
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-promoted palladium-catalyzed β-methylene C–H arylation of primary aldehydes†
Ke
Yang
 a,
Zhi
Li
a,
Chong
Liu
b,
Yunjian
Li
a,
Qingyue
Hu
a,
Mazen
Elsaid
b,
Bijin
Li
b,
Jayabrata
Das
d,
Yanfeng
Dang
*c,
Debabrata
Maiti
*d and
Haibo
Ge
*b
a,
Zhi
Li
a,
Chong
Liu
b,
Yunjian
Li
a,
Qingyue
Hu
a,
Mazen
Elsaid
b,
Bijin
Li
b,
Jayabrata
Das
d,
Yanfeng
Dang
*c,
Debabrata
Maiti
*d and
Haibo
Ge
*b
aJiangsu Key Laboratory of Advanced Catalytic Materials & Technology, School of Petrochemical Engineering, Changzhou University, Jiangsu 213164, China
bDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409-1061. E-mail: Haibo.Ge@ttu.edu
cTianjin Key Laboratory of Molecular Optoelectronic Sciences, Department of Chemistry, School of Science, Tianjin University, Tianjin 300072. E-mail: yanfeng.dang@tju.edu.cn
dDepartment of Chemistry and Interdisciplinary Program in Climate Studies, Indian Institute of Technology Bombay, Mumbai 400076. E-mail: dmaiti@iitb.ac.in
First published on 25th April 2022
Abstract
The transient directing group (TDG) strategy allowed long awaited access to the direct β-C(sp3)–H functionalization of unmasked aliphatic aldehydes via palladium catalysis. However, the current techniques are restricted to terminal methyl functionalization, limiting their structural scopes and applicability. Herein, we report the development of a direct Pd-catalyzed methylene β-C–H arylation of linear unmasked aldehydes by using 3-amino-3-methylbutanoic acid as a TDG and 2-pyridone as an external ligand. Density functional theory calculations provided insights into the reaction mechanism and shed light on the roles of the external and transient directing ligands in the catalytic transformation.
Simple aliphatic functional groups enrich the skeletal backbones of many natural products, pharmaceuticals, and other industrial materials, influencing the utility and applications of these substances and dictating their reactivity and synthetic modification pathways. Aliphatic aldehydes are some of the most ubiquitous structural units in organic materials.1 Their relevance in nature and industry alike, combined with their reactivity and synthetic versatility, attracted much attention from the synthetic organic and medicinal chemistry communities over the years (Fig. 1).2 Efficient means to the functionalization of these molecules have always been highly sought after.
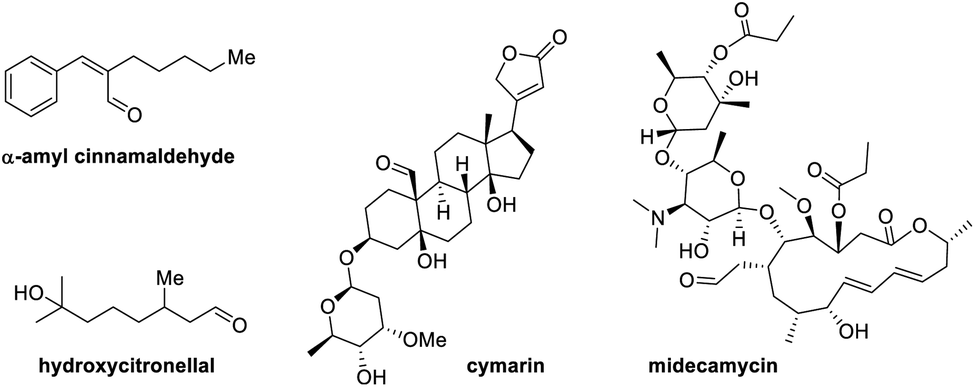 | ||
| Fig. 1 Select aliphatic aldehyde-containing medicines and biologically active molecules. | ||
Traditionally, scientists have utilized the high reactivity of the aldehyde moiety in derivatizing a variety of functional groups by the means of red-ox and nucleophilic addition reactions. The resourceful moiety was also notoriously used to install functional groups at the α-position via condensation and substitution pathways.3 Although β-functionalization is just as robust, it has generally been more restrictive as it often requires the use of α,β-unsaturated aldehydes.4,5 Hence, transition metal catalysis emerged as a powerful tool to access β-functionalization in saturated aldehydes.6 Most original examples of metal-catalyzed β-C–H functionalization of aliphatic aldehydes required the masking of aldehydes into better metal coordinating units since free unmasked aldehydes could not form stable intermediates with metals like palladium on their own.7 Although the masking of the aldehyde moiety into an oxime, for example, enabled the formation of stable 5-membered palladacycles, affording β-functionalized products, this system requires the installation of the directing group prior to the functionalization, as well as the subsequent unmasking upon the reaction completion, compromising the step economy and atom efficiency of the overall process.8 Besides, some masking and unmasking protocols might not be compatible with select substrates, especially ones rich in functional groups. As a result, the development of a one-step direct approach to the β-C–H functionalization of free aliphatic aldehydes was a demanding target for synthetic chemists.
α-Amino acids have been demonstrated as effective transient directing groups (TDGs) in the remote functionalization of o-alkyl benzaldehydes and aliphatic ketones by the Yu group in 2016.9 Shortly after, our group disclosed the first report on the direct β-C–H arylation of aliphatic aldehydes using 3-aminopropanoic acid or 3-amino-3-methylbutanoic acid as a TDG.10 The TDG was found to play a similar role to that of the oxime directing group by binding to the substrate via reversible imine formation, upon which, it assists in the assembly of a stable palladacycle, effectively functionalizing the β-position.11 Since the binding of the TDG is reversible and temporary, it is automatically removed upon functionalization, yielding an efficient and step-economic transformation. This work was succeeded by many other reports that expanded the reaction and the TDG scopes.12–14 However, this system suffers from a significant restriction that demanded resolution; only substitution of methyl C–H bonds of linear aldehydes was made possible via this approach (Scheme 1a–e). The steric limitations caused by incorporating additional groups at the β-carbon proved to compromise the formation of the palladacycle intermediate, rendering the subsequent functionalization a difficult task.12
 | ||
| Scheme 1 Pd-catalyzed β-C–H bond functionalization of aliphatic aldehydes enabled by transient directing groups. | ||
Encouraged by the recent surge in use of 2-pyridone ligands to stabilize palladacycle intermediates,15,16 we have successfully developed the first example of TDG-enabled Pd-catalyzed methylene β-C–H arylation in primary aldehydes via the assistance of 2-pyridones as external ligands (Scheme 1f). The incorporation of 2-pyridones proved to lower the activation energy of the C–H bond cleavage, promoting the formation of the intermediate palladacycles even in the presence of relatively bulky β-substituents.17 This key advancement significantly broadens the structural scopes and applications of this process and promises future asymmetric possibilities, perhaps via the use of a chiral TDG or external ligand or both. Notably, a closely related work from Yu's group was published at almost the same time.18
We commenced our investigation of the reaction parameters by employing n-pentanal (1a) as an unbiased linear aldehyde and 4-iodoanisole (2a) in the presence of catalytic Pd(OAc)2 and stoichiometric AgTFA, alongside 3-amino-3-methylbutanoic acid (TDG1) and 3-(trifluoromethyl)-5-nitropyridin-2-ol (L1) at 100 °C (Table 1). Initial solvent screening results (entries 1–5) suggested that an optimal 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volumetric ratio of HFIP to HOAc (2.0 mL total) afforded the desired product 3a in 47% NMR yield. Further studies indicated that an increased loading of L1 (from 30% to 60%) and TDG1 (from 40% to 60%) improved the NMR yield of 3a to 70% (entries 6–9). The subsequent screening of Pd(II) sources proved Pd(OAc)2 to be the optimal catalyst, while Pd(TFA)2, PdCl2 and PdBr2 provided only moderate yields (entries 10–12). Notably, a significantly lower yield was observed in the absence of the 2-pyridone ligand, and no desired product was isolated altogether in the absence of the TDG (entries 13 and 14). The incorporation of 15 mol% Pd catalyst was deemed necessary after only 55% yield of 3a was obtained when 10 mol% loading of Pd(OAc)2 was instead used (entry 15).
:1 volumetric ratio of HFIP to HOAc (2.0 mL total) afforded the desired product 3a in 47% NMR yield. Further studies indicated that an increased loading of L1 (from 30% to 60%) and TDG1 (from 40% to 60%) improved the NMR yield of 3a to 70% (entries 6–9). The subsequent screening of Pd(II) sources proved Pd(OAc)2 to be the optimal catalyst, while Pd(TFA)2, PdCl2 and PdBr2 provided only moderate yields (entries 10–12). Notably, a significantly lower yield was observed in the absence of the 2-pyridone ligand, and no desired product was isolated altogether in the absence of the TDG (entries 13 and 14). The incorporation of 15 mol% Pd catalyst was deemed necessary after only 55% yield of 3a was obtained when 10 mol% loading of Pd(OAc)2 was instead used (entry 15).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Pd source | L (mol%) | TDG1 (mol%) | Solvent (v/v, mL) | Yield (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), Pd source (15 mol%), AgTFA (0.3 mmol), L1, TDG1, solvent (2.0 mL), 100 °C, 12 h. Yields are based on 1a, determined by 1H-NMR using dibromomethane as an internal standard. b Isolated yield. c Pd(OAc)2 (10 mol%). | |||||
| 1 | Pd(OAc)2 | L1 (30) | TDG1 (40) | HFIP | 30 |
| 2 | Pd(OAc)2 | L1 (30) | TDG1 (40) | AcOH | <5 |
| 3 | Pd(OAc)2 | L1 (30) | TDG1 (40) | HFIP/AcOH (1:1) |
28 |
| 4 | Pd(OAc)2 | L1 (30) | TDG1 (40) | HFIP/AcOH (9:1) |
47 |
| 5 | Pd(OAc)2 | L1 (30) | TDG1 (40) | HFIP/AcOH (1:9) |
<5 |
| 6 | Pd(OAc)2 | L1 (30) | TDG1 (60) | HFIP/AcOH (9:1) |
50 |
| 7 | Pd(OAc)2 | L1 (30) | TDG1 (80) | HFIP/AcOH (9:1) |
25 |
| 8 | Pd(OAc)2 | L1 (60) | TDG1 (60) | HFIP/AcOH (9:1) |
70(68)b |
| 9 | Pd(OAc)2 | L1 (75) | TDG1 (60) | HFIP/AcOH (9:1) |
51 |
| 10 | Pd(TFA)2 | L1 (60) | TDG1 (60) | HFIP/AcOH (9:1) |
60 |
| 11 | PdCl2 | L1 (60) | TDG1 (60) | HFIP/AcOH (9:1) |
52 |
| 12 | PdBr2 | L1 (60) | TDG1 (60) | HFIP/AcOH (9:1) |
54 |
| 13 | Pd(OAc)2 | — | TDG1 (60) | HFIP/AcOH (9:1) |
9 |
| 14 | Pd(OAc)2 | L1 (60) | — | HFIP/AcOH (9:1) |
0 |
| 15c | Pd(OAc)2 | L1 (60) | TDG1 (60) | HFIP/AcOH (9:1) |
55 |

To advance our optimization of the reaction conditions, a variety of 2-pyridones and TDGs were tested (Scheme 2). Originally, pyridine-2(1H)-one (L2) was examined as the external ligand, but it only yielded the product (3a) in 7% NMR yield. Similarly, other mono- and di-substituted 2-pyridone ligands (L3–L10) also produced low yields, fixating L1 as the optimal external ligand. Next, various α- and β-amino acids (TDG1–10) were evaluated, yet TDG1 persisted as the optimal transient directing group. These amino acid screening results also suggest that a [5,6]-bicyclic palladium species is likely the key intermediate in this protocol since only β-amino acids were found to provide appreciable yields, whereas α-amino acids failed to yield more than trace amounts of the product. The supremacy of TDG1 when compared to other β-amino acids is presumably due to the Thorpe–Ingold effect that perhaps helps facilitate the C–H bond cleavage and stabilize the [5,6]-bicyclic intermediate further.
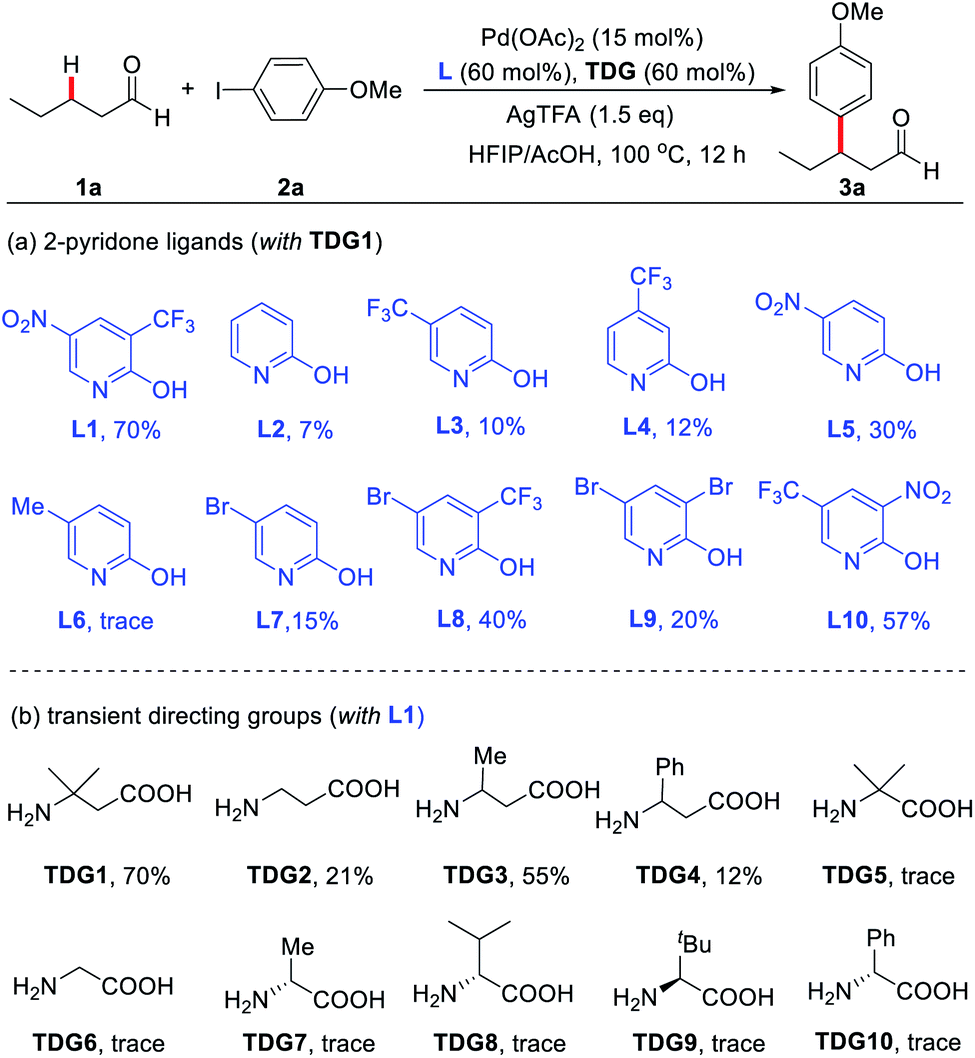 | ||
| Scheme 2 Optimization of 2-pyridone ligands and transient directing groups. | ||
With the optimized reaction conditions in hand, substrate scope study of primary aliphatic aldehydes was subsequently carried out (Scheme 3). A variety of linear primary aliphatic aldehydes bearing different chain lengths provided the corresponding products 3a–e in good yields. Notably, relatively sterically hindered methylene C–H bonds were also functionalized effectively (3f and 3g). Additionally, 4-phenylbutanal gave rise to the desired product 3h in a highly site-selective manner, suggesting that functionalization of the methylene β-C–H bond is predominantly favored over the more labile benzylic C–H bond. It is noteworthy that the amide group was also well-tolerated and the desired product 3j was isolated in 60% yield. As expected, with n-propanal as the substrate, β-mono- (3k1) and β,β-disubstituted products (3k2) were isolated in 22% and 21% yields respectively. However, in the absence of the key external 2-pyridone ligand, β-monosubstituted product (3k1) was obtained exclusively, albeit with a low yield, indicating preference for functionalizing the β-C(sp3)–H bond of the methyl group over the benzylic methylene group.
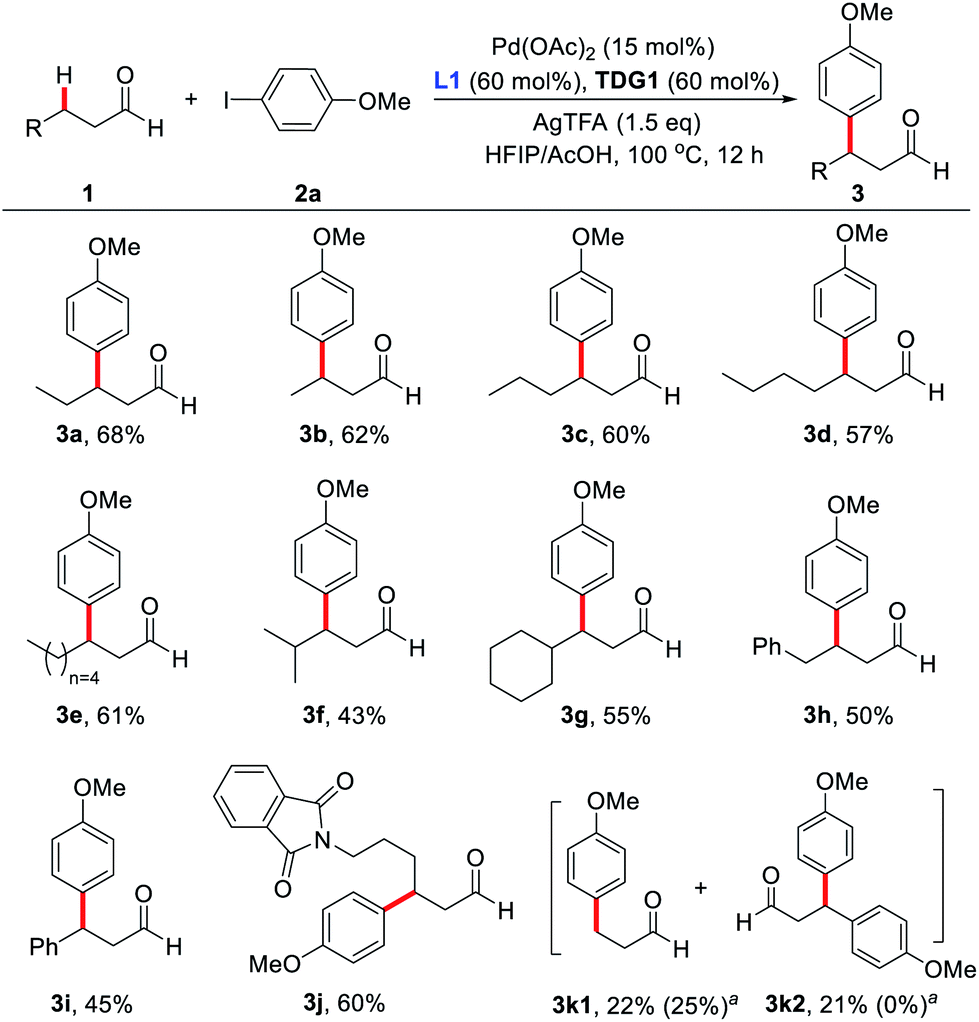 | ||
| Scheme 3 Scope of primary aliphatic aldehydes. Reaction conditions: 1 (0.2 mmol), 2a (0.4 mmol), Pd(OAc)2 (15 mol%), AgTFA (0.3 mmol), L1 (60 mol%), TDG1 (60 mol%), HFIP (1.8 mL), HOAc (0.2 mL), 100 °C, 12 h. Isolated yields. aL1 (60 mol%) was absent and yields are given in parentheses. | ||
Next, substrate scope study on aryl iodides was surveyed (Scheme 4). Iodobenzenes bearing either an electron-donating or electron-withdrawing group at the para-, meta-, or ortho-position were all found compatible with our catalytic system (3l–3ah). Surprisingly, ortho-methyl- and fluoro-substituted aryl iodides afforded the products in only trace amounts. However, aryl iodide with ortho-methoxy group provided the desired product 3ac in a moderate yield. Notably, a distinctive electronic effect pattern was not observed in the process. It should be mentioned that arylated products bearing halogen, ester, and cyano groups could be readily converted to other molecules, which significantly improves the synthetic applicability of the process. Delightfully, aryl iodide-containing natural products like ketoprofen, fenchol and menthol were proven compatible, supplying the corresponding products in moderate yields. Unfortunately, (hetero)aryl iodides including 2-iodopyridine, 3-iodopyridine, 4-iodopyridine and 4-iodo-2-chloropyridine failed to produce the corresponding products. Although our protocol provides a novel and direct pathway to construct β-arylated primary aliphatic aldehydes, the yields of most examples are modest. The leading reasons for this compromise are the following: (1) aliphatic aldehydes are easily decomposed or oxidized to acids; (2) some of the prepared β-arylated aldehyde products may be further transformed into the corresponding α,β-unsaturated aldehydes.
 | ||
| Scheme 4 Scope of aryl iodides. Reaction conditions: 1a (0.2 mmol), 2 (0.4 mmol), Pd(OAc)2 (15 mol%), AgTFA (0.3 mmol), L1 (60 mol%), TDG1 (60 mol%), HFIP (1.8 mL), HOAc (0.2 mL), 100 °C, 12 h. Isolated yields. | ||
Density functional theory (DFT) calculations were performed to help investigate the reaction mechanism and to elucidate the role of the ligand in improving the reactivity (Fig. 2). The condensation of the aliphatic aldehyde 1a with the TDG to form imine-1a was found thermodynamically neutral (ΔG° = −0.1 kcal mol−1). As a result, it was permissible to use imine-1a directly in the calculations. According to the calculations results, the precatalyst [Pd(OAc)2]3, a trimeric complex, initially experiences dissociation and ligand metathesis with imine-1a to generate the Pd(II) intermediate IM1, which is thermodynamically favorable by 21.9 kcal mol−1. Consequently, the deprotonated imine-1a couples to the bidentate ligand to form the stable six-membered chelate complex IM1. Therefore, IM1 is indeed the catalyst resting state and serves as the zero point to the energy profile. We have identified two competitive pathways for the Pd(II)-catalyzed C–H activation starting from IM1, one of which incorporates L1 and another which does not. On the one hand, an acetate ligand substitutes one imine-1a chelator in IM1 to facilitate the subsequent C–H activation leading to IM2, which undergoes C(sp3)–H activation through concerted metalation-deprotonation (CMD) viaTS1 (ΔG‡ = 37.4 kcal mol−1). However, this kinetic barrier is thought to be too high to account for the catalytic activity at 100 °C. On the other hand, the chelate imine-1a could be replaced by two N-coordinated ligands (L1) leading to the Pd(II) complex IM3. This process is endergonic by 6.4 kcal mol−1. To allow the ensuing C–H activation, IM3 dissociates one ligand (L1) producing the active species IM4, which undergoes TS2 to cleave the β-C(sp3)–H bond and form the [5,6]-bicyclic Pd(II) intermediate IM5. Although this step features an energy barrier of 31.2 kcal mol−1, it is thought to be feasible under the experimental conditions (100 °C). Possessing similar coordination ability to that of pyridine, the ligand (L1) effectively stabilizes the Pd(II) center in the C–H activation process, indicating that this step most likely involves a manageable kinetic barrier. This result explicates the origin of the ligand-enabled reactivity (TS2vs.TS1). Additionally, we considered the γ-C(sp3)–H activation pathway viaTS2′ which was found to have a barrier of 35.5 kcal mol−1. The higher energy barrier of TS2′ compared to that of TS2 is attributed to its larger ring strain in the [6,6]-bicyclic Pd(II) transition state, which reveals the motive for the site-selectivity. Reverting back to the supposed pathway, upon the formation of the bicyclic intermediate IM5, it undergoes ligand/substrate replacement to afford intermediate IM6, at which the Ar–I coordinates to the Pd(II) center to enable oxidative addition viaTS3 (ΔG‡ = 27.4 kcal mol−1) leading to the five-coordinate Pd(IV) complex IM7. Undergoing direct C–C reductive elimination in IM7 would entail a barrier of 29.6 kcal mol−1 (TS4). Alternatively, iodine abstraction by the silver(I) salt in IM7 is thermodynamically favorable and irreversible, yielding the Pd(IV) intermediate IM8 coordinated to a TFA ligand. Subsequently, C–C reductive coupling viaTS5 generates the Pd(II) complex IM9 and concludes the arylation process. This step was found both kinetically facile (6.1 kcal mol−1) and thermodynamically favorable (30.7 kcal mol−1). Finally, IM9 reacts with imine-1avia metathesis to regenerate the palladium catalyst IM1 and release imine-3a in a highly exergonic step (21.0 kcal mol−1). Ultimately, imine-3a undergoes hydrolysis to yield the aldehyde product 3a and to release the TDG.
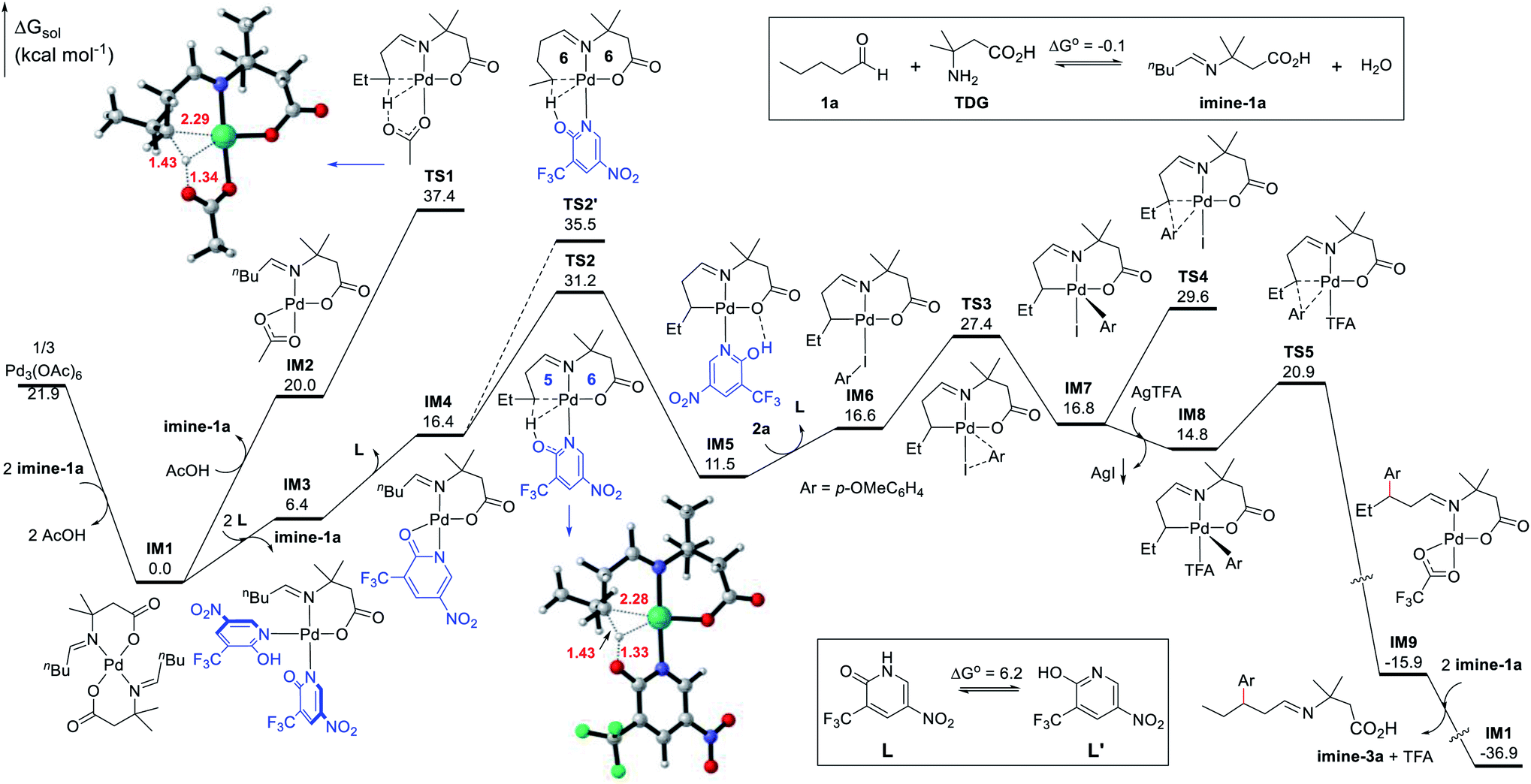 | ||
| Fig. 2 Free energy profiles for the ligand-promoted Pd(II)-catalyzed site-selective C–H activation and C–C bond formation, alongside the optimized structures of the C–H activation transition states TS1 and TS2 (selected bond distances are labelled in Å). Energies are relative to the complex IM1 and are mass-balanced. | ||
Conclusions
In summary, we have developed a palladium-catalyzed direct methylene β-C–H arylation of linear primary aliphatic aldehydes using commercially available 3-amino-3-methylbutanoic acid as a transient directing group and 3-(trifluoromethyl)-5-nitropyridin-2-ol as an external ligand. This reaction circumvents the requirement of having α–β-unsaturation, allowing direct β-C(sp3)–H functionalization and granting much needed access to β-tertiary aldehydes with high site-selectivity and excellent functional group compatibility. In the case of arylation, the newly generated tertiary center is stereogenic, opening the door to future asymmetric synthetic applications via the use of chiral transient directing and external ligands, especially since DFT calculations suggest that both components are bound in the arylation step, promising adequate stereocontrol. Finally, the versatility of the aldehyde moiety and the endless transformation possibilities expand the synthetic utility of this method. Further studies are being conducted in our laboratory to develop new transformations and to explore the means of stereochemistry control over the newly generated chiral center.Data availability
All experimental and computational data associated with this study are available in ESI.†Author contributions
D. M. and H. G. conceived and designed the research. Y. D. performed theoretical calculations. K. Y. planned and performed the experiments. K. Y. also analyzed the experimental data. Z. L., C. L., Y. L., Q. H., B. L. and J. D. performed the experiments. K. Y., M. E. and H. G. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge NSF (CHE-2029932), Robert A. Welch Foundation (D-2034-20200401), NSFC (No. 22073067), and Texas Tech University for financial support. Ke Yang is grateful for financial support from NSFC (No. 21702019) and Advanced Catalysis and Green Manufacturing Collaborative Innovation Center, Changzhou University. Ke Yang also acknowledge the analytical testing support from Analysis and Testing Center, NERC Biomass of Changzhou University.Notes and references
- (a) A. Parenty, A. P. X. Moreau and J.-M. Campagne, Chem. Rev., 2006, 106, 911 CrossRef CAS PubMed; (b) Y. Kim and P. Walsh, Acc. Chem. Res., 2012, 45, 1533 CrossRef PubMed.
- (a) T.-Y. Luh, M.-K. Leung and K.-T. Wong, Chem. Rev., 2000, 100, 3187 CrossRef CAS PubMed; (b) B. I. Roman, N. D. Kimpe and C. V. Stevens, Chem. Rev., 2010, 110, 5914 CrossRef CAS PubMed.
- (a) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed; (b) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC.
- For selected examples of using α,β-unsaturated aldehydes, see: (a) N. A. Paras and D. W. C. MacMillan, J. Am. Chem. Soc., 2001, 123, 4370 CrossRef CAS PubMed; (b) J. F. Austin and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 1172 CrossRef CAS PubMed; (c) S. P. Brown, N. C. Goodwin and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 1192 CrossRef CAS PubMed; (d) Y. K. Chen, M. Yoshida and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 9328 CrossRef CAS PubMed; (e) I. Ibrahem, G. Ma, S. Afewerki and A. Córdova, Angew. Chem., Int. Ed., 2013, 52, 878 CrossRef CAS PubMed.
- For selected examples of using photocatalysis, see: (a) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS PubMed; (b) J. A. Terrett, M. D. Clift and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 6858 CrossRef CAS PubMed.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (c) Z.-K. Chen, B.-J. Wang, J.-T. Zhang, W.-L. Yu, Z.-X. Liu and Y.-H. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed.
- (a) D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 3645 CrossRef CAS PubMed; (b) S. Duttwyler, S. Chen, M. K. Takase, K. B. Wiberg, R. G. Bergman and J. A. Ellman, Science, 2013, 339, 678 CrossRef CAS PubMed.
- (a) C.-L. Sun, N. Liu, B.-J. Li, D.-G. Yu, Y. Wang and Z.-J. Shi, Org. Lett., 2010, 12, 184 CrossRef CAS PubMed; (b) Z. Yu, Y. Zhang, J. Tang, L. Zhang, Q. Liu, Q. Li, G. Gao and J. You, ACS Catal., 2020, 10, 203 CrossRef CAS.
- F. L. Zhang, K. Hong, T. J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed.
- K. Yang, Q. Li, Y.-B. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775 CrossRef CAS PubMed.
- (a) Q. Zhao, T. Polsson, X. Pannecouke and T. Besset, Synthesis, 2017, 49, 4808 CrossRef CAS; (b) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS; (c) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456 RSC; (d) S. St John-Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582 RSC; (e) B. Li, K. Seth, B. Niu, L. Pan, H. Yang and H. Ge, Angew. Chem., Int. Ed., 2018, 57, 3401 CrossRef CAS PubMed; (f) B. Niu, K. Yang, B. Lawrence and H. Ge, ChemSusChem, 2019, 12, 2955 CrossRef CAS PubMed; (g) K. Yang, M. Song, H. Liu and H. Ge, Chem. Sci., 2020, 11, 12616 RSC.
- (a) S. St John Campbell, A. J. P. White and J. A. Bull, Chem. Sci., 2017, 8, 4840 RSC; (b) S. St John Campbell and J. A. Bull, Chem. Commun., 2019, 55, 9172 RSC; (c) B.-B. Gou, H.-F. Liu, J. Chen and L. Zhou, Org. Lett., 2019, 21, 7084 CrossRef CAS PubMed; (d) X.-L. Zhang, G.-F. Pan, X.-Q. Zhu, R.-L. Guo, Y.-R. Gao and Y.-Q. Wang, Org. Lett., 2019, 21, 2731 CrossRef CAS PubMed; (e) C. Dong, L. Wu, J. Yao and K. Wei, Org. Lett., 2019, 21, 2085 CrossRef CAS PubMed.
- (a) K. Hong, H. Park and J.-Q. Yu, ACS Catal., 2017, 7, 6938 CrossRef CAS PubMed; (b) L. Pan, K. Yang, G. Li and H. Ge, Chem. Commun., 2018, 54, 2759 RSC; (c) J. Wang, C. Dong, L. Wu, M. Xu, J. Lin and K. Wei, Adv. Synth. Catal., 2018, 360, 3709 CrossRef CAS.
- (a) Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26 CrossRef CAS; (b) Y. Xu, M. C. Young, C. P. Wang, D. M. Magness and G. B. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084 CrossRef CAS PubMed; (c) Y.-W. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554 CrossRef CAS PubMed; (d) A. Yada, W.-Q. Liao, Y. Sato and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 1073 CrossRef CAS PubMed; (e) H. Lin, C. Wang, T. D. Bannister and T. M. Kamenecka, Chem.–Eur. J., 2018, 24, 9535 CrossRef CAS PubMed; (f) M. Kapoor, D. Liu and M. C. Young, J. Am. Chem. Soc., 2018, 140, 6818 CrossRef CAS PubMed; (g) S. St John-Campbell, A. K. Ou and J. A. Bull, Chem.–Eur. J., 2018, 24, 17838 CrossRef CAS PubMed; (h) Z. Wang, Y. Fu, Q. Zhang, H. Liu and J. Wang, J. Org. Chem., 2020, 85, 7683 CrossRef CAS PubMed.
- (a) Y.-Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 17844 Search PubMed; (b) Y.-Q. Chen, S. Singh, Y. Wu, Z. Wang, W. Hao, P. Verma, J. X. Qiao, R. B. Sunoj and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 9966 CrossRef CAS PubMed; (c) L.-J. Xiao, K. Hong, F. Luo, L. Hu, W. R. Ewing, K.-S. Yeung and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 9594 CrossRef CAS PubMed; (d) P. A. Provencher, J. F. Hoskin, J. J. Wong, X. Chen, J.-Q. Yu, K. N. Houk and E. J. Sorensen, J. Am. Chem. Soc., 2021, 143, 20035 CrossRef CAS PubMed; (e) P. A. Provencher, K. L. Bay, J. F. Hoskin, K. N. Houk, J.-Q. Yu and E. J. Sorensen, ACS Catal., 2021, 11, 3115 CrossRef CAS.
- (a) F. Li, Y. Zhou, H. Yang, Z. Wang, Q. Yu and F.-L. Zhang, Org. Lett., 2019, 21, 3692 CrossRef CAS PubMed; (b) S. St John-Campbell, A. J. P. White and J. A. Bull, Org. Lett., 2020, 22, 1807 CrossRef CAS PubMed; (c) B. Li, B. Lawrence, G. Li and H. Ge, Angew. Chem., Int. Ed., 2020, 59, 3078 CrossRef CAS PubMed.
- (a) D.-H. Wang, K. M. Engle, B.-F. Shi and J. Q. Yu, Science, 2010, 327, 315 CrossRef CAS PubMed; (b) S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888 CrossRef CAS PubMed; (c) T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751 CrossRef CAS PubMed; (d) P. Wang, M. E. Farmer, X. Huo, P. Jain, P.-X. Shen, M. Ishoey, J. E. Bradner, S. R. Wisniewski, M. E. Eastgate and J. Q. Yu, J. Am. Chem. Soc., 2016, 138, 9269 CrossRef CAS PubMed; (e) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K. S. Yeung and J. Q. Yu, Nature, 2017, 551, 489 CrossRef CAS PubMed; (f) R.-Y. Zhu, Z.-Q. Li, H. S. Park, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 3564 CrossRef CAS PubMed; (g) G. Xia, Z. Zhuang, L.-Y. Liu, S. L. Schreiber, B. Melillo and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 7783 CrossRef CAS PubMed; (h) H. S. Park, Z. Fan, R.-Y. Zhu and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 12853 CrossRef CAS PubMed.
- Y.-H. Li, Y. Ouyang, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2022, 144, 4727 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01677j |
| This journal is © The Royal Society of Chemistry 2022 |