
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous achievement of defect passivation and carrier transport promotion by using emerald salt for methylammonium-free perovskite solar cells†
Zhenghui
Fan
a,
Yuan
Yin
b,
Bing
Cai
*a,
Qingshan
Ma
a,
Qianlong
Liu
a,
Xinhang
Liu
a,
Yinhua Lv
 *a and
Wen-Hua
Zhang
*acd
*a and
Wen-Hua
Zhang
*acd
aInstitute of Chemical Materials, China Academy of Engineering Physics, 596 Yinhe Road, Chengdu 610200, China. E-mail: bingcai@caep.cn; qiehahah@163.com; whzhang@caep.cn
bCollege of Physics and Optoelectronic Technology, Baoji University of Arts and Sciences, Baoji 721016, China
cYunnan Key Laboratory of Carbon Neutrality and Green Low-carbon Technologies, School of Materials and Energy, Yunnan University, Kunming 650500, China. E-mail: wenhuazhang@ynu.edu.cn
dJiangsu Collaborative Innovation Center for Photovoltaic Science and Engineering, Changzhou University, Changzhou 213164, P. R. China
First published on 13th August 2022
Abstract
Defect passivation along with promoted charge transport is potentially an effective but seldom exploited strategy for high-performance perovskite solar cells (PSCs). Herein, the in situ defect passivation and carrier transport improvement are simultaneously realized by introducing a conductive polymer (i.e., emerald salt, ES) into the precursor solution of methylammonium (MA)-free perovskites. The interaction between ES and uncoordinated Pb2+ reduces defect density to suppress the non-radiative recombination. Moreover, ES can act as a “carrier driver” to promote the carrier transport due to its conductive feature, resulting in efficient PSC devices with a decent power conversion efficiency (PCE) of 23.0%, which is among the most efficient MA-free PSCs. The ES-based unencapsulated devices show superior stability, retaining 89.1% and 83.8% of their initial PCEs when subjected to 35 ± 5% relative humidity (RH) storage and 85 °C thermal aging for 1000 h, respectively. To further assess the large-area compatibility of our strategy, 5 × 5 cm2 mini modules were also fabricated, delivering an impressive efficiency of 19.3%. This work sheds light on the importance of conductive additives in boosting cell performance by playing multiple roles in passivating defects, retarding the moisture invasion, and enhancing and balancing charge transport.
Introduction
Methylammonium (MA)-free perovskite solar cells have attracted extensive interest owing to their excellent optoelectrical properties and remarkable stability.1,2 To fabricate high-quality MA-free perovskite films for high-performance devices, a variety of processing techniques have been explored, and examples include the interdiffusion method,3 anion-exchange process,4 and multisource vacuum deposition.5 Compositional modulation was also demonstrated to be effective in this regard, and Pb(SCN)2,6,7 Pb(Ac)2,8 PbCl2,9 hydrohalic acids,10 CsCl,11etc. have been involved in the precursor solution to improve the film quality and decrease trap densities of the resultant perovskite films. Nevertheless, to date, MA-free PSCs2,12,13 have always presented device performance inferior to the MA-containing counterparts,14 which is mainly imputed to the varied defects (e.g., I vacancies, undercoordinated Pb2+, and so on15) ineluctably generated during the complex crystallization process. These defects can act not only as the non-radiative charge recombination centers, but also as the degradation sources of perovskite to deteriorate device stability.16–18 Therefore, it is of vital importance to eliminate these defects to achieve excellent efficiency and stability for MA-free PSCs.To date, considerable efforts have been dedicated to defect passivation of perovskite films.19–29 Among them, additive engineering offers a facile and in situ way to mediate the crystallization of solution-processed perovskite films and passivate the defects simultaneously, which indeed has been established in MA-containing high-performance systems.30–35 On the basis of Lewis acid-base chemistry, reagents containing carboxyl groups,36 sulfonic acid groups,37 or amino groups38 are able to react with the undercoordinated Pb2+, thereby passivating the Pb2+ or I− vacancy defects. However, research studies on the passivation role of additives in MA-free devices have been rather limited to date.20,21,27,28 It has been reported that 3-(decyldimethylammonio)-propane-sulfonate inner salt (DPSI) can be adopted to coordinate with the perovskite via the lone unpaired electron in S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, leading to a pinhole-less perovskite film and effective reduction in defect density. A power conversion efficiency (PCE) of 21.1% was consequently delivered.28 A multifunctional molecule, D-penicillamine (PA) with –SH, –COOH, and –NH2 groups, was employed to modify a MA-free perovskite film. The charged traps were effectively passivated, and the crystallinity of perovskite films was improved, yielding an impressive PCE of 22.4%.27 A potassium-salt, KPF6, was also introduced into MA-free perovskite solar cells in order to eliminate the hysteresis and improve the device performance, resulting in a champion PCE of 23.35%.13 Recently, a dual-functional additive (iBA-iBDTC) with an organic ammonium cation and dithiocarbamate anion was employed to regulate the crystallization and defects of the MA-free perovskite films. A certified champion PCE of 23.7% has been achieved with the use of iBA-iBDTC, which is the highest efficiency for MA-free PSCs reported to date.39 Nonetheless, the above additives feature an insulating nature, which possibly endows the resultant perovskite films with unsatisfactory conductivity, which is a disadvantage for cell performance. It is thus highly necessary to study the scenario that the highly conductive additives might be employed in perovskite precursors to boost the performance of MA-free perovskite solar cells.
O, leading to a pinhole-less perovskite film and effective reduction in defect density. A power conversion efficiency (PCE) of 21.1% was consequently delivered.28 A multifunctional molecule, D-penicillamine (PA) with –SH, –COOH, and –NH2 groups, was employed to modify a MA-free perovskite film. The charged traps were effectively passivated, and the crystallinity of perovskite films was improved, yielding an impressive PCE of 22.4%.27 A potassium-salt, KPF6, was also introduced into MA-free perovskite solar cells in order to eliminate the hysteresis and improve the device performance, resulting in a champion PCE of 23.35%.13 Recently, a dual-functional additive (iBA-iBDTC) with an organic ammonium cation and dithiocarbamate anion was employed to regulate the crystallization and defects of the MA-free perovskite films. A certified champion PCE of 23.7% has been achieved with the use of iBA-iBDTC, which is the highest efficiency for MA-free PSCs reported to date.39 Nonetheless, the above additives feature an insulating nature, which possibly endows the resultant perovskite films with unsatisfactory conductivity, which is a disadvantage for cell performance. It is thus highly necessary to study the scenario that the highly conductive additives might be employed in perovskite precursors to boost the performance of MA-free perovskite solar cells.
Polymers have also been exploited as additives in MA-containing PSCs to improve the device performance.40 Owing to abundant functional groups on their long-chain carbon backbones, polymers could have a great influence on both PCE and stability of PSCs. It is worth noting that most of the polymers are intrinsically insulators (such as PMMA,41 PEG,42 PEI,43etc.), and an excess amount of these additives would increase the resistance and retard charge transport in PSCs. Only a few of the conjugated polymers (such as MEH-PPV,44 PTAA,45 polypyrrole,46etc.) with double bonds or aromatic rings show conductive ability, which could promote the charge transport in PSCs. Polyanilines are a group of highly conductive polymers with a coexisting structure of benzene (reduction unit) and quinone (oxidation unit). Polyanilines with different oxide/reduced-states have been explored as additives in PSCs (Table S1, ESI†). In 2018, Wei et al. introduced the self-synthesized polyaniline into MAPbI3 precursor solution to adjust the film crystallization,47 and the resultant PSCs with a carbon electrode presents an efficiency of 9.43%. In 2019, Pan et al. employed emerald imine-state polyaniline as an additive to (FAPbI3)0.85(MAPbBr3)0.15 perovskite,48 resulting in a perovskite film with a better crystal structure, smoother morphology, and enhanced light absorption and charge transfer. The cell with polyanilines showed a power conversion efficiency of 19.09%, significantly higher than the pristine one with an efficiency of 16.96%. In 2020, Liu et al. employed the commercial polyaniline as an additive in all-inorganic CsPbI2Br PSCs, and XPS analysis was performed to reveal the interaction between the polyaniline and perovskite film, which was found to contribute to balancing electron and hole transport and a high Voc of 1.33 V for the resultant cell.49
Although some favorable influence of the highly conductive polyaniline on the PSCs was observed, a comprehensive study of their influence is lacking, and the device performance of PSCs is unsatisfactory. It is highly desirable to supplement in-depth study on the interaction of the polyaniline and perovskite film to gain insight into the working mechanism for the conductive polymer-assisted charge transport in PSCs. Therefore, in this study, we expand the acidified emeraldine-type polyaniline (also called as emerald salt, ES, that shows improved conductivity compared to pristine one) to form a MA-free perovskite, which is inherently more stable. We systematically studied the role of ES: (1) improving the crystalline quality of perovskite films, (2) passivating defects via the Lewis acid-base reaction and hydrogen bond, (3) functioning as a “charge driver” to boost carrier transport, (4) balancing electron and hole mobility, and (5) enhancing hydrophobic characteristics of the resulting perovskite film. A decent PCE of 23.0% is obtained for the n-i-p planar cell with ES modification, one of the highest efficiencies for MA-free PSCs to date. Moreover, both the thermal and moisture stabilities are considerably improved upon ES modification, and the unencapsulated ES modified devices retained 83.8% of their initial efficiency after thermal aging at 85 °C for over 1000 h and kept 89.1% of their initial efficiency upon subjection to 35 ± 5% RH storage for 1000 h. The ES passivation strategy was successfully extended to mini modules (substrate: 5 × 5 cm2; active area: 10.3 cm2) to yield a PCE of 19.3%, apparently superior to the control one with 17.7% PCE. The current work renders a facile method to obtain highly efficient and stable MA-free PSCs via the employment of conductive additives.
Results and discussion
Fig. 1a schematically illustrates the device architecture of glass/ITO (indium doped tin oxide)/SnO2/Cs0.15FA0.85PbI2.85Br0.05Cl0.1/2,2′,7,7′-tetrakis [N,N-di(4-methoxyphenyl) amino]-9,9′-spirobifluorene (spiro – OMeTAD)/Au, in which ES was added into perovskite precursors. The influence of ES concentrations on device performance was systematically investigated, and thirty devices for each ES concentration were assembled under identical conditions. The corresponding statistical distribution of PCEs is shown in Fig. 1b. Notably, the PCEs increase with the increased ES contents, and it peaks for the 30 μg mL−1 ES device. Without specific explanation, the ES-based cell or film is referred to the sample with a concentration of 30 μg mL−1 in the following part. Fig. 1c and d present the J–V curves of the best-performing devices in both reverse and forward scans. The control device exhibits a champion PCE of 21.5% along with an open-circuit voltage (VOC) of 1.14 V, a short-circuit current density (JSC) of 23.9 mA cm−2, and a fill factor (FF) of 78.7%. In contrast, the best ES-based cell delivers a substantially improved PCE of 23.0% (with a VOC of 1.18 V, a JSC of 24.1 mA cm−2, and a FF of 80.6%), which is among the most efficient MA-free PSCs to date (Table S2, ESI†). To further verify the reliability of the J–V results, the external quantum efficiency (EQE) and stabilized power output at the maximum power point (MPP) were measured. The ES-based device shows a slightly higher integrated photocurrent of 22.8 mA cm−2 than that of the control device (22.6 mA cm−2) (Fig. S1, ESI†), both of which are well matched with those obtained from the J–V curves. As presented in Fig. 1e, a higher stabilized PCE of 22.4% was detected for the ES-based device than its counterpart (with a PCE of 20.2%), further confirming the beneficial role of ES in device performance. The significantly improved PCE for ES-based devices is mainly from the increased VOC and FF, as displayed in Fig. 1f and g and summarized in the Table S3 (ESI†). The improvement in VOC could be ascribed to the suppressed recombination, which will be discussed later. The improvement of the FF could be ascribed to the reduction of series resistance (Rs).50 As shown in Fig. S2 (ESI†), the control PSCs show an average Rs of 5.91 Ω cm2, whereas it is only 3.27 Ω cm2 for the ES-based device, indicating that ES additives could significantly improve the charge transport and boost the FF of PSCs. In addition, the hysteresis index,51 defined as (PCEreverse − PCEforward)/PCEreverse, is evidently reduced from 0.093 to 0.052 upon ES addition. It is well-known that the hysteresis behavior is associated with the charge carrier extraction and recombination process at the interface of perovskite/charge transport layers.52 The reduction of HI in this work suggests that ES is conducive to boosting the charge transport and suppressing their recombination.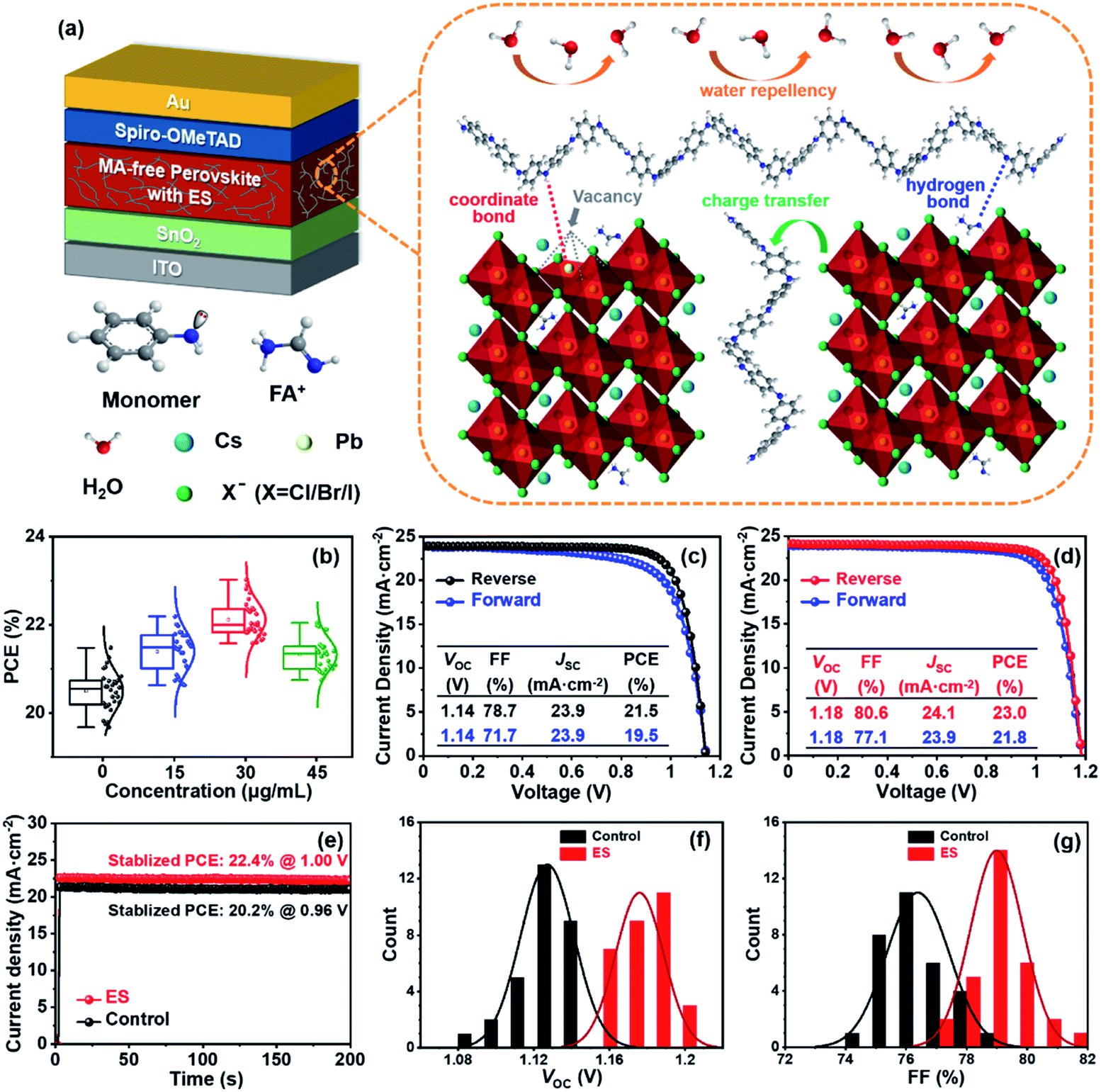 | ||
| Fig. 1 (a) Schematic diagram of a PSC with ES. (b) The statistics of PCEs for devices modified with ES with different concentrations. The best J–V curves for (c) the control and (d) ES-based devices in reverse and forward scan directions. (e) Steady-state output measurements ( 0.96 V for the control device and 1.00 V for the ES-based device). (f) The VOC distribution of PSCs with and without ES. (g) The FF distribution of PSCs with and without ES. | ||
To elucidate the performance improvement upon ES introduction, X-ray diffraction (XRD) patterns were collected to probe the impact of ES on perovskite crystallinity. All the samples in Fig. 2a show the same diffraction peaks at 14.13°, 19.97°, 24.54°,28.33°, and 31.78°, which are assigned to the (110), (112), (202), (220), and (310) crystal planes of α-FAPbI3 without any additional peaks, indicating that pure phase perovskites are obtained with or without the incorporation of ES. Fig. S3a (ESI†) obviously shows that the full width at half maximum (FWHM) value of the (110) plane in the XRD patterns is progressively lowered with addition of ES contents, and the 30 μg mL−1 ES sample exhibits the smallest FWHM (0.122°), suggestive of the highest crystalline quality of perovskite films. Meanwhile, ES-based perovskite films exhibit a remarkably enhanced intensity ratio of the (110)/(112) plane (Fig. S3b, ESI†), especially for perovskite films with 30 μg mL−1 ES, further demonstrating that ES can improve the crystallinity of MA-free based perovskite films.
 | ||
| Fig. 2 (a) The XRD patterns of perovskite films with different amounts of ES. Top-view and cross-sectional SEM images of the perovskite films (b and d) without and (c and e) with ES. | ||
Fig. 2b and c display the top-view scanning electron microscopy (SEM) images of perovskite films. Both samples exhibit pinhole-free and compact surface morphology. Detailed information of grain size for both samples is given in Fig. S4 (ESI†). The average grain size of the ES-based perovskite film is ∼377 nm, which is about 60 nm larger than that of the control. The increased grain size of the ES-based film can also be observed from the cross-sectional SEM images. Compared to the control film with disordered crystal grains (Fig. 2d), the ES-based perovskite film appears with vertically aligned grains and reduced grain boundaries (Fig. 2e). Besides, atomic force microscopy (AFM) (Fig. S5, ESI†) shows that the root-mean-square (RMS) roughness of the ES-based perovskite film is 22.7 nm, apparently lower than that of the control film with RMS = 25.6 nm. Above all, the ES introduction could improve crystallinity, promote uniformity, and reduce roughness of the resultant perovskite films, thus beneficial for the deposition of uniform hole transport materials (HTMs) atop of them.
Fourier transform infrared spectroscopy (FTIR) was performed to gain insight into the interaction between ES and the perovskite film. Fig. 3a clearly shows that both films exhibit signals at 3401 and 3263 cm−1, which are assigned to the stretching vibrations of N–H and C–H of perovskites, respectively;53 while the peak at 1349 cm−1 is attributed to the bending vibration of C–N.54 The pure ES sample shows two characteristic peaks at 1470 and 796 cm−1, which could be assigned to the characteristics of CC stretching and C–H vibration in the benzenoid ring, respectively. In contrast, both characteristic peaks shift to 1480 and 814 cm−1, respectively, for the ES-based perovskite film. These results strongly reveal the interaction between ES and the perovskite film. In addition, the bending vibration of N–H shifts from 1611 cm−1 for the pristine perovskite film to 1628 cm−1 for the ES-based film, implying the formation of hydrogen bonds in such a case.55
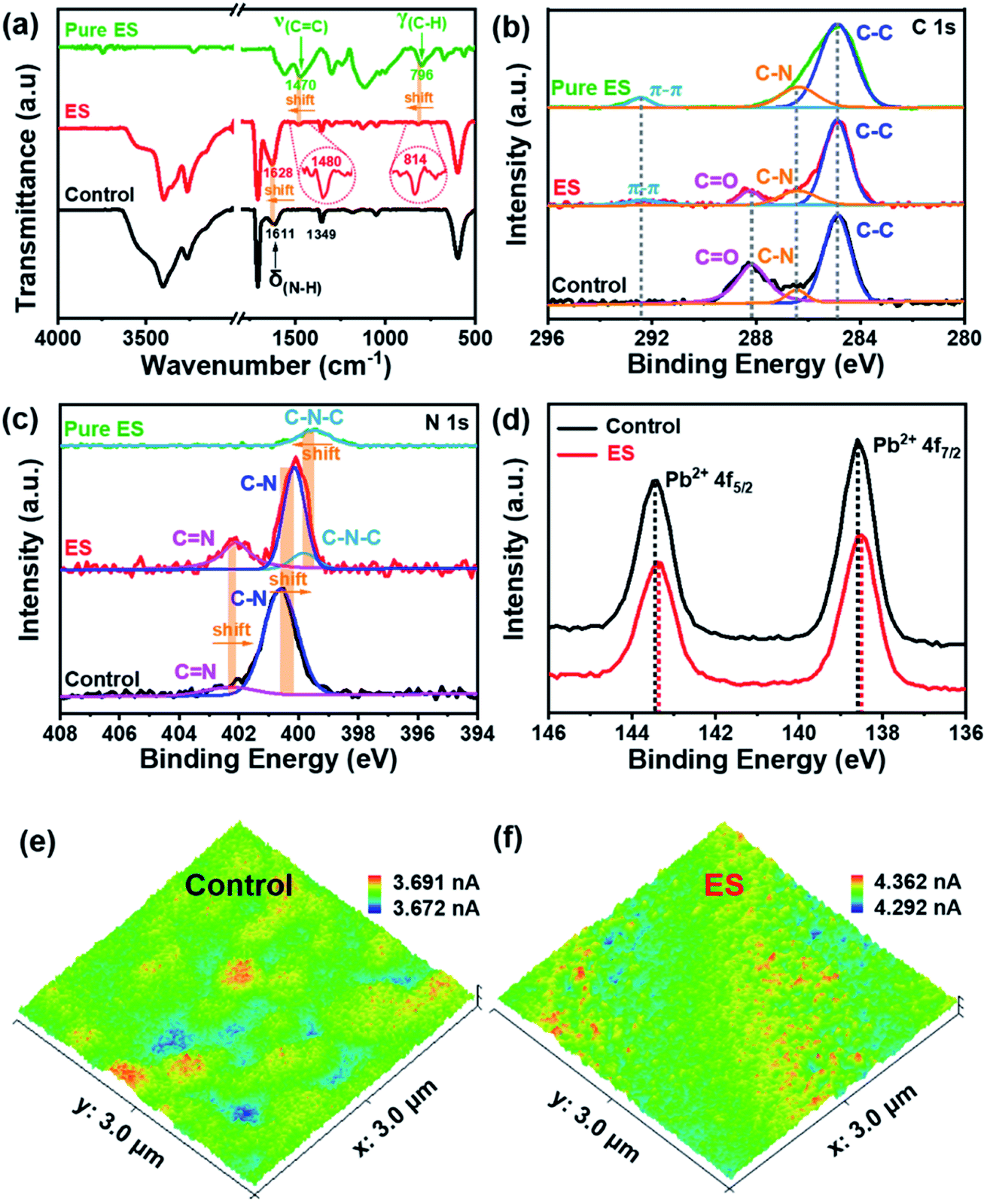 | ||
| Fig. 3 (a) FTIR spectra of the ES molecule, the ES-based perovskite film, and control film. (b–d) High-resolution XPS spectra: (b) C 1s, (c) N 1s and (d) Pb 4f. Current distribution mapping images from c-AFM measurements for (e) control and (f) ES-based devices under a bias voltage of 1.0 V. | ||
X-ray photoelectron spectroscopy (XPS) was utilized to analyse the chemical states, and the survey XPS spectra are shown in Fig. S6 (ESI†). The successful incorporation of ES can be further confirmed by the emerging CC bond (292.4 eV) signal of the C 1s spectra (Fig. 3b) in the ES-based perovskite film. Notably, the peak intensity of CO decreases dramatically in the presence of ES, indicative of the improved stability of the perovskite layer against environmental invasion.56 The N 1s XPS spectra for the control film can be deconvoluted into two peaks (CN at 402.4 eV and C–N at 400.6 eV), whereas it can be deconvoluted into three peaks (CN at 402.1 eV, C–N at 400.1 eV, and C–N–C at 399.8 eV) for the ES-based film (Fig. 3c). This shift may come from the formation of a hydrogen bond between the FA+ and ES.57,58 The newly detectable C–N–C signal originated from the amine in ES (399.5 eV), while the former two signals associated with the FA+ group are bule-shifted upon ES modification. As exhibited in the Pb 4f of XPS spectra (Fig. 3d), two distinct peaks are observed at 138.6 (Pb 4f5/2) and 143.5 eV (Pb 4f7/2) in the control sample, both of which shift towards lower binding energies (138.4 and 143.3 eV) in the presence of ES, indicative of increased electron cloud density of the Pb atom. These results demonstrate that N atoms in ES might afford an unpaired electron to under-coordinated Pb2+ in perovskites to passivate defects.29 To sum up, ES could chemically interact with perovskite materials via either coordinative bonding or hydrogen bonding, which would be effective to suppress the non-radiative recombination and improve the stability of perovskite films.
First-principles calculations were then performed to further understand the defect passivation mechanism at the molecular level (Fig. S7, ESI†). According to the density of states (DOS) distribution, the ES polymer can remove the Pb2+ defect state near the valence band maximum (VBM), hence reducing the Pb2+ defect states. These results further confirm that strong interactions between ES and a perovskite can effectively passivate defects to suppress the non-radiative recombination, which is conducive to improving device performance.
Ultraviolet visible (UV-vis) absorption spectroscopy and ultraviolet photoelectron spectroscopy (UPS) were further conducted to reveal the optical properties and surface energy of the perovskite films in this work. As shown in Fig. S8 (ESI†), both perovskite films show similar band edge absorption with bandgaps of ∼1.58 eV. Consequently, the energy band diagram within devices is determined, as schematically illustrated in Fig. S9 (ESI†). In the presence of ES in the perovskite, the work function (WF) is estimated to be −5.12 eV, up-shifting ∼0.02 eV from the control one (−5.14 eV). The ΔE, defined as the difference between the Fermi-level and VBM, is decreased from 0.41 eV for the control to 0.34 eV for the ES-based perovskite film. The reduction in ΔE indicates that ES modification suggests a more p-typed perovskite film,22,27 enhancing the driving force for hole collection. Except for the difference in ΔE, the ES-based cell exhibits better energy band alignment than the control one, contributing to the improvement in VOC. These results mean that ES in perovskite films could facilitate charge transport, which was evidenced by conductive atomic force microscopy (c-AFM). Fig. 3e and f respectively depict the local dark conductivity mapping in the contact mode under an applied bias of 1.0 V. The control film presents an average value of 3.664 nA, while the ES-based sample shows a higher average current of 4.331 nA, which discloses the increased conductivity of the perovskite film subjected to ES modification. In a word, ES in perovskite films can enhance the match in energy structure alignment and acts as a “carrier driver” to accelerate charge separation and transport, very beneficial for boosting device performance in the final devices.
We then carried out comprehensive analyses to uncover the charge dynamics of our devices. As can be seen from the steady-state photoluminescence (PL) spectra in Fig. 4a, the ES-based perovskite film shows a significantly stronger PL intensity (with peak at 798 nm) with a slightly blue-shifted peak compared to the control sample (with peak as 792 nm), indicating that ES can passivate traps and reduce the non-radiative recombination in perovskite films.59 The time-resolved photoluminescence (TRPL) spectra provide more evidence for the suppressed non-radiative recombination of the ES additive (Fig. 4b), which can be fitted with bi-exponential decays according to the equations below:60–62
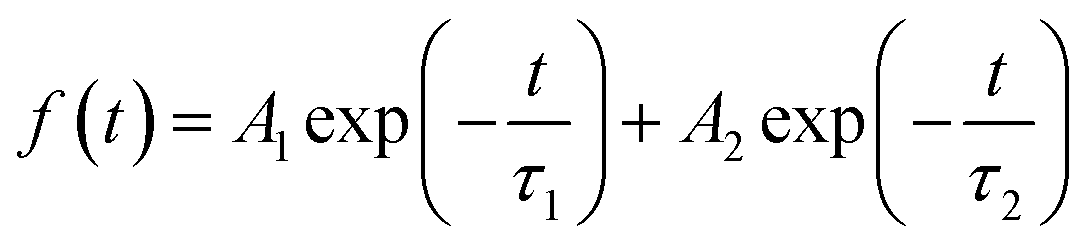 | (1) |
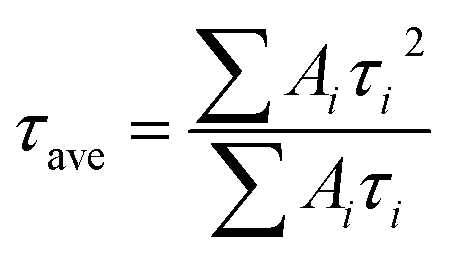 | (2) |
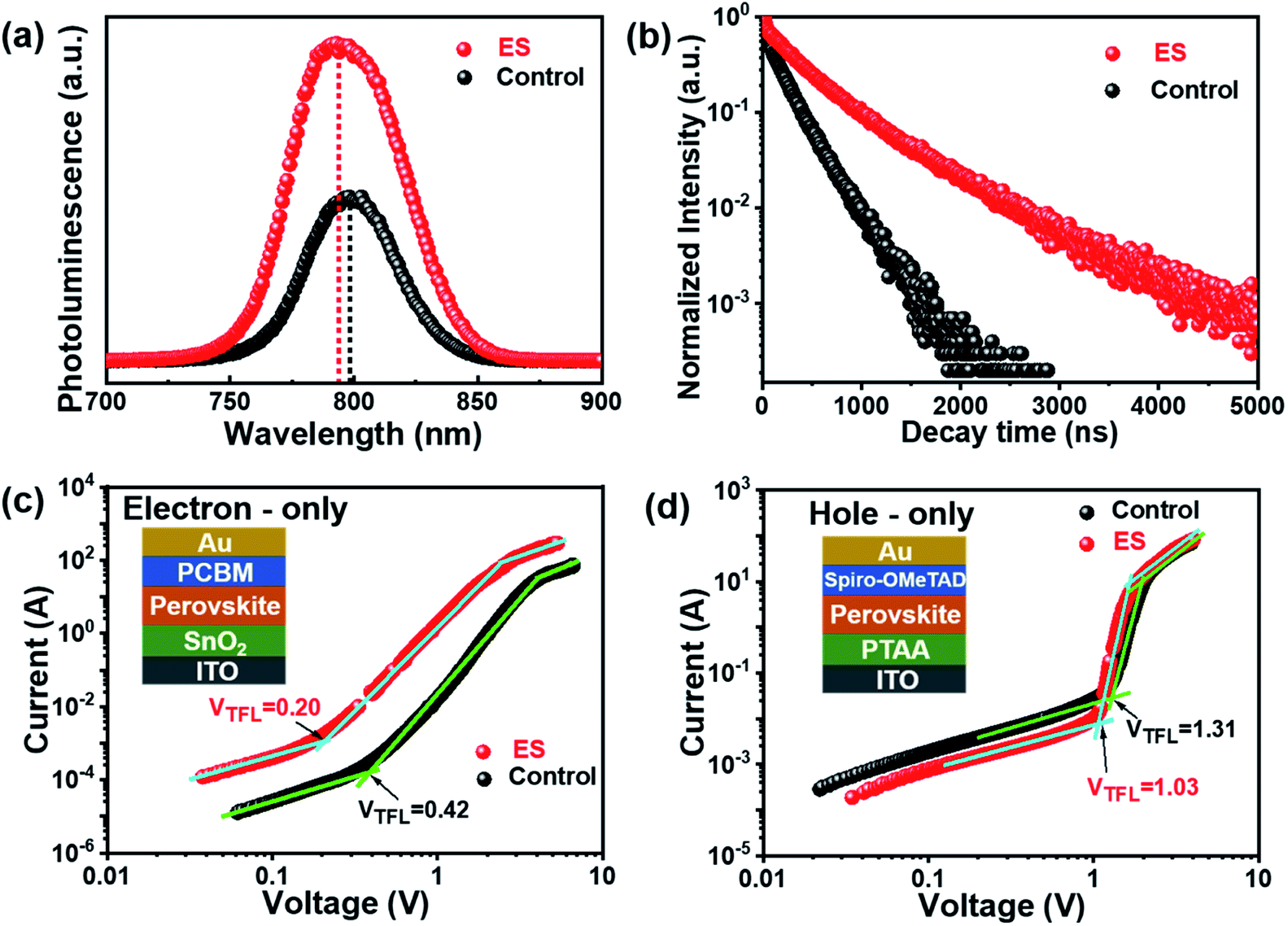 | ||
| Fig. 4 (a) Steady-state PL spectra and (b) TRPL decays. SCLC plots for (c) electron-only and (d) hole-only devices. | ||
The space-charge-limited current (SCLC) curves were recorded to quantitatively evaluate the defect trap densities in perovskite films. Fig. 4c and d illustrate respectively the typical current–voltage (I–V) curves for the electron-only and hole-only architectures in the dark. The defect trap density (Nt) can be calculated by using eqn (3) as follows:2,65,66
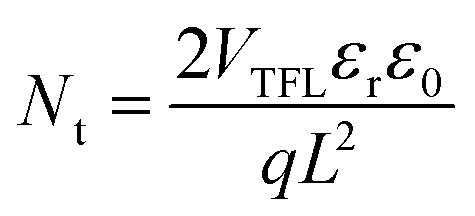 | (3) |
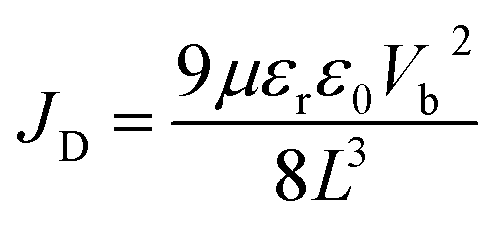 | (4) |
Transient photocurrent decay (TPC, Fig. 5a) and photovoltage decay (TPV, Fig. 5b) measurements performed at the short circuit and open circuit were employed to quantitatively analyse carrier kinetics.68–70 The charge transport (τt) and recombination time constant (τr) are defined as the time gap during which the signal current dampens to 1/e of the its initial value immediately after excitation. The τt derived from TPC via bi-exponential fitting (according to eqn (1) and (2)) was slightly decreased from 2.14 μs for the control cell to 1.44 μs for the ES-modified device. The ES-based device displayed a significantly lowered τt, demonstrating faster charge transport than the control device, which could be attributed to the improvement of film quality and carrier mobility. This result was in accordance with the enhanced JSC and FF. Likewise, the τr can be extracted from the TPV. The ES-based cell has a substantially longer τr of ∼46.1 μs than the unmodified one (τr = ∼21.6 μs), which might be attributed to passivated defects and decreased trap density in the former, which is consistent with the considerable improvement in VOC.71 Electrochemical impedance spectroscopy (EIS) was further performed to probe charge transfer characteristics of the PSCs. Fig. 5c and d display the Nyquist plots for the control and ES-based devices measured under different applied voltages, respectively. Only one semicircle can be clearly observed for each device under high frequency, which can be assigned to the charge transport resistance (Rct), and is associated with carrier transport at the interface with the perovskite. The equivalent circuit model is shown in the inset of Fig. S11 (ESI†).72–74 The fitting parameters are summarized in the Table S6 (ESI†), and the similar dependence of Rct on applied voltage are observed for both devices (Fig. S11, ESI†). Overall, a lower Rct is estimated for the ES-based device than the control one under the same bias voltage. When the bias voltage was increased from 0 to 0.2, 0.4, 0.6, and 0.8 V, the Rct of the ES-modified devices is decreased correspondingly from 3.6 × 104 to 2.5 × 104, 2.0 × 104, 1.6 × 104, and 0.9 × 104 Ω, while it is 8.9 × 104 to 7.2 × 104, 6.1 × 104, 4.5 × 104, and 1.8 × 104 Ω for the control cell without the ES additive. These results provide strong evidence that the ES modification can accelerate carrier transportation, which is in accordance with the increased conductivity and the decreased transport lifetime of the devices discussed above.
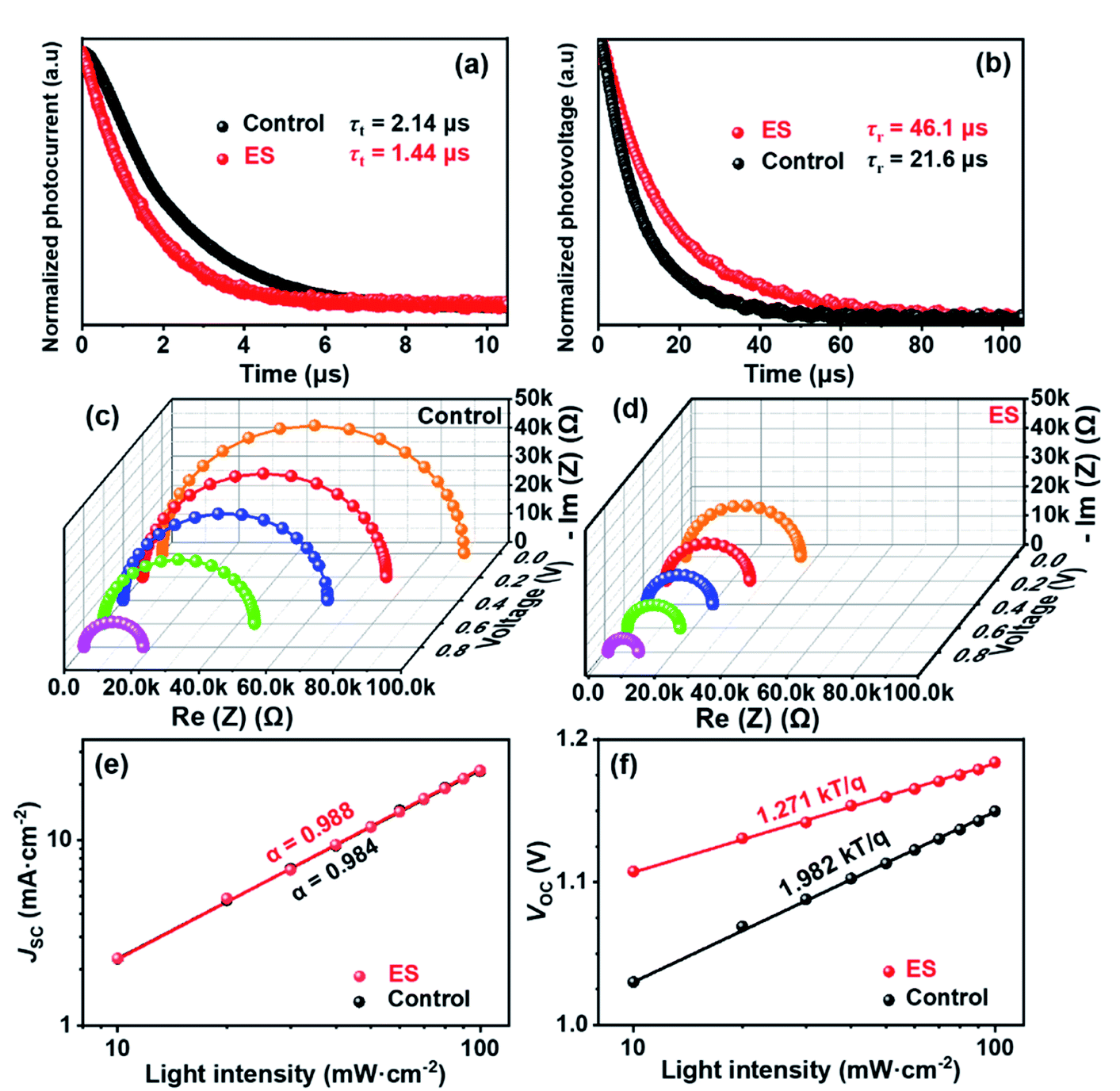 | ||
| Fig. 5 (a) Transient photocurrent (TPC) decay. (b) Transient photovoltage (TPV) decay. EIS plots under varied applied voltages for the device (c) without and (d) with ES. (e) JSC values and (f) VOC values of the corresponding devices vs. light intensities on a seminatural logarithmic scale. | ||
To further investigate the carrier recombination process within devices, the dependence of the PV parameters on the light intensity was thus evaluated. Fig. 5e clearly shows that, for both the control and the ES-based cells, photocurrents exhibit a linear relationship with light intensity, displaying a slope approaching 1. This is indicative of neglectable bimolecular recombination in both devices.75,76 As is well known, the ideality factor (n) can be used to evaluate the recombination process in solar cell devices,77,78 which can be obtained from the dependence of VOC on light intensity (Fig. 5f). The n value between 1 and 2 indicates that Shockley Read Hall (SRH) recombination dominates in the perovskite film. Usually, SRH recombination is inversely proportional to trap density. What's more, both SRH and the interface recombination mechanisms exist when n > 2.79 Calculations shows that the ES-based device has a smaller n of 1.27 than the control device with n = 1.97, suggestive of suppressed SRH recombination for the former one. These results are indicative of the decreased trap density and improved film quality for the ES-based perovskite film, which would be conducive to increasing the VOC, FF, and PCE for the final devices.
At present, the practical application of PSCs is severely impeded by their instability problems, among which the moisture-sensitive nature of perovskite films is an important limiting factor. Water contact angles of the control and the ES-based perovskite films were then measured. As illustrated in Fig. 6a and b, the ES-based perovskite film demonstrates a larger water contact angle of 57.3° than the control one (with a contact angle of 43.6°), owing to the benzenoid and quinonoid units of ES. These results reveal a more hydrophobic surface and a better moisture tolerance for the ES-based perovskite than the control film. The moisture stability for the unencapsulated devices was explored (RH of 35 ± 5%). As shown in Fig. 6dFig. 6c and Table S7 (ESI†), approximately 89.1% of the initial PCE is retained for the ES-based device after 1000 h storage, whereas the control device decays to 34.4% of its original value. As shown in the XRD pattern (Fig. S12a†), the peak intensity of α-FAPbI3 is dramatically reduced for the control films after 500 h aging, whereas the signals for CsPb2X5 (10.0°), δ-phase FAPbI3 (11.9°), PbI2 (12.8°) and δ-phase CsPbI3 (13.1°) emerge. That is to say, the control films undergo decomposition, phase segregation, and phase transformation from the active black phase to the non-active yellow phase. In sharp contrast (Fig. S12b†), the ES-based perovskite film exhibits much stronger peak intensity of α-FAPbI3 and lower signal intensity of all the impurity phases than the control one even after subjection to a humid environment for 1000 h, validating significantly improved moisture stability of MA-free perovskite films. Notably, the phase segregation is restrained with ES modification,80 which might be attributed to the coordination and hydrogen bond formation between ES and perovskites. To further evaluate the potential of the ES-based PSC against moisture, we conducted a stability test under extreme conditions with a high RH of 80 ± 5%. As shown in the XRD patterns (Fig. S13, ESI†), J–V curves (Fig. S14, ESI†), photovoltaic parameters (Table S8, ESI†) and UV-vis spectra (Fig. S15, ESI†), the PCE of the control device decayed to 34.4% of its original value within 48 h storage, whereas the ES-based PSCs retained 65.4% of their initial PCE. When the storage time was further extended to 72 h, the ES-based PSCs still reserved 53.2% of their initial PCE. These results demonstrate that the ES additive can apparently enhance the moisture stability of the resultant cells, probably because their presence within grain boundaries and on the top surface can protect the perovskite from the attack of water. Furthermore, we studied the effect of ES on the thermal stability of MA-free PSCs, in which poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA) was adopted to replace spiro-OMeTAD as HTMs.15,81 As displayed in Fig. 6d and Table S9 (ESI†), the unsealed ES-based device reserves 83.8% of its initial PCE when subjected to thermal aging at 85 °C for 1000 h, while only 42.4% of the initial value was maintained for the control device. The XRD patterns for the control and ES-based perovskite films under 85 °C thermal stress are displayed in Fig. S16 a and b (ESI†), respectively. Both the control films experienced an obvious degradation after thermal aging for 500–1000 h, which was confirmed not only by the reduced intensities in XRD diffractions of α-FAPbI3 but also by the emergence of XRD signal of PbI2 (12.8°). However, upon ES addition, the PbI2 peaks were much lower than that of the control sample under the identical conditions, indicating that the introduction of ES into perovskite films could suppress the decomposition of perovskites under thermal stress. The SEM images for the control and the ES-based perovskite films and devices under 85 °C thermal stress are displayed in Fig. S17 and S18 (ESI†). For the control sample, after heating for 500 h, the upper surface of the perovskite layer decomposed significantly. Upon further extending the storage time to 1000 h, obvious deformation of the bulk perovskite layer could be observed, while for the ES-modified sample, the morphology of the perovskite layers kept almost unchanged after heating for 500 and 1000 h, which confirms the enhanced thermal stability due to the presence of ES. In addition, we also recorded the UV-vis absorption spectra of control and the ES-based perovskite films before and after exposure to 85 °C to evaluate their thermal stability. As displayed in Fig. S19,† the control film suffers from a serious degradation after 1000 h aging at 85 °C owing to the decomposition of the perovskite layer, whereas the ES-based perovskite film displays relatively slow degradation. These results are consistent with the XRD and SEM results in Fig. S16–S18,† indicating that the addition of ES is conducive to the improvement of heat stability. Therefore, the introduction of ES into perovskite films could significantly improve both the moisture and thermal stabilities of MA-free PSCs.
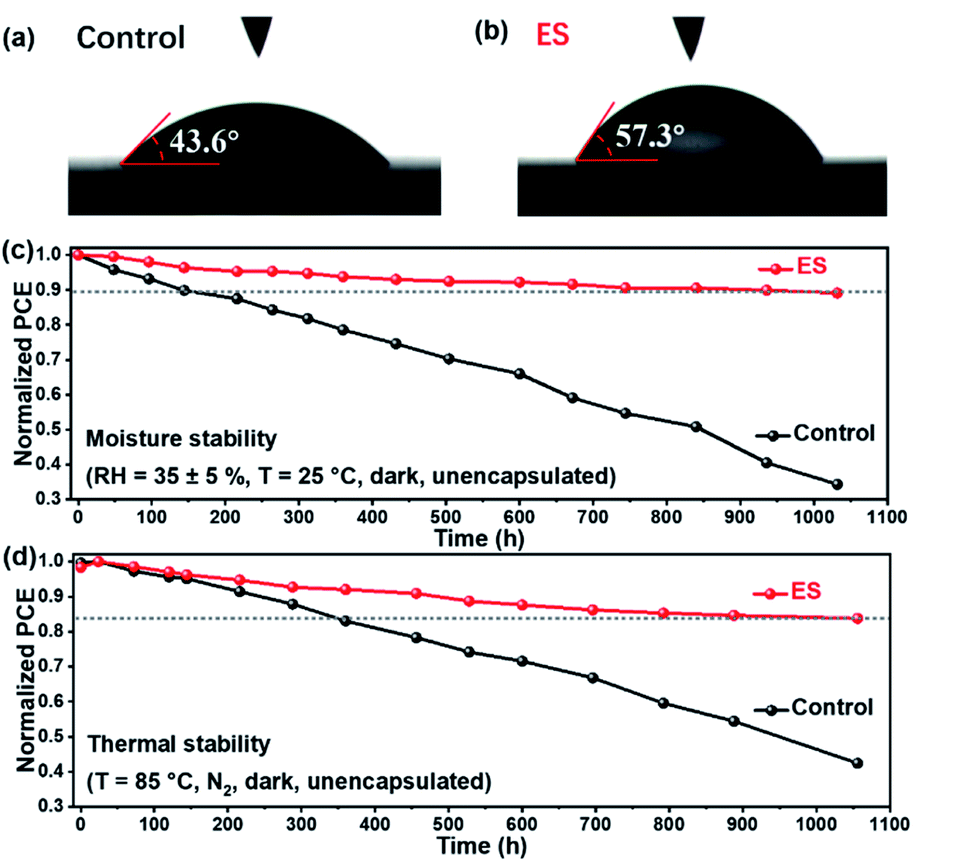 | ||
| Fig. 6 (a and b) The water contact angle tests. The stability of unsealed devices when subjected to (c) 35 ± 5% RH and (d) thermal aging at 85 °C. | ||
Finally, as a proof of concept to corroborate the scalability of ES modification in PSCs, we expanded our study to the mini-PSC modules on a 5 × 5 cm2 FTO substrate. As shown in Fig. 7, each module contains seven single sub-cells with connection in series and shows an overall active area of 10.3 cm2. The control module delivers a PCE of 17.7% together with a VOC of 7.55 V, a JSC of 3.39 mA cm−2, and a FF of 69.1%, while a PCE of 19.3% along with a VOC of 7.92 V, a JSC of 3.41 mA cm−2, and a FF of 71.3% was yielded for the ES-based module. The promoted PV performance is mainly derived from the increase in the VOC and FF, in line with the case in the small-area devices discussed above, solidly verifying the effectiveness of our strategy in scalable PV technology.
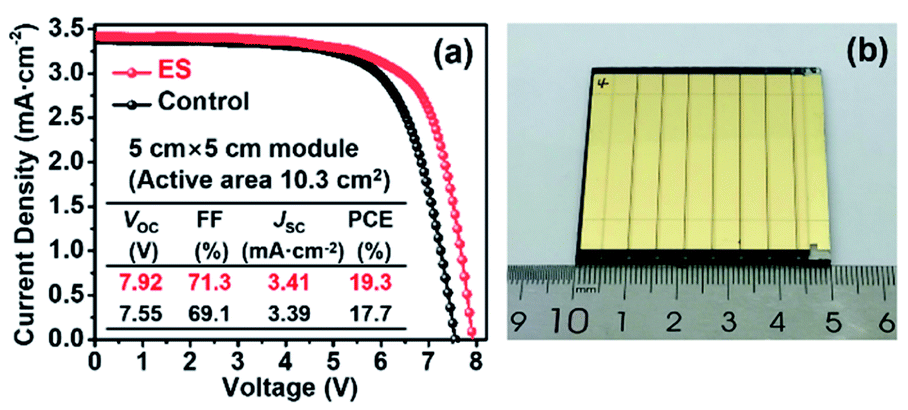 | ||
| Fig. 7 (a) The J–V curves of the control and ES-based modules. (b) Photograph of a module. | ||
Conclusions
In summary, a feasible strategy has been proposed to simultaneously passivate defects and boost charge transport in MA-free perovskite solar cells by incorporating a conductive polymer ES into perovskite precursor solution. The ES molecule can coordinate with Pb2+ through the lone-pair electron of the N atom and form hydrogen bonds with FA+ in perovskite films. The conjugated conductive feature of ES facilitates the carrier transport and enables the balance between electron and hole mobility, yielding an appreciable PCE of 23.0% for the ES-based device. In addition, the hydrophobic nature of ES considerably alleviates the moisture penetration into perovskite films, and unsealed devices retain 89.1% of their initial PCEs under 35 ± 5% RH exposure for 1000 h. The ES-based devices also exhibit excellent thermal-stability, maintaining 83.8% of the original efficiency upon subjection to thermal aging at 85 °C for 1000 h. This work confirms the effectiveness of conductive additives in obtaining highly efficient and stable PSCs, providing hope for further practical application of perovskite technology.Data availability
All data are available in the manuscript and in the ESI.†Author contributions
Z. Fan: investigation, validation, data curation, formal analysis, visualization, writing – original draft. Y. Yin: DFT calculations, visualization, funding acquisition. B. Cai: visualization, supervision, methodology, writing – original draft, writing-review and editing. Q. Ma: investigation, resources. Q. Liu: investigation, resources. X. Liu: investigation, resources. Y. Lv: conceptualization, funding acquisition, methodology, supervision, visualization, writing-original draft, writing – review and editing. W.-H. Zhang: writing-review and editing, resources, supervision, funding acquisition, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the support from the National Natural Science Foundation of China (grant no. 21773218 and 51902005), Key Research and Development Projects of Sichuan province (grant no. 2017GZ0052), China Postdoctoral Science Foundation (grant no. 2019M653485), and Innovation Capability Support Program of Shaanxi Province (grant no.2021KJXX-49).Notes and references
- S.-H. Turren-Cruz, A. Hagfeldt and M. Saliba, Science, 2018, 362, 449–453 CrossRef CAS PubMed.
- Y. Chen, Z. Yang, S. B. Wang, X. J. Zheng, Y. H. Wu, N. Y. Yuan, W. H. Zhang and S. Z. Liu, Adv. Mater., 2018, 30, 1805660 CrossRef PubMed.
- W. Qiu, A. Ray, M. Jaysankar, T. Merckx, J. P. Bastos, D. Cheyns, R. Gehlhaar, J. Poortmans and P. Heremans, Adv. Funct. Mater., 2017, 27, 1700920 CrossRef.
- J. Chen, J. Xu, C. Zhao, B. Zhang, X. Liu, S. Dai and J. Yao, ACS Appl. Mater. Interfaces, 2019, 11, 4597–4606 CrossRef CAS PubMed.
- Y.-H. Chiang, M. Anaya and S. D. Stranks, ACS Energy Lett., 2020, 5, 2498–2504 CrossRef CAS PubMed.
- Y. Yu, C. Wang, C. R. Grice, N. Shrestha, J. Chen, D. Zhao, W. Liao, A. J. Cimaroli, P. J. Roland, R. J. Ellingson and Y. Yan, ChemSusChem, 2016, 9, 3288–3297 CrossRef CAS PubMed.
- Y. Yu, C. Wang, C. R. Grice, N. Shrestha, D. Zhao, W. Liao, L. Guan, R. A. Awni, W. Meng, A. J. Cimaroli, K. Zhu, R. J. Ellingson and Y. Yan, ACS Energy Lett., 2017, 2, 1177–1182 CrossRef CAS.
- J. Du, S. Liu, J. Wu, W. Zhang, W. Zhang, A. Mei, Y. Rong, Y. Hu and H. Han, Sol. RRL, 2020, 4, 2000455 CrossRef CAS.
- J. Yang, Y. Chen, W. Tang, S. Wang, Q. Ma, Y. Wu, N. Yuan, J. Ding and W.-H. Zhang, J. Energy Chem., 2020, 48, 217–225 CrossRef.
- D. P. McMeekin, Z. Wang, W. Rehman, F. Pulvirenti, J. B. Patel, N. K. Noel, M. B. Johnston, S. R. Marder, L. M. Herz and H. J. Snaith, Adv. Mater., 2017, 29, 1607039 CrossRef PubMed.
- X.-X. Gao, W. Luo, Y. Zhang, R. Hu, B. Zhang, A. Züttel, Y. Feng and M. K. Nazeeruddin, Adv. Mater., 2020, 32, 1905502 CrossRef CAS PubMed.
- J. C. Yang, W. J. Tang, R. H. Yuan, Y. Chen, J. Wang, Y. H. Wu, W. J. Yin, N. Y. Yuan, J. N. Ding and W. H. Zhang, Chem. Sci., 2021, 12, 2050–2059 RSC.
- T. Bu, J. Li, H. Li, C. Tian, J. Su, G. Tong, K. Ono Luis, C. Wang, Z. Lin, N. Chai, X.-L. Zhang, J. Chang, J. Lu, J. Zhong, W. Huang, Y. Qi, Y.-B. Cheng and F. Huang, Science, 2021, 372, 1327–1332 CrossRef CAS PubMed.
- R. H. Yuan, B. Cai, Y. H. Lv, X. Gao, J. W. Gu, Z. H. Fan, X. H. Liu, C. Yang, M. Z. Liu and W. H. Zhang, Energy Environ. Sci., 2021, 14, 5074–5083 RSC.
- Y. H. Lv, R. H. Yuan, B. Cai, B. Bahrami, A. H. Chowdhury, C. Yang, Y. H. Wu, Q. Q. Qiao, S. Z. Liu and W. H. Zhang, Angew. Chem., Int. Ed., 2020, 59, 11969–11976 CrossRef CAS PubMed.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 RSC.
- F. Zu, P. Amsalem, M. Ralaiarisoa, T. Schultz, R. Schlesinger and N. Koch, ACS Appl. Mater. Interfaces, 2017, 9, 41546–41552 CrossRef CAS PubMed.
- N. Ahn, K. Kwak, M. S. Jang, H. Yoon, B. Y. Lee, J.-K. Lee, P. V. Pikhitsa, J. Byun and M. Choi, Nat. Commun., 2016, 7, 13422 CrossRef CAS PubMed.
- C. Shen, Y. Wu, S. Zhang, T. Wu, H. Tian, W.-H. Zhu and L. Han, Sol. RRL, 2020, 4, 2000069 CrossRef CAS.
- H. Wang, Y. Song, Y. Kang, S. Dang, J. Feng and Q. Dong, J. Mater. Chem. A, 2020, 8, 7309–7316 RSC.
- J. Hou, F. Deng, Q. Wu, L. Yang, J. Wu, X. Li, Y.-Z. Zheng, N. Li, H. Ding and X. Tao, J. Power Sources, 2020, 449, 227484 CrossRef CAS.
- S. Li, K. Fan, Y. Cui, S. Leng, Y. Ying, W. Zou, Z. Liu, C.-Z. Li, K. Yao and H. Huang, ACS Energy Lett., 2020, 5, 2015–2022 CrossRef CAS.
- S. Li, Z. Liu, Z. Qiao, X. Wang, L. Cheng, Y. Zhai, Q. Xu, Z. Li, K. Meng and G. Chen, Adv. Funct. Mater., 2020, 30, 2005846 CrossRef CAS.
- Q. Yao, Q. Xue, Z. Li, K. Zhang, T. Zhang, N. Li, S. Yang, C. J. Brabec, H.-L. Yip and Y. Cao, Adv. Mater., 2020, 32, 2000571 CrossRef CAS PubMed.
- Y. Chen, J. Yang, S. Wang, Y. Wu, N. Yuan and W.-H. Zhang, iScience, 2020, 23, 100762 CrossRef CAS PubMed.
- J. Chen, J.-Y. Seo and N.-G. Park, Adv. Energy Mater., 2018, 8, 1702714 CrossRef.
- J. Yang, W. Tang, R. Yuan, Y. Chen, J. Wang, Y. Wu, W.-J. Yin, N. Yuan, J. Ding and W.-H. Zhang, Chem. Sci., 2021, 12, 2050–2059 RSC.
- X. Zheng, Y. Deng, B. Chen, H. Wei, X. Xiao, Y. Fang, Y. Lin, Z. Yu, Y. Liu, Q. Wang and J. Huang, Adv. Mater., 2018, 32, 1803428 CrossRef PubMed.
- B. B. Liu, H. Bi, D. M. He, L. Bai, W. Q. Wang, H. K. Yuan, Q. L. Song, P. Y. Su, Z. G. Zang, T. W. Zhou and J. Z. Chen, ACS Energy Lett., 2021, 6, 2526–2538 CrossRef CAS.
- W.-Q. Wu, P. N. Rudd, Q. Wang, Z. Yang and J. Huang, Adv. Mater., 2020, 32, 2000995 CrossRef CAS PubMed.
- X. Feng, R. Chen, Z.-A. Nan, X. Lv, R. Meng, J. Cao and Y. Tang, Adv. Sci., 2019, 6, 1802040 CrossRef PubMed.
- X. Duan, X. Li, L. Tan, Z. Huang, J. Yang, G. Liu, Z. Lin and Y. Chen, Adv. Mater., 2020, 32, 2000617 CrossRef CAS PubMed.
- C. Liang, K. M. M. Salim, P. W. Li, Z. Wang, T. M. Koh, H. Gu, B. Wu, J. M. Xia, Z. P. Zhang, K. Y. Wang, T. H. Liu, Q. Wei, S. S. Wang, Y. X. Tang, G. S. Shao, Y. L. Song, N. Mathews and G. C. Xing, J. Mater. Chem. A, 2020, 8, 5874–5881 RSC.
- C. Liang, D. D. Zhao, Y. Li, X. J. Li, S. M. Peng, G. S. Shao and G. C. Xing, Energy Environ. Mater., 2018, 1, 221–231 CrossRef CAS.
- C. Liang, H. Gu, Y. D. Xia, Z. Wang, X. T. Liu, J. M. Xia, S. W. Zuo, Y. Hu, X. Y. Gao, W. Hui, L. F. Chao, T. T. Niu, M. Fang, H. Lu, H. Dong, H. Yu, S. Chen, X. Q. Ran, L. Song, B. X. Li, J. Zhang, Y. Peng, G. S. Shao, J. P. Wang, Y. H. Chen, G. C. Xing and W. Huang, Nat. Energy, 2021, 6, 38–45 CrossRef CAS.
- L. Zhu, X. Zhang, M. Li, X. Shang, K. Lei, B. Zhang, C. Chen, S. Zheng, H. Song and J. Chen, Adv. Energy Mater., 2021, 11, 2100529 CrossRef CAS.
- K. Xiao, R. Lin, Q. Han, Y. Hou, Z. Qin, H. T. Nguyen, J. Wen, M. Wei, V. Yeddu, M. I. Saidaminov, Y. Gao, X. Luo, Y. Wang, H. Gao, C. Zhang, J. Xu, J. Zhu, E. H. Sargent and H. Tan, Nat. Energy, 2020, 5, 870–880 CrossRef CAS.
- X. Li, M. Ibrahim Dar, C. Yi, J. Luo, M. Tschumi, S. M. Zakeeruddin, M. K. Nazeeruddin, H. Han and M. Grätzel, Nat. Chem., 2015, 7, 703 CrossRef CAS PubMed.
- Y. Li, Z. Chen, B. Yu, S. Tan, Y. Cui, H. Wu, Y. Luo, J. Shi, D. Li and Q. Meng, Joule, 2022, 6, 676–689 CrossRef CAS.
- K. Kim, J. Han, S. Maruyama, M. Balaban and I. Jeon, Sol. RRL, 2021, 5, 2000783 CrossRef CAS.
- D. Q. Bi, C. Y. Yi, J. S. Luo, J. D. Decoppet, F. Zhang, S. M. Zakeeruddin, X. Li, A. Hagfeldt and M. Gratzel, Nat. Energy, 2016, 1, 16142 CrossRef CAS.
- Y. C. Zhao, J. Wei, H. Li, Y. Yan, W. K. Zhou, D. P. Yu and Q. Zhao, Nat. Commun., 2016, 7, 10228 CrossRef CAS PubMed.
- Q. Q. Dong, Z. W. Wang, K. C. Zhang, H. Yu, P. Huang, X. D. Liu, Y. Zhou, N. Chen and B. Song, Nanoscale, 2016, 8, 5552–5558 RSC.
- S. Masi, A. Rizzo, F. Aiello, F. Balzano, G. Uccello-Barretta, A. Listorti, G. Gigli and S. Colella, Nanoscale, 2015, 7, 18956–18963 RSC.
- F. H. Hou, B. Shi, T. T. Li, C. G. Xin, Y. Ding, C. C. Wei, G. C. Wang, Y. L. Li, Y. Zhao and X. D. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 25218–25226 CrossRef CAS PubMed.
- H. Zarenezhad, T. Balkan, N. Solati, M. Halali, M. Askari and S. Kaya, Sol. Energy, 2020, 207, 1300–1307 CrossRef CAS.
- J. W. Wei, F. R. Huang, S. N. Wang, L. Y. Zhou, Y. L. Xin, P. Jin, Z. Cai, Z. D. Yin, Q. Pang and J. Z. Zhang, Mater. Res. Bull., 2018, 106, 35–39 CrossRef CAS.
- H. Zheng, X. Xu, S. Xu, G. Liu, S. Chen, X. Zhang, T. Chen and X. Pan, J. Mater. Chem. C, 2019, 7, 4441–4448 RSC.
- C. Liu, J. He, M. Wu, Y. Wu, P. Du, L. Fan, Q. Zhang, D. Wang and T. Zhang, Sol. RRL, 2020, 4, 2000016 CrossRef CAS.
- D. Y. Li, P. Jiang, W. H. Zhang, J. K. Du, C. Qiu, J. L. Liu, Y. Hu, Y. G. Rong, A. Y. Mei and H. W. Han, Sol. RRL, 2022, 6, 2100554 CrossRef CAS.
- S. N. Habisreutinger, N. K. Noel and H. J. Snaith, ACS Energy Lett., 2018, 3, 2472–2476 CrossRef CAS.
- D. Y. Son, S. G. Kim, J. Y. Seo, S. H. Lee, H. Shin, D. Lee and N. G. Park, J. Am. Chem. Soc., 2018, 140, 1358–1364 CrossRef CAS PubMed.
- A. Solanki, M. M. Tavakoli, Q. Xu, S. S. H. Dintakurti, S. S. Lim, A. Bagui, J. V. Hanna, J. Kong and T. C. Sum, Adv. Mater., 2020, 32, 1907864 CrossRef CAS PubMed.
- Y. Xiao, G. Han, J. Wu and J.-Y. Lin, J. Power Sources, 2016, 306, 171–177 CrossRef CAS.
- J.-W. Lee, Z. Dai, C. Lee, H. M. Lee, T.-H. Han, N. De Marco, O. Lin, C. S. Choi, B. Dunn, J. Koh, D. Di Carlo, J. H. Ko, H. D. Maynard and Y. Yang, J. Am. Chem. Soc., 2018, 140, 6317–6324 CrossRef CAS PubMed.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photon., 2019, 13, 460–466 CrossRef CAS.
- N. X. Li, S. X. Tao, Y. H. Chen, X. X. Niu, C. K. Onwudinanti, C. Hu, Z. W. Qiu, Z. Q. Xu, G. H. J. Zheng, L. G. Wang, Y. Zhang, L. Li, H. F. Liu, Y. Z. Lun, J. W. Hong, X. Y. Wang, Y. Q. Liu, H. P. Xie, Y. L. Gao, Y. Bai, S. H. Yang, G. Brocks, Q. Chen and H. P. Zhou, Nat. Energy, 2019, 4, 408–415 CrossRef CAS.
- F. Li, X. Deng, F. Qi, Z. Li, D. Liu, D. Shen, M. Qin, S. Wu, F. Lin, S.-H. Jang, J. Zhang, X. Lu, D. Lei, C.-S. Lee, Z. Zhu and A. K. Y. Jen, J. Am. Chem. Soc., 2020, 142, 20134–20142 CrossRef CAS PubMed.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- Y. Lv, B. Cai, Y. Wu, S. Wang, Q. Jiang, Q. Ma, J. Liu and W.-H. Zhang, J. Energy Chem., 2018, 27, 951–956 CrossRef.
- Y. Lv, P. Wang, B. Cai, Q. Ma, X. Zheng, Y. Wu, Q. Jiang, J. J. Liu and W.-H. Zhang, Sol. RRL, 2018, 2, 1800133 CrossRef.
- K. Chen, W. Tang, Y. Chen, R. Yuan, Y. Lv, W. Shan and W.-H. Zhang, J. Energy Chem., 2021, 61, 553–560 CrossRef.
- D.-Y. Son, J.-W. Lee, Y. J. Choi, I.-H. Jang, S. Lee, P. J. Yoo, H. Shin, N. Ahn, M. Choi, D. Kim and N.-G. Park, Nat. Energy, 2016, 1, 16081 CrossRef CAS.
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- D. Yang, X. Zhou, R. Yang, Z. Yang, W. Yu, X. Wang, C. Li, S. Liu and R. P. H. Chang, Energy Environ. Sci., 2016, 9, 3071–3078 RSC.
- C. Yang, Z. Wang, Y. Lv, R. Yuan, Y. Wu and W.-H. Zhang, Chem. Eng. J., 2021, 406, 126855 CrossRef CAS.
- Q. Han, S. H. Bae, P. Sun, Y. T. Hsieh, Y. M. Yang, Y. S. Rim, H. Zhao, Q. Chen, W. Shi, G. Li and Y. Yang, Adv. Mater., 2016, 28, 2253–2258 CrossRef CAS PubMed.
- W. Chen, Y. Z. Wu, Y. F. Yue, J. Liu, W. J. Zhang, X. D. Yang, H. Chen, E. B. Bi, I. Ashraful, M. Gratzel and L. Y. Han, Science, 2015, 350, 944–948 CrossRef CAS PubMed.
- W. Z. Xu, Y. Gao, W. J. Ming, F. He, J. Z. Li, X. H. Zhu, F. Y. Kang, J. Y. Li and G. D. Wei, Adv. Mater., 2020, 32, 2003965 CrossRef CAS PubMed.
- A. Maurano, C. C. Shuttle, R. Hamilton, A. M. Ballantyne, J. Nelson, W. M. Zhang, M. Heeney and J. R. Durrant, J. Phys. Chem. C, 2011, 115, 5947–5957 CrossRef CAS.
- S. Wheeler, D. Bryant, J. Troughton, T. Kirchartz, T. Watson, J. Nelson and J. R. Durrant, J. Phys. Chem. C, 2017, 121, 13496–13506 CrossRef CAS.
- M. Tai, Y. Zhou, X. Yin, J. Han, Q. Zhang, Y. Zhou and H. Lin, J. Mater. Chem. A, 2019, 7, 22675–22682 RSC.
- Y. Zhou, X. Yin, Q. Luo, X. Zhao, D. Zhou, J. Han, F. Hao, M. Tai, J. Li, P. Liu, K. Jiang and H. Lin, ACS Appl. Mater. Interfaces, 2018, 10, 31384–31393 CrossRef CAS PubMed.
- C. Ma, D. Shen, T.-W. Ng, M.-F. Lo and C.-S. Lee, Adv. Mater., 2018, 30, 1800710 CrossRef PubMed.
- Y. Liu, Q. Chen, H.-S. Duan, H. Zhou, Y. Yang, H. Chen, S. Luo, T.-B. Song, L. Dou, Z. Hong and Y. Yang, J. Mater. Chem. A, 2015, 3, 11940–11947 RSC.
- M. M. Mandoc, F. B. Kooistra, J. C. Hummelen, B. d. Boer and P. W. M. Blom, Appl. Phys. Lett., 2007, 91, 263505 CrossRef.
- W. Tress, M. Yavari, K. Domanski, P. Yadav, B. Niesen, J. P. Correa Baena, A. Hagfeldt and M. Graetzel, Energy Environ. Sci., 2018, 11, 151–165 RSC.
- J. Chen and N.-G. Park, Adv. Mater., 2019, 31, 1803019 CrossRef CAS PubMed.
- X. J. Zheng, B. Chen, M. J. Yang, C. C. Wu, B. Orler, R. B. Moore, K. Zhu and S. Priya, ACS Energy Lett., 2016, 1, 424–430 CrossRef CAS.
- Z. Yang, J. Dou, S. Kou, J. Dang, Y. Ji, G. Yang, W.-Q. Wu, D.-B. Kuang and M. Wang, Adv. Funct. Mater., 2020, 30, 1910710 CrossRef CAS.
- H. P. Zhou, Q. Chen, G. Li, S. Luo, T. B. Song, H. S. Duan, Z. R. Hong, J. B. You, Y. S. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: detailed experimental parts, EQE curves, FWHM and SEM images, AFM and XPS spectra, theoretical calculation results, UV-vis absorption spectra, UPS spectra, EIS plots, and tables that summarize the polyaniline additives and MA-free PSCs and TRPL, SCLC, EIS and J–V parameters. See https://doi.org/10.1039/d2sc01804g |
| This journal is © The Royal Society of Chemistry 2022 |