
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantiomeric β-sheet peptides from Aβ form homochiral pleated β-sheets rather than heterochiral rippled β-sheets†
Xingyue
Li
 a,
Stephanie E.
Rios
a and
James S.
Nowick
*ab
a,
Stephanie E.
Rios
a and
James S.
Nowick
*ab
aDepartment of Chemistry, University of California Irvine, 4126 Natural Sciences I, Irvine, CA 92697-2025, USA. E-mail: jsnowick@uci.edu
bDepartment of Pharmaceutical Sciences, University of California Irvine, 4126 Natural Sciences I, Irvine, CA 92697-2025, USA
First published on 31st May 2022
Abstract
In 1953, Pauling and Corey postulated “rippled” β-sheets, composed of a mixture of D- and L-peptide strands, as a hypothetical alternative to the now well-established structures of “pleated” β-sheets, which they proposed as a component of all-L-proteins. Growing interest in rippled β-sheets over the past decade has led to the development of mixtures of D- and L-peptides for biomedical applications, and a theory has emerged that mixtures of enantiomeric β-sheet peptides prefer to co-assemble in a heterochiral fashion to form rippled β-sheets. Intrigued by conflicting reports that enantiomeric β-sheet peptides prefer to self-assemble in a homochiral fashion to form pleated β-sheets, we set out address this controversy using two β-sheet peptides derived from Aβ17–23 and Aβ30–36, peptides 1a and 1b. Each of these peptides self-assembles to form tetramers comprising sandwiches of β-sheet dimers in aqueous solution. Through solution-phase NMR spectroscopy, we characterize the different species formed when peptides 1a and 1b are mixed with their respective D-enantiomers, peptides ent-1a and ent-1b. 1H NMR, DOSY, and 1H,15N-HSQC experiments reveal that mixing peptides 1a and ent-1a results in the predominant formation of homochiral tetramers, with a smaller fraction of a new heterochiral tetramer, and mixing peptides 1b and ent-1b does not result in any detectable heterochiral assembly. 15N-edited NOESY reveals that the heterochiral tetramer formed by peptides 1a and ent-1a is composed of two homochiral dimers. Collectively, these NMR studies of Aβ-derived peptides provide compelling evidence that enantiomeric β-sheet peptides prefer to self-assemble in a homochiral fashion in aqueous solution.
Introduction
Do enantiomeric β-sheet peptides prefer to self-assemble in a homochiral fashion or co-assemble in a heterochiral fashion? In the early 1950s, Pauling and Corey introduced the terms “pleated” β-sheets and “rippled” β-sheets to describe two types of β-sheet assembly.1–3 In both a pleated β-sheet and a rippled β-sheet, adjacent peptide strands hydrogen bond through edge-to-edge interactions (Fig. 1). Pleated β-sheets are composed of peptide strands of the same chirality (all L-peptide strands or all D-peptide strands), while peptide strands of opposite chirality (L-peptide strands and D-peptide strands) are required to form rippled β-sheets. In a pleated β-sheet, the side chains of sequential residues are oriented up-down-up-down and those of the adjacent peptide strands are also oriented up-down-up-down (Fig. 1A). In a rippled β-sheet, however, the side chains of sequential residues are oriented up-down-up-down and those of the adjacent peptide strands are oriented down-up-down-up (Fig. 1B). Although pleated β-sheets are a near ubiquitous feature of proteins, rippled β-sheets are not found in nature, because ribosomal proteins and peptides are composed only of L-amino acids.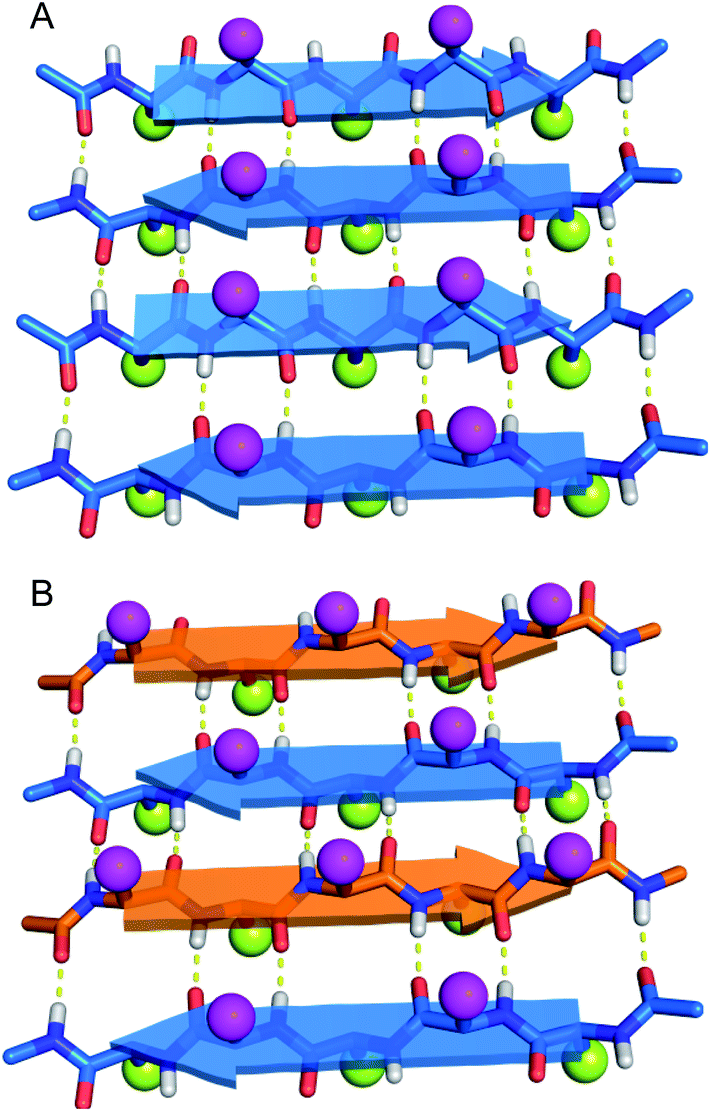 | ||
| Fig. 1 Molecular models of (A) pleated β-sheets formed from L-peptide strands (blue) and (B) rippled β-sheets formed from a mixture of L-peptide and D-peptide strands (orange). Amino acid side chains are depicted as balls, with magenta balls on the top face and yellow balls on the bottom face of the β-sheets. | ||
Rippled β-sheets, formed by mixing D- and L-peptides, have recently attracted considerable interest as biomaterials and for other biomedical applications.4–6 Schneider and co-workers demonstrated the effects of chirality with the hydrogel-forming peptide MAX1. When MAX1 was mixed with an equimolar amount of its enantiomer, the resulting hydrogel showed four times greater rigidity than that of the enantiopure MAX1 peptide.7,8 Nilsson and co-workers demonstrated by isotope-edited IR spectroscopy and FRET studies that mixtures of enantiomeric peptides L-Ac-(FKFE)2-NH2 and D-Ac-(FKFE)2-NH2 form rippled β-sheet fibrils. The authors further demonstrated by isothermal titration calorimetry (ITC) that the resulting heterochiral assembly is more thermodynamically favored than the homochiral assembly.9 In a subsequent paper, the authors reported that the hydrogel formed by the heterochiral rippled β-sheets is stronger and more resistant to proteolytic degradation than the hydrogel formed by the homochiral L-pleated β-sheets.10
The co-assembly of enantiomeric β-sheet peptides is not limited to designed biomaterials, and has also been used to characterize and study fibril formation of Aβ40 and Aβ42. Nilsson and co-workers demonstrated by isotope-edited IR spectroscopy and solid-state NMR spectroscopy that L- and D-Aβ16-22 heptapeptides co-assemble to give rippled β-sheets and showed that the heterochiral assembly is more thermodynamically favorable.11 Raskatov and co-workers observed that mixing D-Aβ42 with L-Aβ42 led to accelerated non-toxic fibril formation and attenuated cytotoxicity by suppressing oligomer formation.12 Raskatov subsequently proposed “Aβ chiral inactivation” as a potential therapeutic strategy for Alzheimer's disease.6,13 Recently, structures of the rippled β-sheet assembly have been reported. Tycko and Raskatov used solid-state NMR spectroscopy to elucidate 15N,13C-labeled D,L-Aβ40 fibril polymorphs in rippled β-sheets consisting of three different registrations in the hydrogen-bonded antiparallel alignment.14 Raskatov and co-workers also reported the X-ray crystallographic structure of a rippled β-sheet formed from a mixture of L- and D-triphenylalanine.15 DFT calculations have further supported a model in which heterochiral rippled β-sheets are favored over homochiral pleated β-sheets.16,17 From these studies, a theory has emerged in which mixtures of enantiomeric β-sheet peptides are thought to prefer to co-assemble in a heterochiral fashion to form rippled β-sheets, rather than self-assemble in a homochiral fashion to form pleated β-sheets.
In 2004, our laboratory reported that enantiomeric β-sheet pentapeptides strongly prefer to form homochiral pleated β-sheet dimers in CDCl3 solution, rather than heterochiral rippled β-sheet dimers, with a selectivity of 3.1–4.2 kcal mol−1.18 Recently Gellman and co-workers have studied homochiral and heterochiral β-sheet formation in aqueous solution using a β-hairpin model system and have found that peptides containing homochiral peptide strands fold to form β-hairpins, while peptides containing heterochiral peptide strands do not.19 Intrigued by the conflicting reports of preferred homochiral and heterochiral β-sheet assembly, we set out to reconcile these findings using a minimal aqueous model system that recapitulates both the edge-to-edge hydrogen-bonding interactions that occur in β-sheet formation and additional face-to-face packing interactions that occur in gel and fibril formation. The model system consists of two well characterized β-sheet peptides derived from Aβ17–23 and Aβ30–36, peptides 1a and 1b.20,21 Peptides 1a and 1b both form tetramers comprising sandwiches of β-sheet dimers. Using NMR spectroscopy, we identify and characterize the different tetramers formed by mixing peptides 1a and 1b with their respective D-enantiomers, ent-1a and ent-1b. Through these studies, we find that homochiral pairing to form pleated β-sheets is preferred over heterochiral pairing to form rippled β-sheets.
Results and discussion
1H NMR spectroscopy shows that mixing peptides 1a and ent-1a gives a new assembly
Enantiomerically pure peptide 1a forms a homochiral tetramer in aqueous solution at millimolar concentrations. Peptide 1a is a macrocyclic β-hairpin peptide containing two heptapeptide strands linked by two δOrn turn units.22 The upper strand of peptide 1a is derived from Aβ17–23, and the lower strand contains a Hao amino acid flanked by two dipeptides to promote solubility and prevent uncontrolled aggregation.23 The tetramer formed by peptide 1a consists of a sandwich of β-sheet dimers. Hydrogen-bonding interactions between the edges of the β-strands stabilize the dimers, and hydrophobic packing of the side chains further stabilizes the tetrameric assembly.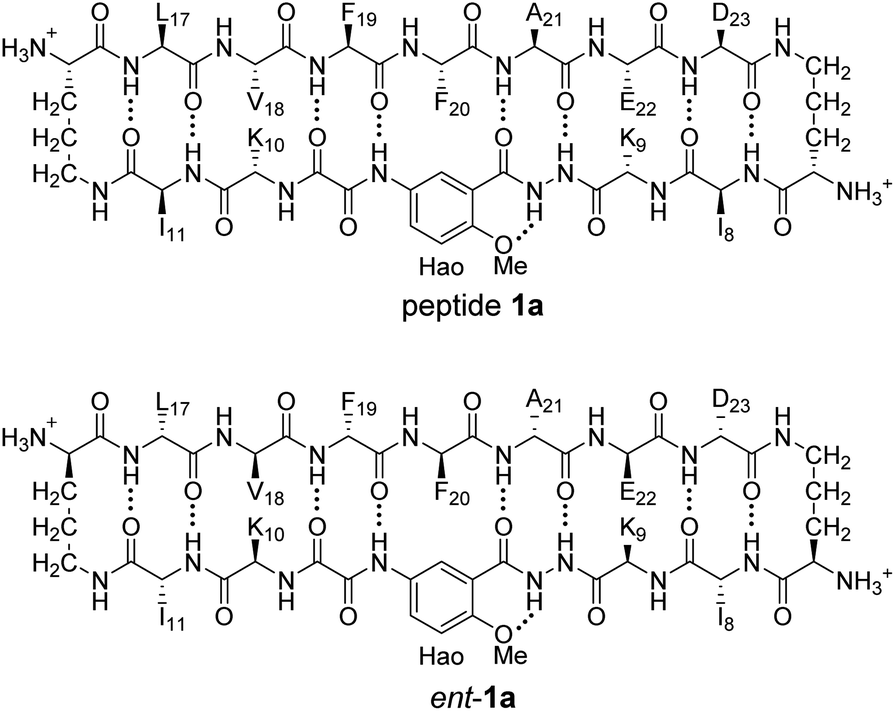
The 1H NMR spectrum of peptide 1a at 8.0 mM in D2O at 298 K displays a predominant set of resonances associated with a homochiral tetramer, and a smaller set of resonances (4%) associated with the monomer. When peptides 1a and ent-1a are mixed in equal concentrations (16.0 mM total), new resonances (29%) emerge that were previously unobserved for each enantiopure peptide (Fig. 2). An EXSY experiment at 328 K shows that these new resonances exchange with the homochiral tetramer and monomer and thus correspond to a new heterochiral assembly (Fig. S1–S4†).24
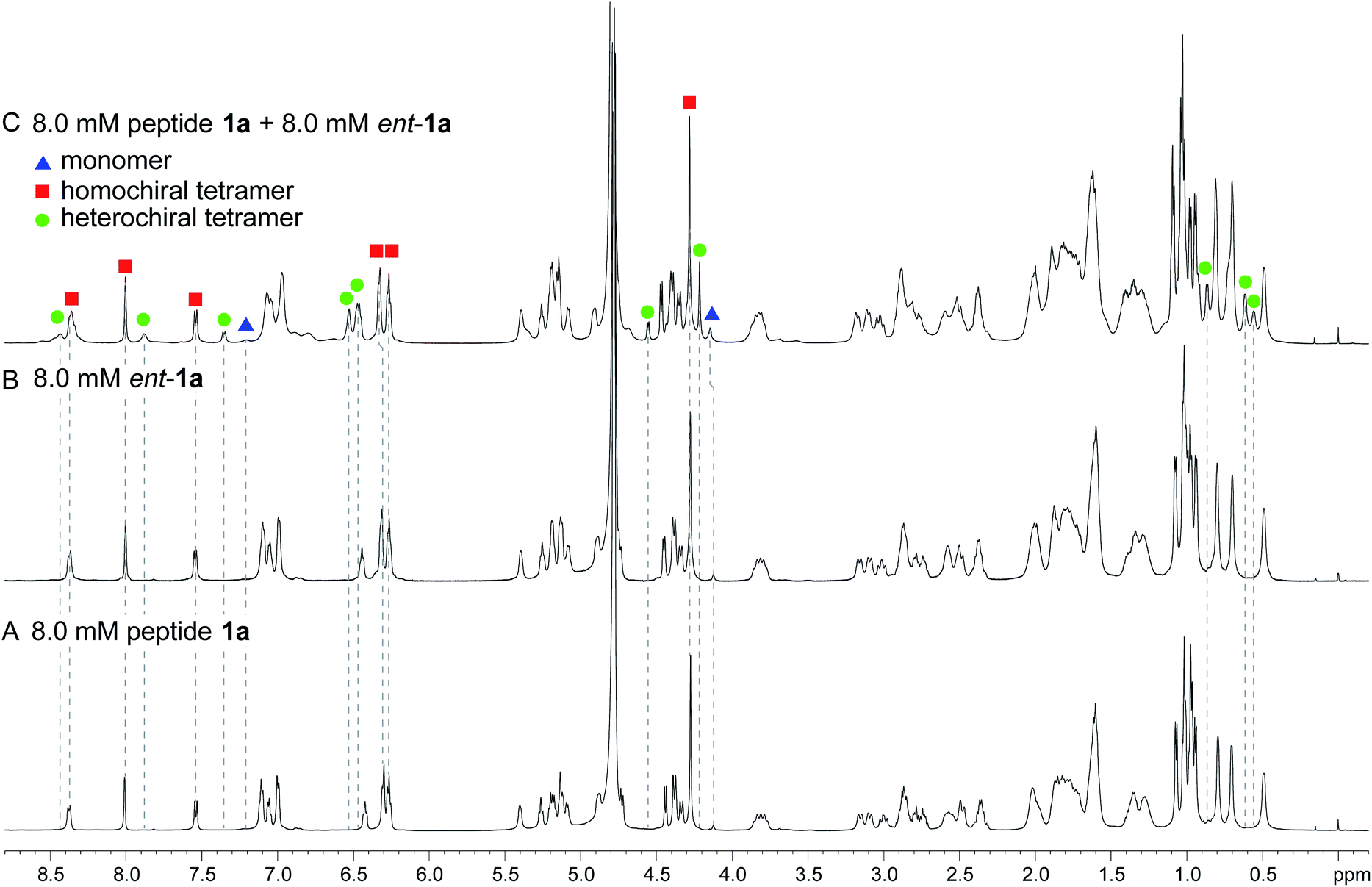 | ||
| Fig. 2 1H NMR spectra of (A) 8.0 mM peptide 1a, (B) 8.0 mM peptide ent-1a, and (C) 8.0 mM peptide 1a and 8.0 mM peptide ent-1a in D2O at 600 MHz and 298 K with 0.06 mM DSA as an internal standard.25 Dashed lines mark key resonances associated with the monomer, homochiral tetramer, and heterochiral tetramer. These resonances are designated as follows: blue triangle, monomer; red square, homochiral tetramer; green circle, heterochiral tetramer. | ||
1H,15N HSQC and DOSY studies reveal a heterochiral tetramer
HSQC studies using 15N isotopic labeling corroborate the formation of the new assembly observed in the 1D 1H NMR spectrum. 1H,15N HSQC experiments give a unique crosspeak for each species containing an 15N isotope, readily allowing the identification of the different isotopically labeled species present.20,21 We thus prepared an isotopologue of peptide 1a containing an 15N label on Phe20—peptide 2a—and studied its mixture with peptide ent-1a by 1H,15N HSQC.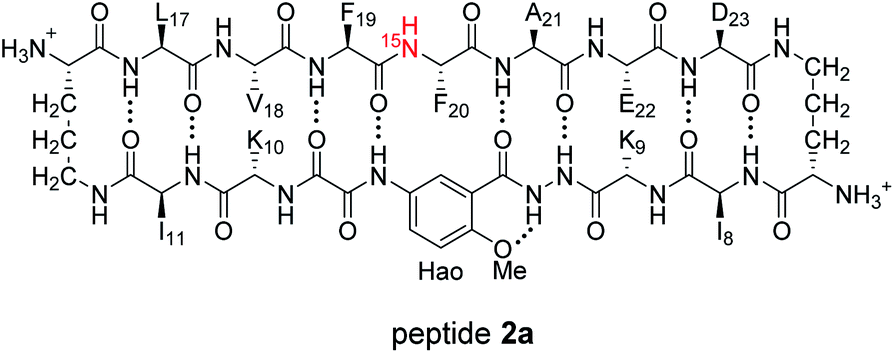
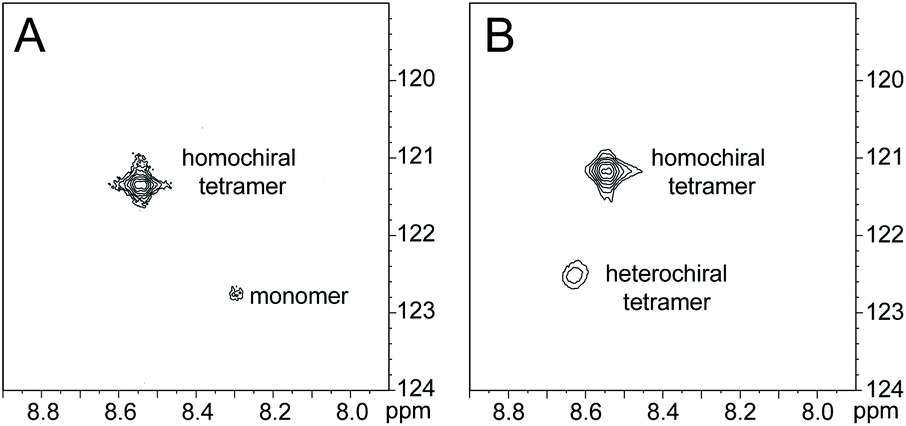
The 1H,15N HSQC spectrum of 8.0 mM peptide 2a in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O–D2O solution shows two crosspeaks—one associated with the homochiral tetramer and the other with the monomer (Fig. 3A).20 The tetramer has a crosspeak that appears at 8.56 ppm in the 1H dimension and 121.4 ppm in the 15N dimension; the monomer has a crosspeak that appears at 8.30 ppm in the 1H dimension and 122.8 ppm in the 15N dimension. When peptide 2a is mixed with peptide ent-1a (16.0 mM total), the crosspeak of the monomer is no longer observed, and a new crosspeak appears at 8.63 ppm in the 1H dimension and 122.5 ppm in the 15N dimension (Fig. 3B). This crosspeak is not observed in the 1H,15N HSQC spectrum of the enantiomerically pure peptide 2a and is thus associated with the formation of a heterochiral species. The weaker intensity of this new crosspeak indicates that the homochiral tetramer forms preferentially under the conditions of the experiment.
:1 H2O–D2O solution shows two crosspeaks—one associated with the homochiral tetramer and the other with the monomer (Fig. 3A).20 The tetramer has a crosspeak that appears at 8.56 ppm in the 1H dimension and 121.4 ppm in the 15N dimension; the monomer has a crosspeak that appears at 8.30 ppm in the 1H dimension and 122.8 ppm in the 15N dimension. When peptide 2a is mixed with peptide ent-1a (16.0 mM total), the crosspeak of the monomer is no longer observed, and a new crosspeak appears at 8.63 ppm in the 1H dimension and 122.5 ppm in the 15N dimension (Fig. 3B). This crosspeak is not observed in the 1H,15N HSQC spectrum of the enantiomerically pure peptide 2a and is thus associated with the formation of a heterochiral species. The weaker intensity of this new crosspeak indicates that the homochiral tetramer forms preferentially under the conditions of the experiment.
 | ||
| Fig. 3
1H,15N HSQC spectra of (A) 8.0 mM peptide 2a and (B) 8.0 mM peptide 1a and 8.0 mM peptide ent-1a in 9:1 H2O–D2O at 500 MHz and 298 K. | ||
Diffusion-ordered spectroscopy (DOSY) studies suggest that the new heterochiral species is a tetramer. The DOSY spectrum of 8.0 mM peptide 1a in D2O shows a diffusion coefficient of 11.6 ± 0.9 × 10−11 m2 s−1 for the tetramer (Fig. S5†). This value is similar to what we have previously reported for peptide 1a at 8.0 mM and 298 K.20 In the DOSY spectrum of the mixture of peptides 1a and ent-1a (8.0 mM each), the resonances corresponding to the homochiral tetramer show a diffusion coefficient of 10.7 ± 0.7 × 10−11 m2 s−1, and the resonances corresponding to the heterochiral tetramer show a diffusion coefficient of 10.2 ± 0.7 × 10−11 m2 s−1 (Fig. S6†). The small differences among the diffusion coefficients may reflect transient non-specific interactions among the tetramers at the higher concentration (16.0 mM total) of the mixing experiment leading to a lower diffusion coefficient.26
Supramolecular assembly of the heterochiral tetramer
Through NOESY studies of peptide 1a, our laboratory previously established that the tetramer formed by peptide 1a consists of sandwiches of β-sheet dimers.20,21 The heterochiral tetramer formed by peptides 1a and ent-1a can adopt a similar structure, in which two dimers form a sandwich-like tetramer. Peptides 1a and ent-1a can come together in two different ways to form heterochiral tetramers in 2:2 stoichiometry—either as an L2D2 topological isomer, in which the sandwich consists of L·L and D·D homochiral dimers, or as an (LD)2 topological isomer, in which the sandwich consists of two L·D heterochiral dimers (Fig. 4). The L2D2 tetramer should give one set of resonances in the 1H NMR spectrum, distinct from those of the L4 and D4 homochiral tetramers, which collectively should give one set of resonances. The (LD)2 tetramer should also give one set of resonances in the 1H NMR spectrum. Peptides 1a and ent-1a could also come together to give heterochiral tetramers in 3:1 and 1:3 stoichiometry, L3D1 and L1D3, which should give four sets of resonances in the 1H NMR spectrum. The observation of a single set of new resonances in the 1H NMR spectra of the mixture thus indicates the formation of a single heterochiral tetramer with a 2:2 stoichiometry as either the L2D2 or the (LD)2 topological isomer.
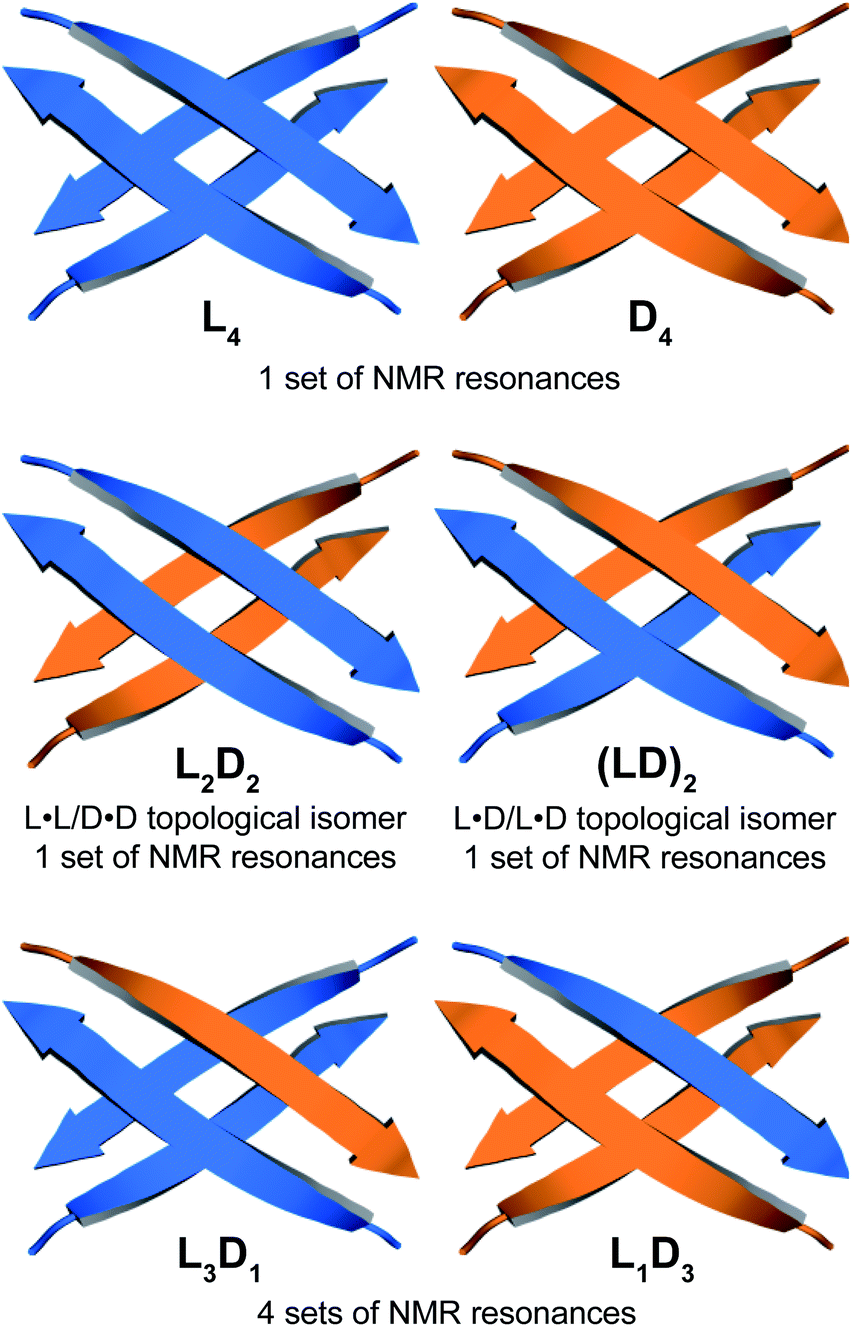 | ||
| Fig. 4 Cartoon illustrations representing the possible combinations of homochiral and heterochiral tetramers formed from a mixture of peptides 1a and ent-1a. Macrocyclic peptide 1a is represented by a blue arrow and macrocyclic peptide ent-1a is represented by an orange arrow. | ||
15N-edited NOESY studies reveal homochiral dimers within the heterochiral tetramer
The 15N-edited NOESY spectrum of 15N-labeled enantiomerically pure peptide 2a shows three NOE crosspeaks associated with close contacts in the homochiral tetramer (Fig. 5B). The Phe2015NH proton of the tetramer shows an interstrand NOE with the Ala21 α-proton in its dimerization partner, as well as a strong intrastrand NOE with the Phe19 α-proton and a weaker intrastrand NOE with the Phe20 α-proton. This pattern of NOEs is characteristic of the proximities observed in β-sheet structure (Fig. 5A).28,29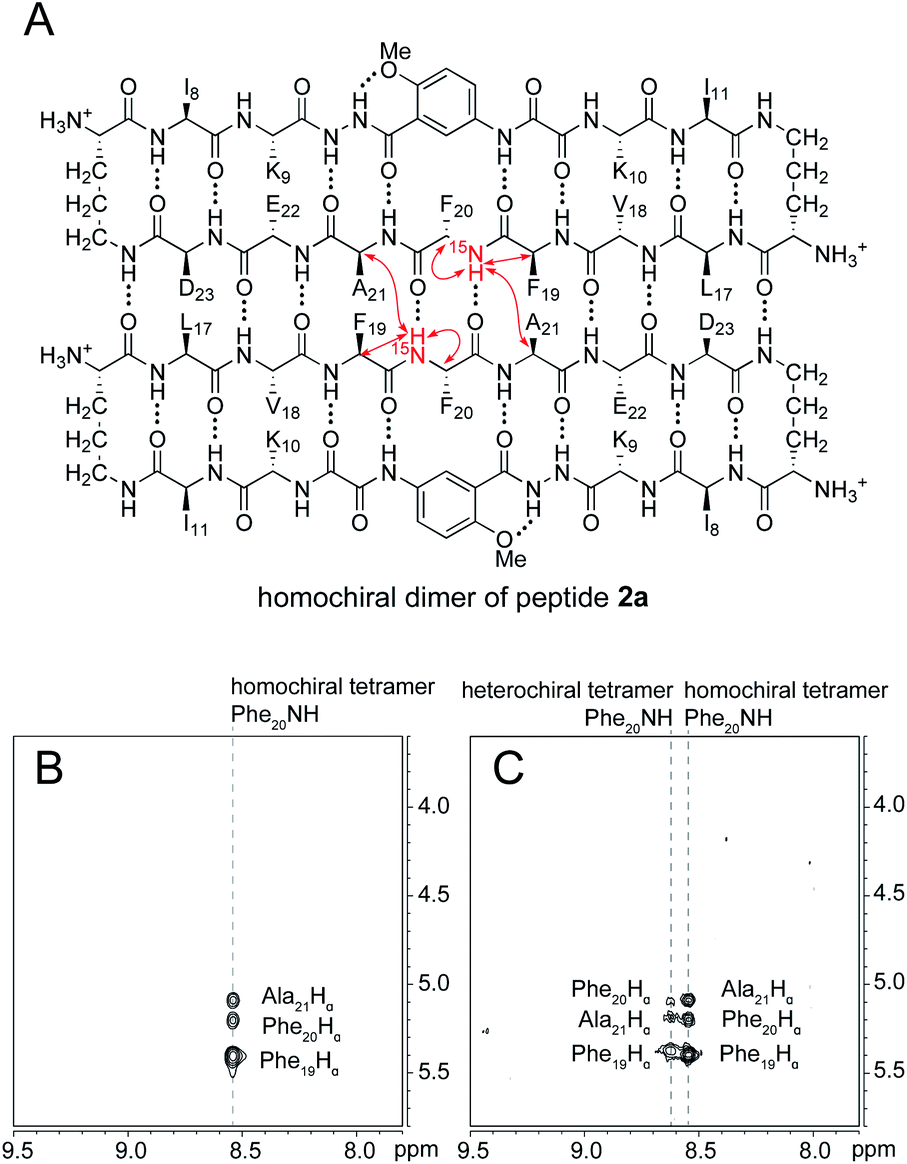 | ||
| Fig. 5 (A) Structure of the homochiral dimer subunit of the tetramer formed from 15N-labeled peptide 2a. Red arrows represent NOEs observed in the 15N-edited NOESY spectrum. (B) 15N-edited NOESY spectrum of 8.0 mM peptide 2a in 9:1 H2O–D2O at 500 MHz and 298 K. (C) 15N-edited NOESY spectrum of 8.0 mM peptide 2a and 8.0 mM peptide ent-1a in 9:1 H2O–D2O at 500 MHz and 298 K. | ||
When 15N-labeled peptide 2a is mixed with unlabeled peptide ent-1a, a new set of weaker NOE crosspeaks associated with the heterochiral tetramer emerges, in addition to the NOE crosspeaks associated with the homochiral tetramer (Fig. 5C). In the set of NOE crosspeaks from the heterochiral tetramer, the Phe2015NH proton shows an interstrand NOE with the Ala21 α-proton in its dimerization partner, as well as a relatively strong intrastrand NOE with the Phe19 α-proton and a weaker intrastrand NOE with the Phe20 α-proton. Although the observation of a new set of crosspeaks establishes the formation of a heterochiral tetramer, it does not distinguish between the L2D2 and the (LD)2 topological isomers.
To differentiate between the L2D2 and the (LD)2 topological isomers, we strategically incorporated two deuterated residues (d8-Phe19 and d4-Ala21) into 15N-labeled peptide 2a, to create peptide 3a, and we studied its interaction with unlabeled ent-1a by 15N-edited NOESY experiments. A homochiral dimer in which peptide 3a is paired with itself should not exhibit an interstrand NOE between the 15NH proton of Phe20 and the α-proton of d4-Ala21, because the α-proton has been replaced with deuterium (Fig. 6A). In contrast, a heterochiral dimer in which peptide 3a is paired with peptide ent-1a should exhibit an interstrand NOE between the 15NH proton of Phe20 in peptide 3a and the α-proton of Ala21 in peptide ent-1a (Fig. 6B).
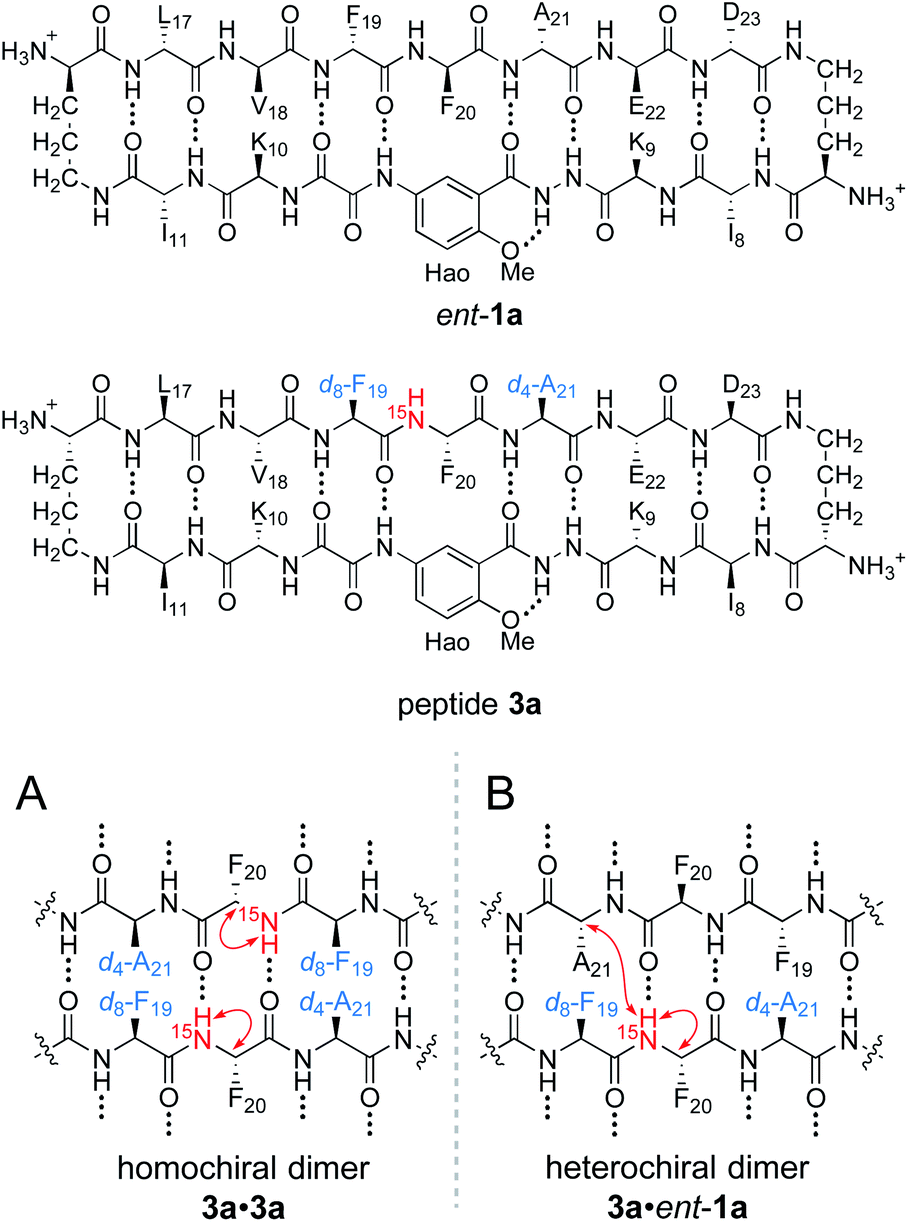 | ||
| Fig. 6 Structures of peptide ent-1a and triply labeled peptide 3a. (A) Expected NOEs in the 15N-edited NOESY spectrum of a homochiral dimer subunit. (B) Expected NOEs in the 15N-edited NOESY spectrum of a heterochiral dimer subunit. | ||
15N-Edited NOESY studies of the mixture of peptides 3a and ent-1a reveal a weak crosspeak associated with a heterochiral tetramer composed of homochiral dimers, in addition to crosspeaks associated with the homochiral tetramer. Enantiomerically pure peptide 3a exhibits the expected intrastrand NOE crosspeak between the 15NH proton and the α-proton of Phe20 and an unexpected weaker NOE crosspeak to the α-proton of d8-Phe19 (Fig. 7A). This weaker NOE results from incomplete deuterium labeling at the α-position of the d8-Phe19.30 In the mixture of peptides 3a and ent-1a, a new NOE crosspeak between the 15NH proton and the α-proton of Phe20 associated with the heterochiral tetramer is observed (Fig. 7B). No additional NOE crosspeaks are observed for the heterochiral tetramer. The presence of only an intrastrand NOE crosspeak in the 15N-edited NOESY spectrum of the mixture indicates that peptides 3a and ent-1a are not dimerization partners and shows that the heterochiral tetramer is composed of two homochiral dimer subunits.
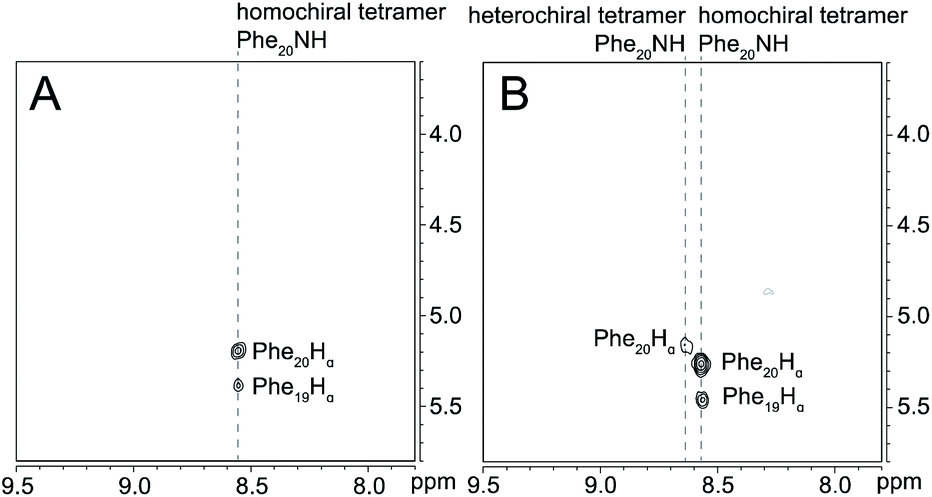 | ||
| Fig. 7
15N-Edited NOESY spectra of (A) 8.0 mM triply labeled peptide 3a and (B) 8.0 mM peptide 3a and 8.0 mM unlabeled peptide ent-1a in 9:1 H2O–D2O at 500 MHz and 298 K. | ||
Collectively the 1D, 1H,15N HSQC, DOSY, and 15N-edited NOESY studies establish that peptides 1a and ent-1a preferentially form the L4 and D4 homochiral tetramers, in addition to a smaller amount of the L2D2 heterochiral tetramer (Fig. 4). The formation of the L2D2 heterochiral tetramer rather than the (LD)2 heterochiral tetramer demonstrates that even within heterochiral assemblies, enantiomeric β-sheet peptides prefer to self-assemble in a homochiral fashion. Thus, the formation of the L2 and D2 pleated β-sheets is preferred over the formation of the LD rippled β-sheets.
1H NMR spectroscopy shows that mixing peptides 1b and ent-1b does not result in heterochiral assembly
To further assess the preferences for homochiral or heterochiral assembly using a different β-sheet peptide, we studied the assembly of peptides 1b and ent-1b.20 Peptide 1b is a homologue of peptide 1a that contains Aβ30–36 instead of Aβ17–23. We had previously found that peptide 1b also assembles to form a tetramer at millimolar concentrations, albeit with an equilibrium that favors the tetramer less strongly.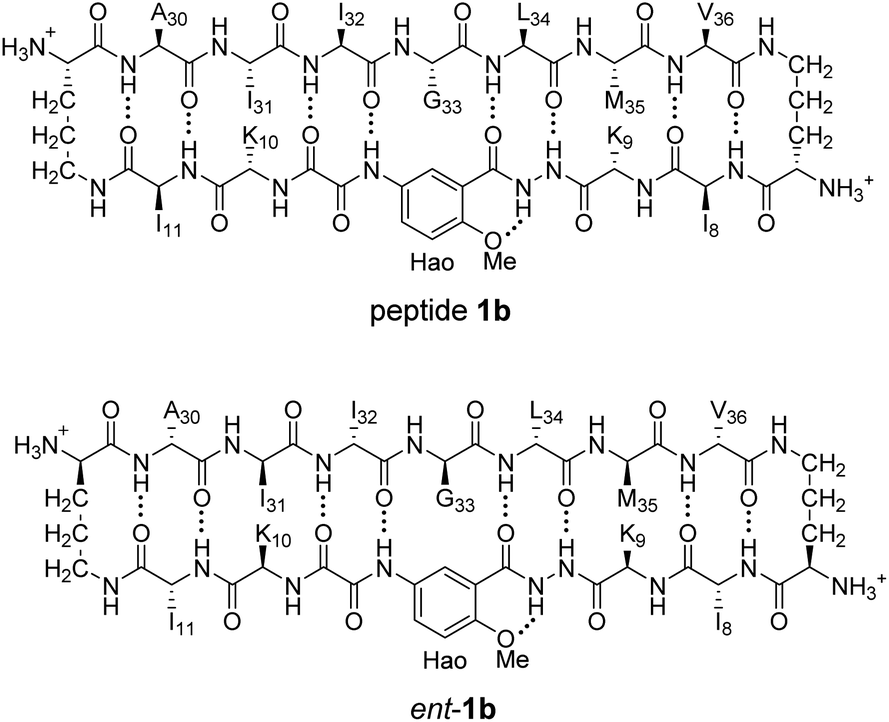
When peptide 1b is mixed with peptide ent-1b, no evidence of heterochiral tetramer formation is observed. The 1H NMR spectrum of peptide 1b at 4.0 mM in D2O at 298 K displays sets of resonances associated with both the monomer and the homochiral tetramer (Fig. 8A). The 4.0 mM 1H NMR spectrum of peptide ent-1b is identical to that of peptide 1b (Fig. 8B). At 8.0 mM, the spectrum of peptide 1b displays a shift in equilibrium toward the tetramer (Fig. 8C). The spectrum broadens slightly, suggesting exchange between the monomer and tetramer on an intermediate timescale (ca. 10−1 s) or additional non-specific interactions.
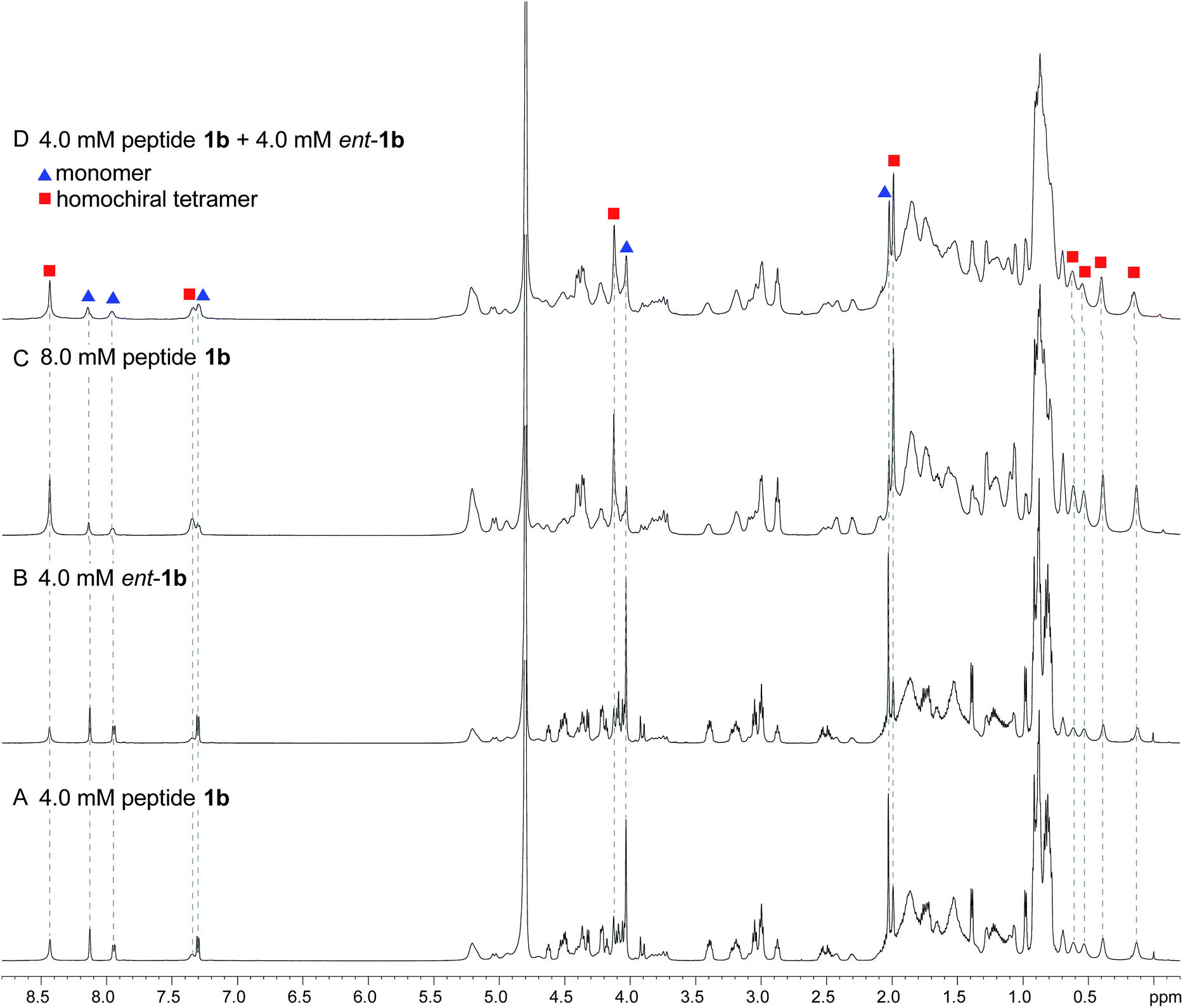 | ||
| Fig. 8 1H NMR spectra of (A) 4.0 mM peptide 1b, (B) 4.0 mM peptide ent-1b, (C) 8.0 mM peptide 1b, and (D) 4.0 mM peptide 1b and 4.0 mM peptide ent-1b in D2O at 600 MHz and 298 K with 0.06 mM DSA.25 (Spectra were referenced against an external DSA standard.) Dashed lines mark key resonances associated with the monomer and homochiral tetramer. These resonances are labeled as follows: blue triangle, monomer; red square, homochiral tetramer. | ||
When peptides 1b and ent-1b are mixed (4.0 mM of each), no new peaks form, and only peaks associated with the monomer and homochiral tetramer are observed (Fig. 8D). As with the 8.0 mM spectrum of peptide 1b, there is slight broadening of the spectrum of the mixture of peptides 1b and ent-1b, suggesting exchange on an intermediate timescale or additional non-specific interactions.
The DOSY spectrum of 1.0 mM peptide 1b in D2O shows a monomer with a diffusion coefficient of 19.5 ± 0.7 × 10−11 m2 s−1 (Fig. S7†). This value is similar to that which we have previously reported for peptide 1b at 1.0 mM and 298 K.20 At 4.0 mM, an additional set of smaller resonances associated with the homochiral tetramer appears (Fig. S8†); the monomer shows a diffusion coefficient of 17.7 ± 0.6 × 10−11 m2 s−1, and the tetramer shows a diffusion coefficient of 13.0 ± 0.4 × 10−11 m2 s−1. The decrease in diffusion coefficient of the monomer, as well as the somewhat higher than expected diffusion coefficient of the tetramer—typically about 0.6× that of the monomer,26ca. 12 × 10−11 m2 s−1—suggest intermediate exchange between the monomer and the tetramer on the 75 ms time scale of the DOSY experiment.
When the concentration of peptide 1b is doubled to 8.0 mM, the resonances associated with the homochiral tetramer predominate (Fig. S9†); the monomer shows a diffusion coefficient of 15.9 ± 0.7 × 10−11 m2 s−1, and the tetramer shows a diffusion coefficient of 12.5 ± 0.4 × 10−11 m2 s−1. The further decrease in diffusion coefficient of the monomer is consistent with intermediate exchange. The DOSY spectrum of the mixture of peptides 1b and ent-1b (4.0 mM of each) shows diffusion coefficients of the monomer and tetramer of 14.4 ± 1.1 × 10−11 m2 s−1 and 10.7 ± 0.8 × 10−11 m2 s−1, respectively (Fig. S10†). The low value of the monomer is consistent with intermediate exchange between the monomer and the tetramer. The value of the tetramer is somewhat lower than expected, suggesting additional transient non-specific interactions among the tetramers.26,27 The absence of any additional new peaks in the spectrum of the mixture, not present in the spectra of the enantiomerically pure peptides, provides good evidence that mixing peptides 1b and ent-1b does not result in any detectable heterochiral assembly.
1H,15N HSQC studies corroborate the presence of only monomer and homochiral tetramer in the mixture of peptides 1b and ent-1b. To identify and confirm the monomer and homochiral tetramer by 1H,15N HSQC, we prepared an isotopologue of peptide 1b containing an 15N label on Gly33—peptide 2b—and studied its mixture with peptide ent-1b. We studied increasing concentrations of enantiomerically pure peptide 2b (1.0 mM, 4.0 mM, and 8.0 mM), and compared the crosspeaks to those found in the mixture of peptides 2b and ent-1b (8.0 mM total).
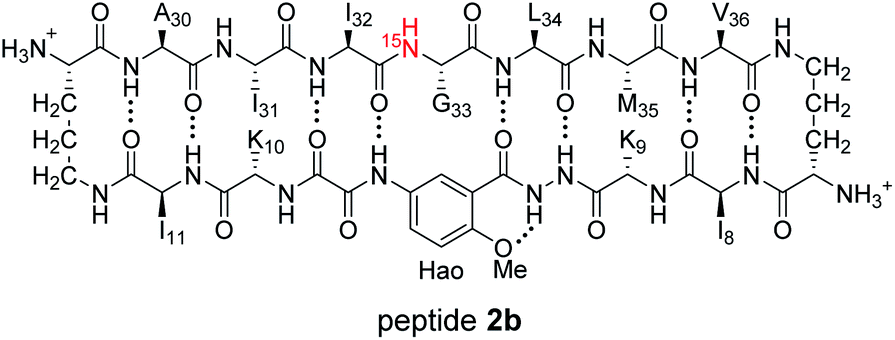
The 1H,15N HSQC spectrum of 1.0 mM peptide 2b in 9:1 H2O–D2O solution shows only a crosspeak associated with the monomer, at 8.35 ppm in the 1H dimension and 112.2 ppm in the 15N dimension (Fig. 9A). At 4.0 mM peptide 2b, the monomer is still present and a crosspeak associated with the homochiral tetramer appears 115.8 ppm in the 15N dimension and 9.31 ppm in the 1H dimension (Fig. 9B).20 When the concentration of peptide 2b is doubled to 8.0 mM, the relative intensity of the tetramer crosspeak increases (Fig. 9C). When peptide 2b is mixed with peptide ent-1b (4.0 mM each), crosspeaks associated with the monomer and homochiral tetramer are still present and no new crosspeaks are observed (Fig. 9D). The lack of new crosspeaks further establishes that enantiomeric β-sheet peptides prefer to self-assemble in a homochiral fashion.
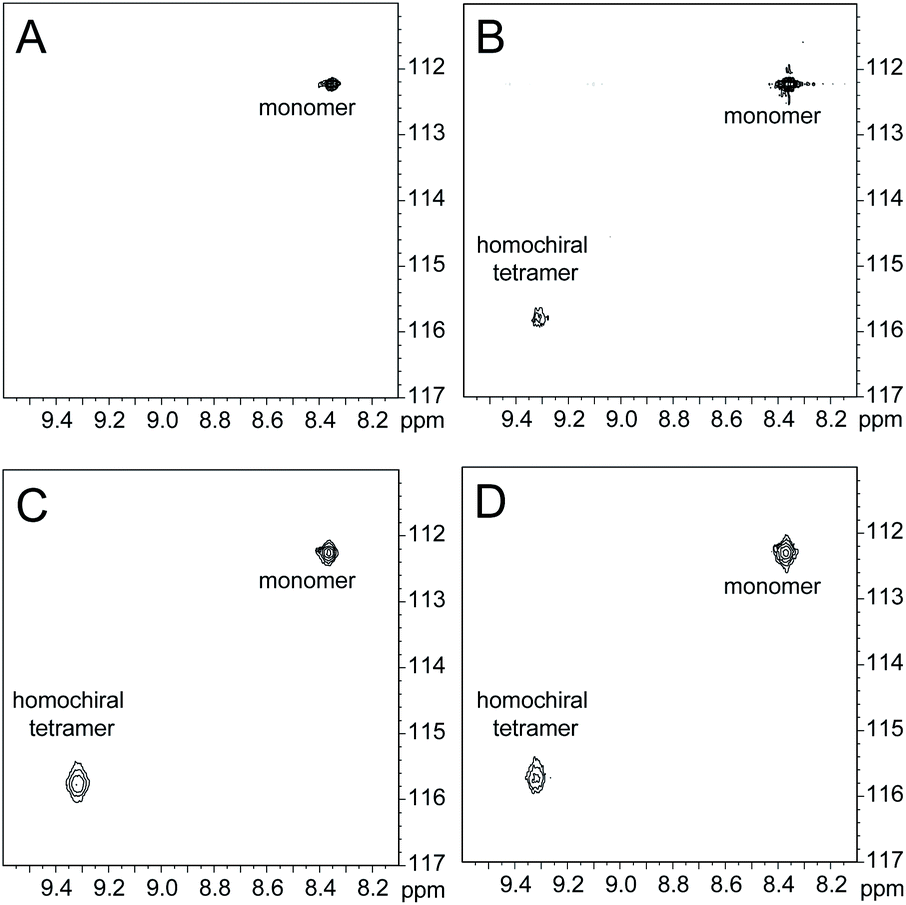 | ||
| Fig. 9
1H,15N HSQC spectra of (A) 1.0 mM peptide 2b, (B) 4.0 mM peptide 2b, (C) 8.0 mM peptide 2b, and (D) 4.0 mM peptide 2b and 4.0 mM peptide ent-1b in 9:1 H2O–D2O at 500 MHz and 298 K. | ||
Conclusion
The solution-phase NMR studies of Aβ-derived peptides 1a and 1b and the corresponding enantiomers and isotopologues provide further evidence that enantiomeric β-sheet peptides prefer to self-assemble in a homochiral fashion. These studies recapitulate our laboratory's findings that small β-sheet peptides strongly prefer to form homochiral dimers in chloroform solution,18 as well as Gellman's findings in homo- and heterochiral β-hairpin systems.19 How then, do we reconcile these findings with the findings of other researchers where heterochiral assembly is preferred?In the studies of Schneider, Nilsson, Raskatov, and Tycko described in the introduction, heterochiral assembly occurs in the solid or gel state.5–17 Heterochiral assembly in the solid state is driven heavily by the packing of molecules, which in addition to edge-to-edge hydrogen bonding, drives the formation of fibrils and crystal lattices. These packing interactions involve not just the side chains within individual β-sheets, but also the packing of β-sheets together. Heterochiral packing is generally preferred over homochiral packing in the crystal state, which leads to denser solids and a preference for racemic crystal formation—a phenomenon known as “Wallach's rule”.31–33 Thus, it appears that packing in the solid state may drive the formation of heterochiral mixtures of β-sheet peptides, and in some cases the formation of rippled β-sheets. In the solution phase, where crystal packing forces are absent, rippled β-sheet formation is strongly disfavored. Thus, no evidence of heterochiral pairing to form rippled β-sheets is observed with peptides 1a and 1b and the corresponding enantiomers.
Data availability
The data supporting this article are available in the ESI.†Author contributions
Xingyue Li synthesized the peptides, performed the experiments, analyzed the results, and prepared the manuscript with James S. Nowick. Stephanie E. Rios assisted with the peptide synthesis. James S. Nowick supervised the project and assisted in the experimental design and writing of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank Dr Nicholas Truex for guidance and samples provided, Dr Philip Dennison and the UCI Department of Chemistry NMR Spectroscopy Facility for assistance with NMR experiments, and Ben Katz, Dr Felix Grun, and the UCI Mass Spectrometry facility for assistance with mass spectrometry experiments. We thank the National Institutes of Health (NIH) for funding (GM097562 and AG072587).Notes and references
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 251–256 CrossRef CAS PubMed.
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 729–740 CrossRef CAS PubMed.
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1953, 39, 253–256 CrossRef CAS PubMed.
- B. Lotz, ChemBioChem, 2022, e202100658 CAS.
- J. A. Raskatov, J. P. Schneider and B. L. Nilsson, Acc. Chem. Res., 2021, 54, 2488–2501 CrossRef CAS PubMed.
- A. R. Foley and J. A. Raskatov, Curr. Opin. Chem. Biol., 2021, 64, 1–9 CrossRef CAS PubMed.
- K. J. Nagy, M. C. Giano, A. Jin, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2011, 133, 14975–14977 CrossRef CAS PubMed.
- K. Nagy-Smith, P. J. Beltramo, E. Moore, R. Tycko, E. M. Furst and J. P. Schneider, ACS Cent. Sci., 2017, 3, 586–597 CrossRef CAS PubMed.
- R. J. Swanekamp, J. T. M. Dimaio, C. J. Bowerman and B. L. Nilsson, J. Am. Chem. Soc., 2012, 134, 5556–5559 CrossRef CAS PubMed.
- R. J. Swanekamp, J. J. Welch and B. L. Nilsson, Chem. Commun., 2014, 50, 10133–10136 RSC.
- J. M. Urban, J. Ho, G. Piester, R. Fu and B. L. Nilsson, Molecules, 2019, 24, 1983 CrossRef CAS PubMed.
- S. Dutta, A. R. Foley, C. J. A. Warner, X. Zhang, M. Rolandi, B. Abrams and J. A. Raskatov, Angew. Chem., Int. Ed., 2017, 56, 11506–11510 CrossRef CAS PubMed.
- J. A. Raskatov, Chem.–Eur. J., 2017, 23, 16920–16923 CrossRef CAS PubMed.
- J. A. Raskatov, A. R. Foley, J. M. Louis, W. M. Yau and R. Tycko, J. Am. Chem. Soc., 2021, 143, 13299–13313 CrossRef CAS PubMed.
- A. J. Kuhn, B. Ehlke, T. C. Johnstone, S. R. J. Oliver and J. A. Raskatov, Chem. Sci., 2022, 13, 671–680 RSC.
- J. A. Raskatov, ChemBioChem, 2020, 21, 2945–2949 CrossRef CAS PubMed.
- J. A. Raskatov, Biopolymers, 2021, 112, e23391 CrossRef CAS PubMed.
- D. M. Chung and J. S. Nowick, J. Am. Chem. Soc., 2004, 126, 3062–3063 CrossRef CAS PubMed.
- X. Liu and S. H. Gellman, ChemBioChem, 2021, 22, 2772–2776 CrossRef CAS PubMed.
- N. L. Truex, Y. Wang and J. S. Nowick, J. Am. Chem. Soc., 2016, 138, 13882–13890 CrossRef CAS PubMed.
- N. L. Truex and J. S. Nowick, J. Am. Chem. Soc., 2016, 138, 13891–13900 CrossRef CAS PubMed.
- J. S. Nowick and J. O. Brower, J. Am. Chem. Soc., 2003, 125, 876–877 CrossRef CAS PubMed.
- J. S. Nowick, D. M. Chung, K. Maitra, S. Maitra, K. D. Stigers and Y. Sun, J. Am. Chem. Soc., 2000, 122, 7654–7661 CrossRef CAS.
- The EXSY experiments were performed with a mixing time of 500 ms. Exchange occurs too slowly to be detected at 298 K but is observed at 328 K, suggesting that exchange occurs on the time scale of seconds at 328 K and tens of seconds at 298 K.
- J. S. Nowick, O. Khakshoor, M. Hashemzadeh and J. O. Brower, Org. Lett., 2003, 5, 3511–3513 CrossRef CAS PubMed.
- O. Khakshoor, B. Demeler and J. S. Nowick, J. Am. Chem. Soc., 2007, 129, 5558–5569 CrossRef CAS PubMed.
- Alternatively, the formation of hexamers in rapid equilibrium with the tetramers cannot be precluded. We have previously observed that β-sheet peptides that form a tetramer comprising two β-sheet dimers in solution can form a hexamer comprising three β-sheet dimers in the crystal state, O. Khakshoor, A. J. Lin, T. P. Korman, M. R. Sawaya, S. C. Tsai, D. Eisenberg and J. S. Nowick, J. Am. Chem. Soc., 2010, 132, 11622–11628 CrossRef CAS PubMed.
- J. S. Nowick, Org. Biomol. Chem., 2006, 4, 3869–3885 RSC.
- K. Wüthrich NMR of Proteins and Nucleic Acids, Wiley, New York, 1986, pp. 125–129 Search PubMed.
- d8-Phenylalanine was purchased from Cambridge Isotope Laboratories and was reported to be 98% isotopic purity. The 1H NMR spectrum of the d8-phenylalanine shows a disproportionate amount of 1H isotopic impurity at the α-position. See Fig. S11† for spectral data.
- C. P. Brock, W. B. Schweizer and J. D. Dunitz, J. Am. Chem. Soc., 1991, 113, 9811–9820 CrossRef CAS.
- T. Friščić, L. Fábián, J. C. Burley, D. G. Reid, M. J. Duer and W. Jones, Chem. Commun., 2008, 1644–1646 RSC.
- P. S. Navare and J. C. MacDonald, Cryst. Growth Des., 2011, 11, 2422–2428 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of peptide synthesis, Fmoc-protection of amino acids, NMR spectroscopic studies, and peptide characterization data. See https://doi.org/10.1039/d2sc02080g |
| This journal is © The Royal Society of Chemistry 2022 |