
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWide bandgaps and strong SHG responses of hetero-oxyfluorides by dual-fluorination-directed bandgap engineering†
Yilei
Hu‡
a,
Xingxing
Jiang‡
 b,
Tianhui
Wu‡
a,
Yanyan
Xue‡
a,
Chao
Wu
*a,
Zhipeng
Huang
a,
Zheshuai
Lin
b,
Jun
Xu
a,
Mark G.
Humphrey
c and
Chi
Zhang
*a
b,
Tianhui
Wu‡
a,
Yanyan
Xue‡
a,
Chao
Wu
*a,
Zhipeng
Huang
a,
Zheshuai
Lin
b,
Jun
Xu
a,
Mark G.
Humphrey
c and
Chi
Zhang
*a
aChina–Australia Joint Research Center for Functional Molecular Materials, School of Chemical Science and Engineering, Tongji University, Shanghai 200092, China. E-mail: chizhang@tongji.edu.cn; wuc@tongji.edu.cn
bTechnical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China
cResearch School of Chemistry, Australian National University, Canberra, ACT 2601, Australia
First published on 27th July 2022
Abstract
A wide bandgap is an essential requirement for a nonlinear optical (NLO) material. However, it is very challenging to simultaneously engineer a wide bandgap and a strong second-harmonic generation (SHG) response, particularly in NLO materials containing second-order Jahn–Teller (SOJT) distorted units. Herein, we employ a bandgap engineering strategy that involves the dual fluorination of two different types of SOJT distorted units to realize remarkably wide bandgaps in the first examples of 5d0-transition metal (TM) fluoroiodates. Crystalline A2WO2F3(IO2F2) (A = Rb (RWOFI) and Cs (CWOFI)) exhibit the largest bandgaps yet observed in d0-TM iodates (4.42 (RWOFI) and 4.29 eV (CWOFI)), strong phase-matching SHG responses of 3.8 (RWOFI) and 3.5 (CWOFI) × KH2PO4, and wide optical transparency windows. Computational studies have shown that the excellent optical responses result from synergism involving the two fluorinated SOJT distorted units ([WO3F3]3− and [IO2F2]−). This work provides not only an efficient strategy for bandgap modulation of NLO materials, but also affords insight into the relationship between the electronic structure of the various fluorinated SOJT distorted units and the optical properties of crystalline materials.
Introduction
Nonlinear optical (NLO) materials, exemplified by commercial β-BaB2O4 (β-BBO), LiB3O5 (LBO), KH2PO4 (KDP), KTiOPO4 (KTP) and AgGaS2 (AGS),1 can effectively expand the spectral range of lasers, and may therefore serve as key materials for the all-solid-state laser devices that are widely applied in modern laser technologies such as photolithography, spectral analysis, tissue imaging, and environmental monitoring.2 A high-performance NLO material should not only possess non-centrosymmetry, but it should also exhibit a large second-harmonic generation (SHG) coefficient, a wide bandgap, sufficient birefringence, and good physicochemical stability.3 Unfortunately, two of the key optical properties, a wide bandgap and a strong SHG coefficient, are two often-competing optical parameters and difficult to engineer simultaneously in a material, owing to their contrasting microstructural requirements.4Modulation of SHG responses can be achieved by incorporating second-order Jahn–Teller (SOJT) distorted cations,5–7 such as octahedrally coordinated d0-transition metal (TM) cations (e.g., Ti4+, V5+, Nb5+, Mo6+, etc.)5 or stereochemically-active lone-pair cations (e.g., Se4+, Te4+, and I5+, etc.),6 although, in general, these microstructural building units promote a red-shift in the absorption edge. A structurally related strategy involves fluorination of the TM cation-centered oxyanions8 in order to blue-shift the absorption edge, but the SHG responses of the resultant materials are usually limited (<1.0 KDP) owing to the often-antiparallel arrangements of the fluorinated TM oxyanions. A complementary approach relies on combination of two types of oxyanions containing SOJT distorted cations; this has been demonstrated by the successful syntheses of a variety of hetero-oxyanion materials with strong SHG responses,9,10 such as ASe2V3O12 (A = Rb, Tl),9a A2(MoO3)3(SeO3)2 (A = Rb, Tl),9b MgTeMoO6,9c AMoO3(IO3) (A = Rb, Cs, NH4) (A = Rb, Cs, NH4),9d,e K(VO)2O2(IO3)3,9f and [C(NH2)3]2Mo2O5(IO3)4·2H2O.9g However, the bandgaps of these materials are relatively narrow owing to the existence of d-orbital electrons in the TM cations, which significantly restricts the optical transparency range in the short-wavelength region and reduces the laser damage threshold.
Nevertheless, of the possible chemical systems, assembly of hetero-oxyanions may still offer an ideal platform for optical property modulation because of their multiple tunable microstructural building units.10 The two mutually exclusive optical properties of bandgap and SHG response are intimately related to the electronic band structure of the material, which can be micro-controlled by the intrinsic properties and spatial arrangements of the building units.11 In this study, we propose a general bandgap engineering approach based on fluorinating two types of oxyanions, both containing SOJT cations. In this approach, a judicious assembly of fluorine-rich units containing heavy cations addresses the problematic competition between bandgap and SHG response in hetero-oxyanion materials. To implement this strategy, heavy 5d0-TM octahedra (e.g., Hf4+, Ta5+ and W6+) are preferred to conventional 3d0/4d0-TM octahedra of the same family because heavy 5d0-TM octahedra are anticipated to form less-covalent bonds with oxygen/fluorine owing to their lower effective electronegativity (referring to the ease of oxygen-to-metal charge transfer for the heavy 5d0-TM octahedra);12 this will increase the bandgap of the resultant materials. The simultaneous fluorination of two different types of oxyanions in the one structure (i.e. d0-TM-centered octahedra and non-metal-centered polyhedra) can induce further significant differences in the band structures and increase the bandgap owing to the highly electronegative fluorine, an outcome observed in recently reported fluorooxoborates13 and fluorophosphates.14 Furthermore, fluorine acting as chemical “scissors” can effectively reduce the dimensionality of the structure, which may be highly beneficial for the enhancement of bandgap.15 Finally, large macroscopic polarization (corresponding to a large SHG effect) may be achieved by stacking the oxyfluorine anions in an additive mode. To demonstrate the effectiveness of our proposed strategy, we disclose the successful synthesis of the first examples of 5d0-TM fluoroiodates A2WO2F3(IO2F2) (A = Rb (RWOFI), Cs (CWOFI)). Their unique lambda (Λ)-shaped [WO2F3(IO2F2)]2− hetero-oxyfluorine anions not only drive the formation of the polar structures that exhibit strong phase-matching SHG responses of 3.8 (RWOFI) and 3.5 (CWOFI) times that of KDP, sufficient birefringences, and wide optical transparency windows from the ultraviolet to the mid-infrared, but more importantly induce very wide bandgaps of 4.42 eV (RWOFI) and 4.29 eV (CWOFI) – both among the largest bandgaps for d0-TM iodates. Herein we report their syntheses, crystal structures, electronic structures, and optical properties, as well as structure–property correlations employing a combination of experiments and first-principles calculations.
Results and discussion
Although considerable research pursuing non-centrosymmetric iodates containing 3d0/4d0-TM cations susceptible to the SOJT effect has been undertaken, there has thus far been only one 5d0-TM iodate reported (K5(W3O9F4)(IO3),16 containing the W6+ cation). The synthetic difficulty lies in the fact that tungsten is a refractory metal, and is extremely difficult to dissolve in common solvents. Unlike the previous synthetic method, in this study hydrofluoric acid was used not only as the solvent but also as a fluorine source for the syntheses of hetero-oxyfluorides A2WO2F3(IO2F2) based on the following reaction equation:| 2A2CO3 + 2WO3 + 2H5IO6 + 10HF → 2A2WO2F3(IO2F2) + O2↑ + 2CO2↑ + 10H2O |
Our efforts to synthesize homologues with other alkali metal cations (e.g., Na+, K+) were unsuccessful, possibly due to the large differences in ionic radii among these alkali metal cations (0.102, 0.138, 0.152, and 0.167 nm for Na+, K+, Rb+, and Cs+, respectively).17a The purities of the two hetero-oxyfluorides were confirmed by comparison of their experimental PXRD patterns with simulated patterns derived from single-crystal X-ray diffraction data (Fig. S1†). The energy dispersive X-ray spectroscopy analyses reveal element distribution maps for RWOFI and CWOFI with molar ratios of 9.38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.01:4.02:26.55 and 8.75:4.78:4.54:25.11 for Rb/Cs, W, I and F, respectively, which are consistent with the molar ratios determined from single-crystal X-ray diffraction (Fig. S2†).
:5.01:4.02:26.55 and 8.75:4.78:4.54:25.11 for Rb/Cs, W, I and F, respectively, which are consistent with the molar ratios determined from single-crystal X-ray diffraction (Fig. S2†).
A2WO2F3(IO2F2) (A = Rb, Cs) are the first examples of 5d0-TM fluoroiodates. They are isostructural and crystallize in the same non-centrosymmetric and polar orthorhombic space group, Cmc21 (no. 36), so only the structure of CWOFI is described in detail here (Tables S1–S5†). Its structure is composed of zero-dimensional (0D) [WO2F3(IO2F2)]2− hetero-oxyfluorine anions and Cs+ cations (Fig. 1b). In the [WO2F3(IO2F2)]2− hetero-oxyfluorine anion, the unique I5+ cation is coordinated by two O atoms and two F atoms, forming an unsymmetrical [IO2F2] tetrahedron (Fig. 1a). The two I–F bond lengths (1.974(8) Å) are longer than the two I–O lengths (1.790(17) and 1.759(14) Å), and the F–I–F bond angle is close to linear (177.8(6)°). Each W6+ cation is octahedrally coordinated with two terminal O atoms, three terminal F atoms, and one bridging O atom from an [IO2F2]− group bound in a unidentate fashion (Fig. 1a). The W–F and W–O bond distances lie in the ranges 1.733(16)–1.953(8) and 1.800(10)–2.263(17) Å, respectively. One [WO3F3] octahedron further connects with one [IO2F2] tetrahedron by sharing O atoms, forming the Λ-shaped [WO2F3(IO2F2)]2− hetero-oxyfluorine anion (Fig. 1a) with a W–O–I angle of 140.78°; this differs from other Λ-shaped iodate-containing species such as cis-[ZrF6(IO3)2],17acis-[VO2F2(IO3)2],17b and [Ga(IO3)2F4].17c The [WO2F3(IO2F2)]2− hetero-oxyfluorine anion is the first example of a TM iodate containing two different types of heavy atom-containing SOJT distorted cations. The Cs+ cations connect the Λ-shaped [WO2F3(IO2F2)]2− hetero-oxyfluorine anions, forming a 3D layer-piled structure along the b-axis (Fig. 1b).
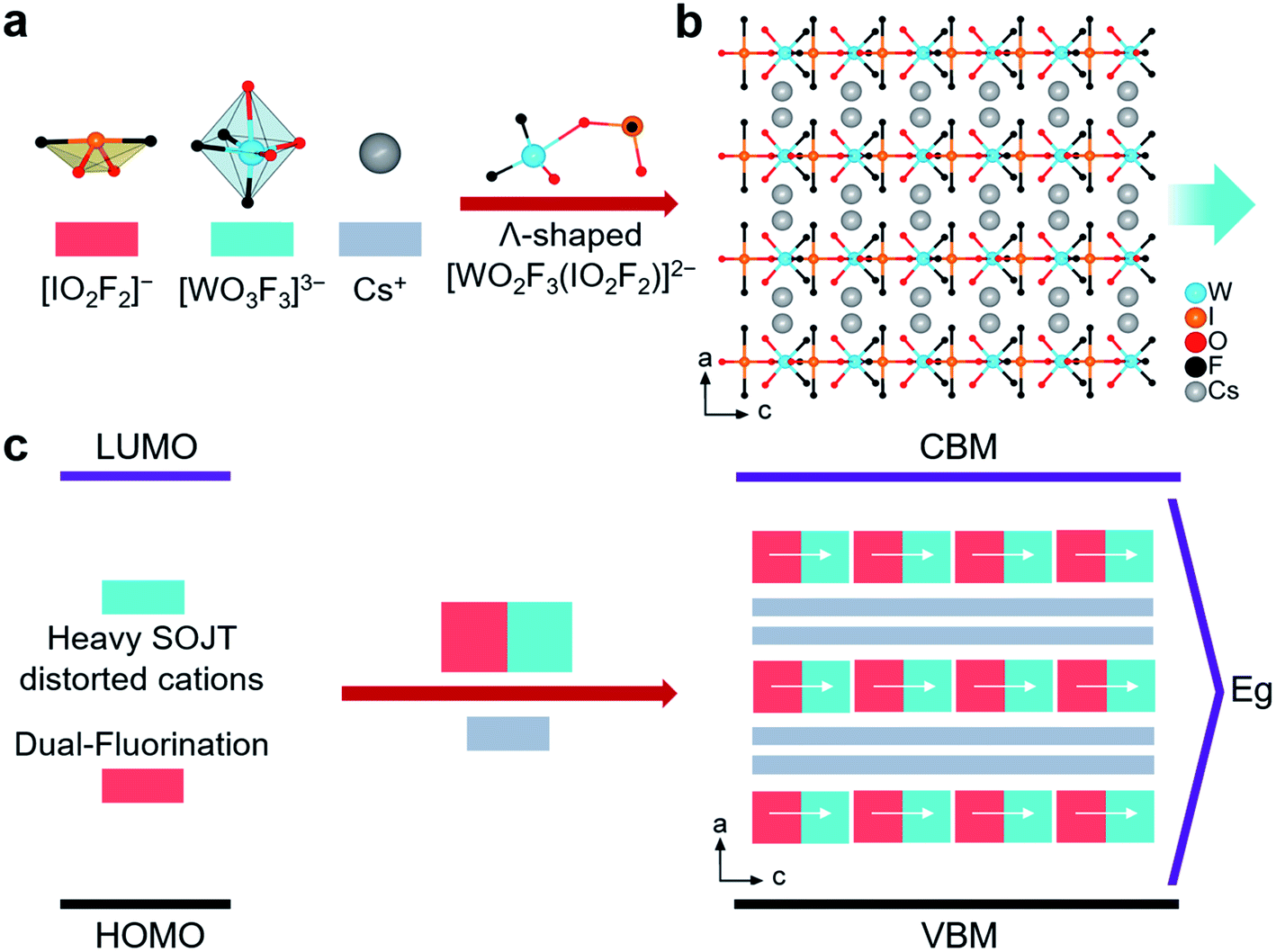 | ||
| Fig. 1 (a) Structural building units in CWOFI. (b) Crystal structure of CWOFI viewed along the b-axis. The blue arrow in the panel represents the net dipole moment of the structure. (c) Structural building unit description of the arrangement of 0D [WO2F3(IO2F2)]2− units and Cs+ cations. The Cs–O/F bonds are omitted for clarity. | ||
The most critical structural feature of CWOFI, however, is the tungsten-centered oxyfluorine anion, which plays a key role in construction of the polar fluoroiodate. The W6+ cation undergoes intra-octahedral distortion toward the terminal fluorine atom, a corner (C4) distortion, with one short [1.733(16) Å], one long [2.263(17) Å], and four normal [1.800(10)–1.953(8) Å] W–O/F bonds (Fig. S3†). The magnitude of the out-of-center distortion (Δd) is 0.84, which corresponds to strong distortion (Δd > 0.8).18 The C4 distorted [WO3F3] octahedron is favorable for creating Λ-shaped units with a fixed polarization orientation. The bridging O(2) with one long bond has the greater residual negative charge, based on BVS calculations (Table S5†), and is therefore predicted to bond in preference to the other O/F terminal ligands; it consequently links to the lone-pair cation I5+ to form the Λ-shaped [WO2F3(IO2F2)]2− hetero-oxyfluorine anion. The W6+ cations are blocked from distorting toward a corner, owing to the unsymmetrical [IO2F2]− oxyfluorine anions, the lone-pair cation I5+ serving to reinforce the direction of the intra-octahedral distortion. The polar orientation of the Λ-shaped [WO2F3(IO2F2)]2− hetero-oxyfluorine anion is fixed and it cannot rotate due to significant steric effects (Fig. 1a). As a result, the [WO2F3(IO2F2)]2− hetero-oxyfluorine anions pack in the crystal structure with their polar orientations arranged in an additive fashion.
Our proposed bandgap engineering strategy involving dual fluorination of two different types of SOJT distorted units has afforded A2WO2F3(IO2F2), the structure of which bears a remarkable resemblance to those of conventional d0-TM iodates. Their structures are constructed by judicious assembly of two different types of SOJT distorted units (Fig. 1), except for three following crucial differences. Firstly, 3d0/4d0-TM cations (e.g., V5+, Ti4+, Nb5+, and Mo6+) are responsible for the bandgaps of the d0-TM iodates, and these bandgaps are generally less than 3.8 eV19 owing to the presence of d–d transitions. By contrast, the bandgaps of the hetero-oxyfluorides RWOFI and CWOFI are expected to blue-shift because the effective electronegativity12 of the heavy 5d0 tungsten cation is lower than that of the 3d0/4d0-TM cations. Secondly, in stark contrast to conventional d0-TM iodates, the dual fluorination of two different types of oxyanions containing SOJT cations may afford structure-building oxyfluorine anions ([WO3F3]3− and [IO2F2]−) with relatively wide HOMO–LUMO gaps,20 and thereby potentially induce a wide bandgap in RWOFI and CWOFI. Thirdly, the abundant fluorine, acting as chemical “scissors”, induces formation of the 0D Λ-shaped [WO2F3(IO2F2)]2− hetero-oxyfluorine anion, which favors the formation of polar structures, and is beneficial for strong SHG responses, owing to the crystal packing along the c-axis.
The optical transmission spectra of RWOFI and CWOFI single crystals suggest that both compounds possess broad optical transparency windows of 0.28–5.28 and 0.289–5.33 μm (Fig. 2a, b, S4a, and b†) that cover the IR atmospheric window (3–5 μm). The optical bandgaps of RWOFI and CWOFI from their UV cutoff wavelengths are 4.42 and 4.29 eV. In contrast to K5(W3O9F4)(IO3) (3.83 eV) with a similar composition, the bandgaps of RWOFI and CWOFI are significantly blue-shifted, which can be attributed to the combination of two different types of SOJT-distorted oxyfluorine anions as well as the reduced 0D structures. The bandgaps of RWOFI and CWOFI are the largest for d0-TM iodates reported to date. IR absorption below 1000 cm−1 was measured on crystalline powder samples of the two materials (Fig. S4c and d†). The absorption bands ranging from 460 to 861 cm−1 can be assigned to the stretching and bending vibrations of the [IO2F2] tetrahedra, while the strong peaks between 950 and 900 cm−1 belong to the vibration frequencies of the [WO3F3] octahedra. From the two-phonon approximation, the IR edges of RWOFI and CWOFI should be larger than 5 μm, with the maximum absorption bands around 977 cm−1 and 966 cm−1, respectively, indicating that RWOFI and CWOFI are mid-IR transparent NLO materials.
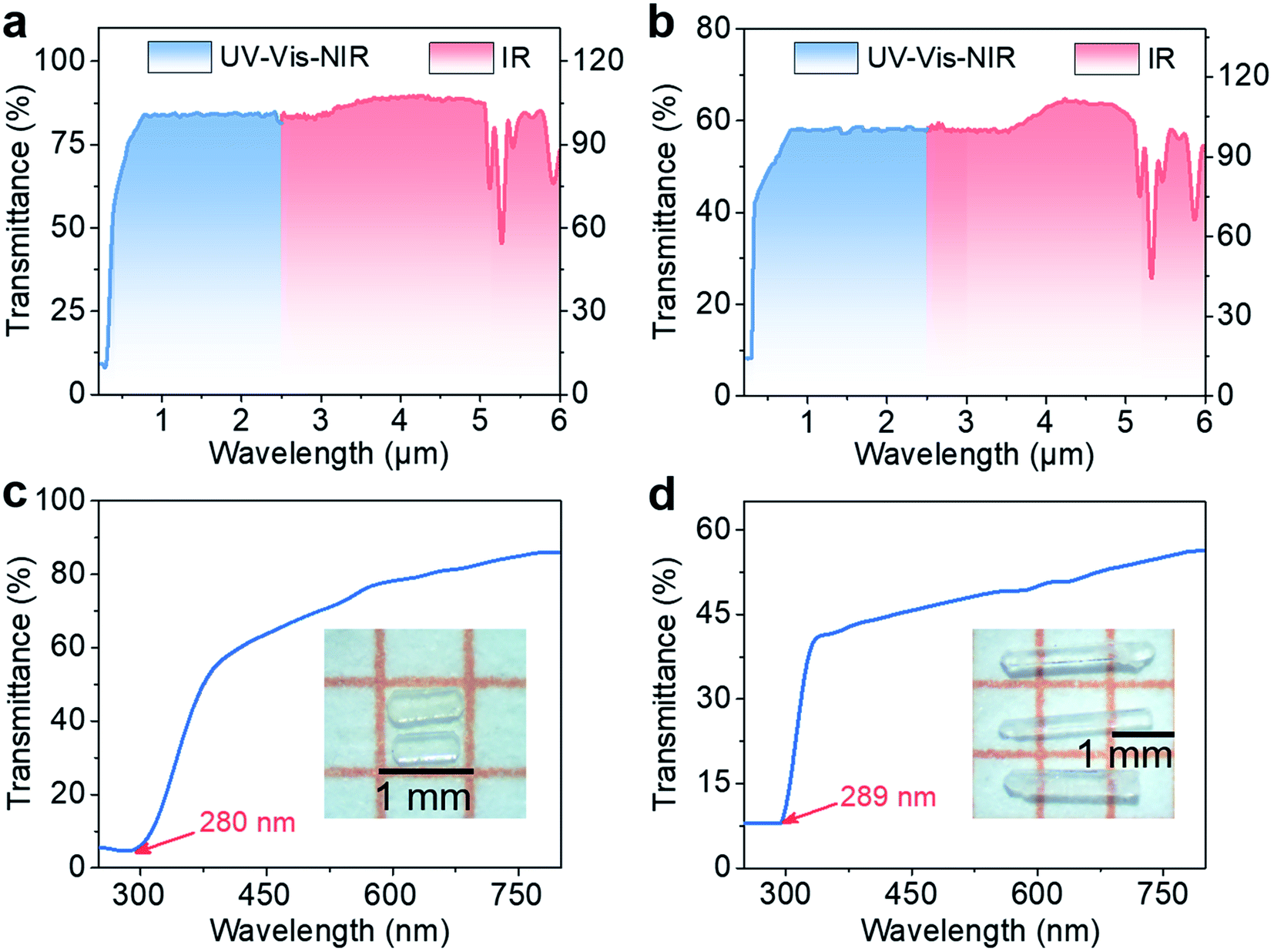 | ||
| Fig. 2 Optical transparency windows (top) and UV-Vis-NIR transmission spectra (bottom) of RWOFI (a and c) and CWOFI (b and d). The insets show the crystals of RWOFI (c) and CWOFI (d) used for measurements. | ||
The thermogravimetric analysis (Fig. S5†) reveals that RWOFI and CWOFI are stable at temperatures up to 288 °C, beyond which a two-step decomposition is observed. The first step, a sharp drop between 288–543 °C, corresponds to a weight loss of 28.20% and 25.31%, assigned to the loss of 0.5 I2 and 1.5 F2 molecules (28.72% and 25.28% for calculated values). The second step between 543 °C and 700 °C can be assigned to the loss of 1 F2 molecule, with experimental weight losses of 5.53% and 4.93% (5.93% and 5.17% for calculated values).
Powder SHG measurements with 1064 nm laser radiation reveal that RWOFI and CWOFI are phase-matchable and display SHG signals of 3.8× and 3.5 × KDP, respectively, in the particle size range 105–150 mm (Fig. 3). The measured SHG signals are significantly larger than those of previously reported crystals with single-fluorinated SOJT distorted units, such as α-BaMoO2F4 (0.7 × α-SiO2),8b NaVOF4(H2O) (1 × α-SiO2),8c Rb2VO(O2)2F (0.8 × KDP),8d KNaNbOF5 (3 × KDP),8a KWO3F (3 × KDP),8e and CsIO2F2 (3 × KDP).21 Dipole moment calculations were performed on RWOFI and CWOFI by the bond valence method,22 the results being summarized in Table S6.† The calculated net dipole moments of their unit cells are similar, consistent with their similar measured SHG results. The dipole moments of the [WO3F3]3− and [IO2F2]− oxyfluorine anions in the a and b direction nearly cancel, the vector sum of the dipole moments of both oxyanions effectively pointing along the c-axis. The dipole moment of the [IO2F2] tetrahedron is larger than that of the [WO3F3] octahedron, indicating that the [IO2F2]− oxyfluorine anions make the major contribution to the SHG responses.
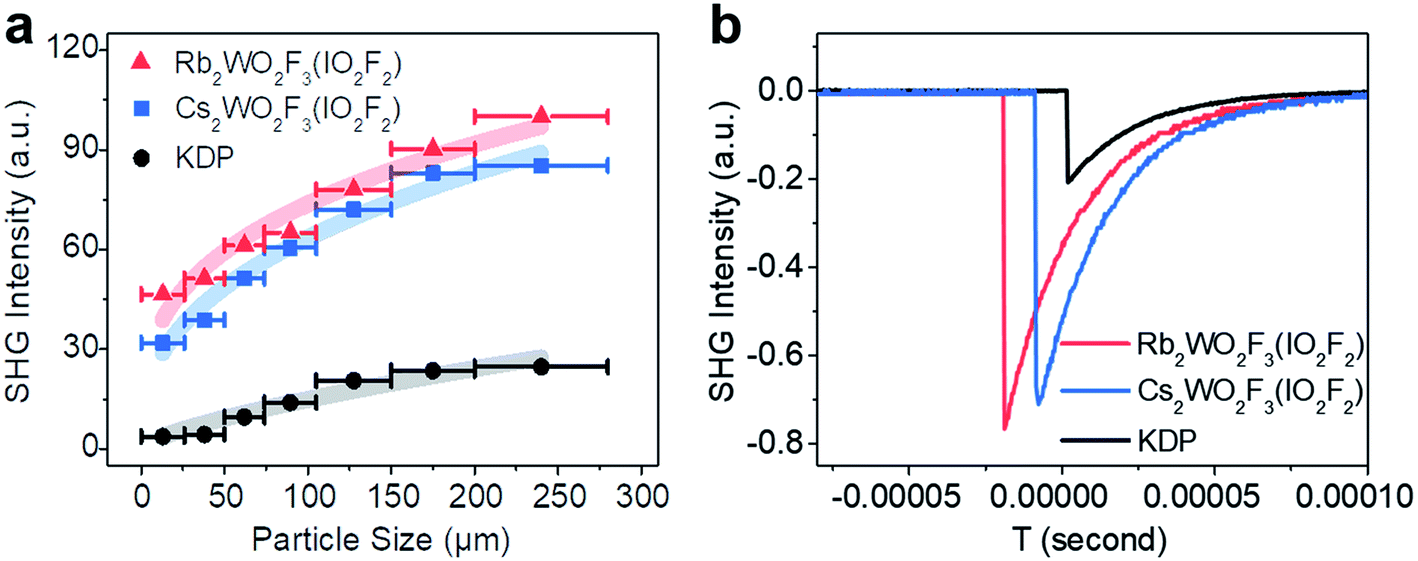 | ||
| Fig. 3 (a) Phase-matching curves of RWOFI and CWOFI with 1064 nm laser radiation. The solid curves are guides for the eye and not fits to the data. (b) Oscilloscope traces of the SHG signals (105–150 μm) for powders of RWOFI and CWOFI at λ = 1064 nm. KDP samples serve as the references. | ||
Electronic structure calculations of RWOFI and CWOFI were undertaken to shed light on the origin of the linear and nonlinear optical properties. The band structures demonstrate that the two compounds are direct bandgap materials with a high efficiency of light utilization. The calculated bandgaps of RWOFI and CWOFI obtained from the band structures are 3.14 and 3.09 eV (Fig. S6†), respectively, smaller than the experimental results of 4.42 and 4.29 eV, a difference that can be ascribed to the discontinuity of exchange–correlation energy in DFT calculations. Examination of the partial and total density of states show that the I 5p and W 5d orbitals overlap with the O 2p and F 2p orbitals over the entire energy range (Fig. 4a and S7a†). The Rb 4p nonbonding orbitals in RWOFI are far from the Fermi level compared to the Cs 5p nonbonding orbitals in CWOFI. Thus, RWOFI shows a slightly larger bandgap, consistent with the trend in experimental results. The contribution to the valence band (VB) close to the Fermi level is essentially O 2p and F 2p in nature, with the latter located at a lower energy because of its larger electronegativity. Both I 5p and O 2p orbitals play key roles in the lower energy regions of the conduction band (CB), consistent with strong I–O/F and W–O/F bonds interactions.
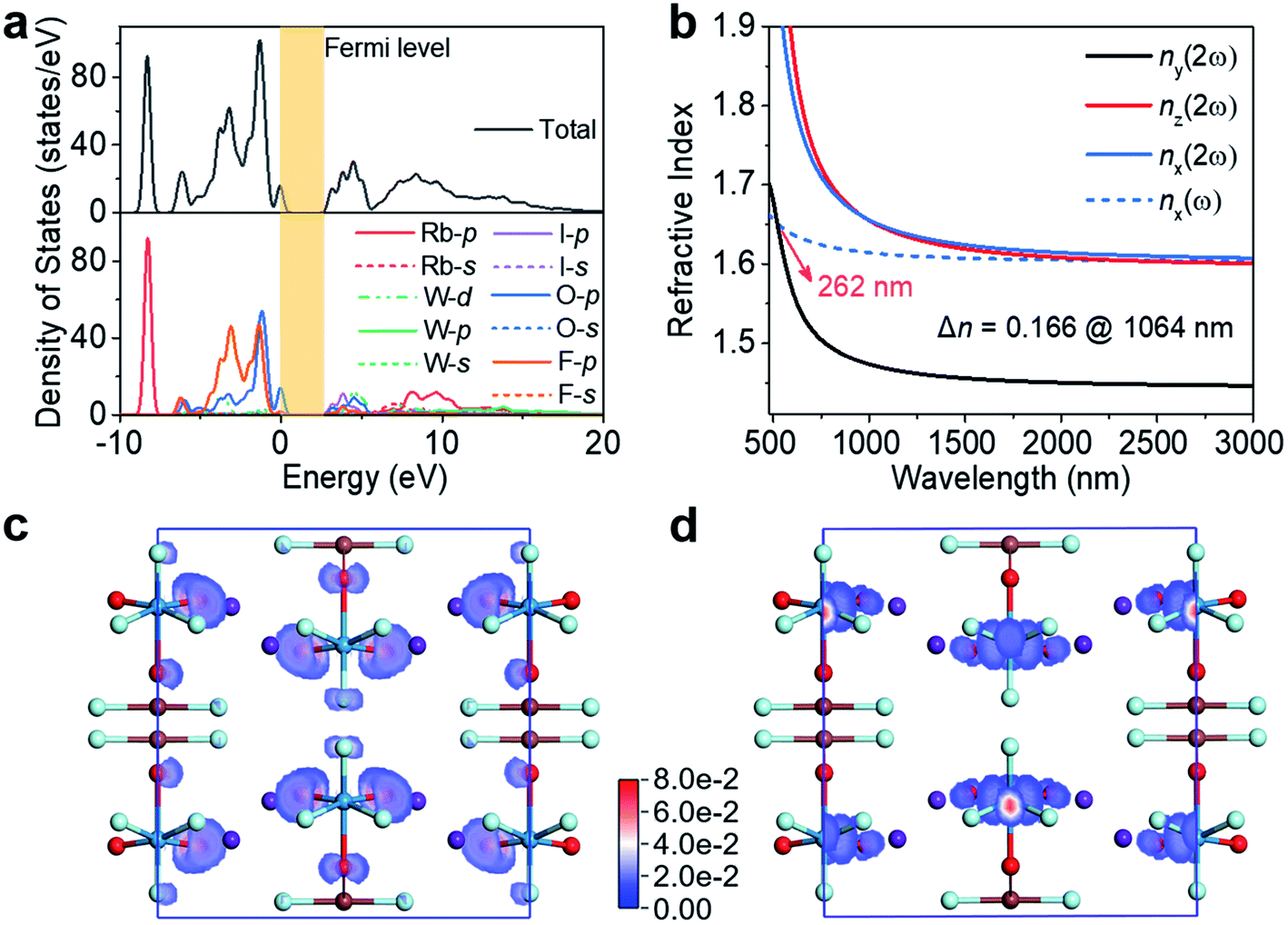 | ||
| Fig. 4 (a) Total and partial density of states projected onto the constituent atoms in RWOFI. (b) Calculated refractive indexes in RWOFI. SHG-weighted densities for (c) occupied and (d) unoccupied electronic states in RWOFI. Color codes: I brown, W green, O red, F light green, Rb purple. | ||
To clearly demonstrate the influences of the two heavy oxyfluorine anions on the bandgaps, we firstly compare the electronic structures23 of RWOFI and CWOFI with K5(W3O9F4)(IO3), which possess similar compositions but exhibit different configurations near the Fermi level. K5(W3O9F4)(IO3) has isolated [IO3]− oxyanions with three terminal oxygen atoms that manifest as non-bonding O 2p orbitals in the electronic structure, while most oxygen atoms in the [IO2F2]− oxyfluorine anion of RWOFI and CWOFI are either further linked to [WO3F3]3− oxyfluorine anions or replaced by highly electronegative F−. The contributions of non-bonding O 2p orbitals in RWOFI and CWOFI are consequently much less important than those in K5(W3O9F4)(IO3). Secondly, in contrast to the interconnected tungsten-centered octahedra in K5(W3O9F4)(IO3), the [WO3F3] octahedra in RWOFI and CWOFI are isolated from each other, which leads to a narrow conduction band width and a significant increase in the bandgap.15 Thirdly, the bottom of the CB in conventional 3d0/4d0-TM iodates mainly consists of d0-TM empty d orbitals and O 2p orbitals; in contrast, the heavy 5d0-W6+ cation has a weaker orbital overlap with O2−/F− anions in RWOFI and CWOFI due to its relatively low effective electronegativity, which results in an upward shift of the bottom of the CB.12 The wide bandgaps observed in RWOFI and CWOFI can clearly be attributed to synergism between the [WO3F3]3− and [IO2F2]− oxyfluorine anions.
Based on the restriction of Kleinman's symmetry, there are three non-zero SHG tensors (d31, d32, d33) for RWOFI and CWOFI (listed in Table S7†). The absolute values of the largest tensor d31 are 1.59 and 1.85 pm /V for RWOFI and CWOFI, respectively, corresponding to approximately 4.1 and 4.7 times that of KDP (d36 = 0.39 pm V−1), and therefore consistent with the experimental values. The birefringences are calculated to be 0.166 (RWOFI) and 0.137 (CWOFI) @ 1064 nm. The shortest phase-matchable wavelength of RWOFI is ca. 262 nm (Fig. 4b), which is shorter than its UV cut-off edge of 280 nm. In comparison, the shortest phase-matchable wavelength of CWOFI is red-shifted to 302 nm (Fig. S7b†), which is attributed to its relatively small birefringence and large chromatic dispersion. These results are consistent with the phase-matching capability of the two crystalline materials shown experimentally at 1064 nm. A real-space atom-cutting analysis24 of the contributions of the constituent oxyfluorine anions [IO2F2]− and [WO3F3]3− to the NLO properties has been undertaken, the resultant data being listed in Table S7.† The [IO2F2]− oxyfluorine anions make the major contributions to the coefficient d31 (66.5 and 70.2%), the [WO3F3]3− oxyfluorine anions account for 27.1 and 27.3%, and the contributions from the alkali-metal cations (6.3 and 2.5%) are negligible, for RWOFI and CWOFI, respectively. In general, the SHG properties are closely related to the relevant virtual (electron and hole) excitations between the states near the Fermi level.25 A wide bandgap usually increases the difficulty of the virtual excitation, resulting in a weak SHG response, making the simultaneous engineering of a wide bandgap and a strong SHG response a key problem in the development of efficient NLO materials.26 SHG-weighted electron densities27 illustrated in Fig. 4c, d, S7c, and d† further confirm the dominant contributions of the 0D [WO2F3(IO2F2)]2− hetero-oxyfluorine anions electron clouds to the SHG responses. Since the SHG properties are closely related to the band structures, the excellent linear and nonlinear optical properties in RWOFI and CWOFI can be assigned to their unique band structures that result from the bandgap engineering by dual fluorination of the two types of SOJT distorted units.
Conclusions
In summary, the first examples of 5d0-TM fluoroiodates, A2WO2F3(IO2F2), have been successfully synthesized by employing a bandgap engineering strategy based on dual fluorination of two different oxyanions. Due to the optimized combination of two types of fluorinated SOJT distorted units, A2WO2F3(IO2F2) exhibit wide bandgaps (4.42 eV (RWOFI), 4.29 eV (CWOFI)), the largest of d0-TM iodates to date. They additionally possess strong SHG responses of 3.8 × KDP (RWOFI) and 3.5 × KDP (CWOFI) at 1064 nm, sufficient birefringence (0.166 (RWOFI) and 0.137 (CWOFI) at 1064 nm), and broad optical transparency windows. Theoretical calculations have elucidated that the wide bandgaps of the two hetero-oxyfluorides originate from collaboration of the [WO3F3]3− and [IO2F2]− oxyfluorine anions. This study provides an efficient bandgap engineering strategy for the development of high-performance NLO materials with an optimized balance of linear and nonlinear optical properties.Data availability
All of the related experimental and computational data are provided in the ESI.†Author contributions
Y. L. H. synthesized the crystals, performed the experiments, and wrote the draft. X. X. J. performed the theoretical calculations. T. H. W. and Y. Y. X. collected the data. W. C. performed SHG measurements and edited the manuscript. Z. P. H., Z. S. L. and M. G. H. reviewed the manuscript. J. X. provided methods for crystal growth. C. Z. supervised the process and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (no. 51432006, 52002276), the Ministry of Education of China for the Changjiang Innovation Research Team (no. IRT14R23), the Ministry of Education and the State Administration of Foreign Experts Affairs for the 111 Project (no. B13025), and the Innovation Program of Shanghai Municipal Education Commission. M. G. H. thanks the Australian Research Council for support (DP170100411).Notes and references
- D. N. Nikogosyan, Nonlinear Optical Crystals: A Complete Survey, Springer, Heidelberg, 2012 Search PubMed.
- (a) C. T. Chen, Y. Wang, B. Wu, K. Wu, W. Zeng and L. Yu, Nature, 1995, 373, 322–324 CrossRef CAS; (b) D. Cyranoski, Nature, 2009, 457, 953–955 CrossRef CAS PubMed; (c) L. Wu, S. Patankar, T. Morimoto, N. L. Nair, E. Thewalt, A. Little, J. G. Analytis, J. E. Moore and J. Orenstein, Nat. Phys., 2017, 13, 350–355 Search PubMed; (d) C. Wu, G. Yang, M. G. Humphrey and C. Zhang, Coord. Chem. Rev., 2018, 375, 459–488 CrossRef CAS.
- (a) K. M. Ok, Acc. Chem. Res., 2016, 49, 2774–2785 CrossRef CAS PubMed; (b) H. H. Cheng, F. M. Li, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2022, 61, e202115669 CAS; (c) G. Peng, N. Ye, Z. S. Lin, L. Kang, S. L. Pan, M. Zhang, C. S. Lin, X. F. Long, M. Luo, Y. Chen, Y. H. Tang, F. Xu and T. Yan, Angew. Chem., Int. Ed., 2018, 57, 8968–8972 CrossRef CAS PubMed; (d) P. S. Halasyamani and J. M. Rondinelli, Nat. Commun., 2018, 9, 2972 CrossRef PubMed; (e) C. Wu, T. H. Wu, X. X. Jiang, Z. J. Wang, H. Y. Sha, L. Lin, Z. S. Lin, Z. P. Huang, X. F. Long, M. G. Humphrey and C. Zhang, J. Am. Chem. Soc., 2021, 143, 4138–4142 CrossRef CAS PubMed.
- (a) G. H. Zou and K. M. Ok, Chem. Sci., 2020, 11, 5404–5409 RSC; (b) Z. Y. Bai, L. H. Liu, D. M. Wang, C. L. Hu and Z. B. Lin, Chem. Sci., 2021, 12, 4014–4020 RSC; (c) H. P. Wu, B. B. Zhang, H. W. Yu, Z. G. Hu, J. Y. Wang, Y. C. Wu and P. S. Halasyamani, Angew. Chem., Int. Ed., 2020, 59, 8922–8926 CrossRef CAS PubMed; (d) C. Wu, X. X. Jiang, Z. J. Wang, H. Y. Sha, Z. S. Lin, Z. P. Huang, X. F. Long, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 14806–14810 CrossRef CAS; (e) S. F. Li, X. M. Jiang, Y. H. Fan, B. W. Liu, H. Y. Zeng and G. C. Guo, Chem. Sci., 2018, 9, 5700–5708 RSC.
- (a) H. G. Kim, T. Tran, W. Choi, T. S. You, P. S. Halasyamani and K. M. Ok, Chem. Mater., 2016, 28, 2424–2432 CrossRef CAS; (b) T. L. Chao, W. J. Chang, S. H. Wen, Y. Q. Lin, B. C. Chang and K. H. Lii, J. Am. Chem. Soc., 2016, 128, 9061–9064 CrossRef PubMed; (c) W. G. Zhang, H. W. Yu, J. Cantwell, H. P. Wu, K. R. Poeppelmeier and P. S. Halasyamani, Chem. Mater., 2016, 28, 4483–4491 CrossRef CAS; (d) C. Wu, X. X. Jiang, L. Lin, Y. L. Hu, T. H. Wu, Z. S. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 22447–22453 CrossRef CAS PubMed.
- (a) H. W. Yu, J. Young, H. P. Wu, W. G. Zhang, J. M. Rondinelli and P. S. Halasyamani, J. Am. Chem. Soc., 2016, 138, 4984–4989 CrossRef CAS PubMed; (b) J. Chen, C. L. Hu, F. F. Mao, B. P. Yang, X. H. Zhang and J. G. Mao, Angew. Chem., Int. Ed., 2019, 58, 11666–11669 CrossRef CAS PubMed; (c) F. G. You, F. Liang, Q. Huang, Z. G. Hu, Y. C. Wu and Z. S. Lin, J. Am. Chem. Soc., 2019, 141, 748–752 CrossRef CAS PubMed; (d) J. Y. Chung, H. Jo, S. Yeon, H. R. Byun, T. S. You, J. I. Jang and K. M. Ok, Chem. Mater., 2020, 32, 7318–7326 CrossRef CAS; (e) C. Wu, L. H. Li, L. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Chem. Mater., 2020, 32, 3043–3053 CrossRef CAS.
- (a) P. S. Halasyamani and K. R. Poeppelmeier, Chem. Mater., 1998, 10, 2753–2769 CrossRef CAS; (b) J. Chen, C. L. Hu, Y. L. Lin, Y. Chen, Q. Q. Chen and J. G. Mao, Chem. Sci., 2022, 13, 454–460 RSC; (c) H. X. Fan, C. S. Lin, K. C. Chen, G. Peng, B. X. Li, G. Zhang, X. F. Long and N. Ye, Angew. Chem., Int. Ed., 2020, 59, 5268–5272 CrossRef CAS PubMed; (d) Y. N. Li, Z. X. Chen, W. D. Yao, R. L. Tang and S. P. Guo, J. Mater. Chem. C, 2021, 9, 8659–8665 RSC; (e) R. B. Fu, Q. R. Shui, Z. J. Ma, Z. Q. Zhou, H. X. Tang and X. T. Wu, Chem.–Eur. J., 2022, 28, e202103687 Search PubMed.
- (a) F. H. Ding, K. J. Griffith, C. P. Kocer, R. J. Saballos, Y. R. Wang, C. Zhang, M. L. Nisbet, A. J. Morris, J. M. Rondinelli and K. R. Poeppelmeier, J. Am. Chem. Soc., 2020, 142, 12288–12298 CrossRef CAS PubMed; (b) M. R. Marvel, J. Lesage, J. Baek, P. S. Halasyamani, C. L. Stern and K. R. Poeppelmeier, J. Am. Chem. Soc., 2007, 129, 13963–13969 CrossRef CAS PubMed; (c) H. Jo, M. H. Lee and K. M. Ok, Chem. Mater., 2021, 33, 1875–1882 CrossRef CAS; (d) G. Peng, Y. Yang, T. Yan, D. Zhao, B. X. Li, G. Zhang, Z. S. Lin and N. Ye, Chem. Sci., 2020, 11, 7396–7400 RSC; (e) S. Liu, X. M. Liu, S. G. Zhao, Y. C. Liu, L. N. Li, Q. R. Ding, Y. Q. Li, Z. S. Lin, J. H. Luo and M. C. Hong, Angew. Chem., Int. Ed., 2020, 59, 9414–9417 CrossRef CAS PubMed; (f) H. X. Tang, R. B. Fu, Z. J. Ma and X. T. Wu, Inorg. Chem., 2021, 60, 17364–17370 CrossRef CAS PubMed.
- (a) H. Y. Chang, S. H. Kim, K. M. Ok and P. S. Halasyamani, Chem. Mater., 2009, 21, 1654–1662 CrossRef CAS; (b) H. Y. Chang, S. W. Kim and P. S. Halasyamani, Chem. Mater., 2010, 22, 3241–3250 CrossRef CAS; (c) J. J. Zhang, Z. H. Zhang, Y. X. Sun, C. Q. Zhang, S. J. Zhang, Y. Liu and X. T. Tao, J. Mater. Chem., 2012, 22, 9921–9927 RSC; (d) R. E. Sykora, K. M. Ok, P. S. Halasyamani and T. E. Albrecht-Schmitt, J. Am. Chem. Soc., 2002, 124, 1951–1957 CrossRef CAS PubMed; (e) Y. H. Li, G. P. Han, H. W. Yu, H. Li, Z. H. Yang and S. L. Pan, Chem. Mater., 2019, 31, 2992–3000 CrossRef CAS; (f) C. F. Sun, C. L. Hu, X. Xu, B. P. Yang and J. G. Mao, J. Am. Chem. Soc., 2011, 133, 5561–5572 CrossRef CAS PubMed; (g) W. Zeng, X. H. Dong, Y. Tian, L. Huang, H. M. Zeng, Z. E. Lin and G. H. Zou, Chem. Commun., 2022, 58, 3350–3353 RSC.
- (a) Y. L. Hu, C. Wu, X. X. Jiang, Z. J. Wang, Z. P. Huang, Z. S. Lin, X. F. Long, M. G. Humphrey and C. Zhang, J. Am. Chem. Soc., 2021, 143, 12455–12459 CrossRef CAS PubMed; (b) C. Wu, X. X. Jiang, Z. J. Wang, L. Lin, Z. S. Lin, Z. P. Huang, X. F. Long, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 3464–3468 CrossRef CAS PubMed; (c) J. Chen, C. L. Hu, X. H. Zhang, B. X. Li, B. P. Yang and J. G. Mao, Angew. Chem., Int. Ed., 2020, 59, 5381–5384 CrossRef CAS PubMed.
- L. Kang, F. Liang, X. X. Jiang, Z. S. Lin and C. T. Chen, Acc. Chem. Res., 2020, 53, 209–217 CrossRef CAS PubMed.
- H. W. Eng, P. W. Barnes, B. M. Auer and P. M. Woodward, J. Solid State Chem., 2003, 175, 94–109 CrossRef CAS.
- (a) M. Mutailipu, M. Zhang, Z. H. Yang and S. L. Pan, Acc. Chem. Res., 2019, 52, 791–801 CrossRef CAS PubMed; (b) G. Q. Shi, Y. Wang, F. F. Zhang, B. B. Zhang, Z. H. Yang, X. L. Hou, S. L. Pan and K. R. Poeppelmeier, J. Am. Chem. Soc., 2017, 139, 10645–10648 CrossRef CAS PubMed.
- J. Lu, J. N. Yue, L. Xiong, W. K. Zhang, L. Chen and L. M. Wu, J. Am. Chem. Soc., 2019, 141, 8093–8097 CrossRef CAS PubMed.
- E. A. Axtell, Y. Park, K. Chondroudis and M. G. Kanatzidis, J. Am. Chem. Soc., 1998, 120, 124–136 CrossRef CAS.
- C. Wu, L. Lin, X. X. Jiang, Z. S. Lin, Z. P. Huang, M. G. Humphrey, P. S. Halasyamani and C. Zhang, Chem. Mater., 2019, 31, 10100–10108 CrossRef CAS.
- (a) L. Lin, X. X. Jiang, C. Wu, Z. S. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Chem. Mater., 2021, 33, 5555–5562 CrossRef CAS; (b) H. W. Yu, M. L. Nisbet and K. R. Poeppelmeier, J. Am. Chem. Soc., 2018, 140, 8868–8876 CrossRef CAS PubMed; (c) J. Chen, C. L. Hu, F. F. Mao, J. H. Feng and J. G. Mao, Angew. Chem., Int. Ed., 2019, 58, 2098–2102 CrossRef CAS PubMed.
- P. S. Halasyamani, Chem. Mater., 2004, 16, 3586–3592 CrossRef CAS.
- J. Chen, C. L. Hu, F. Kong and J. G. Mao, Acc. Chem. Res., 2021, 54, 2775–2783 CrossRef CAS PubMed.
- M. Q. Gai, T. H. Tong, Y. Wang, Z. H. Yang and S. L. Pan, Chem. Mater., 2020, 32, 5723–5728 CrossRef CAS.
- Q. Wu, H. M. Liu, F. C. Jiang, L. Kang, L. Yang, Z. S. Lin, Z. G. Hu, X. G. Chen, X. G. Meng and J. G. Qin, Chem. Mater., 2016, 28, 1413–1418 CrossRef CAS.
- (a) P. A. Maggard, T. S. Nault, C. L. Stern and K. R. Poeppelmeier, J. Solid State Chem., 2003, 175, 27–33 CrossRef CAS; (b) C. Wu, X. X. Jiang, L. Lin, W. Y. Dan, Z. S. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 27151–27157 CrossRef CAS PubMed.
- Z. S. Lin, X. X. Jiang, L. Kang, P. F. Gong, S. Y. Luo and M. H. Lee, J. Phys. D: Appl. Phys., 2014, 47, 253001 CrossRef.
- J. Lin, M. H. Lee, Z. P. Liu, C. T. Chen and C. J. Pickard, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 13380–13389 CrossRef CAS.
- R. He, Z. S. Lin, M. H. Lee and C. T. Chen, J. Appl. Phys., 2011, 109, 103510 CrossRef.
- K. Wu and S. L. Pan, Coord. Chem. Rev., 2018, 377, 191–208 CrossRef CAS.
- M. H. Lee, C. H. Yang and J. H. Jan, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235110 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2164004 and 2164005. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02137d |
| ‡ Y. L. Hu, X. X. Jiang, T. H. Wu and Y. Y. Xue contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |