
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing CO2 electroreduction to CH4 over Cu nanoparticles supported on N-doped carbon†
Yahui
Wu
ab,
Chunjun
Chen
 *ab,
Xupeng
Yan
ab,
Ruizhi
Wu
ab,
Shoujie
Liu
c,
Jun
Ma
a,
Jianling
Zhang
a,
Zhimin
Liu
ab,
Xueqing
Xing
f,
Zhonghua
Wu
f and
Buxing
Han
*abde
*ab,
Xupeng
Yan
ab,
Ruizhi
Wu
ab,
Shoujie
Liu
c,
Jun
Ma
a,
Jianling
Zhang
a,
Zhimin
Liu
ab,
Xueqing
Xing
f,
Zhonghua
Wu
f and
Buxing
Han
*abde
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: hanbx@iccas.ac.cn; chenchunjun@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cChemistry and Chemical Engineering of Guangdong Laboratory, Shantou 515063, China
dPhysical Science Laboratory, Huairou National Comprehensive Science Center, Beijing 101400, China
eShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
fInstitute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
First published on 5th July 2022
Abstract
The electroreduction of CO2 to CH4 has attracted extensive attention. However, it is still a challenge to achieve high current density and faradaic efficiency (FE) for producing CH4 because the reaction involves eight electrons and four protons. In this work, we designed Cu nanoparticles supported on N-doped carbon (Cu-np/NC). It was found that the catalyst exhibited outstanding performance for the electroreduction of CO2 to CH4. The FE toward CH4 could be as high as 73.4% with a high current density of 320 mA cm−2. In addition, the mass activity could reach up to 6.4 A mgCu−1. Both experimental and theoretical calculations illustrated that the pyrrolic N in NC could accelerate the hydrogenation of *CO to the *CHO intermediate, resulting in high current density and excellent selectivity for CH4. This work conducted the first exploration of the effect of N-doped species in composites on the electrocatalytic performance of CO2 reduction.
Introduction
The electrochemical CO2 reduction reaction (CO2RR) is a promising approach to achieve carbon neutrality,1–6 which can convert CO2 into valuable chemicals and fuels using renewable energy.7–11 Among all these products, CH4 is a highly desired product because it holds the highest heating value of 55.5 MJ kg−1.12 However, CO2 electroreduction to CH4 still suffers from high overpotential and low activity and selectivity.13–16 Designing highly efficient and robust electrocatalysts is crucial to solve this problem.Cu-based catalysts have been proven to be the most promising electrocatalysts for producing hydrocarbon products from the CO2RR. In recent years, many methods have been applied to enhance the activity of the CO2RR over Cu-based catalysts, including alloying,17 doping,18,19 modifying with other compounds,20,21 changing the shape and size,22–24 and building an interface and defects.25–27 However, it is still challenging to achieve high selectivity for CH4 at a high current density.28–33 This is because the generation of CH4 involves eight electrons and four protons, which easily bifurcates to give broad product distributions.34–36 According to a previous report,37 the co-adsorption of *CO and *H played an important role in the production of CH4, and the selectivity of CH4 can be enhanced by a high surface *H coverage, due to sufficient *H supply for the hydrogenation of intermediates.37 However, the high surface *H coverage could result in the undesired hydrogen evolution reaction (HER) and hinder the adsorption of intermediates. Thus, it is necessary to find a method to break the linear scale relationship of the single catalyst sites, which will enhance the selectivity of CH4 and decrease the yield of H2 simultaneously.
To solve the above problem, here we proposed the idea to introduce additional catalytic sites, which can enhance the activation of H2O and hydrogenation of intermediates but not cause excessive production of H2. According to a previous report, N-doped carbon (NC) catalysts could promote the activation of H2O.38–40 In addition, the adsorption of *H can be regulated by changing the type of N-doped species.41–43 Thus we can assume that NC would be a potential platform for tuning the activity and selectivity of Cu-based catalysts. Although NC has been used to modify Cu-based catalysts, the products were ethanol and ethylene.41 In addition, the role of the N-doped species in the composites is not clear.
Herein, we used NC with different contents of N-doped species as another component to modify Cu nanoparticles (Cu-np/NC). In this strategy, the selectivity of CH4 over Cu-np could be enhanced by introducing NC with rich pyrrolic N species, and the faradaic efficiency (FE) of CH4 could reach up to 73.4% with a current density of 320 mA cm−2. Especially, the mass activity reached up to 6.4 A mgCu−1. An in situ surface enhanced Raman spectroscopy (SERS) study demonstrated that the formation of the *CHO intermediate could be promoted over Cu-np/NC, which is an important intermediate for producing CH4. Experimental and DFT studies indicated that the pyrrolic N in NC could accelerate the hydrogenation of intermediates, resulting in excellent selectivity for CH4.
Results and discussion
Cu-np and NC were prepared respectively according to previous reports.44,45 The size of the obtained Cu-np was about 40 nm (Fig. S1†). NC(x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y) was prepared using arginine and melamine,45 and x:y represents the ratio of arginine and melamine in the preparation process. All of the NC(x:y) exhibited a nanosheet morphology (Fig. S2†). Cu-np/NC(x:y) was prepared by blending Cu-np and NC(x:y) (see the Methods for experimental details).
:y) was prepared using arginine and melamine,45 and x:y represents the ratio of arginine and melamine in the preparation process. All of the NC(x:y) exhibited a nanosheet morphology (Fig. S2†). Cu-np/NC(x:y) was prepared by blending Cu-np and NC(x:y) (see the Methods for experimental details).
The electrocatalytic performance of Cu-np/NC(1:4) was first studied, and the other Cu-np/NC(x:y) catalysts will be studied in the following section. The electrocatalytic performance of the CO2RR was evaluated in a flow cell, as reported in our previous report.34,41 The catalysts were sprayed on hydrophobic carbon paper as the cathode, and Ni foam was used as the anode.34,41,46,47 1 M KOH solution was used as the electrolyte. The gaseous and liquid products were analyzed by gas chromatography (GC) and nuclear magnetic resonance (NMR) spectroscopy, respectively.
Over Cu-np, CO2 could be reduced to various products, such as CO, CH4, C2H4 and C2H5OH (Fig. 1A and S3†). However, the selectivity of a single product is low (less than 45%). It is noted that the selectivity of CH4 was significantly enhanced by adding NC(1:4) and increased with the content of NC(1:4) (Fig. 1A and S4†). The selectivity of CH4 reached the highest when the content of NC(1:4) was 99 wt% (Cu-np/NC(1:4, 99 wt%)). The FE of CH4 could reach up to 73.4%, which is much higher than that of Cu-np. When the content of NC(1:4) was 99.5 wt%, the selectivity of CH4 decreased with increasing yield of H2, which may have originated from the insufficient Cu sites. For NC(1:4), only small amounts of CO were detected (Fig. S5†). Consequently, it can be assumed that the Cu sites were the active sites and the selectivity of CH4 was enhanced by the addition of NC(1:4). The partial current density of CH4 over Cu-np/NC(1:4, 99 wt%) was 234 mA cm−2 at −1.1 V vs. RHE, which is about 4.1 times higher than that over Cu-np (Fig. S6†). In the meantime, the CH4-to-other ratio was enhanced from 0.25 on Cu-np to 20.2 on Cu-np/NC(1:4, 99 wt%) (Fig. S7†), indicating that the selectivity of CH4 was enhanced and the other products were suppressed. Compared with the state-of-the-art catalysts, Cu-np/NC(1:4, 99 wt%) performed as one of the best catalysts in FE, current density and overpotential for CH4 (Fig. 1B and Table S1†). In addition, based on such a low content (1 wt%) of Cu-np in Cu-np/NC(1:4, 99 wt%), the mass activity could be as high as 6.4 A mgCu−1. In the following discussion, the content of NC was fixed at 99 wt% in Cu-np/NC(x:y).
 | ||
| Fig. 1 (A) The distribution of products at −1.1 V vs. RHE over Cu-np and Cu-np/NC(1:4). (B) Comparison of the overpotential (η), FE and CH4 partial current density of Cu-np/NC(1:4, 99 wt%) with those of state-of-the-art Cu-based catalysts. (C) The distribution of products over Cu-np/NC(1:4, 99 wt%) at different cell voltages in a MEA system. (D) The current density and FE of CH4 on Cu-np/NC(1:4, 99 wt%) at −4 V in 50 hour potentiostatic electrolysis tests. | ||
The electrolysis of the CO2RR was also carried out using membrane electrode assembly-based reactors (Fig. S8†). Cu-np/NC(1:4) was used as the cathode catalyst for the CO2RR and an iridium oxide-based catalyst as the anode for the oxygen evolution reaction. The selectivity of CH4 was 60% with a current density of 230 mA cm−2 at a cell voltage of −4 V (Fig. 1C and S9†). Furthermore, the stability of Cu-np/NC(1:4) was investigated at a cell voltage of −4 V. H2O was injected into the cathode flow channel to prevent salt accumulation in the gas diffusion layer (GDL) micropores. The selectivity of CH4 and the current density had no obvious change for 50 h (Fig. 1D), indicating that Cu-np/NC(1:4) exhibited excellent stability.
The electrocatalytic performance of the CO2RR over other Cu-np/NC(x:y) was also evaluated in a flow cell. As shown in Fig. S10,† the selectivity of CH4 over Cu-np/NC(x:y) varied with the x:y value. The highest FEs of CH4 over Cu-np/NC(1:2) and Cu-np/NC(1:8) were 65.8% and 59.6% respectively. Thus, we can deduce that NC(x:y) played an important role in the selectivity of CH4. According to a previous report,45 the intrinsic properties of NC were mainly attributed to the N-doped species. The correlation between CH4 selectivity and the type of N-doped species was investigated (Fig. S11, S12 and Table S2†). The FE of CH4 increased with increasing pyrrolic N content, whereas no regularity can be found for pyridinic N and graphitic N. These results indicated that the pyrrolic N in the NC(x:y) may play a crucial role in the enhancement of selectivity of CH4.
In addition, the electrochemically active surface areas (ECSAs) and Nyquist plots of Cu-np/NC(x:y) were measured. The charge transfer resistance (Rct) for the different Cu-np/NC(x:y) was similar (Fig. S13†). Although the ECSAs of Cu-np/NC(x:y) varied slightly with the different NC(x:y) (Fig. S14†), the normalized partial current densities for CH4 by ECSAs were similar to the geometric partial current density (Fig. S15†). These results indicated that different CO2RR performances of Cu-np/NC(x:y) with different x:y values were not originated from the slight change of the Rct and ECSAs.
Cu-np/NC(1:4) was characterized by transmission electron microscopy (TEM), and we can observe that Cu-np was dispersed on NC(1:4), as shown in Fig. 2A and B. In addition, the lattice distance of Cu(111) was observed by high-resolution transmission electron microscopy (HR-TEM) (Fig. 2C), which is consistent with that in Cu-np. However, the characteristic peaks of Cu cannot be observed on Cu-np/NC(1:4) in XRD patterns, and this is because the content of Cu is too low (Fig. S16†).
 | ||
| Fig. 2 (A and B) The TEM images of Cu-np/NC(1:4). (C) The HR-TEM image of Cu-np/NC(1:4). (D) The operando XANES spectra at the Cu K-edge for Cu-np and Cu-np/NC(1:4) at −0.7 V vs. RHE during the CO2RR. (E) The corresponding Fourier transform FT(k3w(k)) for Cu-np and Cu-np/NC(1:4) at −0.7 V vs. RHE during the CO2RR. | ||
X-ray photoelectron spectroscopy (XPS) and operando X-ray absorption spectroscopy (XAS) were carried out to monitor the valence state and coordinate environment of Cu during the CO2RR by the method used in our previous study.34,41,48 From XPS, we can observe that the Cu valence state in Cu-np and Cu-np/NC(1:4) was similar, which is attributed to Cu0 (Fig. S17†). As shown in X-ray absorption near edge structure (XANES) spectroscopy (Fig. 2D), the pre-edge peaks of Cu-np and Cu-np/NC(1:4) were close to Cu foil before reaction. When the potential (−0.7 V vs. RHE) was applied, the spectra of Cu-np and Cu-np/NC(1:4) were still similar to that of metallic Cu. According to extended X-ray absorption fine structure (EXAFS) spectroscopy (Fig. 2E and S18†), only a peak corresponding to the Cu–Cu bond was observed, indicating that the metallic Cu was the active site for Cu-np and Cu-np/NC(1:4) during the CO2RR. Furthermore, the Cu–Cu coordination number of Cu-np and Cu-np/NC(1:4) during the CO2RR were quantified by using the ARTEMIS programs of IFEFFIT (Fig. S19, S20 and Table S3†).The Cu–Cu coordination number and bond distance in Cu-np/NC(1:4) were close to that in Cu-np during the CO2RR. These results indicate that the addition of NC did not change the coordination properties of Cu during the CO2RR.
DFT calculations were then carried out to gain insights into the effect of the N-doped species in Cu-np/NC on the selectivity of CH4. Cu(111) was used to represent Cu-np (Fig. S21†), which is in accordance with the results of HR-TEM. Cu(111) was located on a layer of N-doped graphene (NG) to represent the model of Cu-np/NC (Fig. S22†). From the results above, the pyrrolic N species played a crucial role in the selectivity of CH4. Then, the reaction energy diagrams of CO2 reduction to CH4 were first characterized over Cu(111) and Cu(111) on pyrrolic N-doped graphene (Cu(111)/pyrrolic N).
As shown in Fig. 4A, S23 and S24,† CO2 was first reduced to *CO through the *COOH intermediate, and then *CO was further reduced to CH4 through the *CHO intermediate. On Cu(111), the hydrogenation of *CO to *CHO shows the highest energy barrier (0.71 eV), which is considered as the rate-limiting step for producing CH4. The hydrogenation of CO2 to *COOH and hydrogenation of *CO to *CHO were promoted over Cu(111)/pyrrolic N. Although the hydrogenation of *CO to *CHO still shows the highest energy barrier over Cu(111)/pyrrolic N, it was only 0.30 eV, which was much lower than that over Cu(111). These results suggested that the reduction of CO2 to CH4 over Cu(111) can be significantly enhanced by combining with pyrrolic N doped NC. Furthermore, the reaction energy diagrams were characterized at −0.5 V applied potential (Fig. S25†), and Cu(111)/pyrrolic N also is more favorable for producing CH4 than Cu(111).
In addition, the hydrogenation of *CO to *CHO was also studied over Cu (111)/NC with different N-doped species (Fig. 3B and S26–S29†), and H2O was used as the donor of hydrogen, because 1 M KOH solution was used as the electrolyte in the CO2RR. Compared with Cu(111), the formation of *CHO and *OH from *CO and *H2O can be enhanced over Cu(111)/graphitic N, Cu(111)/pyrrolic N and Cu (111)/pyridinic N. It is noted that Cu(111)/pyrrolic N exhibited the lowest energy barrier (−0.16 eV), indicating that the pyrrolic N played the main role in the outstanding activity and selectivity of CH4, which was consistent with the experimental results.
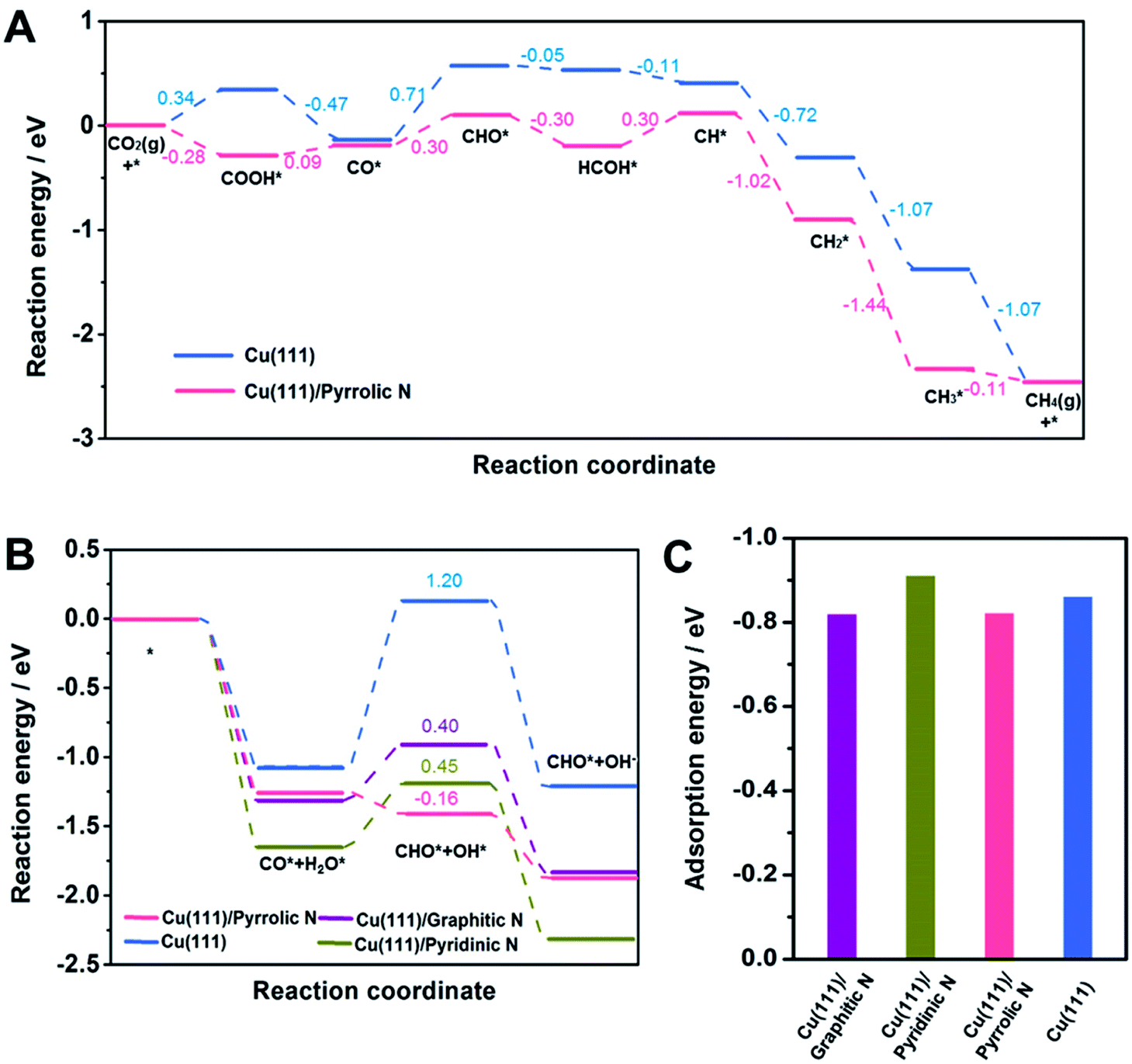 | ||
| Fig. 3 (A) A reaction energy diagram for the CO2RR to CH4 over Cu(111) and Cu(111)/pyrrolic N. (B) A reaction energy diagram for *CO hydrogenation to *CHO on Cu(111), Cu(111)/graphitic N, Cu(111)/pyridinic N and Cu(111)/pyrrolic N. (C) The adsorption energy of *CO on different models. | ||
According to a previous report,49 the adsorption of *CO played an important role in the selectivity of products. Thus, the adsorption of *CO on different models was studied. The adsorption of *CO on Cu(111), Cu(111)/graphitic N, Cu(111)/pyrrolic N and Cu(111)/pyridinic N was comparable (Fig. 3C), indicating that the adsorption of *CO was not changed by adding NC. Thus we can assume that the outstanding performance for CH4 over Cu-np/NC was attributed to the activation of H2O by pyrrolic N.
From the results of DFT calculations above, we can know that the pyrrolic N in NC can enhance the activation of H2O and accelerate the hydrogenation of intermediates. To explore the effect of H2O activation on the generation of CH4, the kinetic isotopic effect (KIE) of H/D over Cu-np/NC(x:y) catalysts was measured (Fig. 4A and S30†). The KIEs of H/D are defined as the ratio of CH4 formation rates in H2O and D2O. It has been reported that the reaction is considered to be controlled by the primary isotope effect when the KIE value is greater than 2.49,50 The KIE value was 2.9 over Cu-np, suggesting that the activation of H2O was involved in the rate-determining step. The KIE value over Cu-np/NC(x:y) decreased with the increase of the content of pyrrolic N in NC, suggesting that the dissociation of H2O can be enhanced by the pyrrolic N. For Cu-np/NC(1:4), the content of pyrrolic N reached the highest, and the KIE value was about 1.6, indicating that the dissociation of H2O was no longer involved in the rate-determining step, which is consistent with the results of DFT. Thus it can be deduced that the pyrrolic N in NC can accelerate H2O activation.
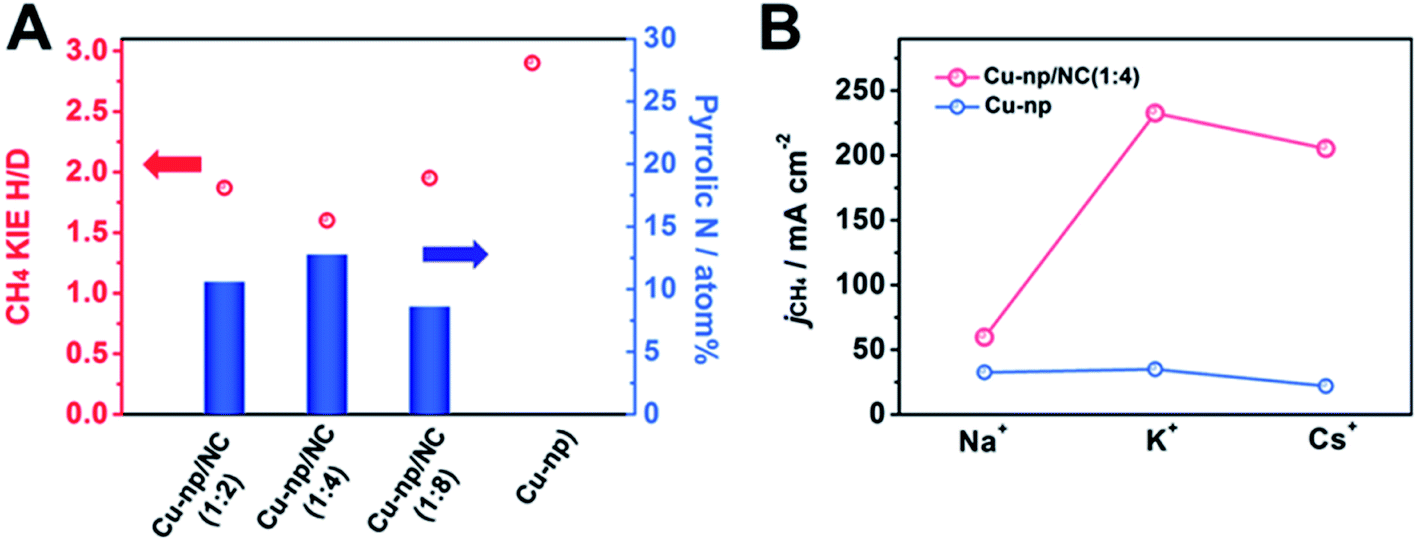 | ||
| Fig. 4 (A) Kinetic isotopic effect (KIE) of H/D on the CO2RR to CH4 at −1.1 V versus RHE. (B) Effect of alkali metal cations in the MOH (M = Na+, K+ and Cs+) electrolyte on the CO2RR to CH4 at −1.1 V versus RHE over Cu-np and Cu-np/NC(1:4) catalysts. | ||
The role of H2O activation in the generation of CH4 was further studied by investigating the effect of alkali metal (M) cations in a MOH electrolyte. It is known that the cation can combine with H2O to form a hydrated cation of M+(H2O)n, and the value of n was 13, 7 and 6 for Na+, K+ and Cs+, respectively.49 The radii of M+(H2O)n decrease in the order of Na+ > K+ > Cs+.51 The smaller n and radii of M+(H2O)n enable a greater ability to dissociate H2O.49,51 It can be known that the formation rate of CH4 was improved markedly over Cu-np/NC(1:4) by changing the cation from Na+ to K+ (Fig. 4B). The formation rate of CH4 in CsOH was smaller than that in KOH. This may be because the generation of H2 was also enhanced (Fig. S31†). For Cu-np, the formation rate of CH4 increased slightly when changing the cation from Na+ to K+. Thus we can assume that NC can enhance CH4 formation by promoting H2O activation through interaction with hydrated cations.
Furthermore, the reaction intermediates during the CO2RR were traced by in situ surface-enhanced Raman spectroscopy (SERS).34,41 At open-circuit potential (OCP), no CuxO species were observed on Cu-np and Cu-np/NC(1:4), which was consistent with the results of XPS and XAS. The peaks located at 1336 cm−1 and 1580 cm−1 were observed on NC and Cu-np/NC(1:4), which were assigned to the D band and G band of graphene, respectively.52–54 Weak peaks were also observed on Cu-np, which may be from the carbon paper. It is noted that a new Raman peak located at 526 cm−1 appeared on Cu-np and Cu-np/NC(1:4) at −0.3 V vs. RHE, which was attributed to the adsorption of preliminary intermediates (such as *CO2 or *OCO−) on the Cu surface.55 These results indicated that the activation of CO2 occurred on Cu sites. For Cu-np, a new Raman band located at 1895 cm−1 appeared at −0.3 V vs. RHE, which corresponded to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching on Cu.41 In contrast, no adsorption of *CO was observed on Cu-np/NC(1:4). This may be because the obtained *CO can be consumed quickly. Compared with Cu-np, Cu-np/NC(1:4) showed three other new peaks located at 567 cm−1, 1430 cm−1 and 1660 cm−1 at −0.3 V vs. RHE, which may be attributed to the adsorption of *OH, *HCOH and *CHO on the Cu surface.50,54,55 Thus we can deduce that the reaction *CO + *H2O → *CHO + *OH can be accelerated over Cu-np/NC and thus the selectivity of CH4 can be enhanced, which was consistent with the experimental and calculation results (Fig. 5).
O stretching on Cu.41 In contrast, no adsorption of *CO was observed on Cu-np/NC(1:4). This may be because the obtained *CO can be consumed quickly. Compared with Cu-np, Cu-np/NC(1:4) showed three other new peaks located at 567 cm−1, 1430 cm−1 and 1660 cm−1 at −0.3 V vs. RHE, which may be attributed to the adsorption of *OH, *HCOH and *CHO on the Cu surface.50,54,55 Thus we can deduce that the reaction *CO + *H2O → *CHO + *OH can be accelerated over Cu-np/NC and thus the selectivity of CH4 can be enhanced, which was consistent with the experimental and calculation results (Fig. 5).
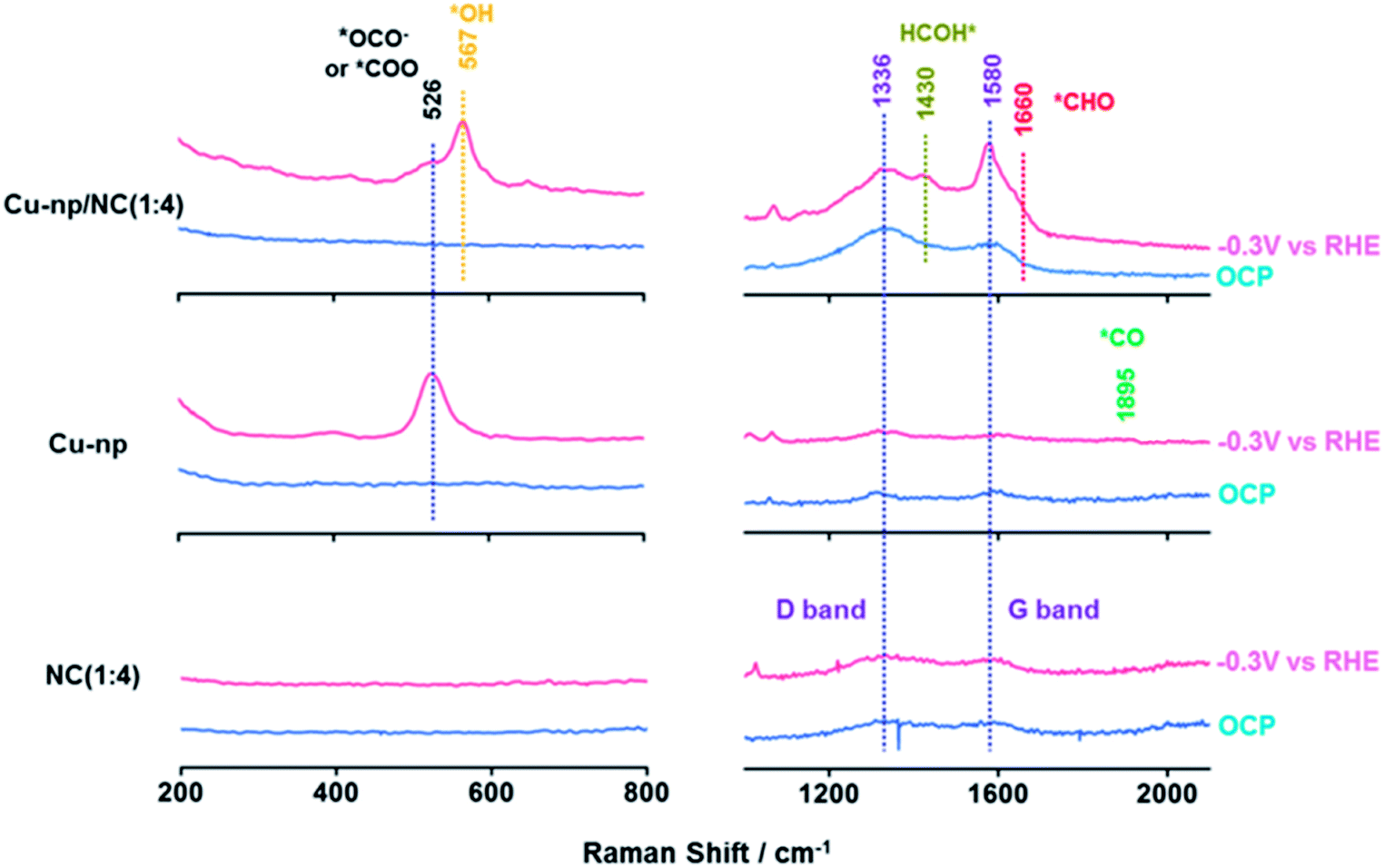 | ||
| Fig. 5 The in situ surface-enhanced Raman spectra over Cu-np, Cu-np/NC(1:4) and NC(1:4) at −0.3 V vs. RHE during the CO2RR. | ||
Conclusions
In summary, Cu-np/NC composites were designed to enhance the electroreduction of CO2 to CH4. The selectivity of CH4 reached 73.4%, with a current density of 320 mA cm−2. Based on experimental and theoretical studies, the effect of the N-doped species on the electrocatalytic performance of CO2 reduction was elucidated. The pyrrolic N in NC can enhance the activation of H2O, which could accelerate the hydrogenation of intermediates, resulting in excellent selectivity for CH4 and high current density. We believe that this work opens a way for the design of efficient catalysts for the electroreduction of CO2 to CH4.Data availability
The data that support the findings of this study are available within the article and its ESI.†Author contributions
Y. H. W., C. J. C., and B. X. H. proposed the project, designed the experiments, and wrote the manuscript; Y. H. W. performed the whole experiments; X. P. Y., R. Z. W., S. J. L., J. M., J. L. Z., Z. M. L., X. Q. X., Z. H. W. performed the analysis of experimental data; C. J. C. and B. X. H. supervised the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Key Research and Development Program of China (2017YFA0403102, 2017YFA0403003, and 2017YFA0403101), National Natural Science Foundation of China (22003070, 21890761, 21733011, and 22121002) and Chinese Academy of Sciences (QYZDY-SSW-SLH013). The operando X-ray absorption spectroscopy (XAS) measurements were performed using a modified flow cell at the 1W1B, 1W2B and 4B9A beamlines at the Beijing Synchrotron Radiation Facility (BSRF), China.Notes and references
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C.-W. Pao, J. Xiao, C. Xia and J. Zeng, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS PubMed.
- Y. Xu, F. Li, A. Xu, J. P. Edwards, S.-F. Hung, C. M. Gabardo, C. P. O'Brien, S. Liu, X. Wang, Y. Li, J. Wicks, R. K. Miao, Y. Liu, J. Li, J. E. Huang, J. Abed, Y. Wang, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 2932 CrossRef CAS.
- W. Wang, L. Shang, G. Chang, C. Yan, R. Shi, Y. Zhao, G. I. N. Waterhouse, D. Yang and T. Zhang, Adv. Mater., 2019, 31, e1808276 CrossRef PubMed.
- J. Santos-Lorenzo, R. San José-Velado, J. Albo, G. Beobide, P. Castaño, O. Castillo, A. Luque and S. Pérez-Yáñez, Microporous Mesoporous Mater., 2019, 284, 128–132 CrossRef CAS.
- J. Albo and A. Irabien, J. Catal., 2016, 343, 232–239 CrossRef CAS.
- J. Albo, M. Perfecto-Irigaray, G. Beobide and A. Irabien, J. CO2 Util., 2019, 33, 157–165 CrossRef CAS.
- J. Albo, G. Beobide, P. Castaño and A. Irabien, J. CO2 Util., 2017, 18, 164–172 CrossRef CAS.
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen, J. Zhong, H. Sun, Y. Lian, B. Yang, X. Yuan, H. Yu, Y. Liu, X. Yang, J. Guo, M. H. Rümmeli, Y. Jiao and Y. Peng, Adv. Mater., 2021, 33, 2101741 CrossRef CAS.
- S. Chen, Y. Su, P. Deng, R. Qi, J. Zhu, J. Chen, Z. Wang, L. Zhou, X. Guo and B. Y. Xia, ACS Catal., 2020, 10, 4640–4646 CrossRef CAS.
- Q. Hu, Z. Han, X. Wang, G. Li, Z. Wang, X. Huang, H. Yang, X. Ren, Q. Zhang, J. Liu and C. He, Angew. Chem., Int. Ed., 2020, 59, 19054–19059 CrossRef CAS PubMed.
- Y. Li, A. Xu, Y. Lum, X. Wang, S.-F. Hung, B. Chen, Z. Wang, Y. Xu, F. Li, J. Abed, J. E. Huang, A. S. Rasouli, J. Wicks, L. K. Sagar, T. Peng, A. H. Ip, D. Sinton, H. Jiang, C. Li and E. H. Sargent, Nat. Commun., 2020, 11, 6190 CrossRef CAS PubMed.
- S.-N. Sun, J.-N. Lu, Q. Li, L.-Z. Dong, Q. Huang, J. Liu and Y.-Q. Lan, Chem. Catal., 2021, 1, 1133–1144 CrossRef.
- K. Ye, A. Cao, J. Shao, G. Wang, R. Si, N. Ta, J. Xiao and G. Wang, Sci. Bull., 2020, 65, 711–719 CrossRef CAS.
- S. G. Han, D. D. Ma and Q. L. Zhu, Small Methods, 2021, 5, e2100102 CrossRef PubMed.
- H. Yang, L. Shang, Q. Zhang, R. Shi, G. I. N. Waterhouse, L. Gu and T. Zhang, Nat. Commun., 2019, 10, 4585 CrossRef PubMed.
- J. Albo, A. Sáez, J. Solla-Gullón, V. Montiel and A. Irabien, Appl. Catal., B, 2015, 176–177, 709–717 CrossRef CAS.
- M. Perfecto-Irigaray, J. Albo, G. Beobide, O. Castillo, A. Irabien and S. Pérez-Yáñez, RSC Adv., 2018, 8, 21092–21099 RSC.
- I. Merino-Garcia, J. Albo, P. Krzywda, G. Mul and A. Irabien, Catal. Today, 2020, 346, 34–39 CrossRef CAS.
- J. Albo, D. Vallejo, G. Beobide, O. Castillo, P. Castaño and A. Irabien, ChemSusChem, 2017, 10, 1100–1109 CrossRef CAS PubMed.
- I. Merino-Garcia, J. Albo and A. Irabien, Nanotechnology, 2017, 29, 014001 CrossRef PubMed.
- I. Merino-Garcia, J. Albo, J. Solla-Gullón, V. Montiel and A. Irabien, J. CO2 Util., 2019, 31, 135–142 CrossRef CAS.
- Z. Li, R. Wu, L. Zhao, P. Li, X. Wei, J. Wang, J. S. Chen and T. Zhang, Nano Res., 2021, 14, 3795–3809 CrossRef CAS.
- L. Lv, X. He, J. Wang, Y. Ruan, S. Ouyang, H. Yuan and T. Zhang, Appl. Catal., B, 2021, 298, 120531 CrossRef CAS.
- S. Kunze, P. Grosse, M. Bernal Lopez, I. Sinev, I. Zegkinoglou, H. Mistry, J. Timoshenko, M. Y. Hu, J. Zhao, E. E. Alp, S. W. Chee and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2020, 59, 22667–22674 CrossRef CAS.
- W. Zhu, L. Zhang, P. Yang, X. Chang, H. Dong, A. Li, C. Hu, Z. Huang, Z.-J. Zhao and J. Gong, Small, 2018, 14, 1703314 CrossRef.
- S. J. Raaijman, M. P. Schellekens, P. J. Corbett and M. T. M. Koper, Angew. Chem., Int. Ed., 2021, 60, 21732–21736 CrossRef CAS PubMed.
- Y. F. Lu, L. Z. Dong, J. Liu, R. X. Yang, J. J. Liu, Y. Zhang, L. Zhang, Y. R. Wang, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2021, 60, 26210–26217 CrossRef CAS PubMed.
- G. Zhang, Z.-J. Zhao, D. Cheng, H. Li, J. Yu, Q. Wang, H. Gao, J. Guo, H. Wang, G. A. Ozin, T. Wang and J. Gong, Nat. Commun., 2021, 12, 5745 CrossRef CAS PubMed.
- X. Yan, C. Chen, Y. Wu, S. Liu, Y. Chen, R. Feng, J. Zhang and B. Han, Chem. Sci., 2021, 12, 6638–6645 RSC.
- D. Yang, Q. Zhu and B. Han, The Innovation, 2020, 1, 100016 CrossRef PubMed.
- Y. Zhang, L.-Z. Dong, S. Li, X. Huang, J.-N. Chang, J.-H. Wang, J. Zhou, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2021, 12, 6390 CrossRef CAS.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed.
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang, J. Luo, Y. Zhu, M. Gu, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS PubMed.
- S. Liu, H. Yang, X. Huang, L. Liu, W. Cai, J. Gao, X. Li, T. Zhang, Y. Huang and B. Liu, Adv. Funct. Mater., 2018, 28, 1800499 CrossRef.
- H. Wang, Y.-K. Tzeng, Y. Ji, Y. Li, J. Li, X. Zheng, A. Yang, Y. Liu, Y. Gong, L. Cai, Y. Li, X. Zhang, W. Chen, B. Liu, H. Lu, N. A. Melosh, Z.-X. Shen, K. Chan, T. Tan, S. Chu and Y. Cui, Nat. Nanotechnol., 2020, 15, 131–137 CrossRef CAS PubMed.
- C. Chen, X. Yan, S. Liu, Y. Wu, Q. Wan, X. Sun, Q. Zhu, H. Liu, J. Ma, L. Zheng, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 59, 16459–16464 CrossRef CAS PubMed.
- Y. Mun, S. Lee, K. Kim, S. Kim, S. Lee, J. W. Han and J. Lee, J. Am. Chem. Soc., 2019, 141, 6254–6262 CrossRef CAS PubMed.
- J.-Y. Kim, D. Hong, J.-C. Lee, H. G. Kim, S. Lee, S. Shin, B. Kim, H. Lee, M. Kim, J. Oh, G.-D. Lee, D.-H. Nam and Y.-C. Joo, Nat. Commun., 2021, 12, 3765 CrossRef CAS PubMed.
- C. Choi, T. Cheng, M. Flores Espinosa, H. Fei, X. Duan, W. A. Goddard III and Y. Huang, Adv. Mater., 2019, 31, 1805405 CrossRef PubMed.
- L. Li, C. Tang, Y. Zheng, B. Xia, X. Zhou, H. Xu and S.-Z. Qiao, Adv. Energy Mater., 2020, 10, 2000789 CrossRef CAS.
- C. Liu, M. Zhang, J. Li, W. Xue, T. Zheng, C. Xia and J. Zeng, Angew. Chem., Int. Ed., 2021, 143, 3245–3255 Search PubMed.
- Z.-Z. Niu, L.-P. Chi, R. Liu, Z. Chen and M.-R. Gao, Energy Environ. Sci., 2021, 14, 4169–4176 RSC.
- Y. Wu, C. Chen, X. Yan, S. Liu, M. Chu, H. Wu, J. Ma and B. Han, Green Chem., 2020, 22, 6340–6344 RSC.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, M. Chevrier Daniel, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–800 CrossRef CAS PubMed.
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 892 CrossRef.
- R. Song, Y. Dong, D. Zhang, J. Sheng, S. Yang and H. Song, Energy Technol., 2021, 9, 2100309 CrossRef CAS.
- M. Sun, S. Yun, J. Shi, Y. Zhang, A. Arshad, J. Dang, L. Zhang, X. Wang and Z. Liu, Small, 2021, 17, 2102300 CrossRef CAS PubMed.
- C. Chen, X. Yan, Y. Wu, S. Liu, X. Sun, Q. Zhu, R. Feng, T. Wu, Q. Qian, H. Liu, L. Zheng, J. Zhang and B. Han, Chem. Sci., 2021, 12, 5938–5943 RSC.
- W. Shan, R. Liu, H. Zhao, Z. He, Y. Lai, S. Li, G. He and J. Liu, ACS Nano, 2020, 14, 11363–11372 CrossRef CAS PubMed.
- N. Bodappa, M. Su, Y. Zhao, J.-B. Le, W.-M. Yang, P. Radjenovic, J.-C. Dong, J. Cheng, Z.-Q. Tian and J.-F. Li, J. Am. Chem. Soc., 2019, 141, 12192–12196 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02222b |
| This journal is © The Royal Society of Chemistry 2022 |