield of product 3a, while Yb(OTf)3 gave 3a in 92% yield but 13% ee. To our delight, in the presence of the Mg(OTf)2/L3-PiPr2 complex, the reaction occurred smoothly to generate the desired product 3a in 73% yield with 78% ee (entry 4). Encouraged by these results, various chiral N,N′-dioxide ligands were investigated in cooperation with Mg(OTf)2, including changes in the length of the linker, backbones of chiral amino acids, and substituents on the aromatic amide group (entries 5–10). It was found that L3-PiMe2 derived from 2,6-dimethyl aniline could improve the result dramatically, providing the product 3a in 97% yield with 88% ee (entry 10). The screening of other solvents suggested that CHCl3 could further increase the enantioselectivity to 95% ee (entry 11). Notably, when the catalyst loading was reduced to 5 mol%, there was no obvious effect on the outcomes (entry 12). A further decrease to 2 mol% still demonstrated excellent reactivity and a slight deterioration of the enantioselectivity (99% yield with 93% ee, entry 13).|
|
|||||
|---|---|---|---|---|---|
| Entry | Lewis acid | Ligand | t (h) | Yieldb (%) | eec (%) |
a Unless otherwise noted, all reactions were carried out with 1a (0.10 mmol), 2a (0.15 mmol), Lewis acid/ligand (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mol%) in DCE (0.5 mL) at 35 °C.
b NMR yield detected by using CH2Br2 as an internal standard.
c Enantiomeric excess determined by HPLC analysis on a chiral stationary phase.
d Carried out in CHCl3 (0.5 mL).
e Mg(OTf)2/L3-PiMe2 (1:1, 5 mol%).
f Mg(OTf)2/L3-PiMe2 (1:1, 2 mol%). DCE = 1,2-dichloroethane, Tf = trifluoromethanesulfonyl. :1, 10 mol%) in DCE (0.5 mL) at 35 °C.
b NMR yield detected by using CH2Br2 as an internal standard.
c Enantiomeric excess determined by HPLC analysis on a chiral stationary phase.
d Carried out in CHCl3 (0.5 mL).
e Mg(OTf)2/L3-PiMe2 (1:1, 5 mol%).
f Mg(OTf)2/L3-PiMe2 (1:1, 2 mol%). DCE = 1,2-dichloroethane, Tf = trifluoromethanesulfonyl.
|
|||||
| 1 | Sc(OTf)3 | L3-PiPr2 | 24 | Trace | — |
| 2 | In(OTf)3 | L3-PiPr2 | 24 | Trace | — |
| 3 | Yb(OTf)3 | L3-PiPr2 | 3 | 92 | 13 |
| 4 | Mg(OTf)2 | L3-PiPr2 | 3 | 73 | 78 |
| 5 | Mg(OTf)2 | L2-PiPr2 | 12 | 99 | 68 |
| 6 | Mg(OTf)2 | L3-PrPr2 | 12 | 97 | 69 |
| 7 | Mg(OTf)2 | L3-RaPr2 | 12 | 99 | 79 |
| 8 | Mg(OTf)2 | L3-PiAd | 17 | 91 | 12 |
| 9 | Mg(OTf)2 | L3-PiEt2 | 6 | 95 | 82 |
| 10 | Mg(OTf)2 | L3-PiMe2 | 3 | 97 | 88 |
| 11d | Mg(OTf)2 | L3-PiMe2 | 3 | 99 | 95 |
| 12e | Mg(OTf)2 | L3-PiMe2 | 3 | 99 | 95 |
| 13f | Mg(OTf)2 | L3-PiMe2 | 12 | 99 | 93 |
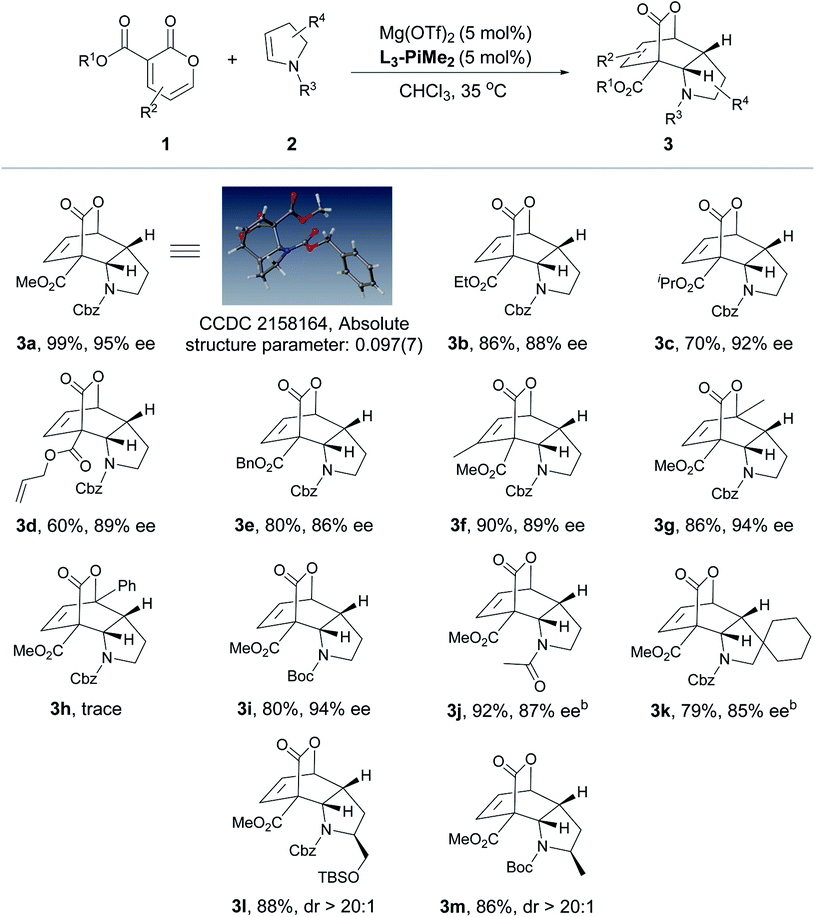
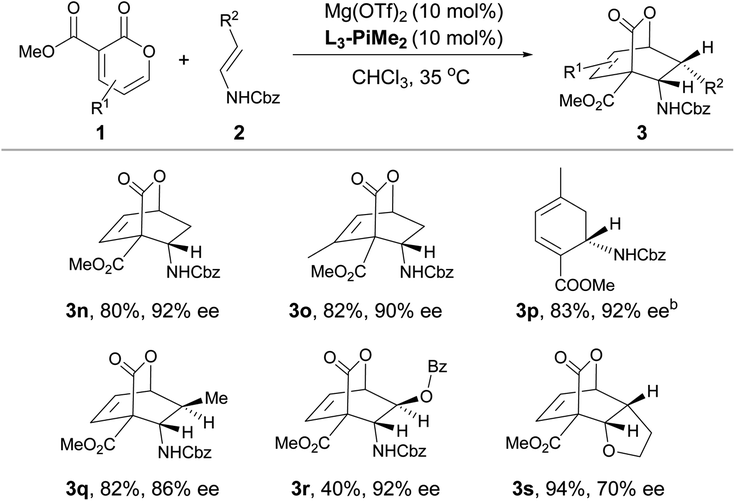
After the optimal reaction conditions were established, the substrate scope of this transformation was further investigated (Table 2 and 3). It was found that 2-pyrones bearing various ester groups such as methyl, ethyl, isopropyl, allyl and benzyl groups were well tolerated, affording 3a−3e in good yields with excellent enantioselectivities (60–99% yields and 86–95% ee). Meanwhile, the absolute configuration of product 3a was determined unambiguously by X-ray crystallography analysis. 2-Pyrone with a methyl group at the C4 or C6 position was also compatible, providing 3f and 3g in excellent yields (90% and 86% yields) and ee values (89% and 94% ee). Unfortunately, when 2-pyrone contained a phenyl group at the C6 position, the IEDDA reaction did not occur, probably due to the steric effect. Next, the scope with respect to the substituted cyclic enamines was also examined. By changing different N-protecting groups of enamines, both Boc- and acetyl-protected cyclic enamines were confirmed to be well tolerated, affording 3i and 3j in good yields (80% and 92%) with high enantioselectivities (94% and 87% ee). The spiro-cyclic enamine 2k was reactive as well to give desired product 3k with moderate enantioselectivity. In addition, the chiral enamines 2l and 2m underwent the diastereoselective IEDDA reaction very well, delivering an exclusive diastereoisomer 3l and 3m, respectively. Furthermore, the scope of acyclic enamines was also evaluated. As summarized in Table 3, terminal enamine 2n reacted smoothly with 2-pyrone 1a to afford the corresponding chiral bridged cyclolactone 3n in 80% yield with 92% ee. C4- or C6-methyl substituted 2-pyrones were also tolerated (3o and 3p), while cyclohexadiene 3p was obtained in one pot through the tandem Diels–Alder reaction and in situ retro-[4 + 2] extrusion of CO2 at an elevated temperature. We found that the methyl and benzyloxy substituted (E)-enamines 2q and 2r afforded the desired products in moderate to good yields and enantioselectivities. Gratifyingly, the IEDDA reaction of 2,3-dihydrofuran and 2-pyrone also occurred smoothly to provide 3s in good yield but with a moderate ee (94% yield with 70% ee).
|
a All reactions were carried out with 1 (0.10 mmol), 2 (0.15 mmol), Mg(OTf)2/L3-PiMe2 (1:1, 10 mol%) in CHCl3 (0.5 mL) at 35 °C. Isolated yield. Enantiomeric excess was determined by HPLC on a chiral stationary phase.
b The reaction was conducted at 35 °C for 36 h, and then heated at 110 °C for 2 h.
|
|---|
|
To illustrate the potential utility of the methodology, a scale-up synthesis of 3a proceeded under the standard conditions. As shown in Scheme 2a, 2 mmol of compound 1a reacted smoothly with 3 mmol of 2a, furnishing the desired product 3a in 82% yield with 96% ee after recrystallization. Meanwhile, several postcatalytic derivatizations were also conducted using enantiomerically pure product 3a. By treatment with diazomethane and a catalytic amount of palladium acetate, the stereospecific cyclopropanation of the alkene motif in 3a was accomplished, thus generating a complex polycyclic product 3t in 68% yield with 96% ee and >20:1 dr. Complete extrusion of CO2via retro-Diels–Alder reaction led to the formation of a cis-tetrahydroindole structure 3u without epimerization in chlorobenzene under reflux. Subsequent regioselective and stereoselective 1,6-Michael addition with MeLi and CuI afforded the corresponding cis-hexahydroindole derivative 3v bearing multiple stereocenters in moderate yield with maintained enantioselectivity.
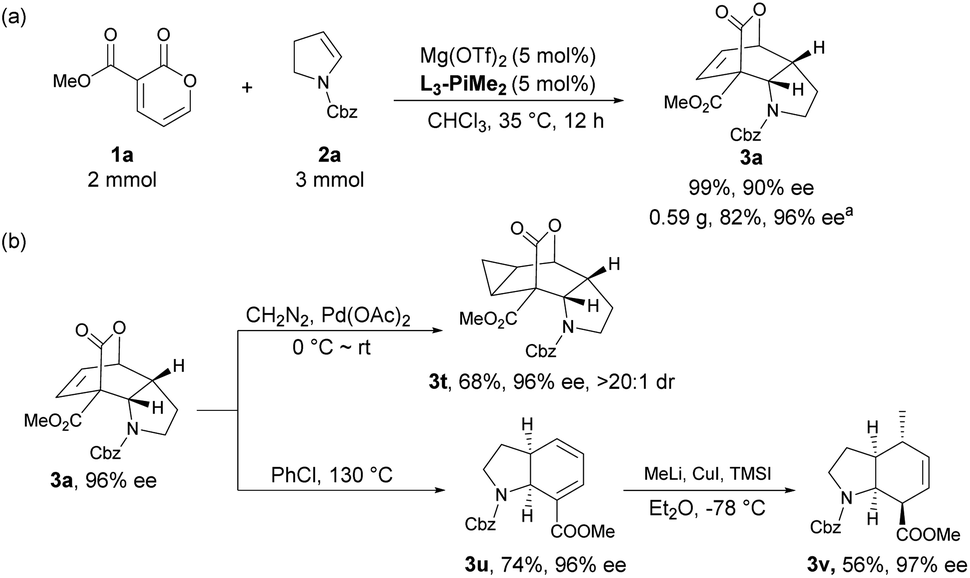 | ||
| Scheme 2 (a) Scale-up synthesis; (b) further transformation of the product. a Yield and enantiomeric excess were determined after recrystallization. | ||
Natural product minovincine,23 characterized by a spiroindoline pentacyclic framework with contiguous stereocenters, is considered as a “biogenetic turntable” between the vindolinine and kopsinine classes of isolates.24 Intrigued by its fascinating structural features and potential biological activities, minovincine has long attracted considerable interest within the chemical synthesis community.25,26 However, there are few examples in the literature for the enantioselective total synthesis of (−)-minovincine.27–29 Based on our present approach and interest in synthesis of natural alkaloids,30 the cis-hydroindole scaffold present in minovincine inspired us to develop a concise synthetic route for the enantioselective formal total synthesis of the naturally occurring enantiomer (+)-minovincine. As shown in Scheme 3, the enantiomerically pure product 3a was readily reduced to 4 in 92% yield with 97% ee by treatment with 5 mol% Crabtree's catalyst under a hydrogen atmosphere. The hydrolysis of the tricyclic lactone 4 was then accomplished by using KOH, followed by extrusion of CO2 and methyl esterification by using TMSCH2N2 to afford the cis-hydroindole derivative 5 in 91% overall yield. Protection of the hydroxyl of 5 with tert-butyldimethylsilyl chloride (TBSCl) furnished 6 in almost quantitative yield. Subsequent deprotection of the benzyloxycarbonyl (Cbz) group of 6 (H2, Pd/C, EtOH, rt) led to 7 in good yield. Further N-alkylation of 7 with 1,3-diiodopropane produced 8 in 52% yield, then 8 was treated with LDA and DMPU to generate 9 bearing a key tricyclic framework. Exposure of 9 to aq. HCl (1 M) gave the corresponding alcohol, which was further oxidized by PySO3 to form the common-core structure 10 in 70% yield over two steps. The absolute configuration of 10 was determined to be opposite to that reported by Soós, and then it could be converted into (+)-minovincine in three steps according to a known procedure.29
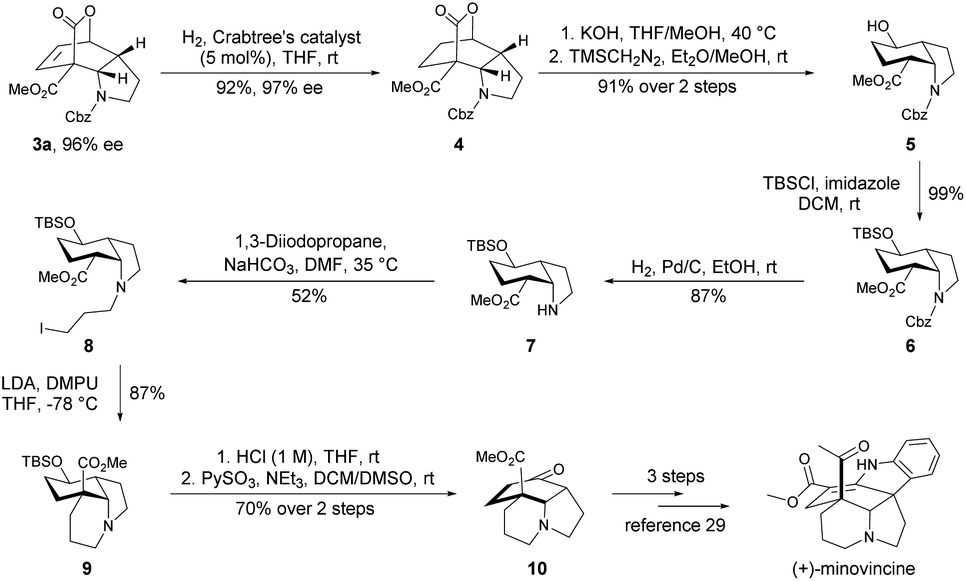 | ||
| Scheme 3 Enantioselective formal total synthesis of (+)-minovincine. TBSCl = tert-butyldimethylsilyl chloride, LDA = lithium diisopropylamide, DMPU = 1,3-dimethyl-tetrahydropyrimidin-2(1H)-one. | ||
Based on the crystal structures of chiral N,N′-dioxide-metal complexes22a and the absolute configuration of this IEDDA reaction product, we proposed a putative stereochemical model to rationalize the stereoselectivity shown in Fig. 1. The coordination of the chiral N,N′-dioxide ligand L3-PiMe2 with Mg(OTf)2 in a tetradentate manner generates an octahedral structure. Then 2-pyrone 1a coordinates tightly to the Lewis acid catalyst through the two carbonyl groups of the ester motif, resulting in a decrease of its LUMO level to accelerate the IEDDA reaction. Simultaneously, due to the steric hindrance of the bulky amide group of the ligand, cyclic enamine 2a prefers to attack from the Si-face of 2-pyrone to give endo-adduct (3aR,4R,7S,7aS)-3a with excellent stereoselectivity.
 | ||
| Fig. 1 Proposed stereochemical model. | ||
Conclusions
In conclusion, we have developed a novel strategy for the highly enantioselective synthesis of the cis-hydroindole motif, involving a chiral N,N′-dioxide/Mg(OTf)2 complex catalyzed asymmetric IEDDA reaction of 2-pyrones and cyclic enamines. A range of cis-hydroindole derivatives were obtained in good yields with high stereoselectivities under mild reaction conditions (up to 99% yield, and 95% ee). This protocol was also compatible for acyclic enamines and 2,3-dihydrofuran. Meanwhile, the scale-up synthesis and further postcatalytic derivatizations were conducted to measure the synthetic potential of the method. Particularly, an alternative and facile access to efficient formal total synthesis of (+)-minovincine was demonstrated by employing the transformations. Further investigations of this reaction in total synthesis of other bioactive natural products are ongoing in our laboratory.Data availability
All experimental and characterization data in this manuscript are available in the ESI.† Crystallographic data for compound (3aR,4R,7S,7aS)-3a has been deposited at the Cambridge Crystallographic Data Center and assigned number 2158164.Author contributions
F. Q. Z. performed the experiments and prepared the ESI† and paper. B. T. R. performed some experiments. Y. Q. Z. helped with resolving the X-ray crystallographic data. Y. B. L. and X. M. F. conceived the concept, directed the project and helped with modifying the paper and ESI†.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from National Natural Science Foundation of China (No. 22001177) and Shenzhen Bay Laboratory (No. S201100003 and S211101001-1).Notes and references
- M. E. Amer, M. Shamma and A. J. Freyer, J. Nat. Prod., 1991, 54, 329–363 CrossRef CAS.
- J. Leonard, Nat. Prod. Rep., 1999, 16, 319–338 RSC.
- S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC.
- K. Ersmark, J. R. Del Valle and S. Hanessian, Angew. Chem., Int. Ed., 2008, 47, 1202–1223 CrossRef CAS PubMed.
- R. A. Pilli, G. B. Rosso and M. d. C. F. de Oliveira, Nat. Prod. Rep., 2010, 27, 1908–1937 RSC.
- J. Khazir, B. A. Mir, S. A. Mir and D. Cowan, J. Asian Nat. Prod. Res., 2013, 15, 764–788 CrossRef CAS.
- Y. Wang, F. Xie, B. Lin, M. Cheng and Y. Liu, Chem.–Eur. J., 2018, 24, 14302–14315 CrossRef CAS PubMed.
- J. M. Saya, E. Ruijter and R. V. A. Orru, Chem.–Eur. J., 2019, 25, 8916–8935 CrossRef CAS PubMed.
- M. Hurst and B. Jarvis, Drugs, 2001, 61, 867–896 CrossRef CAS PubMed.
- F. J. Sayago, P. Laborda, M. I. Calaza, A. I. Jiménez and C. Cativiela, Eur. J. Org. Chem., 2011, 2011–2028 CrossRef CAS.
- E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed.
- For chiral pool approaches, see: (a) P. Wipf, Y. Kim and D. M. Goldstein, J. Am. Chem. Soc., 1995, 117, 11106–11112 CrossRef CAS; (b) J. Bonjoch, J. Catena, E. Isábal, M. López-Canet and N. Valls, Tetrahedron: Asymmetry, 1996, 7, 1899–1902 CrossRef CAS; (c) S. Hanessian and M. Tremblay, Org. Lett., 2004, 6, 4683–4686 CrossRef CAS PubMed; (d) B. M. Ruff, S. Zhong, M. Nieger, M. Sickert, C. Schneider and S. Bräse, Eur. J. Org. Chem., 2011, 6558–6566 CrossRef CAS; (e) S. Hanessian, S. Dorich and H. Menz, Org. Lett., 2013, 15, 4134–4137 CrossRef CAS PubMed.
- For asymmetric metal-catalyzed reactions, see: (a) C. S. Schindler, S. Diethelm and E. M. Carreira, Angew. Chem., Int. Ed., 2009, 48, 6296–6299 CrossRef CAS PubMed; (b) B. M. Trost, T. Kaneko, N. G. Andersen, C. Tappertzhofen and B. Fahr, J. Am. Chem. Soc., 2012, 134, 18944–18947 CrossRef CAS PubMed; (c) Z. Sun, M. Zhou, X. Li, X. Meng, F. Peng, H. Zhang and Z. Shao, Chem.–Eur. J., 2014, 20, 6112–6119 CrossRef CAS PubMed; (d) D. Dailler, G. Danoun and O. Baudoin, Angew. Chem., Int. Ed., 2015, 54, 4919–4922 CrossRef CAS PubMed.
- For aminocatalysis, see: L. Pantaine, V. Coeffard, X. Moreau and C. Greck, Org. Lett., 2015, 17, 3674–3677 CrossRef CAS PubMed.
- For chiral Brönsted acid catalysts, see: (a) Y. Han, B. Zheng and Y. Peng, Adv. Synth. Catal., 2015, 357, 1136–1142 CrossRef CAS; (b) C. Parra, C. Bosch, E. Gómez-Bengoa, J. Bonjoch and B. Bradshaw, J. Org. Chem., 2016, 81, 10172–10179 CrossRef CAS PubMed.
- For selected reviews, see: (a) H. B. Kagan and O. Riant, Chem. Rev., 1992, 92, 1007–1019 CrossRef CAS; (b) J. D. Winkler, Chem. Rev., 1996, 96, 167–176 CrossRef CAS PubMed; (c) E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650–1667 CrossRef CAS; (d) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS; (e) K.-i. Takao, R. Munakata and K.-i. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed; (f) J. Han, A. X. Jones and X. Lei, Synthesis, 2015, 47, 1519–1533 CrossRef CAS; (g) M. S. Xie, L. L. Lin and X. M. Feng, Chem. Rec., 2017, 17, 1184–1202 CrossRef CAS PubMed.
- For selected examples about the Diels–Alder reaction from our group, see: (a) M. S. Xie, X. H. Chen, Y. Zhu, B. Gao, L. L. Lin, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2010, 49, 3799–3802 CrossRef CAS PubMed; (b) H. P. Hu, Y. B. Liu, J. Guo, L. L. Lin, Y. L. Xu, X. H. Liu and X. M. Feng, Chem. Commun., 2015, 51, 3835–3837 RSC; (c) X. Y. Hao, L. L. Lin, F. Tan, C. K. Yin, X. H. Liu and X. M. Feng, ACS Catal., 2015, 5, 6052–6056 CrossRef CAS; (d) H. F. Zheng, X. H. Liu, C. R. Xu, Y. Xia, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 10958–10962 CrossRef CAS PubMed; (e) Y. Lu, Y. H. Zhou, L. L. Lin, H. F. Zheng, K. Fu, X. H. Liu and X. M. Feng, Chem. Commun., 2016, 52, 8255–8258 RSC; (f) Y. H. Zhou, Y. Lu, X. Y. Hu, H. J. Mei, L. L. Lin, X. H. Liu and X. M. Feng, Chem. Commun., 2017, 53, 2060–2063 RSC.
- For an important application of cyclic enamines, see: L. Bai, Y. Ma and X. Jiang, J. Am. Chem. Soc., 2021, 143, 20609–20615 CrossRef CAS PubMed.
- For selected reviews, see: (a) K. Afarinkia, V. Vinader, T. D. Nelson and G. H. Posner, Tetrahedron, 1992, 48, 9111–9171 CrossRef CAS; (b) X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515–5546 CrossRef CAS PubMed; (c) Q. Cai, Chin. J. Chem., 2019, 37, 946–976 CrossRef CAS; (d) X.-G. Si, Z.-M. Zhang and Q. Cai, Synlett, 2021, 32, 947–954 CrossRef CAS; (e) G. Huang, C. Kouklovsky and A. de la Torre, Chem.–Eur. J., 2021, 27, 4760–4788 CrossRef CAS PubMed.
- For selected examples, see: (a) G. H. Posner, J.-C. Carry, J. K. Lee, D. S. Bull and H. Dai, Tetrahedron Lett., 1994, 35, 1321–1324 CrossRef CAS; (b) I. E. Markó, G. R. Evans and J.-P. Declercq, Tetrahedron, 1994, 50, 4557–4574 CrossRef; (c) G. H. Posner, H. Dai, D. S. Bull, J.-K. Lee, F. Eydoux, Y. Ishihara, W. Welsh, N. Pryor and S. Petr, J. Org. Chem., 1996, 61, 671–676 CrossRef CAS PubMed; (d) I. E. Markó, I. Chellé-Regnaut, B. Leroy and S. L. Warriner, Tetrahedron Lett., 1997, 38, 4269–4272 CrossRef; (e) Y. Hashimoto, R. Abe, N. Morita and O. Tamura, Org. Biomol. Chem., 2018, 16, 8913–8916 RSC; (f) Y. Zhou, Z. Zhou, W. Wu and Y. Chen, Acta Chim. Sin., 2018, 76, 382–386 CrossRef CAS; (g) X.-W. Liang, Y. Zhao, X.-G. Si, M.-M. Xu, J.-H. Tan, Z.-M. Zhang, C.-G. Zheng, C. Zheng and Q. Cai, Angew. Chem., Int. Ed., 2019, 58, 14562–14567 CrossRef CAS PubMed; (h) C. J. F. Cole, L. Fuentes and S. A. Snyder, Chem. Sci., 2020, 11, 2175–2180 RSC; (i) X.-G. Si, Z.-M. Zhang, C.-G. Zheng, Z.-T. Li and Q. Cai, Angew. Chem., Int. Ed., 2020, 59, 18412–18417 CrossRef CAS PubMed; (j) M.-M. Xu, X.-Y. You, Y.-Z. Zhang, Y. Lu, K. Tan, L. Yang and Q. Cai, J. Am. Chem. Soc., 2021, 143, 8993–9001 CrossRef CAS PubMed; (k) M.-M. Xu, L. Yang, K. Tan, X. Chen, Q.-T. Lu, K. N. Houk and Q. Cai, Nat. Catal., 2021, 4, 892–900 CrossRef CAS; (l) Y. Lu, M.-M. Xu, Z.-M. Zhang, J. Zhang and Q. Cai, Angew. Chem., Int. Ed., 2021, 60, 26610–26615 CrossRef CAS PubMed.
- (a) M. Feng and X. Jiang, Chem. Commun., 2014, 50, 9690–9692 RSC; (b) N. Wang, S. Du, D. Li and X. Jiang, Org. Lett., 2017, 19, 3167–3170 CrossRef CAS PubMed; (c) N. Wang, J. Liu, C. Wang, L. Bai and X. Jiang, Org. Lett., 2018, 20, 292–295 CrossRef CAS PubMed.
- For selected reviews about the properties of chiral N,N′-dioxide-Lewis acid complexes, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298–302 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621–2631 CrossRef CAS PubMed; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791–797 CrossRef CAS; (e) M. Y. Wang and W. Li, Chin. J. Chem., 2021, 39, 969–984 CrossRef CAS; (f) S. X. Dong, X. H. Liu and X. M. Feng, Acc. Chem. Res., 2022, 55, 415–428 CrossRef CAS PubMed.
- For isolation of minovincine, see: (a) M. Plat, J. LeMen, M.-M. Janot, H. Budzikiewicz, J. M. Wilson, L. J. Durham and C. Djerassi, Bull. Chem. Soc. Chim. Fr., 1962, 2237–2241 CAS; (b) M. P. Cava, S. S. Tjoa, Q. A. Ahmed and A. I. Da Rocha, J. Org. Chem., 1968, 33, 1055–1059 CrossRef CAS PubMed.
- W. Zi, Z. Zuo and D. Ma, Acc. Chem. Res., 2015, 48, 702–711 CrossRef CAS PubMed.
- For a racemic total synthesis, see: (a) M. E. Kuehne and W. G. Earley, Tetrahedron, 1983, 39, 3707–3714 CrossRef CAS; (b) M. E. Kuehne and W. G. Earley, Tetrahedron, 1983, 39, 3715–3717 CrossRef CAS; (c) G. Kalaus, I. Juhász, I. Greiner, M. Kajtár-Peredy, J. Brlik, L. Szabó and C. Szántay, J. Org. Chem., 1997, 62, 9188–9191 CrossRef CAS; (d) G. Kalaus, L. Léder, I. Greiner, M. Kajtár-Peredy, K. Vékey, L. Szabó and C. Szántay, Tetrahedron, 2003, 59, 5661–5666 CrossRef CAS.
- For a semisynthesis, see: N. Langlois and R. Z. Andriamialisoa, J. Org. Chem., 1979, 44, 2468–2471 CrossRef CAS.
- B. N. Laforteza, M. Pickworth and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2013, 52, 11269–11272 CrossRef CAS PubMed.
- T. Morikawa, S. Harada and A. Nishida, J. Org. Chem., 2015, 80, 8859–8867 CrossRef CAS PubMed.
- S. Varga, P. Angyal, G. Martin, O. Egyed, T. Holczbauer and T. Soós, Angew. Chem., Int. Ed., 2020, 59, 13547–13551 CrossRef CAS PubMed.
- (a) J. Xu, Z. W. Zhong, M. Y. Jiang, Y. Q. Zhou, X. H. Liu and X. M. Feng, CCS Chem., 2021, 3, 1894–1902 CrossRef CAS; (b) J. Xu, R. Z. Li, N. Xu, X. H. Liu, F. Wang and X. M. Feng, Org. Lett., 2021, 23, 1856–1861 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2158164. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01458k |
| This journal is © The Royal Society of Chemistry 2022 |