
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscrete palladium clusters that consist of two mutually bisecting perpendicular planes†
Naoya
Kojima
a,
Misaki
Kato
a and
Yusuke
Sunada
 *ab
*ab
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 4-6-1, Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: sunada@iis.u-tokyo.ac.jp
bInstitute of Industrial Science, The University of Tokyo, 4-6-1, Komaba, Meguro-ku, Tokyo 153-8505, Japan
First published on 15th June 2022
Abstract
The construction of novel molecules with unprecedented alignments of the constituent elements has revolutionized the field of functional materials. The arrangement of two or more planar subunits in a mutually perpendicular fashion is a frequently encountered approach to produce novel functional materials. Previous examples of such materials can be categorized into two well-investigated families: spiro-conjugated and dumbbell-shaped structures, wherein the two planes are aligned orthogonally via a single atom or an axis, respectively. This article describes a third family: reaction of [Pd(CNtBu)2]3 with Sn3Me8 or Ge6Me12 afforded a Pd7Sn4 cluster and a Pd8Ge6 cluster that consist of two mutually bisecting perpendicular planes. In the Pd7Sn4 cluster, the two equivalent Pd5Sn2 planes share three palladium atoms that include a dihedral angle of 85.6°.
Introduction
The precise alignment of the constituent elements in a molecule is arguably the most fundamental and essential strategy to develop novel functional molecules. Reflecting the increasing interest in two-dimensional (2D) materials1 such as graphene2 and metal chalcogenide nanosheets,3 the construction of planar molecules in which all the constituent elements are arranged in a 2D fashion has recently attracted extensive attention in both organic and inorganic chemistry on account of their potential to revolutionize the field of functional materials due to their inherent chemical and physical properties. The arrangement of planar subunits in a twisting fashion is also a well-investigated method to obtain molecules with unique characteristics.4 Möbius-strip-shaped molecules,5 negatively curved polyaromatics,6 warped nanographene,7 and nanobelt-shaped molecules8 are recent representative examples of molecules that consist of novel twisted planar structures, and they are often constructed via the appropriate connection of the planar subunits. The arrangement of planar subunits in a perpendicular fashion is an extreme approach to constructing a twisted structure. This produces several unique chemical characteristics in the resulting molecules; for instance, orbital and/or electronic interactions among the planar subunits are effectively interrupted, which contributes to the creation of novel functional materials.9 Thus, several molecules that consist of mutually perpendicular planes have been developed. Previously reported structures in which the planar subunits are arranged in an orthogonal manner are classified into the two categories shown in Fig. 1, i.e., spiro-conjugated10 ((a) in Fig. 1) and dumbbell-shaped11 ((b) in Fig. 1) structures, wherein two planar subunits are linked via a single atom or an axis, respectively. Herein, we report the synthesis and structural characterization of a new class of molecules, which consist of two mutually perpendicular planes that bisect each another ((c) in Fig. 1). | ||
| Fig. 1 Conceptual illustration of the two most common families of molecules that consist of mutually perpendicular planes ((a) and (b)), and this work ((c)). | ||
Recently, the construction of some planar metal-sheet molecules has been realized via the aid of organic molecules that can assemble multiple metal atoms in a planar fashion. For instance, Murahashi and co-workers have revealed that (poly)cyclic aromatic hydrocarbons can act as a template for the arrangement of Pd atoms in a 2D sheet structure.12 Our group has recently discovered that oligosilanes with pseudoplanar structures are good precursors for the planar arrangement of metal atoms.13 For instance, a bicyclic ladder polysilane with a bent planar structure, decaisopropylbicyclo[2.2.0]hexasilane, is a good template to afford Pd11 clusters with “folding” metal nanosheet structures via treatment with the palladium(0) isocyanide [Pd(CNtBu)2]3 (1).13a Similarly, planar Pd4, Pd5, Pd6, and Pd7 clusters were obtained when cyclotetrasilanes or a cyclopentasilane were used instead of the bicyclic ladder polysilane.13b,c As heavier group-14 elements such as tin and germanium tend to adopt hypercoordinated structures such as five- or six-coordinated structures,14 we hypothesized that the use of organotin and organogermanium compounds with E–E (E = Sn, Ge) bonds could lead to an expansion of the architectures of the obtained clusters via linking of the subunits through hypercoordination. In this paper, we wish to report that tristannane or cyclohexagermane contribute to the formation of two palladium clusters, 2 and 3, which consist of two planar palladium subunits that bisect one another with a dihedral angle of ca. 90°. To the best of our knowledge, 2 and 3 represent the first examples of discrete molecules with mutually bisecting perpendicular planes that have been characterized crystallographically and spectroscopically.
Results and discussion
Synthesis of Pd7Sn4 cluster 2 that consists of two mutually bisecting perpendicular planes
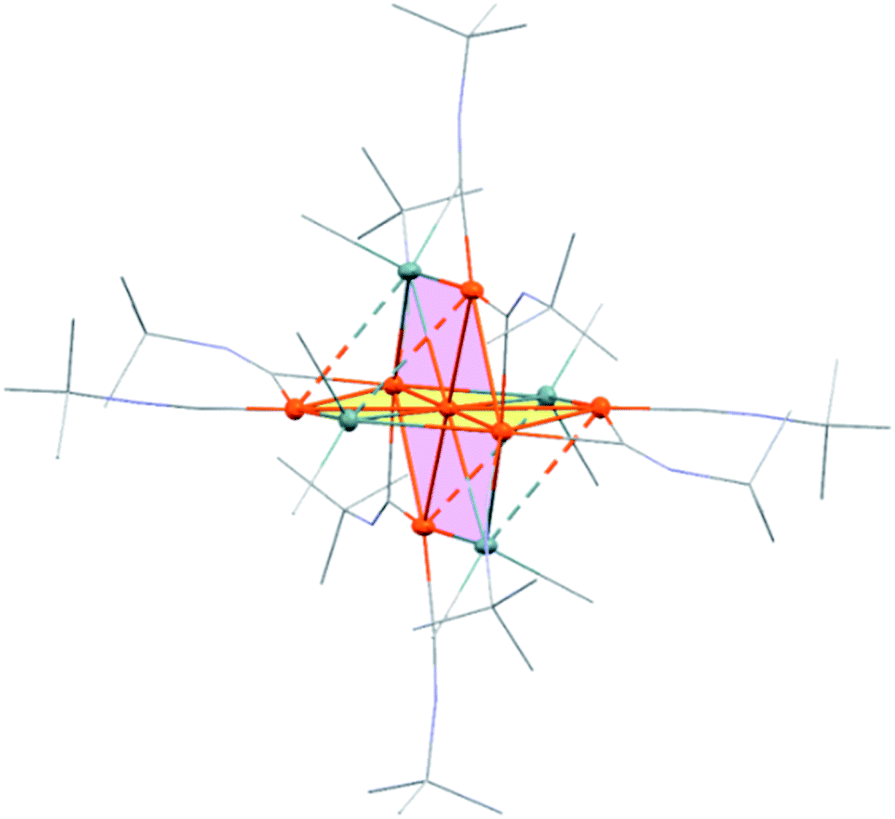
Treatment of octamethyltristannane (Sn3Me8, 4), with 7/3 equivalents of [Pd(CNtBu)2]3 (1) in toluene at room temperature resulted in the formation of dark red crystals of Pd7(SnMe2)4(CNtBu)10 (2) in 74% yield based on Pd (Scheme 1). This reaction concomitantly furnished SnMe4, presumably via the migration of a methyl group.15 The surprising structure of 2, which consists of two mutually bisecting perpendicular planes, was determined using a single-crystal X-ray diffraction (XRD) analysis. The molecular structure of 2 and the core Pd7Sn4 structure of 2 are shown in Fig. 2, S17 and S18 in the ESI,† respectively. Selected bond lengths and angles are summarized in Table S1 in the ESI.† As shown in Fig. 3, the molecular structure of 2 contains seven palladium atoms and four tin atoms that construct the mutually bisecting perpendicularly planar skeleton. In other words, there are two structurally equivalent planes that consist of five palladium atoms and two Sn atoms, which are designated as Pd5Sn2 sheet I and Pd5Sn2 sheet II in Fig. 3. These two planes mutually bisect one another with a dihedral angle of 85.6°. The atom Pd(1) is located at an inversion center. Each Pd5Sn2 sheet contains two stannylene moieties (SnMe2) that reside in the plane of the Pd5 sheet. The three linearly assembled Pd atoms, Pd(1), Pd(2), and Pd(2)*, are shared by the two Pd5Sn2 planes. | ||
| Scheme 1 Synthesis of Pd7(SnMe2)4(CNtBu)10 (2). | ||
 | ||
| Fig. 2 ORTEP drawing of 2 with thermal ellipsoids at 50% probability. All carbon atoms and nitrogen atoms are shown in wireframe style; all hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 3 The core Pd7Sn4 structure of 2 (left). Front view of sheet I (right). | ||
As the structures of the two Pd5Sn2 subunits are almost equivalent, the array of Pd and Sn atoms in sheet I that is depicted in Fig. 3 can be considered as representative. All the Pd and Sn atoms are arranged in a highly planar fashion, and the deviation of the five palladium and two tin atoms from the least square plane defined by all atoms is less than 0.07 Å. As shown in Fig. 3, four Pd atoms (Pd(2), Pd(2)*, Pd(3), and Pd(3)*) and two Sn atoms construct a hexagonal framework, wherein Pd(1) is located at the center of gravity of this hexagon. In each sheet, five palladium atoms are arranged to form a planar bow-tie-type structure. In sheet I, the Pd(2)–Pd(1)–Pd(3), Pd(1)–Pd(2)–Pd(3), and Pd(1)–Pd(3)–Pd(2) angles are 58.323(11)°, 60.185(10)°, and 61.492(9)°, respectively, which indicates that these three palladium atoms form an almost equilateral triangle, and that two triangles are connected by sharing the Pd(1) atom to form a bow-tie structure.16 The radial Pd–Pd bonds (sheet I: Pd(1)–Pd(2) = 2.8005(3) Å; Pd(1)–Pd(3) = 2.7650(3) Å; sheet II: Pd(1)–Pd(2) = 2.8005(3) Å; Pd(1)–Pd(4) = 2.7714(5) Å) are somewhat elongated compared to the Pd–Pd bonds on the edge (sheet I: Pd(2)–Pd(3) = 2.7121(5) Å; sheet II: Pd(2)–Pd(4) = 2.6908(5) Å). It is also noteworthy that a similar elongation of the radial Pd–Pd bonds has been reported for the pentanuclear palladium cluster [Pd5(μ3-selenophene)4(CH3CN)4][BF4]4, which contains a distorted bow-tie-type Pd5 framework.16a These Pd–Pd bond distances are comparable to those found in previously reported polynuclear palladium clusters.17
In sheet I, two Sn atoms (Sn(1) and Sn(1)*) bridge two Pd atoms (Pd(2)* and Pd(3); Pd(2) and Pd(3)*) with bond distances of 2.8300(3) Å and 2.6586(5) Å, respectively. Although the former is significantly elongated, the latter is comparable to that found in a previously reported dipalladium complex with a bridging stannylene ligand.18 The relatively short bonds between Sn(1) or Sn(1)* and the central Pd(1) atom (2.6216(4) Å) indicate the presence of a bonding interaction (vide infra). Similarly, the Pd(1)–Sn(2) bond (2.6067(4) Å) in sheet II is relatively short compared to the Pd(2)*–Sn(2) (2.7979(5) Å) and Pd(4)–Sn(2) (2.7028(5) Å) bonds. Thus, all the Sn atoms adopt pentacoordinated structures in the planar Pd5Sn2 subunits.
It is also worth noting that the Pd(1) atom is located on the Ci inversion center of 2, which results in a unique bonding situation. Namely, Pd(1) engages in bonding interactions with four surrounding SnMe2 units with bond distances of 2.6216(4) and 2.6067(4) Å. In addition, the bond separations between Pd(1) and the six surrounding Pd atoms (2.7650(3)–2.8005(3) Å) fall into the range expected for Pd–Pd bonding interactions.17 This unique coordination environment of the central Pd(1) atom might play a crucial role to produce the unique structure of 2, which consists of two mutually bisecting perpendicular planes. Hypercoordination of the tin atoms of the SnMe2 unit on one Pd5Sn2 sheet with palladium atoms located on the other Pd5Sn2 sheet, i.e., inter-plane coordination, might be another important factor to form this cluster architecture. The observed inter-plane bond distances of Sn(2)–Pd(3)* (3.1830(3) Å) and Sn(1)*–Pd(4) (3.1893(3) Å) are somewhat lengthened compared to those of the intra-plane Pd–Sn distances discussed above. However, the results of theoretical calculations suggest the presence of weak inter-plane bonding interactions between Sn(2)–Pd(3)* and Sn(1)*–Pd(4), which might contribute to the formation of the unique structure of cluster 2, which consists of two mutually bisecting perpendicular planes (vide infra).
To elucidate the bonding interactions in 2, density functional theory (DFT) calculations were carried out using the PBE0, B3PW91, M06, and B3LYP functionals. Although some of the Pd–Pd and Pd–Sn bond distances were slightly overestimated, the optimized structural parameters obtained using the PBE0 functional were in good agreement with the data obtained from the X-ray diffraction (XRD) analysis (for details, see Table S1 and Fig. S15 in the ESI†). As mentioned above, the length of the Pd–Pd bonds in sheet I are in the range of bonding interactions, and the Wiberg bond index (WBI) values for the Pd–Pd bonds in sheet I were found to be 0.21–0.29, which supports the presence of bonding interactions.12a As mentioned above, the central palladium atom (Pd(1)) adopts a unique coordination environment that involves the surrounding six palladium atoms as well as four tin atoms. The WBI values between the central palladium atom (Pd(1)) and the surrounding six palladium atoms were estimated to be Pd(1)–Pd(2) = 0.29, Pd(1)–Pd(3) = 0.21 and Pd(1)–Pd(4) = 0.22. These parameters supports the presence of Pd–Pd bonding interactions. The WBI values between Pd(1) and the surrounding tin atoms were found to be Pd(1)–Sn(1) = 0.55 and Pd(1)–Sn(2) = 0.57, which further corroborates the presence of sufficiently strong bonding interactions. In addition, the WBIs for Pd(2)*–Sn(1) and Pd(3)–Sn(1) were estimated to be 0.26 and 0.34, respectively. These values indicate that the Sn atoms effectively bridge the Pd atoms located on both the edges and center of the hexagonal structure in sheet I.
In contrast to the short Pd–Pd bonds in sheet I (and sheet II), the inter-plane separation between Pd(3) and Pd(4) is significantly elongated to 3.2925(7) Å. It is worth noting here that the WBIs for the Pd(4)–Sn(1)* and Pd(3)*–Sn(2) inter-plane interactions were found to be 0.10 and 0.09, respectively. In addition, the HOMO−2 is located on the Pd(4)–Sn(1)* and Pd(3)*–Sn(2) bonds (Fig. S15 in the ESI†). These theoretical results imply the presence of weak bonding interactions between Pd(4) and Sn(1)*, as well as Pd(3)* and Sn(2). It could thus be suggested that the propensity of Sn atoms to adopt hypercoordinated structures helps to make this weak binding interaction possible. In other words, the framework of 2 with two perpendicularly arranged Pd5Sn2 planes is effectively stabilized by the four hypercoordinated Sn atoms through inter-plane interactions.
Contour diagrams of the representative molecular orbitals in 2 are shown in Fig. S13–S15 in the ESI.† The LUMO (−0.83 eV) and LUMO+1 (−0.69 eV) are located at similar energy levels; they consist of essentially identical molecular orbitals, but are arranged perpendicularly to one another. Namely, the LUMO is delocalized over the Pd and Sn atoms of sheet I only, whereas the LUMO+1 is delocalized only over sheet II (Fig. S13 in the ESI†).
The structure of 2 in solution was investigated using 1H, 13C, 1H DOSY, 1H–13C HSQC, and 1H–13C HMBC NMR spectroscopy in C6D6. The 1H DOSY spectrum of 2 in C6D6 indicates the presence of a single species, which was confirmed by a single diffusion coefficient (D = 3.7 × 10−10 m2 s−1). In the 1H NMR spectrum, five singlets corresponding to the tBu groups appeared at δ = 1.09, 1.27, 1.32, 1.41, and 1.75 ppm with an integral ratio of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18:18:18:18. In general, the Me signals of terminal and bridging CNtBu ligands on group-10 metal clusters have been reported to fall into a relatively wide range of chemical 1H NMR shifts.13a,c,19 Four magnetically inequivalent Me signals assignable to the SnMe2 moieties were observed at δ = 0.77, 1.04, 1.25, and 1.31 ppm with an integral ratio of 6:6:6:6. These results are consistent with the molecular structure determined using XRD considering the Ci symmetric structure of 2. This assignment is also supported by the 1H–13C HSQC, and 1H–13C HMBC spectra. For instance, five singlets appear at 29.82, 30.29, 30.35, 30.74, and 31.01 ppm in the 13C NMR spectrum of 2 in C6D6, and these peaks are correlated to the 1H NMR signals at 1.09, 1.27, 1.32, 1.41, and 1.75, respectively, in the 1H–13C HSQC NMR spectrum. In addition, the 1H–13C HSQC spectrum of 2 contains four cross-peaks at δ = 0.77/63.13, 1.04/66.82, 1.25/67.10, and 1.31/69.07, which are derived from the four magnetically inequivalent SnMe2 moieties.
:18:18:18:18. In general, the Me signals of terminal and bridging CNtBu ligands on group-10 metal clusters have been reported to fall into a relatively wide range of chemical 1H NMR shifts.13a,c,19 Four magnetically inequivalent Me signals assignable to the SnMe2 moieties were observed at δ = 0.77, 1.04, 1.25, and 1.31 ppm with an integral ratio of 6:6:6:6. These results are consistent with the molecular structure determined using XRD considering the Ci symmetric structure of 2. This assignment is also supported by the 1H–13C HSQC, and 1H–13C HMBC spectra. For instance, five singlets appear at 29.82, 30.29, 30.35, 30.74, and 31.01 ppm in the 13C NMR spectrum of 2 in C6D6, and these peaks are correlated to the 1H NMR signals at 1.09, 1.27, 1.32, 1.41, and 1.75, respectively, in the 1H–13C HSQC NMR spectrum. In addition, the 1H–13C HSQC spectrum of 2 contains four cross-peaks at δ = 0.77/63.13, 1.04/66.82, 1.25/67.10, and 1.31/69.07, which are derived from the four magnetically inequivalent SnMe2 moieties.
The 1H–13C HMBC cross peak between one of the five tBu groups (δ = 1.09) and two carbon signals at 29.82 (–C(CH3)3) and 55.04 (–C(CH3)3) indicate that these signals can be assigned to one of the five CNtBu ligands. Similarly, a combination of the 1H and 13C NMR signals due to other four CNtBu ligands were assigned via the 1H–13C HMBC spectrum as follows; 1H: 1.27 ppm/13C: 30.29 and 54.07 ppm; 1H: 1.32 ppm/13C: 30.35 and 54.85 ppm; 1H: 1.41 ppm/13C: 30.74 and 55.13 ppm; 1H: 1.75 ppm/13C: 31.01 and 56.63 ppm. These NMR spectra indicate that the Pd7Sn4 structure with two mutually bisecting perpendicular planes in 2 is maintained in solution.
Synthesis of Pd8Ge6 cluster 3 that consists of two mutually bisecting perpendicular planes supported by germylene ligands
The use of an organogermanium compound with multiple Ge–Ge bonds also led to the formation of a palladium cluster that consists of two perpendicularly arranged palladium nanosheets. Namely, the reaction of [Pd(CNtBu)2]3 (1) with dodecamethylcyclohexagermane, (Ge6Me12, 5), took place instantly in cyclopentylmethylether (CPME), even at −20 °C, to afford palladium cluster Pd8(GeMe2)6(CNtBu)10 (3), which was isolated in the form of black crystals in 49% yield (based on Pd) (Scheme 2). A single-crystal XRD analysis revealed that 3 also comprises two mutually perpendicular planes that bisect each another with a dihedral angle of 88.6°, albeit that the Pd4 and Pd6 planes are not identical (Fig. 4 and S20 in the ESI†). The Pd4 and Pd6 planes were defined as sheet III and sheet IV in Fig. 4. In both the Pd planes, the palladium atoms are supported by the germylene (GeMe2) units in the plane, i.e., the Pd4 plane (sheet III) consists of four palladium atoms supported by two GeMe2 units, whereas the Pd6 plane (sheet IV) includes four GeMe2 units. Sheets III and IV are intersected by two palladium atoms (Pd(3) and Pd(3)*). | ||
| Scheme 2 Synthesis of Pd8(GeMe2)6(CNtBu)10 (3). | ||
 | ||
| Fig. 4 ORTEP drawing of 3 with thermal ellipsoids at 50% probability. All carbon atoms and nitrogen atoms are shown in wireframe style; all hydrogen atoms are omitted for clarity (upper). The core Pd8Ge6 structure of 3 (lower). | ||
Upon examining the molecular structure of 3 in more detail, we realized that Pd4 sheet III has a hexagonal Pd4Ge2 structure that is similar to that in sheet I and II in cluster 2, with the only difference being the absence of the central palladium atom. The Pd(3)–Pd(4) and Pd(3)*–Pd(4)* bond distances in sheet III are 2.5947(5) Å, which is ca. 0.1 Å shorter than those found in sheet I and II in 2. The synthesis of hexanuclear palladium clusters that include a similar hexagonal Pd4Ge2 subunit, consisting of four palladium and two germylene moieties, has already been reported by Osakada et al.20 However, the Pd4Ge2 planes in that cluster adopt a distorted planar structure, wherein the four palladium atoms deviate from the Pd4Ge2 plane by ca. 0.05–0.29 Å. This contrasts sharply with the highly planar Pd4Ge2 subunit shown in cluster 3, wherein the deviation is less than 0.03 Å for all the palladium atoms.
In contrast, sheet IV consists of six palladium atoms, and the four palladium atoms (Pd(1), Pd(1)*, Pd(3), and Pd(3)*) are arranged in a rhombic fashion with Pd(1)–Pd(3) and Pd(1)–Pd(3)* distances of 2.8182(4) and 2.7975(3) Å, respectively, and the bond distance of Pd(1)–Pd(1)* is 2.6920(3) Å. The Pd(1)–Pd(3)–Pd(1) and Pd(3)–Pd(1)–Pd(3)* angles are 57.287(9)° and 122.713(13)°, respectively. The additional two palladium atoms (Pd(2) and Pd(2)*) are positioned outside the Pd4 rhombus with Pd(1)–Pd(2) (and Pd(1)*–Pd(2)*) bond distances of 2.7771(4) Å. These bond distances are similar to those of the radial Pd–Pd bonds shown in cluster 2. In sheet IV, the six palladium atoms reside in an almost perfect plane, whereas the four germanium atoms are located out of the plane defined by six palladium atoms with deviations of 0.37–0.73 Å.
The four GeMe2 units on sheet IV act as the edges to bridge over two palladium atoms, e.g., Ge(1) bridges Pd(2) and Pd(3)* with bond distances of 2.5578(4) and 2.6783(5) Å, respectively. In addition, four GeMe2 moieties in sheet IV engage in bonding interactions with the inward palladium atoms (Pd(1) and Pd(1)*) as Pd–Ge radials, and the radial Pd(1)–Ge(1) bond distance was found to be 2.4074(4) Å. Thus, these radial Pd–Ge bond distances are significantly shorter than the edge Pd–Ge bond lengths. A similar trend was confirmed in the Pd5Sn2 sheet structures shown in cluster 2.
Similar to the feature observed in cluster 2, the palladium atom (Pd(3) and Pd(3)*) that is shared by sheets III and IV engages in bonding interactions with a higher number of GeMe2 moieties compared to the other palladium atoms. Namely, Pd(3) and Pd(3)* engage in bonding interactions with the surrounding three GeMe2 moieties, whereas the other palladium atoms only engage in one or two Pd–Ge bonding interactions. The bond distances between Pd(3) and the surrounding three germanium atoms (Ge(1)*, Ge(2), and Ge(3); 2.5100(3)–2.7590(6) Å) support the presence of bonding interactions (vide infra). In addition, the bond separations between the Pd(3) or Pd(3)* atoms and the surrounding three palladium atoms (Pd(1), Pd(1)* and Pd(4)) are 2.5947(5), 2.7975(3) and 2.8182(4) Å, respectively, and thus in the range of previously reported bonding interactions.17 Thus, Pd(3) and Pd(3)* adopt a unique coordination environment, which might contribute to the unique architecture of cluster 3. Moreover, similar to cluster 2, the presence of inter-plane bonding interactions between hypercoordinated germanium and palladium atoms was suggested. Namely, bond lengths of 2.8531(6) and 3.2720(3) Å were found for Pd(1)–Ge(3) and Pd(4)–Ge(1)*, respectively. Although the latter is relatively long compared to the intra-plane Pd–Ge bonds, theoretical calculations suggested the presence of weak inter-plane bonding interactions between both the Pd(1)–Ge(3) and Pd(4)–Ge(1)* bonds (vide infra). Thus, the hypercoordination of the germylene moieties might contribute to the molecular structure of cluster 3, which consists of two mutually bisecting perpendicular planes.
The bonding interaction in cluster 3 was also theoretically investigated using the DFT calculations based on the PBE0 functional (Table S3 and Fig. S16 in the ESI†). The presence of intra-plane Pd–Pd bonding interactions was supported by the WBIs (0.17–0.19). As mentioned above, the two palladium atoms that are shared by sheets III and IV (Pd(3) and Pd(3)*) are located sufficiently close to the surrounding three palladium atoms (Pd(1), Pd(1)*, and Pd(4) for Pd(3); Pd(1), Pd(1)*, and Pd(4)* for Pd(3)*). The estimated WBI values among these palladium atoms (0.17–0.19) support the presence of Pd–Pd bonding interactions around Pd(3) and Pd(3)*. In addition, the estimated WBIs for Pd(3)–Ge(2) and Pd(3)–Ge(1)* (∼0.24) indicates the presence of weak Pd–Ge bonds. In sheet IV, the WBIs for the radial Pd–Ge bonds (Pd(1)–Ge(1)/Ge(2)) were estimated to be ca. 0.59, whereas those of the edge Pd–Ge bonds (Pd(2)–Ge(1)/Ge(2)) were ca. 0.39.
Importantly, the presence of an inter-plane interaction was also suggested in cluster 3 by the WBI analysis. Namely, the WBI for Pd(1)–Ge(3) was found to be 0.30, which is comparable to that found for the intra-plane Pd–Ge interaction (Pd(2)–Ge(1)/Ge(2)), which indicates the presence of a moderately strong bonding interaction. A relatively small WBI value (0.11) was obtained for the Pd(4)–Ge(1)* bond, which implies the presence of a weak inter-plane bonding interaction. Similar to cluster 2, these inter-plane bonding interactions between palladium and germanium atoms could be a key to the construction of a unique structure that consists of two perpendicularly arranged palladium nanosheets that bisect one another.
In contrast to 2, cluster 3 is thermally highly unstable in solution, which led to rapid decomposition during the measurement of the NMR spectrum. When isolated cluster 3 was dissolved in C6D6 and kept at room temperature for 1 h, the formation of a complex mixture was observed. However, a relatively pure 1H NMR spectrum for cluster 3 was recorded at lower temperature; five singlets were observed in its 1H NMR spectrum in toluene-d8 at −30 °C at δ = 0.99, 1.32, 1.38, 1.73, and 1.87 ppm with an integral ratio of 36:36:12:24:18; this spectral feature is consistent with the solid-state structure.
Conclusions
In conclusion, we have shown that two palladium clusters that consist of two mutually bisecting perpendicular planes, 2 and 3, can be synthesized with the aid of bridging SnMe2 and GeMe2 units. Although clusters 2 and 3 both consist of two perpendicularly arranged planes, their structural characteristics differ. Namely, Pd7Sn4 cluster 2 consists of two equivalent Pd5Sn2 planes (sheet I and II; Fig. 3), whereas Pd8Ge6 cluster 3 is composed of two inequivalent Pd6 and Pd4 nanosheets. The pseudosymmetric structure of 2 gives rise to two pairs of essentially equivalent molecular orbitals for the LUMO and LUMO+1; the LUMO is delocalized over the palladium and tin atoms in sheet I, whereas the LUMO+1 is delocalized over sheet II. As demonstrated by recent developments in nanocarbon science, the construction of novel molecules with unprecedented alignment of the constituent elements has revolutionized the field of functional materials. We anticipate that the discovery of methodologies to arrange metal atoms in unprecedented manners, such as that shown in this article, will help to advance further developments in nanoscience.Data availability
All experimental and theoretical data are provided in the ESI.†Author contributions
N. Kojima and M. Kato conducted all experiments. All authors analysed the data. Y. Sunada supervised this study and wrote the manuscript. All authors discussed the results and contributed to the preparation of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST, PRESTO Grant Number JPMJPR20A9, Japan.Notes and references
- (a) C. Tan, X. Cao, X. -J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G. -H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed; (b) R. Dong, T. Zhang and X. Feng, Chem. Rev., 2018, 118, 6189–6235 CrossRef CAS PubMed.
- (a) A. K. Geim, Angew. Chem., Int. Ed., 2011, 50, 6967–6985 CrossRef PubMed; (b) K. S. Novoselov, Angew. Chem., Int. Ed., 2011, 50, 6986–7002 CrossRef CAS PubMed; (c) C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed.
- (a) G. H. Han, D. L. Duong, D. H. Keum, S. J. Yun and Y. H. Lee, Chem. Rev., 2018, 118, 6297–6336 CrossRef CAS PubMed; (b) S. Bertolazzi, M. Gobbi, Y. Zhao, C. Backes and P. Samorı, Chem. Soc. Rev., 2018, 47, 6845–6888 RSC.
- (a) S. Sasaki, G. P. C. Drummen and G. -i. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2747 RSC; (b) W. Rettig, Angew. Chem., Int. Ed. Engl., 1986, 25, 971–988 CrossRef; (c) K. Kokado and K. Sada, Angew. Chem., Int. Ed., 2019, 58, 8632–8639 CrossRef CAS PubMed; (d) M. Rickhaus, M. Mayor and M. Juriček, Chem. Soc. Rev., 2016, 45, 1542–1556 RSC.
- (a) R. Herges, Chem. Rev., 2006, 106, 4820–4842 CrossRef CAS PubMed; (b) N. Jux, Angew. Chem., Int. Ed., 2008, 47, 2543–2546 CrossRef CAS PubMed.
- S. Matsubara, Y. Koga, Y. Segawa, K. Murakami and K. Itami, Nat. Catal., 2020, 3, 710–718 CrossRef CAS.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- (a) G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, Science, 2017, 356, 172–175 CrossRef CAS PubMed; (b) T. Matsuno, Y. Ohtomo, M. Someya and H. Isobe, Nat. Commun., 2021, 12, 1575 CrossRef CAS PubMed.
- (a) A. Ito, M. Urabe and K. Tanaka, Angew. Chem., Int. Ed., 2003, 24, 921–924 CrossRef PubMed; (b) C. Wentrup, M. J. Regimbald-Krnel, D. Mgller and P. Comba, Angew. Chem., Int. Ed., 2016, 55, 14600–14605 CrossRef CAS PubMed; (c) P. Maslak, M. P. Augustine and J. D. Burkey, J. Am. Chem. Soc., 1990, 112, 5359–5360 CrossRef CAS.
- (a) T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed; (b) C. Poriel, L. Sicard and J. Rault-Berthelot, Chem. Commun., 2019, 55, 14238–14254 RSC; (c) S. Liu, D. Xia and M. Baumgarten, ChemPlusChem, 2021, 86, 36–48 CrossRef CAS PubMed.
- (a) L. -S. Cui, S. -B. Ruan, F. Bencheikh, R. Nagata, L. Zhang, K. Inada, H. Nakanotani, L. -S. Liao and C. Adachi, Nat. Commun., 2017, 8, 2250 CrossRef PubMed; (b) J. Yang, M. -C. Yoon, H. Yoo, P. Kim and D. Kim, Chem. Soc. Rev., 2012, 41, 4808–4826 RSC; (c) N. Aratani, D. Kim and A. Osuka, Acc. Chem. Res., 2009, 42, 1922–1934 CrossRef CAS PubMed.
- (a) T. Murahashi, M. Fujimoto, M. Oka, Y. Hashimoto, T. Uemura, Y. Tatsumi, Y. Nakao, A. Ikeda, S. Sakaki and H. Kurosawa, Science, 2006, 313, 1104–1110 CrossRef CAS PubMed; (b) T. Ishikawa, A. Kawamura, T. Sugawa, R. Moridaira, K. Yamamoto and T. Murahashi, Angew. Chem., Int. Ed., 2019, 58, 15318–15323 CrossRef CAS PubMed; (c) Y. Ishikawa, K. Yamamoto and T. Murahashi, Angew. Chem., Int. Ed., 2017, 56, 1346–1350 CrossRef CAS PubMed.
- (a) Y. Sunada, R. Haige, K. Otsuka, S. Kyushin and H. Nagashima, Nat. Commun., 2013, 4, 3014 CrossRef PubMed; (b) C. Yanagisawa, S. Yamazoe and Y. Sunada, ChemCatChem, 2021, 13, 167–173 Search PubMed; (c) K. Shimamoto and Y. Sunada, Chem. Commun., 2021, 57, 7649–7652 RSC; (d) K. Shimamoto and Y. Sunada, Chem. –Eur. J., 2019, 25, 3761–3765 CrossRef CAS PubMed; (e) R. Usui and Y. Sunada, Inorg. Chem., 2021, 60, 15101–15105 CrossRef CAS PubMed.
- Y. I. Baukov and S. N. Tandura, Hypervalent Compounds of Organic Germanium, Tin and Lead Derivatives, in The Chemistry of Organic Germanium, Tin and Lead Compounds, ed. Z. Rappoport, John Wiley & Sons, 2002, pp. 963–1239 Search PubMed.
- (a) A. G. Osborne and F. G. A. Stone, J. Chem. Soc. A, 1966, 1143–1146 RSC; (b) E. W. Abel and S. Moorhouse, Inorg. Nucl. Chem. Lett., 1971, 7, 905–908 CrossRef CAS.
- (a) K. Yamamoto, J. Sawada and T. Murahashi, Chem. –Eur. J., 2020, 26, 8388–8392 CrossRef CAS PubMed; (b) D. Fenske, H. Fleischer, H. Krautscheid, J. Magull, C. Oliver and S. Weisgerber, Z. Naturforsch., B: J. Chem. Sci., 1991, 46, 1384–1394 CrossRef CAS; (c) Z. Tang, Y. Nomura, S. Kuwata, Y. Ishii, Y. Mizobe and M. Hidai, Inorg. Chem., 1998, 37, 4909–4920 CrossRef CAS PubMed; (d) A. A. Pasynskii, I. L. Eremenko, B. Orazsakhatov, G. Sh. Gasanov, V. E. Shklover and Yu. T. Struchkov, J. Organomet. Chem., 1984, 269, 147–153 CrossRef CAS; (e) G. Longoni, M. Manassero and M. Sansoni, J. Am. Chem. Soc., 1980, 102, 3242–3244 CrossRef CAS.
- (a) K. Osakada, Y. Tsuchido and M. Tanabe, Coord. Chem. Rev., 2020, 412, 213195 CrossRef CAS; (b) T. Murahashi and H. Kurosawa, Coord. Chem. Rev., 2002, 231, 207–228 CrossRef CAS.
- J. Henoch, A. Auch, F. Diab, K. Eichele, H. Schubert, P. Sirsch, T. Block, R. Pöttgen and L. Wesemann, Inorg. Chem., 2018, 57, 4135–4145 CrossRef CAS PubMed.
- (a) V. W. Day, R. O. Day, J. S. Kristoff, F. J. Hirsekorn and E. L. Muetterties, J. Am. Chem. Soc., 1975, 97, 2571–2573 CrossRef CAS; (b) A. J. Touchton, G. Wu and T. W. Hayton, J. Chem. Phys., 2021, 154, 1/211102–211105/211102 CrossRef PubMed; (c) M. Green, J. A. K. Howard, M. Murray, J. L. Spencer and F. G. A. Stone, J. Chem. Soc. Dalton. Trans., 1977, 1509–1514 RSC.
- T. -a. Koizumi, Y. Tsuchido and K. Osakada, Eur. J. Inorg. Chem., 2020, 2253–2259 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2087548 and 2087549. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02302d |
| This journal is © The Royal Society of Chemistry 2022 |