
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper catalyzed borocarbonylation of benzylidenecyclopropanes through selective proximal C–C bond cleavage: synthesis of γ-boryl-γ,δ-unsaturated carbonyl compounds†
Li-Miao
Yang
,
Hui-Hui
Zeng
,
Xin-Lian
Liu
,
Ai-Jun
Ma
 and
Jin-Bao
Peng
*
and
Jin-Bao
Peng
*
School of Biotechnology and Health Sciences, Wuyi University, Jiangmen, Guangdong 529020, People's Republic of China. E-mail: pengjb_05@126.com
First published on 30th May 2022
Abstract
A copper catalyzed borocarbonylation of BCPs via proximal C–C bond cleavage for the synthesis of γ-boryl-γ,δ-unsaturated carbonyl compounds has been developed. Using substituted benzylidenecyclopropanes (BCPs) and chloroformates as starting material, a broad range of γ-boryl-γ,δ-unsaturated esters were prepared in moderate to excellent yields with excellent regio- and stereoselectivity. Besides, when aliphatic acid chlorides were used in this reaction, γ-boryl-γ,δ-unsaturated ketones could be produced in excellent yields. When substituted BCPs were used as substrates, the borocarbonylation occurred predominantly at the proximal C–C bond trans to the phenyl group in a regio- and stereoselective manner, which leads to the Z-isomers as the products. This efficient methodology involves the cleavage of a C–C bond and the formation of a C–C bond as well as a C–B bond, and provides a new method for the proximal C–C bond difunctionalization of BCPs.
Introduction
Organoboron compounds have emerged as versatile building blocks in organic synthesis owing to their unique reactivity and divergent synthetic applications.1 They have also found wide utility in materials science2 and medicinal chemistry.3 Thus, continued research efforts have been made for their synthesis,4 while the most prevalent synthetic route for their preparation proceeds via metal-catalyzed borylation of organohalides, hydroboration of alkenes, and borylation of C–H, C–Het and C–C bonds. Recently, impressive progress has been made in copper catalyzed borylative difunctionalization of π-systems, which provides a promising strategy for rapid generation of molecular complexity to install a boron and unique groups across π-systems.5 These processes are particularly valuable since the resulting C–B bond could be transformed into various functional groups via C–C and C–heteroatom bond-forming reactions through stereospecific transformations.6 The key step in these reactions is the addition of Cu–Bpin species to C–C π-bonds to give the corresponding β-borylalkylcopper intermediates, which could be captured by various electrophiles to give the difunctionalized products (Scheme 1a). A range of π-systems including alkenes, allenes, dienes, and alkynes as well as some carbon–heteroatom systems such as ketones, aldehydes and imines have been examined and found to react with a copper–boryl species. These reactions employed boro-metalation of π-bonds to give 1,2- or 1,4-difunctionalized products. However, borylative difunctionalization via C–C σ-bond cleavage has rarely been reported.7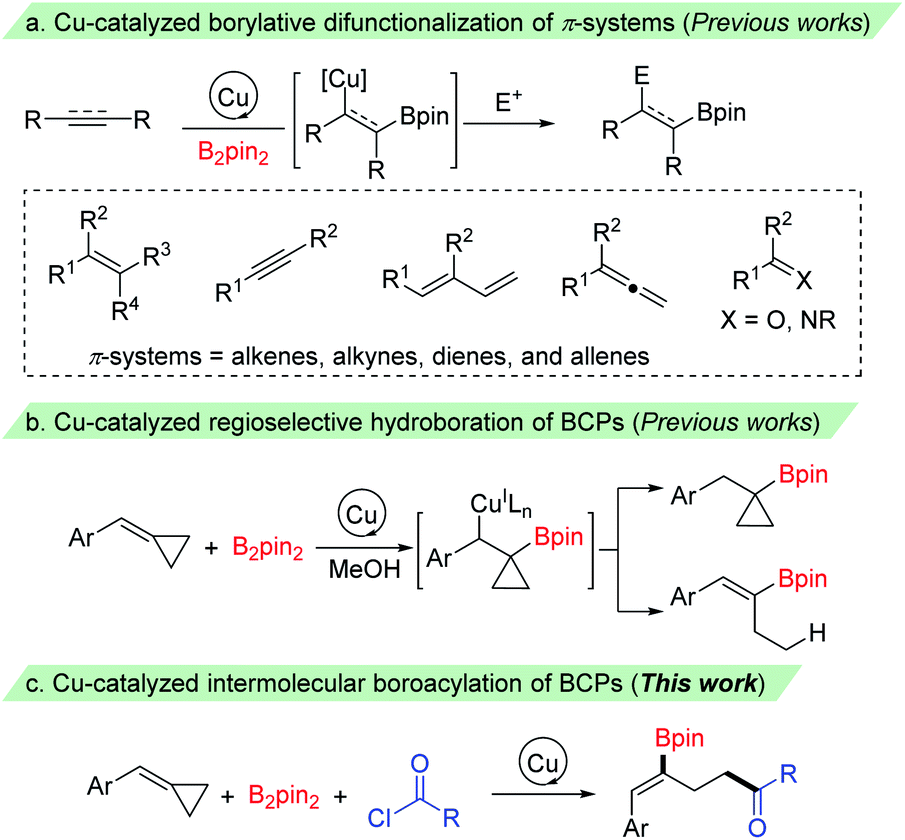 | ||
| Scheme 1 Difunctionalization of π-systems and BCPs. | ||
The introduction of strained small rings, which can make σ-bonds behave like π-bonds, as molecular building blocks has recently emerged as a powerful tool in organic synthesis.8 Among them, methylenecyclopropanes (MCPs) and benzylidenecyclopropanes (BCPs), containing a highly strained cyclopropane ring with an exo-methylene group, are valuable building blocks in organic synthesis owing to their unique structure and high reactivity.9 The reactions of MCPs and BCPs under transition metal catalysis have been extensively explored in the past few decades, and have been frequently used in a range of reactions, such as cycloaddition reactions, cycloisomerizations and ring-opening reactions.10 The cleavage of a proximal or a distal C–C single bond of the cyclopropane ring accompanied by the addition of an E1–E2 bond (such as X–H,11a–j B–B,11k,l B–Si11m–r and other σ bonds11s,t) to give 1,3-difunctionalized products has also been investigated. Recently, McAlpine, Liu and Engle reported an elegant copper-catalyzed hydroboration of BCPs with B2pin2 and MeOH. The benzylcopper intermediate, formed by the addition of Cu–Bpin species to the C–C double bond, underwent a β-carbon elimination/protodecupration process and provided ring-opening hydroboration product alkenylboronates selectively through the choice of a suitable phosphine ligand (Scheme 1b).11j However, to the best of our knowledge, the capture of the Cu–C bond from such β-carbon elimination of BCPs with a carbon electrophile other than a proton has been less reported.12 Herein, we report a copper catalyzed borocarbonylation13 of BCPs with B2pin2 and chloroformates (or acyl chlorides) through the cleavage of a proximal C–C bond of the cyclopropane ring for the synthesis of γ-boryl-γ,δ-unsaturated carbonyl compounds (Scheme 1c).14 Notable features of our study include: (1) regio- and stereoselective boroacylation of BCPs through selective proximal C–C bond cleavage, (2) preparation of 1,3-difunctionalized products with γ-boryl-γ,δ-unsaturated carbonyl compounds, and (3) broad substrate scopes as well as good functional group tolerance.
Results and discussion
Initially, 2-(cyclopropylidenemethyl)naphthalene 1a and phenyl chloroformate 2a were selected as model substrates to react with B2pin2 to evaluate the feasibility of the borocarbonylation reaction. To our delight, using IMesCuCl as a catalyst, PPh3 as the ligand and LiOtBu as a base, when a solution of 1a, B2pin2 and 2a in toluene was stirred at 90 °C for 12 h, the desired borocarbonylation product 3aa was successfully obtained in 58% yield (Table 1, entry 1). This reaction turned out to be highly regio- and stereoselective, and only the Z-isomer was obtained. Then, we examined a range of copper catalysts for this reaction. When the precatalyst was substituted with IPrCuCl, a much reduced conversion was observed and a decreased yield of 26% was obtained (Table 1, entry 2). No reaction was observed when CuBr was used (Table 1, entry 3); other copper catalysts such as CuTc and Cu(CH3CN)4PF6 lead to the formation of the hydroboration products (see details in the ESI†).11j The phosphine ligands were reported to be able to affect the β-carbon elimination step.11j Thus, we then screened a series of ligands. Indeed, a phosphine or a pyridyl ligand is essential for this reaction. Only a trace amount of product 3aa was observed in the absence of ligand (Table 1, entry 4). The yield of 3aa was improved to 67% when BINAP was used (Table 1, entry 5). Other ligands such as PCy3, BuPAd2 and dtbbpy were also effective and produced the desired product 3aa (Table 1, entry 6, see details in the ESI†). The ratio of the ligand to the catalyst also played an important role in this reaction. Decreased yield was obtained when the reaction was performed at a lower ligand/Cu ratio (Table 1, entry 7). A ligand/Cu ratio of 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was found to be optimal and produced 3aa in 69% yield (Table 1, entry 8). Then, a series of bases were tested and it was found that alkali metal alkoxides including KOtBu and NaOMe were effective and provided the desired product 3aa in 58% and 42% yields, respectively (Table 1, entries 9 and 10). The bisborylated product of 1a and small amounts of hydroborylation products were observed as the by-products in these reactions. However, when NaOtBu was used as a base, no desired product 3aa was obtained, only the bisborylated product was observed.11k Other bases such as K2CO3, Na2CO3 and Et3N were ineffective and no reaction was observed (see details in the ESI†). The yield of 3aa could be further improved to 89% when 3.5 equivalents of phenyl chloroformate 2a were used (Table 1, entry 11, see details in the ESI†). Screening of the solvent revealed that toluene is the optimal solvent. When PhCl and MeCN were used as the solvents, the yields decreased to 76% and 43%, respectively (Table 1, entries 12 and 13). Finally, the reaction temperature affected the efficiency of this reaction as well, and an excellent yield of 91% was obtained when the reaction was performed at 100 °C (Table 1, entry 14).
:1 was found to be optimal and produced 3aa in 69% yield (Table 1, entry 8). Then, a series of bases were tested and it was found that alkali metal alkoxides including KOtBu and NaOMe were effective and provided the desired product 3aa in 58% and 42% yields, respectively (Table 1, entries 9 and 10). The bisborylated product of 1a and small amounts of hydroborylation products were observed as the by-products in these reactions. However, when NaOtBu was used as a base, no desired product 3aa was obtained, only the bisborylated product was observed.11k Other bases such as K2CO3, Na2CO3 and Et3N were ineffective and no reaction was observed (see details in the ESI†). The yield of 3aa could be further improved to 89% when 3.5 equivalents of phenyl chloroformate 2a were used (Table 1, entry 11, see details in the ESI†). Screening of the solvent revealed that toluene is the optimal solvent. When PhCl and MeCN were used as the solvents, the yields decreased to 76% and 43%, respectively (Table 1, entries 12 and 13). Finally, the reaction temperature affected the efficiency of this reaction as well, and an excellent yield of 91% was obtained when the reaction was performed at 100 °C (Table 1, entry 14).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cu | Ligand | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), B2pin2 (0.15 mmol), 2a (0.15 mmol), catalyst (5 mol%), ligand (10 mol% for monodentate ligand, 5 mol% for bidentate ligand), base (1.5 equiv.), solvent (1 mL), 90 °C, 12 h. b Isolated yields. c BINAP (2.5 mol%). d BINAP (7.5 mol%). e 2a (0.35 mmol). f 100 °C. | |||||
| 1 | IMesCuCl | PPh3 | LiOtBu | Toluene | 58 |
| 2 | IPrCuCl | PPh3 | LiOtBu | Toluene | 26 |
| 3 | CuBr | PPh3 | LiOtBu | Toluene | No reaction |
| 4 | IMesCuCl | — | LiOtBu | Toluene | Trace |
| 5 | IMesCuCl | BINAP | LiOtBu | Toluene | 67 |
| 6 | IMesCuCl | PCy3 | LiOtBu | Toluene | 29 |
| 7c | IMesCuCl | BINAP | LiOtBu | Toluene | 57 |
| 8d | IMesCuCl | BINAP | LiOtBu | Toluene | 69 |
| 9d | IMesCuCl | BINAP | KOtBu | Toluene | 58 |
| 10d | IMesCuCl | BINAP | NaOMe | Toluene | 42 |
| 11d,e | IMesCuCl | BINAP | LiOtBu | Toluene | 89 |
| 12d,e | IMesCuCl | BINAP | LiOtBu | PhCl | 76 |
| 13d,e | IMesCuCl | BINAP | LiOtBu | MeCN | 43 |
| 14 , , | IMesCuCl | BINAP | LiO t Bu | Toluene | 91 |
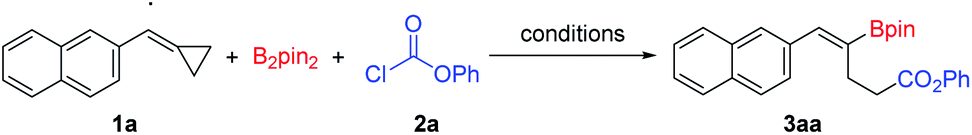
With the optimized reaction conditions in hand (Table 1, entry 14), we began to investigate the substrate scope of this borocarbonylation reaction with emphasis being placed first on BCPs (Scheme 2). A wide range of substituted BCPs were tested and delivered the corresponding products in moderate to excellent yields with excellent regio- and stereoselectivities. When substituted BCPs were used as substrates, the borocarbonylation occurred selectively at the proximal C–C bond trans to the aryl group, leading to the formation of Z-isomers selectively. Only Z-isomers were obtained in these reactions. Both electron-donating (3ca–3ma) and electron-withdrawing groups (3na–3qa) were tolerated. Functional groups such as ether (3ja, 3ka and 3ma), thioether (3la), fluoro- (3na), chloro- (3oa) and cyano (3qa) groups were compatible in this reaction. In addition, heteroaryl-substituted BCPs were tolerated as well. For example, 3-thiophenyl and 5-benzofuranyl substituted substrates reacted successfully with B2pin2 and phenyl chloroformate 2a and provided the corresponding products 3ra and 3sa in 70% and 75% yields, respectively. When a 1-naphthyl substituted substrate was used in this reaction, the corresponding product 3ta was produced in 34% yield. Moreover, ferrocene can be tolerated as well, and the corresponding product 3ua was isolated in 57% yield. However, when diphenyl substituted methylenecyclopropane was used in this reaction, the desired product 3va was obtained in a low yield of 24%, which might have resulted from the steric effect.
 | ||
| Scheme 2 Substrate scope of the BCPs. Reaction conditions: 1 (0.1 mmol), B2pin2 (0.15 mmol), 2a (0.35 mmol), IMesCuCl (5 mol%), BINAP (7.5 mol%), LiOtBu (3.5 equiv.), toluene (1 mL), 100 °C, 12 h, isolated yields. | ||
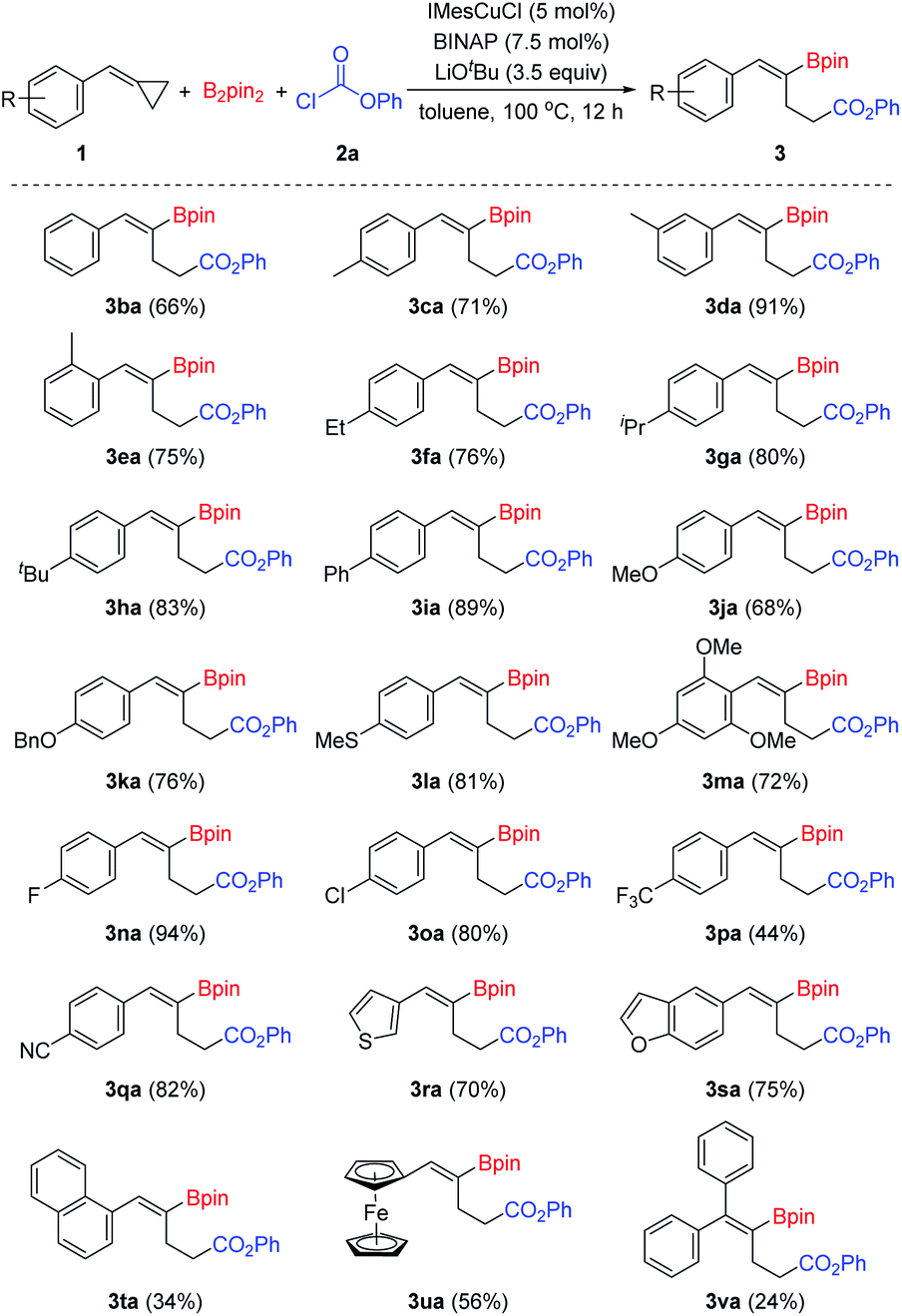
Then, we began to explore the substrate scope of this borocarbonylation reaction with a series of chloroformates. As shown in Scheme 3, phenyl chloroformates with substitution of either electron-withdrawing (3ab and 3ac) or electron-donating groups (3ad) on the phenyl ring reacted smoothly to provide the corresponding products in good yields. Alkyl chloroformates were also suitable substrates: primary and secondary alkyl chloroformates reacted smoothly and afforded the corresponding γ-boryl-γ,δ-unsaturated esters in good to excellent yields (3ae–3am). Moreover, when chloroformate 2n derived from natural cholesterol was treated with B2pin2 and 1a under the standard conditions, the target product 3an was obtained in 33% yield. However, when tert-butyl chloroformate was used in this reaction, no desired borocarbonylation product was obtained, only the bisborylated product was observed as the main by-product.11k
 | ||
| Scheme 3 Substrate scope of chloroformates. Reaction conditions: 1a (0.1 mmol), B2pin2 (0.15 mmol), 2 (0.35 mmol), IMesCuCl (5 mol%), BINAP (7.5 mol%), LiOtBu (3.5 equiv.), toluene (1 mL), 100 °C, 12 h, isolated yields. | ||
In addition to chloroformates, acid chlorides were also tested in this reaction (Scheme 4). Aliphatic acid chlorides were tolerated well in this reaction. Although low yields were obtained when primary alkanoyl chlorides were used under the standard conditions, the desired products could be obtained in acceptable yields at a lower reaction temperature.15 When n-butyryl chloride and n-dodecanoyl chloride were used in this reaction, the desired products 3ao and 3ap were obtained in 47% and 52% yields, respectively. Secondary and tertiary alkanoyl chlorides showed good reactivity and provided the desired products in good to excellent yields. For instance, iso-butyryl chloride, cyclohexanecarbonyl chloride, pivaloyl chloride and 1-adamantanecarbonyl chloride reacted with B2pin2 and 2-(cyclopropylidenemethyl)naphthalene 1a under the standard conditions and produced the corresponding γ-boryl-γ,δ-unsaturated ketones 3ar–3at in 86–93% yields. However, aromatic acid chlorides failed to give the desired products, inseparable mixtures of by-products were obtained when aroyl chlorides were used.
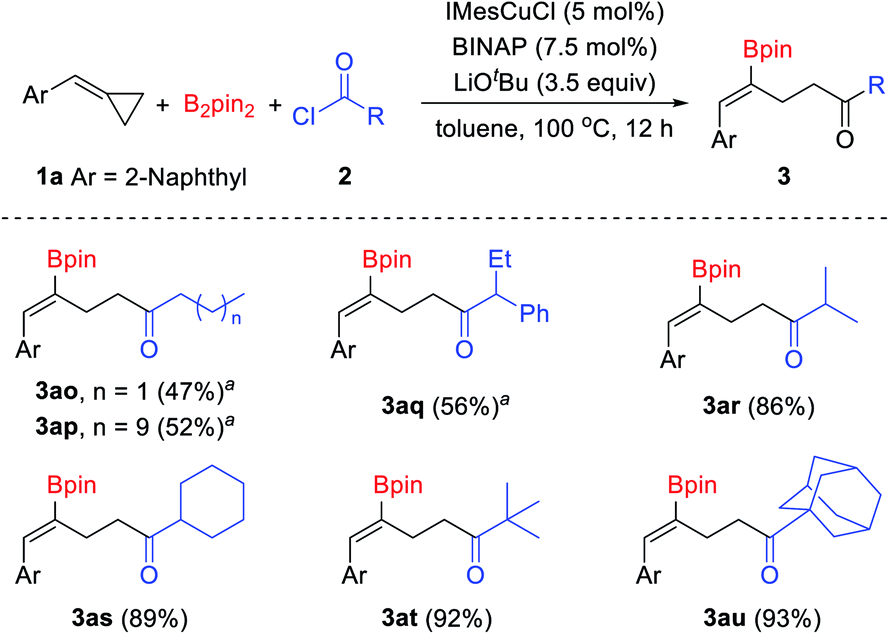 | ||
| Scheme 4 Substrate scope of acid chlorides. Reaction conditions: 1a (0.1 mmol), B2pin2 (0.15 mmol), 2 (0.35 mmol), IMesCuCl (5 mol%), BINAP (7.5 mol%), LiOtBu (3.5 equiv.), toluene (1 mL), 100 °C, 12 h, isolated yields. a60 °C, 24 h. | ||
To understand the reaction pathway and to illustrate the synthetic versatility of the borocarbonylated products, the following reactions were conducted. First, when cyclopropyl substituted substrate 1w was treated with B2pin2 and phenyl chloroformate 2a under the optimal reaction conditions, a 1,5-borocarbonylated product (4) was obtained in 62% yield. This observation indicates that the reaction proceeds via an addition of Cu–Bpin to the C–C double bond followed by a β-C elimination and a carboxylation process (Scheme 5a). As mentioned above, the borocarbonylated products could be used in various transformations. For example, the C–B bond could be easily oxidized by NaBO3·4H2O to give the 1,4-diketone 5 in 95% yield from 3as (Scheme 5b). In another instance, treatment of 3aa with aqueous KHF2 delivered the corresponding potassium trifluoroborate 6 in 87% yield (Scheme 5c). Moreover, the Suzuki–Miyaura cross-coupling reaction of 3aa with iodobenzene worked smoothly to produce 7 in 92% yield (Scheme 5d).
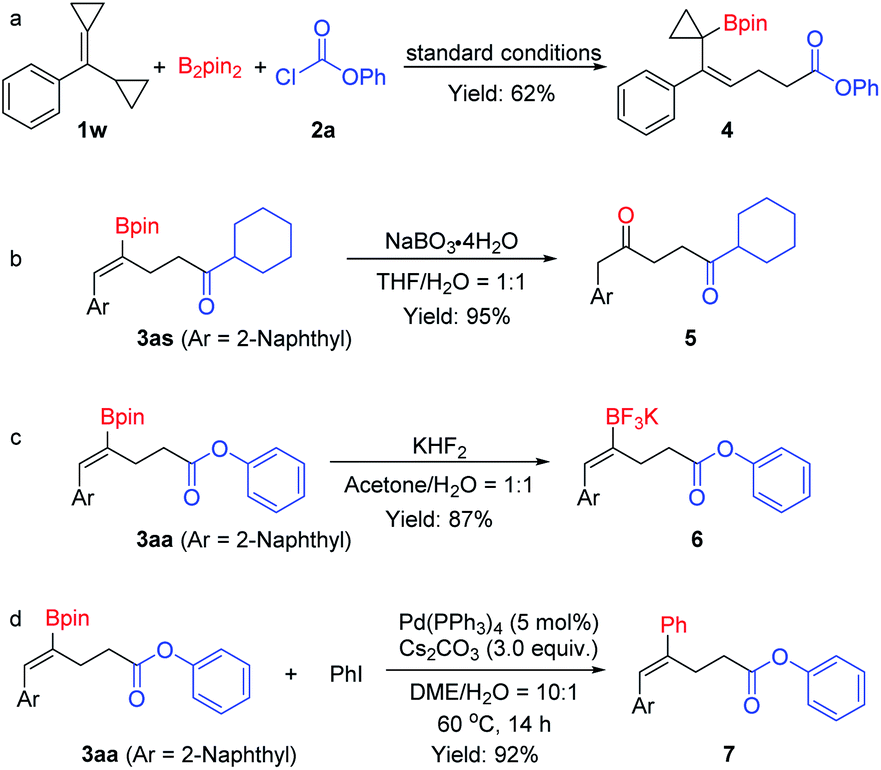 | ||
| Scheme 5 (a) Borocarbonylation of cyclopropyl substituted substrate. (b–d) Derivatization of the borocarbonylation products. | ||
On the basis of the experimental results and the previous literature,11j a plausible catalytic cycle is proposed in Scheme 6. Firstly, the precatalyst IMesCuCl reacts with LiOtBu to give an active (L)Cu–OtBu complex, which reacts with B2pin2 to generate the key (L)Cu–Bpin species. Then, the copper–boryl intermediates react with BCPs via syn-1,2-migratory insertion into the C–C double-bond to afford the borylcuprated intermediate A, which undergoes a β-carbon elimination to generate a homoallylic copper(I) complex (B). The β-carbon elimination is the stereo-determining step.16 A σ-bond rotation should occur prior to the β-C elimination step to avoid the steric repulsion, thus favoring the formation of Z isomers. Subsequently, intermediate B reacts with acyl chloride to give Cu(III) complex Cvia oxidative addition. Finally, reductive elimination of C releases the product 3 and regenerates (L)CuX, which is used for the next catalytic cycle. However, the direct nucleophilic attack of acyl chloride by the homoallylic copper(I) complex B to afford product 3 cannot be excluded.
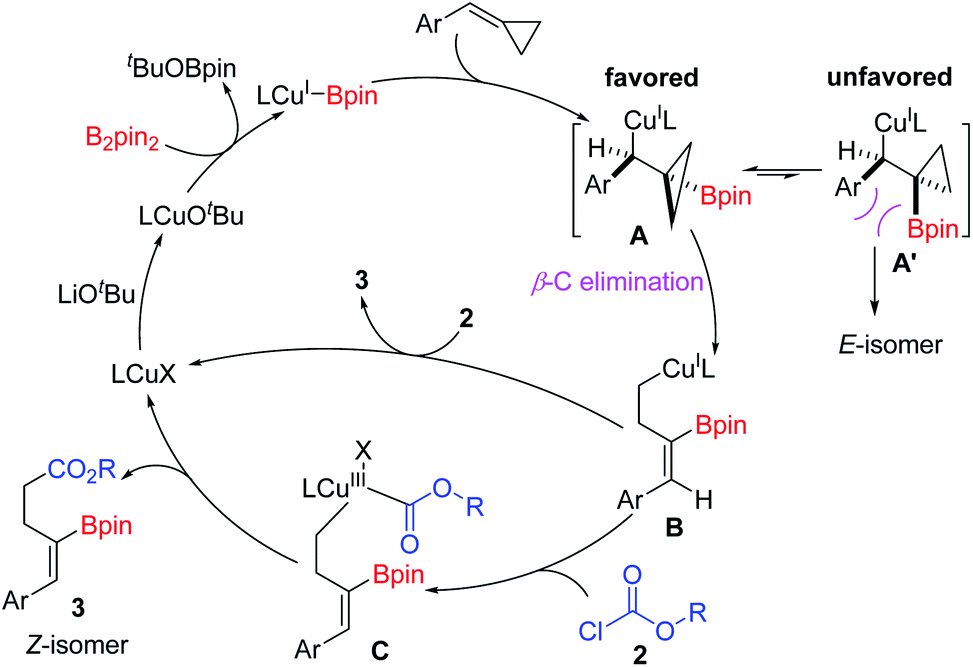 | ||
| Scheme 6 Proposed catalytic cycle. | ||
Conclusions
In summary, we have developed a copper catalyzed borocarbonylation of BCPs via proximal C–C bond cleavage for the synthesis of γ-boryl-γ,δ-unsaturated carbonyl compounds. Using substituted benzylidenecyclopropanes (BCPs) and chloroformates as starting material, a broad range of γ-boryl-γ,δ-unsaturated esters were prepared in moderate to excellent yields with excellent regio- and stereoselectivity. Besides, when aliphatic acid chlorides were used in this reaction, γ-boryl-γ,δ-unsaturated ketones could be produced in excellent yields. When substituted BCPs were used as substrates, the borocarbonylation occurred predominantly at the proximal C–C bond trans to the phenyl group in a regio- and stereoselective manner, which leads to the Z-isomers as the products. Control experiments indicate that the reaction proceeds via a mechanism involving migratory insertion of Cu–Bpin species to the C–C double bond of BCP and subsequent β-C elimination and acylation of the intermediary homoallylic Cu(I) complex. This efficient methodology involves the cleavage of a C–C bond and the formation of a C–C bond as well as a C–B bond, and provides a new method for the proximal C–C bond difunctionalization of BCPs.Data availability
All experimental data and detailed procedures are available in the ESI.†Author contributions
J.-B. P. conceived and directed the project. L.-M. Y. performed the experiments. H.-H. Z., X.-L. L. and A.-J. M. participated in substrate synthesis and discussions. L.-M. Y. and J.-B. P. wrote the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (NSFC; 21801225), Wuyi University (2019td02 and 2018TP018), the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2020AL015), and the Department of Education of Guangdong Province (2020KCXTD036) is gratefully acknowledged. The authors appreciate the general support provided by Prof. Shu-Yu Zhang of SJTU.Notes and references
- (a) E. R. Burkhardt and K. Matos, Chem. Rev., 2006, 106, 2617–2650 CrossRef CAS PubMed; (b) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- (a) P. C. Trippier and C. McGuigan, MedChemComm, 2010, 1, 183–198 RSC; (b) R. Smoum, A. Rubinstein, V. M. Dembitsky and M. Srebnik, Chem. Rev., 2012, 112, 4156–4220 CrossRef CAS PubMed; (c) D. M. Volochnyuk, A. O. Gorlova and O. O. Grygorenko, Chem.–Eur. J., 2021, 27, 15277–15326 CrossRef CAS PubMed.
- (a) K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179–1191 CrossRef CAS; (b) C. M. Vogels and S. A. Westcott, Curr. Org. Chem., 2005, 9, 687–699 CrossRef CAS; (c) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (d) S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Chem. Soc. Rev., 2020, 49, 4564–4591 RSC; (e) M. Wang and Z. Shi, Chem. Rev., 2020, 120, 7348–7398 CrossRef CAS PubMed; (f) J. Hu, M. Ferger, Z. Shi and T. B. Marder, Chem. Soc. Rev., 2021, 50, 13129–13188 RSC.
- For selected reviews, see: (a) D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC; (b) K. Semba and Y. Nakao, Tetrahedron, 2019, 75, 709–719 CrossRef CAS; (c) W. Xue and M. Oestreich, ACS Cent. Sci., 2020, 6, 1070–1081 CrossRef CAS PubMed; (d) A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS; (e) G. J. P. Perry, T. Jia and D. J. Procter, ACS Catal., 2020, 10, 1485–1499 CrossRef CAS; (f) C. Yang and X. Jia, Chin. J. Org. Chem., 2021, 41, 2280–2289 CrossRef; (g) S. K. Dorn and M. K. Brown, ACS Catal., 2022, 12, 2058–2063 CrossRef CAS.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- (a) M. Murakami and N. Chatani, Cleavage of Carbon–Carbon Single Bonds by Transition Metals, Wiley, New York, 2015 CrossRef; (b) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed; (c) M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed.
- R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241–246 CrossRef CAS PubMed.
- (a) A. Brandi and A. Goti, Chem. Rev., 1998, 98, 589–636 CrossRef CAS PubMed; (b) I. Nakamura and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111–129 CrossRef CAS; (c) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213–1270 CrossRef CAS; (d) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed.
- For some selected reviews, see: (a) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213–1270 CrossRef CAS PubMed; (b) H. Pellissier, Tetrahedron, 2003, 66, 8341–8375 CrossRef; (c) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed; (d) A. Masarwa and I. Marek, Chem.–Eur. J., 2010, 16, 9712–9721 CrossRef CAS PubMed; (e) L. Yu and R. Guo, Org. Prep. Proced. Int., 2011, 43, 209–259 CrossRef CAS; (f) B.-L. Lu, L. Dai and M. Shi, Chem. Soc. Rev., 2012, 41, 3318–3339 RSC; (g) M. Shi, J.-M. Lu, Y. Wei and L.-X. Shao, Acc. Chem. Res., 2012, 45, 641–652 CrossRef CAS PubMed; (h) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317–7420 CrossRef CAS PubMed; (i) L. Yu, M. Liu, F. Chen and Q. Xu, Org. Biomol. Chem., 2015, 13, 8379–8392 RSC; (j) W. Fang and M. Shi, Chem.–Eur. J., 2018, 24, 9998–10005 CrossRef CAS PubMed.
- For reactions with Sn–H, see: (a) M. Lautens, C. Meyer and A. Lorenz, J. Am. Chem. Soc., 1996, 118, 10676–10677 CrossRef CAS ; For reactions with Si–H, see: ; (b) A. G. Bessmertnykh, K. A. Blinov, Y. K. Grishin, N. A. Donskaya, E. V. Tveritinova, N. M. Yur'eva and I. P. Beletskaya, J. Org. Chem., 1997, 62, 6069–6076 CrossRef CAS ; For reactions with C–H, see:; (c) N. Tsukada, A. Shibuya, I. Nakamura and Y. Yamamoto, J. Am. Chem. Soc., 1997, 119, 8123–8124 CrossRef CAS; (d) I. Nakamura, S. Saito and Y. Yamamoto, J. Am. Chem. Soc., 2000, 122, 2661–2662 CrossRef CAS; (e) H. Taniguchi, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2009, 131, 11298–11299 CrossRef CAS PubMed; (f) K. Ogata, Y. Atsuumi and S. Fukuzawa, Org. Lett., 2010, 12, 4536–4539 CrossRef CAS PubMed ; For reactions with N–H, see: ; (g) I. Nakamura, H. Itagaki and Y. Yamamoto, J. Org. Chem., 1998, 63, 6458–6459 CrossRef CAS; (h) E. Smolensky, M. Kapon and M. S. Eisen, Organometallics, 2005, 24, 5495–5498 CrossRef CAS; (i) E. Smolensky, M. Kapon and M. S. Eisen, Organometallics, 2007, 26, 4510–4527 CrossRef CAS ; For reactions with B–H, see: ; (j) J. M. Medina, T. Kang, T. G. Erbay, H. Shao, G. M. Gallego, S. Yang, M. Tran-Dubé, P. F. Richardson, J. Derosa, R. T. Helsel, R. L. Patman, F. Wang, C. P. Ashcroft, J. F. Braganza, I. McAlpine, P. Liu and K. M. Engle, ACS Catal., 2019, 9, 11130–11136 CrossRef CAS ; For reactions with B–B, see: ; (k) T. Ishiyama, S. Momota and N. Miyaura, Synlett, 1999, 1999, 1790–1792 CrossRef; (l) Q. Chen, X. Zhang, S. Su, Z. Xu, N. Li, Y. Li, H. Zhou, M. Bao, Y. Yamamoto and T. Jin, ACS Catal., 2018, 8, 5901–5906 CrossRef CAS ; For reactions with B–Si, see: ; (m) M. Suginome, T. Matsuda and Y. Ito, J. Am. Chem. Soc., 2000, 122, 11015–11016 CrossRef CAS; (n) T. Pohlmann and A. de Meijere, Org. Lett., 2000, 2, 3877–3879 CrossRef CAS; (o) T. Ohmura, H. Taniguchi, Y. Kondo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 3518–3519 CrossRef CAS PubMed; (p) T. Ohmura, H. Taniguchi and M. Suginome, Org. Lett., 2009, 11, 2880–2883 CrossRef CAS PubMed; (q) Y. Akai, T. Yamamoto, Y. Nagata, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2012, 134, 11092–11095 CrossRef CAS PubMed; (r) T. Ohmura, H. Taniguchi and M. Suginome, ACS Catal., 2015, 5, 3074–3077 CrossRef CAS ; For reactions with other σ-bonds, see: ; (s) N. Chatani, T. Takeyasu and T. Hanafusa, Tetrahedron Lett., 1988, 29, 3979–3982 CrossRef CAS; (t) R. Yu and X. Fang, Org. Lett., 2020, 22, 594–597 CrossRef CAS PubMed.
- (a) K. Ogata, D. Shimada, S. Furuya and S. Fukuzawa, Org. Lett., 2013, 15, 1182–1185 CrossRef CAS PubMed; (b) A. Masarwa, D. Didier, T. Zabrodski, M. Schinkel, L. Ackermann and I. Marek, Nature, 2014, 505, 199–203 CrossRef CAS PubMed.
- (a) F.-Y. Yang, M.-Y. Wu and C.-H. Cheng, J. Am. Chem. Soc., 2000, 15, 7122–7123 CrossRef; (b) F.-Y. Yang, M. Shanmugasundaram, S.-Y. Chuang, P.-J. Ku, M.-Y. Wu and C.-H. Cheng, J. Am. Chem. Soc., 2003, 125, 12576–12583 CrossRef CAS PubMed; (c) T. Fujihara, A. Sawada, T. Yamaguchi, Y. Tani, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2017, 56, 1539–1543 CrossRef CAS PubMed; (d) A. Boreux, K. Indukuri, F. Gagosz and O. Riant, ACS Catal., 2017, 7, 8200–8204 CrossRef CAS; (e) Y. Huang, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2017, 56, 13314–13318 CrossRef CAS PubMed; (f) A. Sawada, T. Fujihara and Y. Tsuji, Adv. Synth. Catal., 2018, 360, 2621–2625 CrossRef CAS; (g) A. Whyte, K. I. Burton, J. Zhang and M. Lautens, Angew. Chem., Int. Ed., 2018, 57, 13927–13930 CrossRef CAS PubMed; (h) L.-J. Cheng and P. Mankad, Angew. Chem., Int. Ed., 2018, 57, 10328–10332 CrossRef CAS PubMed; (i) J. Han, W. Zhou, P.-C. Zhang, H. Wang, R. Zhang, H.-H. Wu and J. Zhang, ACS Catal., 2019, 9, 6890–6895 CrossRef CAS; (j) A. Torelli, A. Whyte, L. Polishchuk, J. Bajohr and M. Lautens, Org. Lett., 2020, 22, 7915–7919 CrossRef CAS PubMed; (k) F.-P. Wu, Y. Yuan, C. Schünemann, P. C. J. Kamer and X.-F. Wu, Angew. Chem., Int. Ed., 2020, 59, 10451–10455 CrossRef CAS PubMed; (l) Y. Yuan, F.-P. Wu, J.-X. Xu and X.-F. Wu, Angew. Chem., Int. Ed., 2020, 59, 17055–17061 CrossRef CAS PubMed; (m) Z. Su, Y. Feng, R. Zou, X. Qiu, J. Wang and C. Tao, Chem. Commun., 2020, 56, 7483–7486 RSC; (n) D. Fiorito, Y. Liu, C. Besnard and C. Mazet, J. Am. Chem. Soc., 2020, 142, 623–632 CrossRef CAS PubMed; (o) Z. Yang, P. Li, H. Lu and G. Li, J. Org. Chem., 2021, 86, 4616–4624 CrossRef CAS PubMed.
- When we were preparing the manuscript, Wu and a co-worker reported a borocarbonylation of benzylidenecyclopropanes with Cu/Pd catalysis. F.-P. Wu and X.-F. Wu, Chem. Sci., 2022, 13, 4321–4326 RSC.
- A ring-opening hydrochlorination product of BCP was obtained at 100 °C as the main by-product, which might have resulted from reaction with HCl generated from the decarbonylative decomposition of acid chlorides. B. Xu and M. Shi, Org. Lett., 2003, 5, 1415–1418 CrossRef CAS PubMed.
- M. E. O'Reilly, S. Dutta and A. S. Veige, Chem. Rev., 2016, 116, 8105–8145 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and copies of NMR spectra. See https://doi.org/10.1039/d2sc01992b |
| This journal is © The Royal Society of Chemistry 2022 |