
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalysed hydroalumination of internal alkynes†
Wen-Tao
Li
a,
Meng-Yang
Hu
a,
Jun-Wen
Xiong
a,
Xin-Yu
Zhang
a and
Shou-Fei
Zhu
 *ab
*ab
aFrontiers Science Center for New Organic Matter, State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China
bHaihe Laboratory of Sustainable Chemical Transformations, Tianjin 300192, China. E-mail: sfzhu@nankai.edu.cn
First published on 13th June 2022
Abstract
Although research on iron-catalysed reactions has recently achieved significant progress, the activity and selectivity of iron catalysts are generally inferior to those of noble-metal catalysts. The development of new iron-catalysed reactions, especially those in which iron catalysts exhibit superior activity or selectivity to other catalysts, is the key to promote iron catalysis. Herein, we report the first protocol for iron-catalysed hydroalumination of internal alkynes. Specifically, in the presence of iron catalysts bearing 2,9-diaryl-1,10-phenanthroline ligands, internal alkynes were stereo- and regioselectively hydroaluminated with the commercially available reagent diisobutylaluminum hydride. Compared with other metal-catalysed alkyne hydroalumination reactions reported in the literature, the iron-catalysed protocol has the following advantages: unusual amino-group-directed regioselectivity, a wide substrate scope, good functional group tolerance, high selectivity, and mild reaction conditions. The alkenylaluminum products prepared in this way could undergo a diverse array of transformations, and were used for the synthesis of bioactive compounds. The current study expands the scope of iron catalysis, provides a new efficient access to alkenylaluminum, discloses the origin of the superiority of iron catalysts, and thus may inspire further studies in related fields.
Introduction
Iron catalysis attracts intensive attention mainly because of two reasons. First, because iron, which is the most abundant transition metal in the Earth's crust, is inexpensive and biocompatible, iron-catalysed organic reactions meet the requirements for green and sustainable chemistry. Second, the abundant valence states and variable spin states of iron provide opportunities to develop new reactions. Although research on iron-catalysed reactions has recently achieved significant progress, the activity and selectivity of iron catalysts are generally inferior to those of noble-metal catalysts.1 The development of new iron-catalysed reactions, especially those in which iron catalysts exhibit superior activity or selectivity to other catalysts, is the key to promote iron catalysis.Our group has a long-standing interest in the study of iron-catalysed reactions.2 As part of this ongoing work, we herein report the first protocol for iron-catalysed hydroalumination reactions of alkynes with DIBAL-H (Scheme 1). By using 2,9-diaryl-1,10-phenanthroline iron complexes as catalysts, we realized the hydroalumination of a diverse array of internal alkynes with specificity for cis-addition and with high regioselectivity. By subsequently employing the rich chemistry of the alkenylaluminum products,3 we efficiently synthesized a number of functionalized and unfunctionalized trisubstituted alkenes. The following aspects of this protocol distinguish it from previously reported catalytic methods of alkyne hydroalumination.4–6 First, the chemo- and regioselectivity were markedly better than those for previously reported catalysts under identical reaction conditions. Second, the protocol exhibited good functional group tolerance: substrates with alkenyl, halogen, hydroxy, amino, and acetal groups were successfully hydroaluminated. Third, using this protocol, we realized unprecedented amino-directed hydroalumination of internal alkynes, which enabled, for the first time, the selective cis-addition of aluminum to the alkyl side of aryl alkyl alkynes. Fourth, we achieved highly selective hydroalumination of conjugated alkynes to generate various new types of conjugated alkenylaluminums.
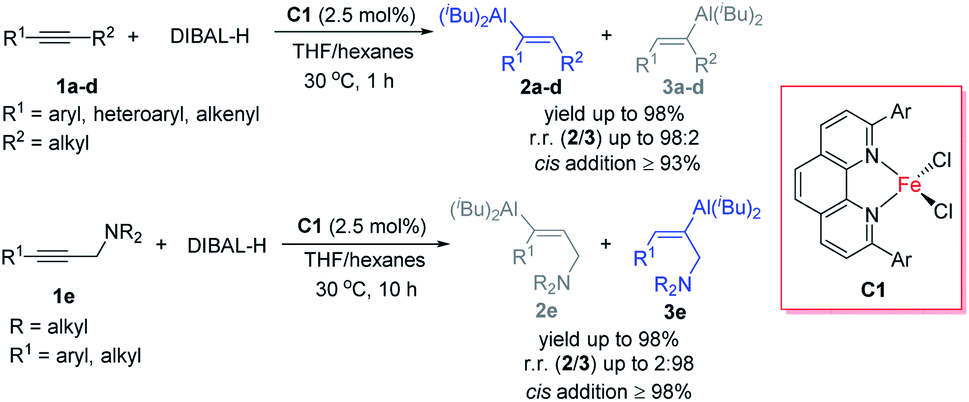 | ||
| Scheme 1 Iron-catalysed hydroalumination of internal alkynes. | ||
Results and discussion
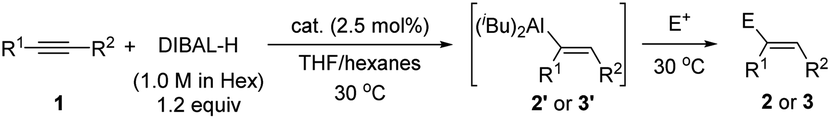
We initiated our study by using hex-1-yn-1-ylbenzene (1aa) as a model substrate and commercial DIBAL-H (1.0 M in hexanes) as the hydroalumination reagent (Table 1). Because of the high activity of the alkenylaluminum products, we determined the isolated yields and regioselectivities after hydrolysis with D2O. First, we investigated various catalysts. When we used some transition-metal catalysts reported in the literature for hydroalumination of other alkynes, the reactions proceeded smoothly to achieve full conversion of the alkyne 1aa, but the product yields and regioselectivities were unsatisfactory (entries 1–4). We attributed the poor yields mainly to two side reactions: over-reduction and double bond isomerization. Next, we explored various iron catalysts. Although FeCl2 exhibited low activity (entry 5), the addition of N(sp2) ligands markedly improved both the conversion and the regioselectivity. Systematic evaluation of ligands (entries 6–18) revealed that 2,9-diaryl-1,10-phenanthroline iron complexes effectively catalysed the reactions, with high selectivity for cis-addition products and moderate to high regioselectivity. When 2,9-dianthryl-substituted phenanthroline ligand L1g or 2,9-diphenanthrenyl-substituted phenanthroline ligand L1h was used, the regioselectivity reached 90% (entries 12 and 13). Because the coordinative THF may passivate DIBAL-H,7 the reaction without catalyst was totally inactive in the current study (entry 19).|
|
|||||
|---|---|---|---|---|---|
| Entrya | Cat. | Conv. (%) | Yield (%) | r.r. (2aa/3aa) | Z/E (2aa) |
| a Reaction conditions: 1aa (0.2 mmol), DIBAL-H (1.0 M in hexanes, 0.24 mmol), catalyst (2.5 mol%) in THF (1 mL) at rt (∼30 °C). Conversion, yield, r.r. (regioisomeric ratio), and Z/E were determined by 1H NMR with 1,3,5-trimethoxybenzene as an internal standard after quenching with D2O (30 °C, 30 min). ND, not detected. b 5 mol% catalyst was used. c The reaction was performed at rt and under reflux conditions. | |||||
| 1b | Cp2TiCl2 | 45 | 40 | 81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 :19 |
98:2 |
| 2b | Cp2ZrCl2 | 11 | 8 | 67:33 |
94:6 |
| 3b | Ni(PPh3)2Cl2 | 100 | 69 | 65:35 |
98:2 |
| 4b | Ni(dppp)Cl2 | 64 | 38 | 67:33 |
94:6 |
| 5 | FeCl2 | 7 | 5 | 56:44 |
88:12 |
| 6 | C1a | 26 | 21 | 75:25 |
98:2 |
| 7 | C1b | 52 | 36 | 76:24 |
98:2 |
| 8 | C1c | 99 | 69 | 69:31 |
98:2 |
| 9 | C1d | 100 | 94 | 73:27 |
97:3 |
| 10 | C1e | 100 | 84 | 71:29 |
98:2 |
| 11 | C1f | 100 | 97 | 85:15 |
98:2 |
| 12 | C1g | 100 | 81 |
91:9 |
97:3 |
| 13 | C1h | 100 | 91 |
90:10 |
98:2 |
| 14 | C1i | 70 | 54 | 88:12 |
87:13 |
| 15 | C2 | 30 | 21 | 65:35 |
87:13 |
| 16 | C3 | 11 | 5 | 55:45 |
92:8 |
| 17 | C4a | 100 | 94 | 69:31 |
97:3 |
| 18 | C4b | 81 | 48 | 60:40 |
94:6 |
| 19c | None | Trace | ND | ND | ND |
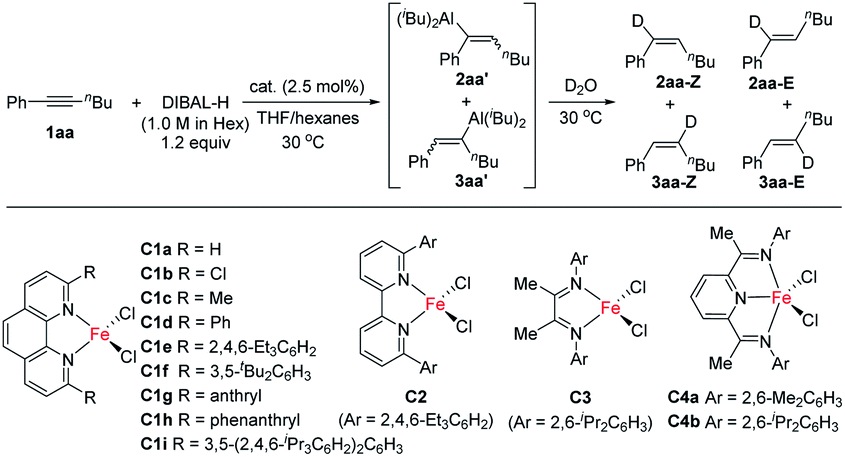
We also systematically evaluated other reaction parameters (ESI Tables S2–S7†). When the iron catalyst, the ligand, or FeCl2 was omitted under otherwise standard conditions (Table 1, entry 12), no reaction occurred, even at elevated temperature (Table S2†). More detailed ligand evaluation indicated that the monodentate phosphorus ligands, bidentate phosphorus ligands, and N-heterocyclic carbene ligands were inactive in the iron-catalysed alkyne hydroalumination; the other bidentate or tridentate N-ligands gave poor regioselectivities (Table S3†). These data clearly demonstrate the superiority of phenanthroline based ligands in this reaction. The conversion and regioselectivity were low when DIBAL-H was replaced with another hydroalumination reagent [Red-Al, LiAlH4, LiAlH(OtBu)3, HAlCl2·2THF, Al(iBu)3, Al(iBu)2Cl, or AlEt2Cl, Table S4†]. The addition of organometallic reagents or a base as an additive exhibited a negative impact on yields and selectivities (Table S5†). The choice of solvent had an obvious effect on the reaction outcome. Only THF and other ether solvents gave good results, while all the other tested solvents decreased both yield and the regioselectivity (Table S6†). We suspect that THF was involved in the formation of the C–Al bond. When iron was replaced with cobalt, nickel, copper, manganese, gold, and palladium the yield or regioselectivity of the reaction dropped considerably (Table S7†).
Under the optimal conditions (Table 1, entry 12), we systematically investigated the substrate scope of the reaction (Scheme 2). First, we studied the effect of the substituent on the phenyl group of various aryl alkyl alkynes 1. The tested substrates gave high yields of alkenes 2aa–2ap in high regioselectivity with aluminum added at the aryl side of the alkynes (r.r. = 87:13 to 96:4) and excellent stereoselectivity (Z/E = 93:7 to >98:2). Substituents at the meta and para positions of the phenyl ring had little effect on the yield or selectivity, but substrates with an electron-donating methoxy group gave the highest regioselectivities (2af and 2am). Substrates with ortho-substituents (2an–2ap) showed slightly better regioselectivities than most of the corresponding meta- or para-substituted compounds. When the phenyl ring was replaced with a fused aromatic ring system (naphthyl, 2ba), a heteroaromatic ring (2-furanyl, 2bb; 2-thiophenyl, 2bc; 5-indolyl, 2bd), or a ferrocenyl moiety (2be), the reaction also proceeded smoothly and afforded satisfactory results. Next, we investigated the effect of changing the alkyl group (R2) with the R1 group being a para-methoxyphenyl (2ca–2cf) or a phenyl (2cg–2cm), respectively. The steric bulk of this group had little effect on the yield and selectivity of the reaction, which indicates that the regioselectivity was determined mainly by the aryl groups of the alkynes.
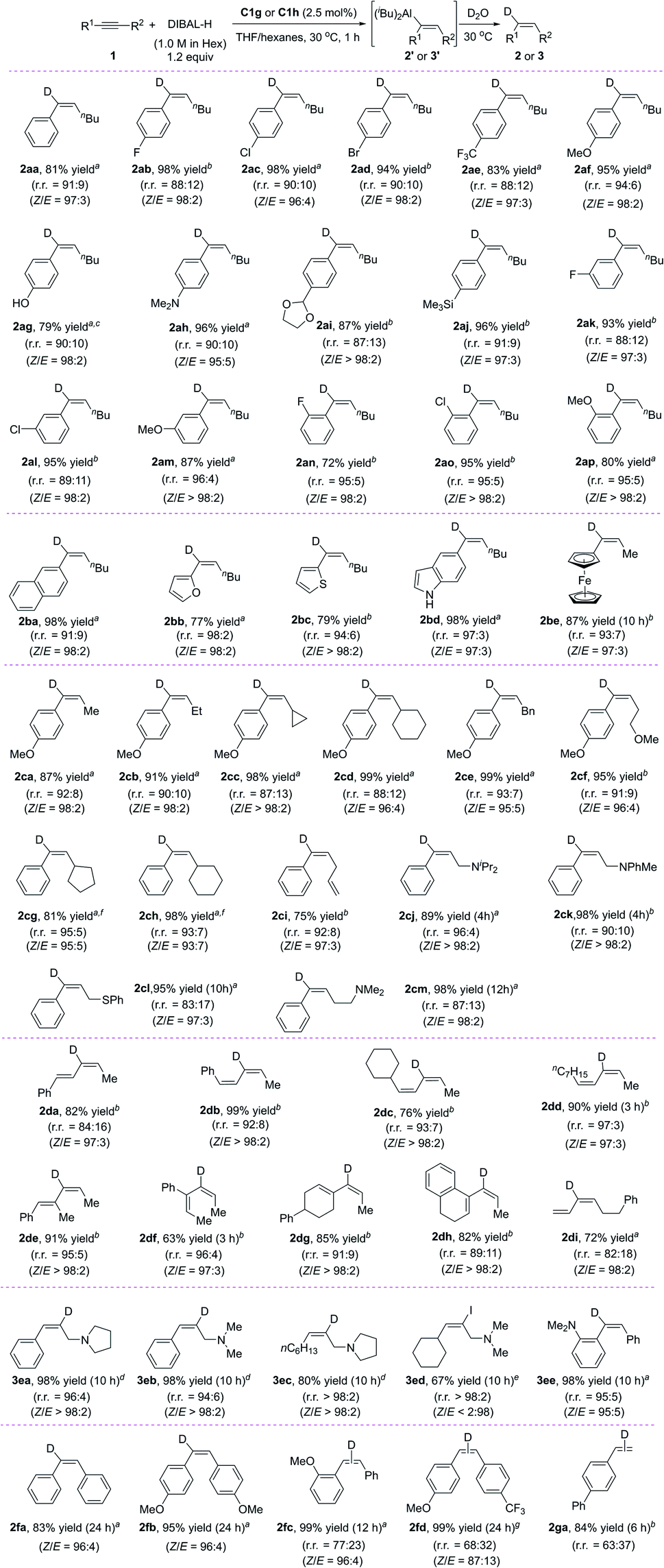 | ||
| Scheme 2 Iron-catalysed hydroalumination of internal alkynes: substrate scope. Reaction conditions: 1 (0.2 mmol), C1 (0.005 mmol, 2.5 mol%), DIBAL-H (1.0 M in hexanes, 0.24 mmol) in THF (1 mL) at 30 °C. Isolated yields were given. The r.r. and Z/E were determined by 1H NMR after quenching with D2O or I2 (30 °C, 30 min). aC1g was used as catalyst. bC1h was used as catalyst. c 2.2 equiv. of DIBAL-H were used. dC1i was used as catalyst. eC1f was used as catalyst, and quenched with I2. f HAlEt2 (1.0 M in toluene, 0.24 mmol) in toluene (1 mL) at 30 °C. | ||
The reaction conditions were compatible with a range of functional groups: fluoride (2ab, 2ak, 2an), chloride (2ac, 2al, 2ao), bromide (2ad), trifluoromethyl (2ae), methoxy (2af, 2am, 2ap, 2ca–2cf), hydroxy (2ag), acetal (2ai), amino (2ah, 2cj, 2ck, 2cm), silyl (2aj) and thiophenyl (2cl). However, the functional groups of ester, amide, and nitro groups at the phenyl of the alkyne failed to give the desired products (data not shown). Notably, the chemoselectivity of the iron catalyst was very good: reaction of a substrate bearing an alkenyl group (2ci) was selective for the triple bond, leaving the double bond untouched. Taking advantage of this feature, we synthesized a series of novel conjugated dienyl aluminums 2da′–2di′ from the corresponding conjugated enynes. As far as we know, the catalytic hydroalumination of conjugated enynes has not previously been reported. Noncatalytic hydroalumination reactions of conjugated enynes give poor yields8 or the products undergo further transformation under the reaction conditions.9
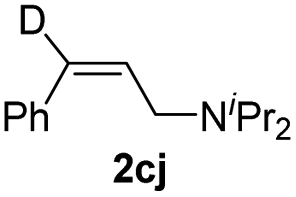
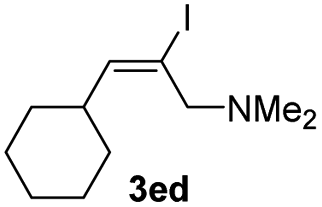
Interestingly, amino groups with little steric bulk and strong basicity could be used as directing groups to control the regioselectivity (3ea–3ee). For example, hydroalumination reactions of aryl- or alkyl-substituted propargylic amines bearing a tetrahydropyrrole or dimethylamino directing group specifically gave cis-addition products generated by addition of aluminum to the end of the triple bond attached to the amino group (3ea–3ee). This selectivity is completely different from that reported in the literature.5e,10 By using our hydroalumination protocol, we were able to add aluminum either to the alkyl end of aryl alkyl alkynes to afford 3ea and 3eb or to the amino-substituted alkyl end of alkyl alkyl alkynes to afford 3ec and 3ed. Similarly, by introducing a dimethylamino group at the ortho position of one of the aryl rings of a diaryl alkyne, we could selectively add aluminum to the amino-substituted aryl side of the triple bond to afford 3ee. Because a dimethylamino group can easily be transformed into other functional groups,11 this protocol is expected to be useful for the synthesis of alkenes with three different aryl substituents. Using C1g as the catalyst, we also evaluated several symmetric and non-symmetric diaryl alkynes (1fa–1fd). The reactions of symmetric diaryl internal alkynes afford cis-addition products (2fa and 2fb) with high yields. The reactions of asymmetric diaryl internal alkynes also gave cis-addition products (2fc and 2fd) with high yields; however, only moderate regioselectivities were observed. In addition, the reaction with a terminal alkyne afforded a cis-addition product (2ga) with good yield but moderate regioselectivity.
As we did for aryl alkyl alkyne 1aa (Table 1, entries 1–4), we evaluated some typical Ti, Zr, and Ni catalysts3–5 in hydroalumination reactions of some of the other typical internal alkynes used in this study, including a vinyl alkyne (1ci), amino alkynes (1cj and 1ed), and a conjugated alkenyl alkyne (1de), and we compared the results with those obtained with our protocol (Table 2). These control experiments clearly showed that the iron catalyst was superior from activity, regioselectivity, and stereoselectivity aspects, perhaps because of the unique steric and electronic structure of the phenanthroline iron complexes. These findings once again demonstrate that when an appropriate ligand is selected, iron catalysts can show activity and selectivity superior to that of other catalysts. Although the hydroalumination of alkynes with DIBAL-H could proceed in refluxing hexanes without catalysts,12 the non-catalytic reactions of the above alkynes with DIBAL-H in refluxing hexanes generally exhibited poor yields and regioselectivities with opposite stereoselectivities (trans-addition as the major) compared to the iron catalysis (Table 2, bottom column).
|
|
||||
|---|---|---|---|---|
| Cat. |
|
|
|
|
| Yield | Yield | Yield | Yield | |
| r.r. | r.r. | r.r. | r.r. | |
| Z/E | Z/E | Z/E | Z/E | |
| a Reaction conditions: 1 (0.2 mmol), catalyst (0.005 mmol, 2.5 mol%), DIBAL-H (1.0 M in hexanes, 0.24 mmol) in THF (1 mL) at 30 °C. Isolated yields were given. The r.r. and Z/E were determined by 1H NMR after quenching with D2O or I2 (30 °C, 30 min). [Fe] = C1g, C1h, or C1i, [Ti] = Cp2TiCl2, [Zr] = Cp2ZrCl2, [Ni1] = Ni(PPh3)2Cl2, [Ni2] = Ni(dppp)Cl2. ND = not detected. b Heated in hexanes at 50 °C for 12 h. | ||||
| [Fe] | 75% | 89% | 91% | 67% |
| 92:8 |
96:4 |
95:5 |
>98:2 |
|
| 97:3 |
>98:2 |
>98:2 |
>98:2 |
|
| [Ti] | 19% | ND | 17% | 63% |
| 83:17 |
64:36 |
71:29 |
||
| 98:2 |
98:2 |
98:2 |
||
| [Zr] | ND | 21% | ND | ND |
| 63:37 |
||||
| >98:2 |
||||
| [Ni1] | 93% | 30% | 79% | 60% |
| 64:36 |
23:77 |
51:49 |
98:2 |
|
| 97:3 |
65:35 |
81:19 |
84:16 |
|
| [Ni2] | 79% | 29% | 50% | 58% |
| 53:47 |
9:91 |
60:40 |
98:2 |
|
| 98:2 |
>98:2 |
95:5 |
60:40 |
|
| Noneb | Messy | 77% | 25% | 55% |
| 14:86 |
71:29 |
97:3 |
||
| <2:98 |
20:80 |
3:97 |
||


We were able to isolate alkenylaluminum–THF adduct 2aa′-THF in a glovebox and to determine its structure by NMR spectroscopy (Scheme 3a). In addition, 2aa′-THF could exchange the ligand with DMAP to generate alkenylaluminum–DMAP adduct 2aa′-DMAP, the structure of which was also determined by NMR spectroscopy.
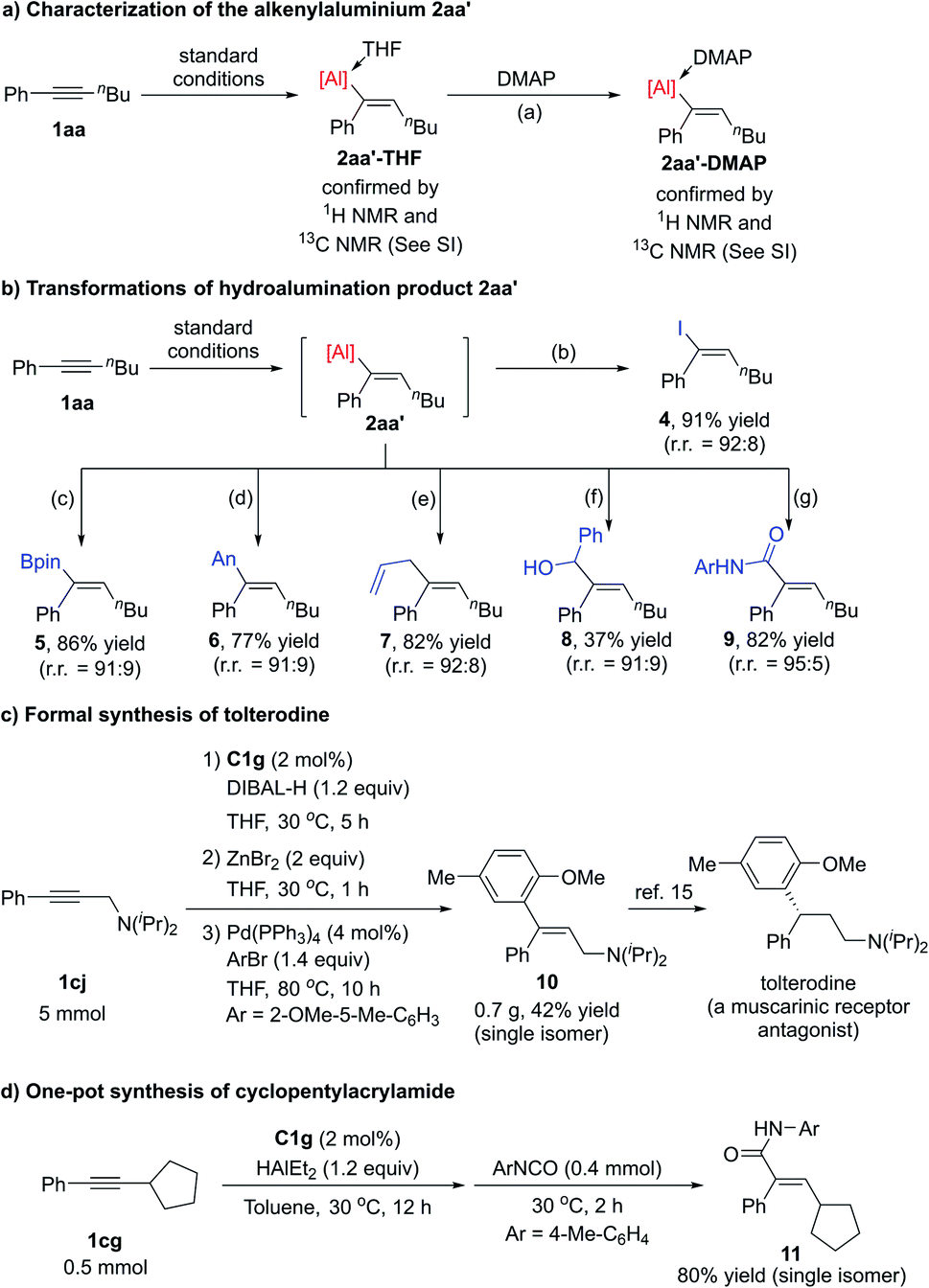 | ||
| Scheme 3 Characterization of a hydroalumination product and synthetic applications of iron-catalysed hydroalumination. Reaction conditions: a 1 equiv. of DMAP, 30 °C, 6 h. b 3 equiv. of I2, 30 °C, 2 h. c 3 equiv. of iPrOBpin, 80 °C, 24 h. d 1.1 equiv. of ZnBr2, 1 h, 30 °C, then 4 mol% Pd(PPh3)4, 1.2 equiv. of 4-bromoanisole, 80 °C, 10 h. e 5 mol% CuCl, 3 equiv. of allylbromide, 30 °C, 10 h. f 0.67 equiv. of PhCHO, 30 °C, 2 h. g 0.8 equiv. of ArNCO (p-tolyl isocyanate), 30 °C, 2 h. | ||
Alkenylaluminum compounds are useful because their C–Al bonds readily undergo a variety of transformations. To demonstrate the utility of our protocol, we synthesized a number of trisubstituted alkenes by means of a one-pot procedure that combined iron-catalysed hydroalumination of internal alkyne 1aa with functionalization of the C–Al bond of alkenylaluminum product 2aa′ (Scheme 3b). The alkenylaluminum 2aa′ could easily transform into trisubstituted vinyl iodine 4 by capturing with iodine, and into vinylboronate 5 by capturing with iPrOBpin. Alkenylaluminum 2aa′ could also be easily converted into a vinylzinc compound, which underwent the Negishi coupling with 4-bromoanisole to give trisubstituted alkene 6. A copper-catalysed allylation reaction of 2aa′ afforded diene 7.13 Alkenylaluminum 2aa′ could be transformed into allylic alcohols 8 by carbonyl addition reactions with PhCHO and could be captured by p-tolyl isocyanate to afford α,β-unsaturated alkenylamide 9.14 In all these transformations, the olefin configurations were maintained.
We also successfully used this hydroalumination protocol for the formal synthesis of a pharmaceutical compound (Scheme 3c). A C1g-catalysed gram-scale hydroalumination of propargylic amine 1cj following by trapping of the alkenylaluminum product with ZnBr2 afforded trisubstituted vinylzinc, which underwent the Negishi coupling in one pot to generate a single isomer of trisubstituted allylic amines 10 in 42% overall yield. The alkene 10 is a key intermediate in the synthesis of the antimuscarinic drug tolterodine.15 We further prepared cyclopentylacrylamide 11 in one pot with high overall yield as a single isomer through the iron-catalysed hydroalumination of internal alkyne 1cg and a following addition with isocyanate (Scheme 3d). The acrylamide 11 could be transformed into a glucokinase activator according to the reported procedure.16
It is worth mentioning that, compared with the synthetic routes reported in the literature, our hydroalumination protocol is more effective in synthesis of α,β-unsaturated alkenylamides and trisubstituted iodoallylamine with defined configuration (Scheme 4). The α,β-unsaturated alkenylamide is a useful building block for many bioactive compounds.16 The synthetic route to these compounds starting with ethyl propiolate comprises a four-step process of carbometalation, iodization, cross coupling, and amidation. In contrast, the iron-catalysed hydroalumination of easily available aryl-alkyl acetylene followed by addition of an isocyanate enabled a one-pot method to these important compounds (Scheme 4a). The trisubstituted iodoallylamine with defined configuration could be used to synthesize bioactive compounds, including the natural toxin product allopumiliotoxin-267A.17 However, the synthesis route of iodoallylamines in the literature involves multiple transformations of hydrostannation, iodization, reduction, bromination, and amination. The use of toxic organotin reagent and poor overall yield make this route less efficient. However, the protocol developed in this study enables a one-pot synthesis of trisubstituted iodoallylamine with high yield and high regioselectivity (Scheme 4b).
 | ||
| Scheme 4 Synthetic advantages of this protocol. | ||
We next conducted a series of control experiments to gain some insight into the catalytic mechanism (Scheme 5). The reaction of alkyne 1aa with DIBAL-D afforded deuterated products 2aa and 3aa in 98% yield (Scheme 5a), which clearly shows that the hydride of DIBAL-H other than the hydrogen of the isobutyl group transfers to the product. When HAlEt2 was used, the chemo- and regioselectivity are similar to those of the reaction of DIBAL-H (Scheme 5b), which implies that the Al component might not be involved in the selectivity-determining step. The deuterium labelling experiment using DAlEt2 gave similar results to those of DIBAL-D (Scheme 5c). The inverse KIE (kH/kD = 0.33) obtained in the competition experiments between DAlEt2 and HAlEt2 (Scheme 5d) indicates that the C–H bond formation is likely a fast-reversible step. At the end of the reaction of 1aa and DIBAL-H, the addition of another internal alkyne 1af could not afford crossover product 2af (Scheme 5e), indicating that the whole reaction is irreversible. The addition of a radical scavenger, 1,1-diphenylethylene, had a neglectable effect on the outcomes of the model reaction of 1aa and DIBAL-H (Scheme 5f), indicating that a radical pathway might not be an operative pathway.
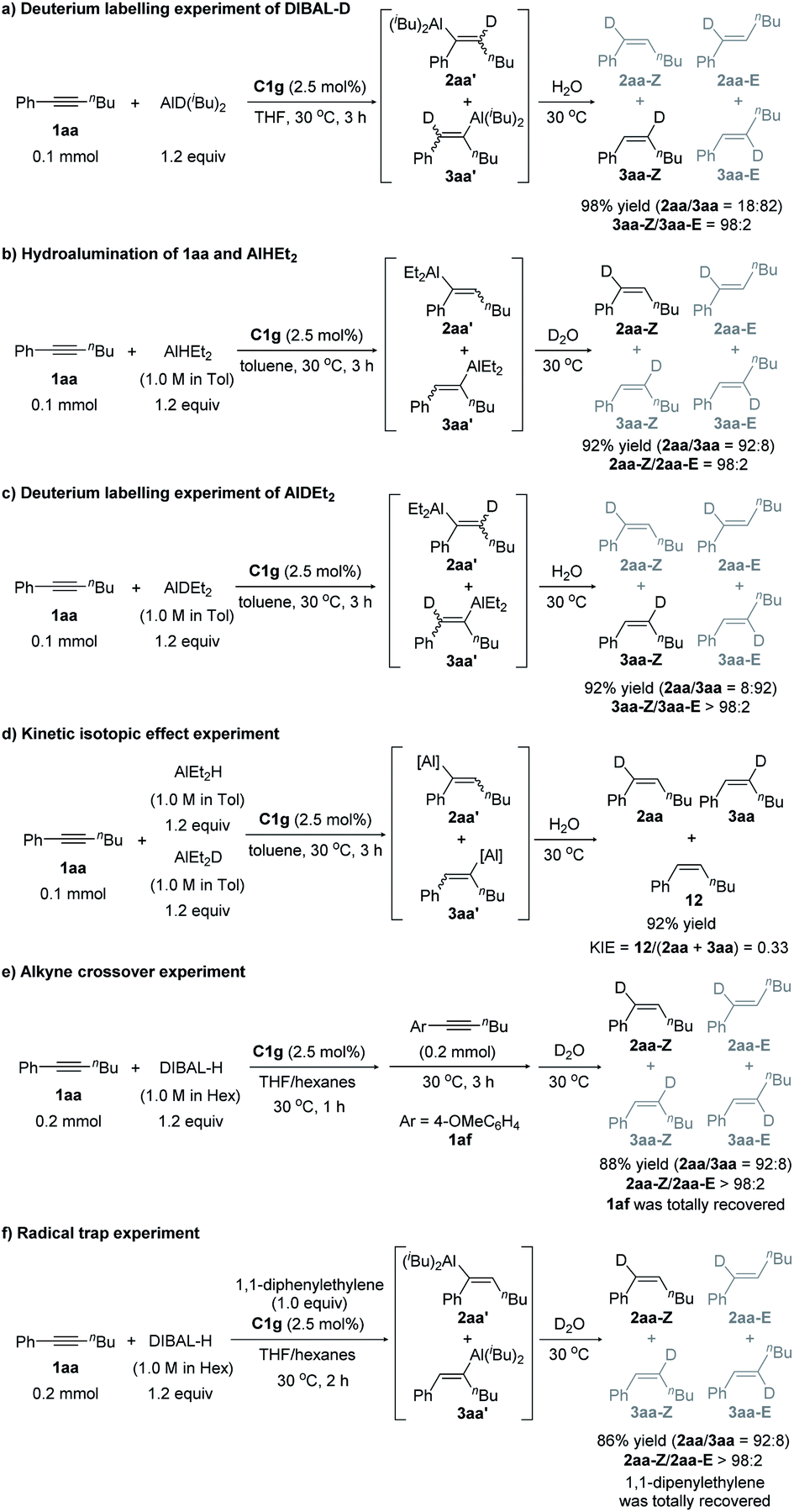 | ||
| Scheme 5 Control experiments. | ||
Based on the above control experiments and by analogy to the related literature,1,2 we proposed a catalytic cycle of iron-catalysed internal alkyne hydroalumination as shown in Scheme 6. Active Fe(II)–H complex I is generated by reducing a 2,9-diaryl-1,10-phenanthroline iron(II) complex with DIBAL-H through a similar pathway to that reported in the literature using other iron complexes.18 The migratory insertion of the C–C triple bond of alkyne 1aa into the Fe–H bond through a transition state (TS) affords the alkenyl iron intermediate III. At the same time, the cis-addition can be expected in this inner-sphere migratory insertion step, which determines the stereoselectivity of the resulting alkene. The following transmetalation of alkenyl iron intermediate III with DIBAL-H produces the desired hydroalumination product and regenerated Fe–H catalyst I. The fact that the two aluminum hydride reagents, DIBAL-H and HAlEt2, give almost the same regional selectivity (Scheme 5b) indicates that the aluminum reagent might not participate in the step of hydride transfer (II–III). The remarkable ether solvent effect on the regioselectivity (Table S5†) indicates that these solvents might participate in the hydride transfer step by coordinating with the iron catalyst. Similar to our previous finding in an iron-catalysed hydrosilylation of olefins,2g there may be significant π–π interaction between the backbone of the 1,10-phenanthroline ligand and the aryl of the alkyne substrate, which leads the hydride to transfer to the alkyl substituent side of alkynes (as shown in the II-A model), and results in the observed regioselectivity. For the reactions with propargylic amines bearing a tetrahydropyrrole or dimethylamino as a directing group, the iron catalyst may coordinate with the amino group, direct the hydride transfer to the triple bond, and exhibit a unique regioselectivity (as shown in the II-B model). However, we cannot rule out the other pathways, including an Fe(0)–Fe(II) catalytic cycle, at the current stage. The more detailed mechanistic studies are still underway in our laboratory.
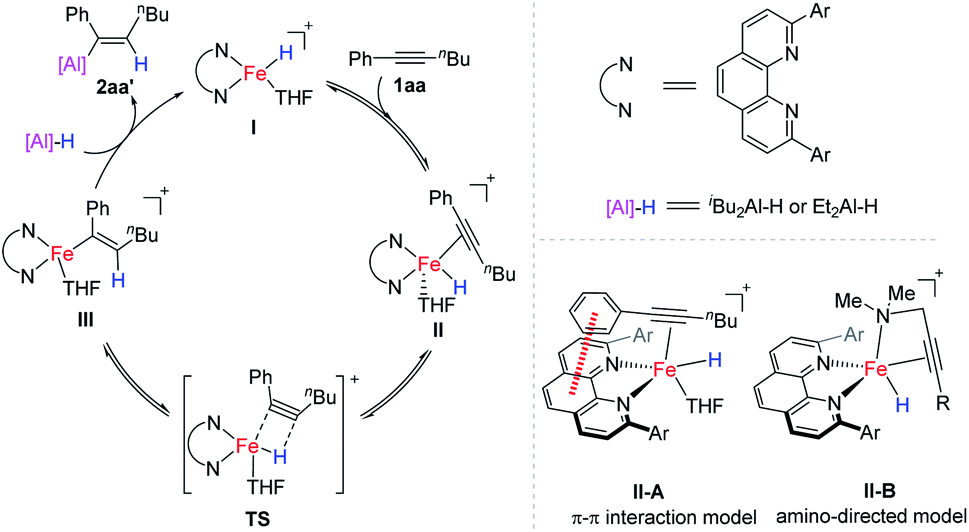 | ||
| Scheme 6 A plausible catalytic cycle and regioselectivity determining models. | ||
Conclusions
In conclusion, we have described herein the first protocol for iron-catalysed hydroalumination of internal alkynes. This mild, efficient, highly selective protocol is operationally simple and has a wide substrate scope and good functional group tolerance. Using commercially available DIBAL-H as the hydroalumination reagent and readily available alkynes as substrates, we synthesized various new alkenylaluminum compounds, which could be easily converted to trisubstituted alkenes, including key intermediates in the synthesis of pharmaceutical compounds. Compared with catalysts reported in the literature, the iron catalysts developed in this study show superior activity, chemoselectivity, and regioselectivity, and they enabled unprecedented amino-directed hydroalumination reactions of internal alkynes. Our findings clearly demonstrate the potential of iron catalysts not only to replace precious metals but also, more importantly, to show superior performance.Author contributions
S.-F. Z. supervised this study. S.-F. Z. and W.-T. L. conceived this work, designed the experiments, analyzed the data, and wrote the manuscript. W.-T. L., M.-Y. H., J.-W. X. and X.-Y. Z. conducted the experiments or analyzed the data. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the National Key R&D Program of China (2021YFA1500200), National Natural Science Foundation of China (21971119, 92156006, and 22001129), the “111” project (B06005) of the Ministry of Education of China, and Haihe Laboratory of Sustainable Chemical Transformations for financial support.Notes and references
- (a) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (b) I. Bauer and H. J. Knolker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (c) A. Fürstner, ACS Cent. Sci., 2016, 2, 778 CrossRef PubMed; (d) R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086 CrossRef CAS PubMed; (e) Q.-M. Liang and D.-T. Song, Chem. Soc. Rev., 2020, 49, 1209 RSC.
- (a) Q. Huang, Y.-X. Su, W. Sun, M.-Y. Hu, W.-N. Wang and S.-F. Zhu, J. Am. Chem. Soc., 2022, 144, 515 CrossRef CAS PubMed; (b) Q. Huang, W.-N. Wang and S.-F. Zhu, ACS Catal., 2022, 12, 2581 CrossRef CAS; (c) W. Sun, M.-P. Li, L.-J. Li, Q. Huang, M.-Y. Hu and S.-F. Zhu, Chem. Sci., 2022, 13, 2721 RSC; (d) S.-F. Zhu, Chin. J. Chem., 2021, 39, 3211 CrossRef CAS; (e) M.-Y. Hu, P. He, T.-Z. Qiao, W. Sun, W.-T. Li, J. Lian, J.-H. Li and S.-F. Zhu, J. Am. Chem. Soc., 2020, 142, 16894 CrossRef CAS PubMed; (f) M.-Y. Hu, J. Lian, W. Sun, T.-Z. Qiao and S.-F. Zhu, J. Am. Chem. Soc., 2019, 141, 4579 CrossRef CAS PubMed; (g) M.-Y. Hu, Q. He, S.-J. Fan, Z.-C. Wang, L.-Y. Liu, Y.-J. Mu, Q. Peng and S.-F. Zhu, Nat. Commun., 2018, 9, 221 CrossRef PubMed; (h) H. Xu, Y.-P. Li, Y. Cai, G.-P. Wang, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 7697 CrossRef CAS PubMed.
-
(a)
M. Dahlmann and M. Lautens, Catalytic Heterofunctionalization, Wiley-VCH Verlag GmbH, 2001, vol. 2, pp. 47–73 Search PubMed;
(b)
A. C. M. Baroni, C. C. P. Arruda and D. B. Carvalho, Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd, 2016, pp. 2–23 Search PubMed;
(c)
M. Zaidlewicz, A. Wolan and M. Budny, Hydrometallation of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C
C and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C Bonds, Elsevier Ltd, 2014, vol. 8.24.2, pp. 937–948 Search PubMed;
(d) U. M. Dzhemilev and A. G. Ibragimov, Russ. Chem. Rev., 2000, 69, 121 CrossRef CAS;
(e) J.-H. Chen, J. Guo and Z. Lu, Chin. J. Chem., 2018, 36, 1075 CrossRef CAS.
C Bonds, Elsevier Ltd, 2014, vol. 8.24.2, pp. 937–948 Search PubMed;
(d) U. M. Dzhemilev and A. G. Ibragimov, Russ. Chem. Rev., 2000, 69, 121 CrossRef CAS;
(e) J.-H. Chen, J. Guo and Z. Lu, Chin. J. Chem., 2018, 36, 1075 CrossRef CAS. - For Ni catalysts, see: (a) J. J. Eisch and M. W. Foxton, J. Organomet. Chem., 1968, 12, 33 CrossRef; (b) F. Gao and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10961 CrossRef CAS PubMed.
- For Ti catalysts, see: (a) E. Winterfeldt, Synthesis, 1975, 9, 617 CrossRef; (b) E. C. Ashby and S. A. Noding, Tetrahedron Lett., 1977, 18, 4579 CrossRef; (c) E. C. Ashby and S. R. Noding, J. Organomet. Chem., 1979, 177, 117 CrossRef CAS; (d) E. C. Ashby and S. A. Noding, J. Org. Chem., 1980, 45, 1035 CrossRef CAS; (e) R.-H. Sun, J. Liu, S. Yang, M. Chen, N. Sun, H.-Y. Chen, X. Xie, X. You, S. Li and Y.-H. Liu, Chem. Commun., 2015, 51, 6426 RSC.
- For Zr catalysts, see: T. Taapken and S. Blechert, Tetrahedron Lett., 1995, 36, 6659 CAS.
- J. J. Eisch and S.-G. Rhee, J. Am. Chem. Soc., 1974, 96, 7276 CrossRef CAS.
- I. Knox, S.-C. Chang and A. H. Andrist, J. Org. Chem., 1977, 42, 3981 CrossRef CAS.
- H. Kinoshita, T. Ishikawa and K. Miura, Org. Lett., 2011, 13, 6192 CrossRef CAS PubMed.
- W. Granitzer and A. Stütz, Tetrahedron Lett., 1979, 20, 3145 CrossRef.
- B. Xu, M.-L. Li, X.-D. Zuo, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2015, 137, 8700 CrossRef CAS PubMed.
- (a) J. J. Eisch and W. C. Kaska, J. Am. Chem. Soc., 1966, 88, 2213 CrossRef CAS; (b) G. Zweifel and J. A. Miller, Org. React., 1984, 32, 375 CAS.
- R. A. Lynd and G. Zweifel, Synthesis, 1974, 9, 658–659 CrossRef.
- H. Lee, S. Cho, Y. Lee and B. Jung, J. Org. Chem., 2020, 89, 12024 CrossRef PubMed.
- Z. Wu, S. D. Laffoon and K. L. Hull, Nat. Commun., 2018, 9, 1185 CrossRef PubMed.
- (a) W. L. Corbett, R. Sarabu and A. Sidduri, WO2001044216, 2001; (b) Y. Fukuda, Y. Asahina, M. Takadoi and M. Yamamoto, WO2009133687, 2009.
- (a) S. Aoyagi, T.-C. Wang and C. Kibayashi, J. Am. Chem. Soc., 1993, 115, 11393 CrossRef CAS; (b) B. Wang, Z. Zhong and G.-Q. Lin, Org. Lett., 2009, 11, 2011 CrossRef CAS PubMed.
- (a) J. M. Smith, R. J. Lachicotte and P. L. Holland, J. Am. Chem. Soc., 2003, 125, 15752 CrossRef CAS PubMed; (b) Y. Yu, A. R. Sadique, J. M. Smith, T. R. Dugan, R. E. Cowley, W. W. Brennessel, C. J. Flaschenriem, E. Bill, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2008, 130, 6624 CrossRef CAS PubMed; (c) M. Oishi, T. Endo, M. Oshima and H. Suzuki, Inorg. Chem., 2014, 53, 5100 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02160a |
| This journal is © The Royal Society of Chemistry 2022 |