
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the timescale of the structural photo-response within oriented metal–organic framework films†
Sumea
Klokic
 a,
Denys
Naumenko
a,
Benedetta
Marmiroli
a,
Francesco
Carraro
b,
Mercedes
Linares-Moreau
b,
Simone Dal
Zilio
c,
Giovanni
Birarda
d,
Rupert
Kargl
e,
Paolo
Falcaro
b and
Heinz
Amenitsch
*a
a,
Denys
Naumenko
a,
Benedetta
Marmiroli
a,
Francesco
Carraro
b,
Mercedes
Linares-Moreau
b,
Simone Dal
Zilio
c,
Giovanni
Birarda
d,
Rupert
Kargl
e,
Paolo
Falcaro
b and
Heinz
Amenitsch
*a
aInstitute of Inorganic Chemistry, Graz University of Technology, 8010 Graz, Austria. E-mail: heinz.amentisch@tugraz.at
bInstitute of Physical and Theoretical Chemistry, Graz University of Technology, 8010 Graz, Austria
cIOM-CNR, Laboratorio TASC, S.S. 14, 163.5 km, Basovizza, Trieste 34149, Italy
dElettra Sincrotrone Trieste – SISSI Bio Beamline, S.S. 14, 163.5 km, Basovizza, Trieste 34149, Italy
eInstitute of Chemistry and Technology of Bio-Based Systems, Graz University of Technology, 8010 Graz, Austria
First published on 27th September 2022
Abstract
Fundamental knowledge on the intrinsic timescale of structural transformations in photo-switchable metal–organic framework films is crucial to tune their switching performance and to facilitate their applicability as stimuli-responsive materials. In this work, for the first time, an integrated approach to study and quantify the temporal evolution of structural transformations is demonstrated on an epitaxially oriented DMOF-1-on-MOF film system comprising azobenzene in the DMOF-1 pores (DMOF-1/AB). We employed time-resolved Grazing Incidence Wide-Angle X-Ray Scattering measurements to track the structural response of the DMOF-1/AB film upon altering the length of the azobenzene molecule by photo-isomerization (trans-to-cis, 343 nm; cis-to-trans, 450 nm). Within seconds, the DMOF-1/AB response occurred fully reversible and over several switching cycles by cooperative photo-switching of the oriented DMOF-1/AB crystallites as confirmed further by infrared measurements. Our work thereby suggests a new avenue to elucidate the timescales and photo-switching characteristics in structurally responsive MOF film systems.
Introduction
Stimuli-responsive materials change their properties as an immediate response towards an external trigger and have attracted great attention for their technological applicability as actuators, sensors or storage systems.1–3 The key to their outstanding functional versatility arises from their structure, which comprises the proper relationship between crystallinity and flexibility.4–6 A prominent example of stimuli-responsive materials is metal–organic frameworks (MOFs),6–10 which are built by a highly ordered porous network of metal ions and organic linker molecules, where both constituents allow chemically tuning of the flexibility of the framework's backbone.6,7,11–13 The structural response of most flexible MOFs can be reversibly triggered by a chemical or physical stimulus,8,14 which led to the ever-growing class of switchable MOFs.13,15,16 Herein, the term ‘switchable’ refers to MOFs that undergo a substantial and reversible change in the unit cell dimensions under the full retention of the framework's connectivity and crystallinity,10 as it was shown by molecular simulations exemplarily for MIL-53(Al),17 DMOF-1 (ref. 11) or CAU-13.18 Although the chemistry and the related design strategies regarding stimuli-responsive MOFs is enormously rich in the literature,6–9,13,19–21 little is known about the temporal response characteristics of the MOF structure towards the incoming stimulus.3,7,22 Yet, only by understanding the MOF response over the time course of its stimuli-induced switch, the design of MOF systems with the desired response kinetics becomes feasible.22,23 This idea of controlling the intrinsic timescale of dynamic structural transformations in MOFs has recently led to the concept of four-dimensional MOFs (4D-MOFs).22 In 4D-MOFs, the time-axis is considered as the fourth dimension and represents the temporal evolution of a stimuli-induced structural transformation in a flexible MOF system. As pointed out by Evans et al.,22 Ehrling et al.16 and Haldar et al.,3 an open challenge is the design of switchable MOF systems with precise temporal responses. However, we note that this goal can only be achieved by the development of a methodological approach that elucidates the time-axis of the structural response in light-responsive MOF systems.It has been recently outlined in numerous reviews that amongst the flexible MOFs,3,8,20,22,24 especially for photo-switchable MOF film systems the structural dynamics remain largely unexplored,25–28 which is mainly because the MOF films employed in previous studies were structurally non-responsive.25,29–33
In this work, we demonstrate the example of a photo-switchable, azobenzene infiltrated DMOF-1/AB film system (DMOF-1 = Zn2BDC2DABCO, BDC = terephthalic acid, DABCO = 1,4-diazabicyclo[2.2.2]octane; AB = azobenzene),28,34–36 and that the photo-triggered temporal response of the DMOF-1/AB structure can be deliberately accelerated by 3 orders of magnitude simply by choosing the oriented MOF-on-MOF film-fabrication strategy37–39 over the bulk system.34 The oriented film growth is ensured by matching the lattice constants of the upper DMOF-1 structure with the lower Cu2BDC2-on-Cu(OH)2 MOF layer,37,38 where the latter is grown in a first step with its (001) plane oriented parallel to the glass substrate.38,40,41 The absorption of ultraviolet light provokes the infiltrated azobenzene molecule3,36,42 in the DMOF-1 pores34,43 to isomerize between its trans-to-cis configuration (ON), whilst visible light triggers the reversed cis-to-trans transition (OFF).3,8,44 Our findings show that within seconds this photo-induced isomerization process forces the DMOF-1 film structure to adapt,28,34,42a thus switching between the ON and OFF state. Yet, although various techniques have been demonstrated to monitor the stimuli-induced structural response over time in switchable bulk MOF systems, such as spectroscopy,23,45 X-ray diffraction46 and similar ones,47 there is still an open quest for experimental techniques which are applicable for MOF film structures.16 To this aim, we herein further report a methodological approach to study the photo-switching behavior of the DMOF-1/AB film system by time-resolved Grazing Incidence Wide Angle X-Ray Scattering (GIWAXS) using synchrotron radiation in combination with laser irradiation sources.48 Moreover, GIWAXS is a powerful tool for real-time monitoring of the structural responses where both, the DMOF-1/AB film surface and the buried Cu2BDC2-on-Cu(OH)2 layer can be resolved with high resolution.49 Hence, based on GIWAXS and infrared spectroscopic data, we found that the azobenzene isomerization channels the entire DMOF-1/AB film in a cooperative lattice movement16,50 to adapt to the size of the infiltrated azobenzene molecule and demonstrate that this ON and OFF behavior of the DMOF-1/AB film is fully reversible over several cycling repetitions.
To the best of our knowledge, an oriented MOF film which undergoes a photo-induced switch of both the azobenzene and the MOF-cage, as defined in this study, has not been reported so far. The great interest in photo-switchable MOFs is mainly because light is arguably one of the most attractive stimuli,21 as it provokes non-destructively the material's response which can be remotely controlled over certain distances.8,26,33,51 Hence, characterizing the timescale of dynamic transitions in photo-switchable MOF films will especially aid in their development for applications21,30,42 in sensing,52 energy storage28 or cargo uptake and release.21,53
Results and discussion
The procedure for the fabrication of the DMOF-1-on-MOF film system is illustrated in Fig. 1(a). The lower MOF layer comprises the oriented Cu2BDC2 structure grown on the Cu(OH)2 nanobelt film, which is fabricated by following the secondary heteroepitaxial growth strategy38 based on an automated nanobelt deposition procedure.41 The subsequent DMOF-1 growth occurs by matching its crystal lattice with the lower Cu2BDC2-on-Cu(OH)2 layer (Fig. 1(b and c))38 and is obtained by following a two-step method: Firstly, the Zn(acetate)2 nodes are covalently attached to the loose carboxylate groups of the Cu2BDC2-on-Cu(OH)2 layer, which is then followed by converting the film in a methanolic linker solution (H2BDC = terephthalic acid, DABCO = 1,4-diazabicyclo[2.2.2]octane). Thusly, the heteroepitaxial Cu2BDC2 MOF film was used for the fabrication of the oriented DMOF-1 system by following the MOF-on-MOF approach.40a The orientation of the fabricated DMOF-1 film system was confirmed by GIWAXS and XRD (X-ray diffraction) measurements (Fig. S1(a and b)†), with the data for the guest-free DMOF-1 system being consistent with the previously reported tetragonal P4/mmm structure (Fig. S1(c)†).43 Considering the lattice parameters of the DMOF-1 and Cu2BDC2-on-Cu(OH)2 systems (Cu2BDC2: P4 space group, a = 10.61 Å, b = 5.80 Å, c = 10.61 Å,38DMOF-1: P4/mmm space group, a = 10.93 Å, b = 10.93 Å, c = 9.61 Å),43 we expected the crystal lattice match to occur between the a and b axes of the DMOF-1 and the Cu2BDC2 structure, respectively (Fig. 1(b and c)). The theoretically calculated lattice mismatch between the two MOF systems accounts for 2.9% for the a axis, and 5.7% for the b axis of the DMOF-1 system, which is in the range of epitaxial growth.37,38,40,54 Based on X-ray data, the (100) diffraction signal of DMOF-1 was observed for the two in-plane XRD geometries with the X-ray incident angle set parallel and perpendicular to the long axis of the Cu(OH)2 nanobelts (Fig. S1(a–c)†).38 In the out-of-plane direction, the (001) diffraction signal of DMOF-1 and Cu2BDC2-on-Cu(OH)2 is observed (Fig. S1(c)†). Hence, these findings indicate that the (h00) and (0k0) faces of the DMOF-1 structure are oriented perpendicular to the substrate. Moreover, the (100) DMOF-1 and the (100) Cu2BDC2 diffraction peaks show a clear angular dependence with the two maxima coinciding at 90° and 270° (Fig. S1(d and e)†). These results support our scientific hypothesis and demonstrate a crystal lattice matching between the a and b axes of DMOF-1 and Cu2BDC2-on-Cu(OH)2 (Fig. 1(b and c)). The nascent framework grows therefore with the aid of the pillaring agent DABCO in the c axis direction with the DMOF-1 pores being available for the introduction of azobenzene as the photo-active molecule (see Fig. 1(d)).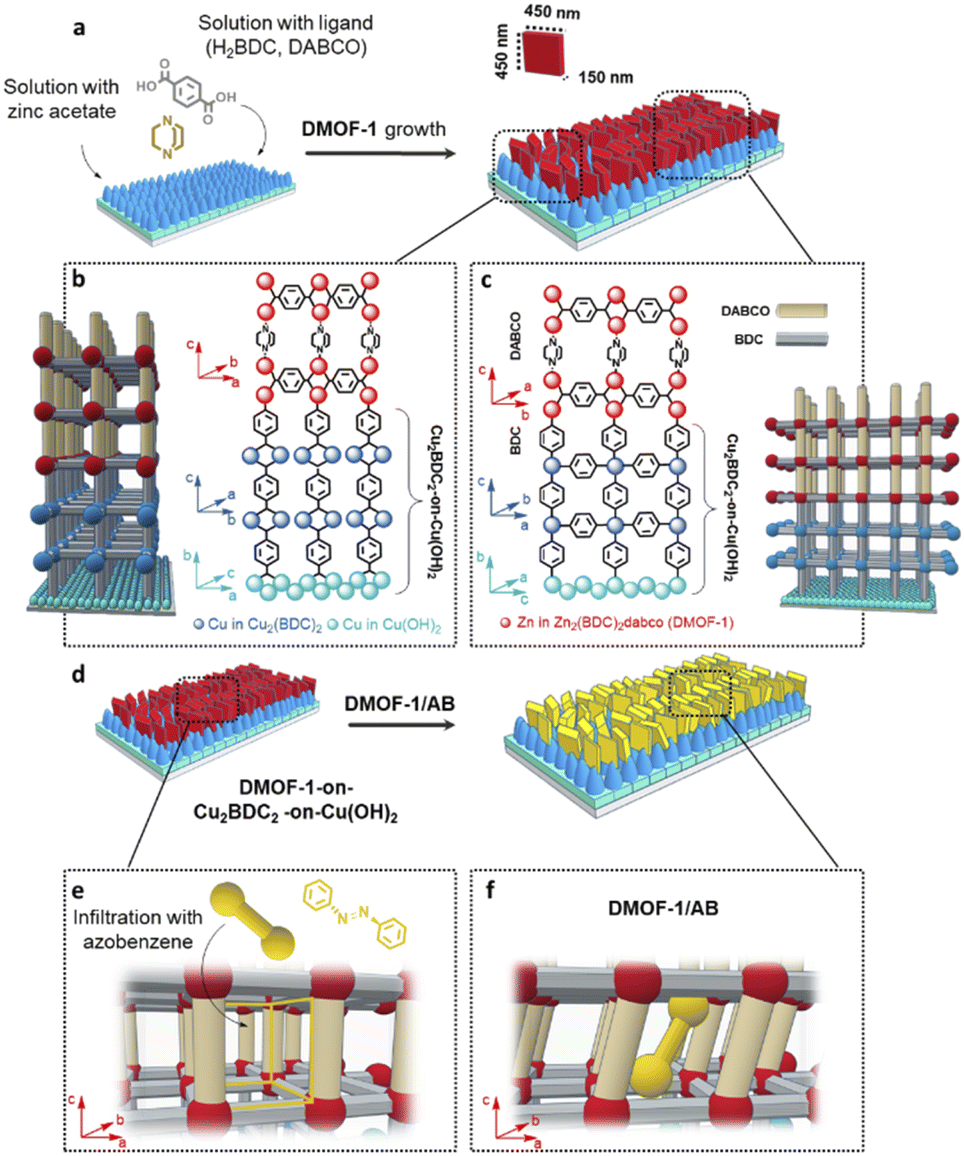 | ||
| Fig. 1 Growth of DMOF-1 via heteroepitaxial growth and subsequent infiltration by azobenzene. (a) Synthetic scheme for the growth of the DMOF-1 film, starting from the heteroepitaxial oriented Cu2BDC2-on-Cu(OH)2 system. The Cu2BDC2-on-Cu(OH)2 layer is first immersed in an ethanolic zinc acetate solution, then in the methanolic linker solution of DABCO and H2BDC. (b and c) Magnified image on the interface of the heteroepitaxially grown DMOF-1 (red cubes) on Cu2BDC2-on-Cu(OH)2 along the parallel and perpendicular directions of Cu(OH)2 nanobelts. (d) Synthetic scheme for the infiltration of DMOF-1 films with azobenzene. (e) Schematic of the infiltration process: the azobenzene molecule enters the vacant MOF pores (yellow cubes) and (f) the flexible DMOF-1 structure is forced to adapt to the size of the infiltrated azobenzene molecule resulting in the DMOF-1/AB assembly. | ||
The morphology of the DMOF-1-on-MOF film was assessed by SEM after each step of the film fabrication process. In Fig. 2, the top-view SEM images show the first MOF layer comprising oriented Cu(OH)2 nanobelts (Fig. 2(a)) and after their conversion to Cu2BDC2 (Fig. 2(b)).38 The growth of the DMOF-1 system on the oriented Cu2BDC2-on-Cu(OH)2 film yields cuboid-shaped crystallites assembled as the final MOF-on-MOF film (Fig. 2(c and d)). The lateral size of the DMOF-1 crystallites was determined to be ∼450 nm with a width of ∼150 nm (see the inset in Fig. 1(a)), whilst the Cu2BDC2-on-Cu(OH)2 film accounts for ∼500 nm.38 The SEM micrographs show a preferential out-of-plane orientation of the DMOF-1 crystallites with respect to the substrate. Detailed analysis of the XRD data was performed to estimate the degree of oriented DMOF-1 crystallites (see the ESI, Fig. S2†).55 Results showed that from the DMOF-1 crystallites a fraction of 45% is preferentially oriented towards the out-of-plane orientation with a degree of alignment of 18° as determined with the azimuthal full width half maximum of the (100) reflection in the azimuthal cut (Fig. S2(d)†).41
 | ||
| Fig. 2 SEM micrographs for the stepwise fabrication of DMOF-1 films. (a) The film preparation is performed starting from an oriented Cu(OH)2 nanobelt film, (b) which is converted to Cu2BDC2. (c and d), Subsequent growth of DMOF-1 shows a preferential out-of-plane alignment of the crystals in the magnified image. | ||
To evaluate the porosity, the DMOF-1-on-Cu2BDC2-on-Cu(OH)2 film was deposited on a quartz crystal microbalance (QCM), where the resonance frequency change of each MOF layer was measured upon exposure to methanol vapor (Fig. S3(a)†). Owing to the porosity of the film, the DMOF-1 structure takes up significantly more methanol compared to the Cu2BDC2 and the bare reference crystal (Fig. S3(b)†). The methanol uptake of DMOF-1 demonstrates the pore accessibility by guest molecules. Accordingly, in the next step, we infiltrated the DMOF-1 pores with azobenzene by a vapor-phase infiltration process yielding the DMOF-1/AB film system (Fig. S4†). Considering the flexibility of the DMOF-1 structure,34,36 we expected the DMOF-1 cage (7.5 Å × 7.5 Å) to adapt to the infiltrated azobenzene (9.0 Å) (see Fig. 1(e and f)).28,34,36 This expected shrinkage of the DMOF-1 cage was confirmed by GIWAXS measurements and is evidenced by the splitting of the (100) reflection at q = 5.85 nm−1, which accounts for a change of Δd = 0.12 Å in the d-spacing (Fig. 3(a and b)). This shrinkage results in the coexistence of two crystalline tetragonal phases (Fig. 3(b)), which has been previously reported to arise from the guest-induced breathing of the infiltrated DMOF-1 bulk structures.28,34 Importantly, the orientation of the DMOF-1 layer is not lost despite a clear structural change (Fig. S5(a)†). Moreover, the position of the X-ray reflection corresponding to the Cu2BDC2 remains unaffected (Fig. 3(b)), which implies that although the DMOF-1 structure was rearranged, no additional stress is induced to the underlying structure.
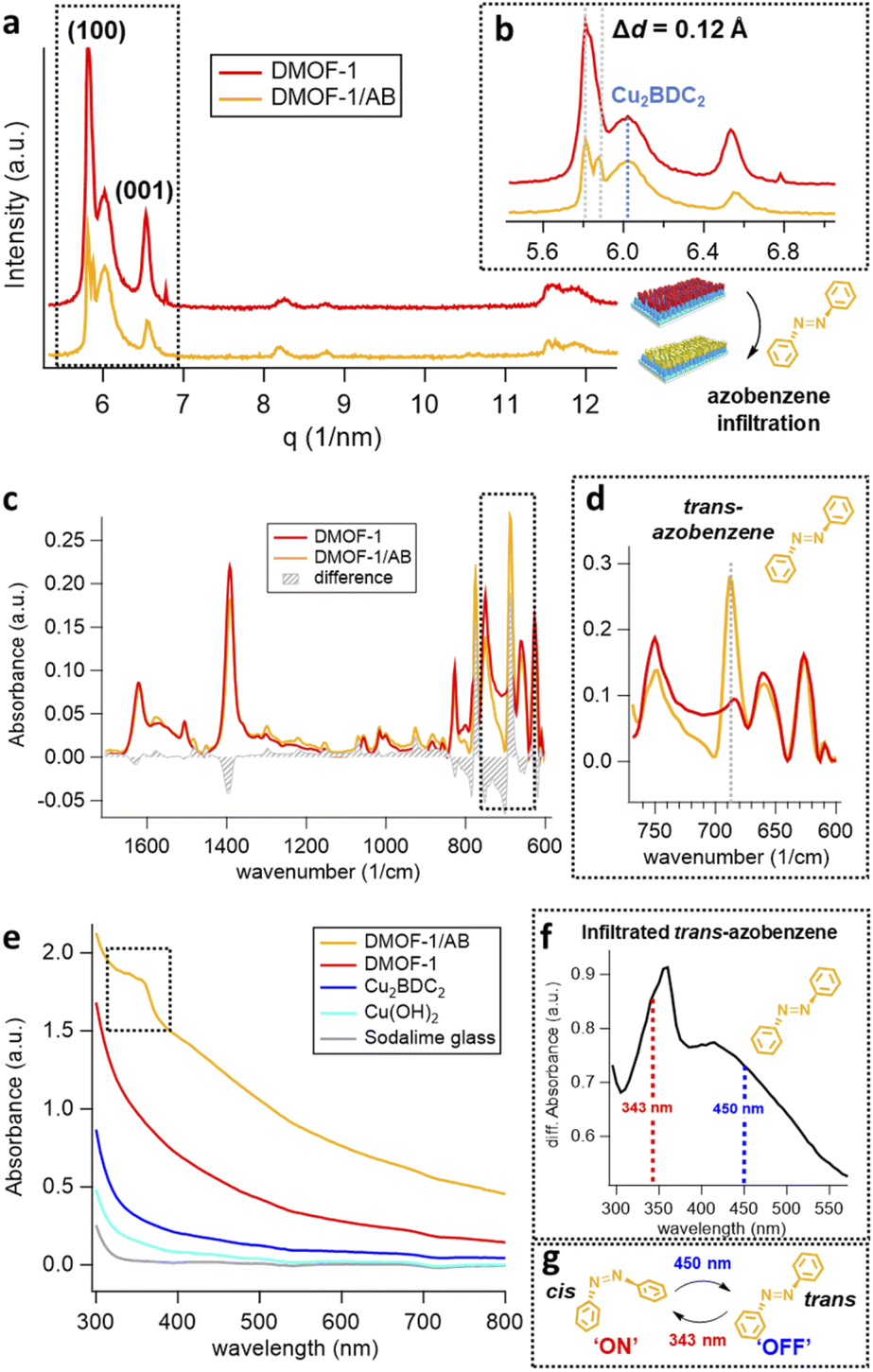 | ||
| Fig. 3 Characterization of the DMOF-1/AB films on glass substrates. (a) GIWAXS pattern of the DMOF-1-on-Cu2BDC2-on-Cu(OH)2 film structure (red trace) and upon infiltration by azobenzene (yellow trace). (b) The shrinkage of the DMOF-1 structure by Δd = 0.12 Å is not affecting the Cu2BDC2-on-Cu(OH)2 system. (c) ATR measurements prior and after infiltration of the DMOF-1 film show the presence of trans-azobenzene as indicated by the (d) intense infrared band at 687 cm−1. (e) UV/Vis absorbance spectra of the DMOF-1/AB film system comprising (from bottom to top), the substrate (sodalime glass), Cu(OH)2 nanobelts, their conversion to Cu2BDC2, DMOF-1 and the infiltrated DMOF-1/AB. (f) Differential absorption spectrum of the infiltrated azobenzene in DMOF-1/AB with the absorption band of the π–π* electronic transition located at ∼360 nm and the n–π* electronic transition at ∼420 nm. The dotted lines mark the wavelengths used to photo-switch the infiltrated azobenzene (g) between its cis-isomer in the ON state (343 nm) and the trans-conformer in the OFF state (450 nm). | ||
The DMOF-1/AB crystallites comprise azobenzene molecules in the trans-configuration which is evidenced by the IR absorption band located at 687 cm−1 (Fig. 3(d)).34 The reduced methanol uptake of azobenzene infiltrated DMOF-1 films indicates the occupation of the DMOF-1 pores with the photo-active molecules (Fig. S3(b)†).42b Moreover, we determined that the infiltration process can be repeated by washing the DMOF-1/AB system with ethanol to remove the azobenzene without destroying the crystallinity and orientation of the MOF structure (Fig. S5(b and c)†).
Time-resolved GIWAXS measurements for the photo-switching of DMOF-1/AB
To evaluate the excitation wavelengths for the photo-isomerization process, the oriented DMOF-1/AB films were purposefully grown on glass substrates to consent UV-Vis characterization. The spectra depicted in Fig. 3(e) show a continuous increase in absorbance at <400 nm, due to the introduction of additional metal-nodes when assembling the DMOF-1/AB-on-Cu2BDC2-on-Cu(OH)2 system.30 The absorption spectrum of the infiltrated azobenzene provided in Fig. 3(f) comprises a broad absorption band located at ∼350 nm (π–π* transition) and one very weak band around ∼440 nm (n–π* transition), which is consistent with the trans-azobenzene in solution (Fig. S6†).2,33 Hence, to successfully trigger the photo-switch of the azobenzene molecule, we prosecuted the forward switch (trans-to-cis, ON) with an excitation wavelength of 343 nm, and reversed the photo-switch (cis-to-trans, OFF) with 450 nm (Fig. 3(g)).To track the time-resolved structural changes of the DMOF-1/AB system during the photo-switching experiments, we performed GIWAXS measurements as schematically depicted in Fig. 4(a) and the protocol outlined in Fig. 4(b). In the beginning (t ∼ 0 s), the DMOF-1/AB structure is relaxed and the infiltrated azobenzene is in its trans-configuration (OFF). Upon excitation with 343 nm, the isomerization of the chromophore to its cis conformer is provoked (forward switch) and consequently, DMOF-1/AB converts to the ON state. Relaxation of the structure when exposed to 450 nm is initiated by the re-isomerization of azobenzene, and the OFF state is reached again (backward switch, see Fig. 3(g)). Typically, one photo-switch requires 100 s to reach the steady state, and to demonstrate the stability of the system we have repeated the ON/OFF photo-switch for 7 cycles. Between the OFF and ON state, we found that the most pronounced changes occurred for the (001) reflection (upper inset, Fig. 5(a)), which implies that the DMOF-1 cage moves in the direction of the pillaring DABCO linker.28 This property is supported by polarization-angle dependent absorption measurements39 at 343 nm wavelength, as the cis-azobenzene molecules were found to align preferentially along the DMOF-1c-axis direction (Fig. S7†). Hence, the photo-switch results mainly in a structural change along the c-axis direction (inset, Fig. 5(a)), which is further confirmed by the lack of significant structural changes in the in-plane direction (Fig. S8†). Closer inspection of the (001) reflection in the out-of-plane direction shows its downshift by Δq = 0.013 nm−1 between the OFF and ON state which channels a structural change by Δd = 0.02 Å (Fig. S9(a)†). This is due to the larger trans-azobenzene molecule (9.0 Å) compared to its cis-conformer (5.5 Å),8 which urges the DMOF-1 pores to adapt between the two configurations (see schematics in Fig. 5(a)). Moreover, the structural stability of the crystalline DMOF-1/AB structure throughout the photo-cycling is confirmed as all reflections were found to remain unaltered over the entire experiment.
 | ||
| Fig. 4 Time-resolved GIWAXS measurements. (a) Schematic GIWAXS set-up for the photo-switching of DMOF-1/AB film structures with the sample-to-detector distance (SD) chosen to track the time-resolved evolution of the (100) and (001) reflections. Spatial overlap of the synchrotron X-ray beam with the excitation light sources for the forward (343 nm) and back isomerization (450 nm) is depicted in the inset. (b) Experimental protocol for the DMOF-1/AB photo-switching, where at t ∼ 0 s DMOF-1/AB is relaxed (OFF). Photoexcitation with 343 nm light radiation (time zero, tON) induces the forward switch (ON) and is evidenced by a sudden drop in the scattering intensity due to the structural changes of DMOF-1/AB. Back-switching of the structure to OFF is performed by excitation with 450 nm (time zero, tOFF), which is accompanied by an increase in the scattered intensity to the initial state (see the inset). | ||
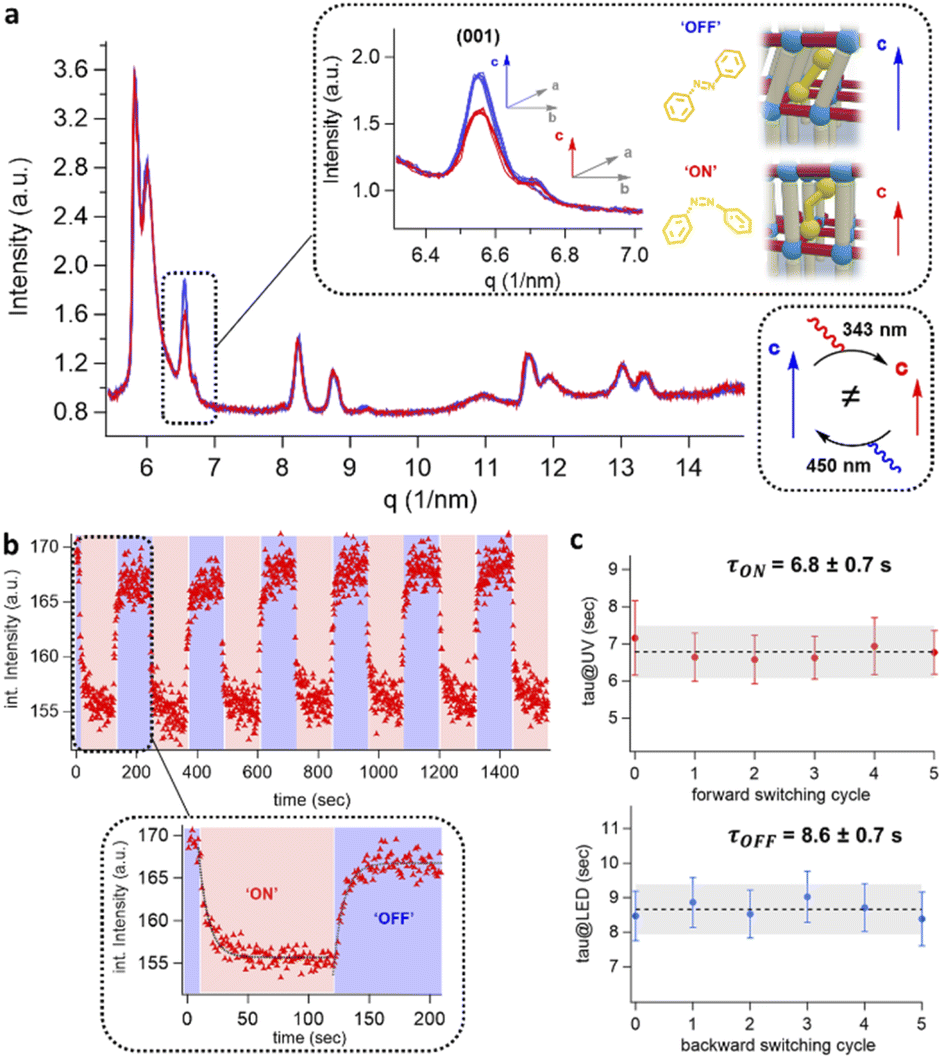 | ||
| Fig. 5 Solid-state photo-switching of the DMOF-1/AB system by time-resolved photoexcitation GIWAXS measurements. (a) GIWAXS radial integrated data of the OFF DMOF-1/AB structure (blue traces) and the ON structure (red traces) on glass substrates. The inset displays the (001) reflection corresponding to the c lattice axis with the most prominent structural changes. (b) The DMOF-1/AB structure was reversibly cycled between the OFF and ON state. The change in peak intensity of the (001) reflection is displayed as integrated intensity as a function of time. The black lines in the inset are the fitting functions to evaluate the switching kinetics. (c) Forward switching to the ON state at 343 nm with an average time constant τON = 6.8 ± 0.7 s and backward switching to the OFF state at 450 nm with an average time constant τOFF = 8.6 ± 0.7 s. | ||
To deduce the switching kinetics of DMOF-1/AB, we performed exponential fits on the integrated intensity of the (001) reflection for the forward and backward switch (Fig. 5(b), inset), which are expressed as
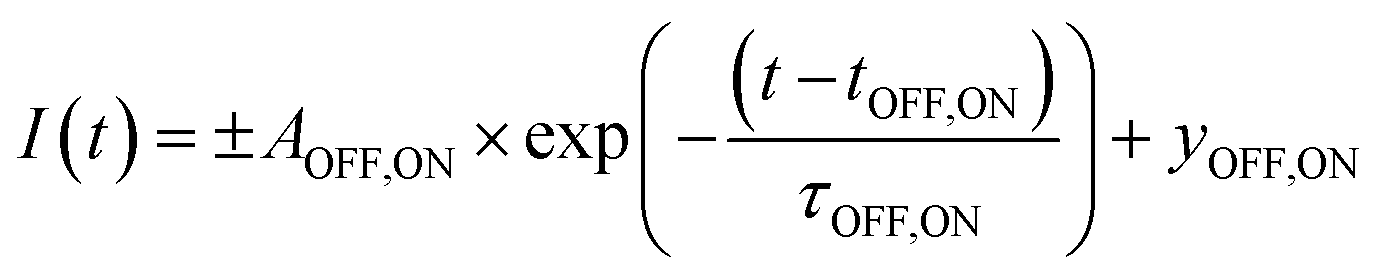
The initial photo-excitation process is evoked by the electronic transitions of the chromophore upon UV exposure, which typically occurs on the time scale of ∼1 ps.60 The structural changes related to the photo-isomerization of azobenzene in solvents were reported to require 0.5–1 ps,60,61 and up to 180–190 ps62 or even seconds in liquid crystals.63 However, photo-switching in solid azobenzene systems such as functionalized azo-adlayers on Au-surfaces strongly decelerates the switch and was found to require 16 s,64 whilst azobenzene-based films that undergo a photo-induced structural phase transition from crystalline to a disordered system switch on the time-scale of ∼220 s.56 In comparison, the photo-response of the herein proposed DMOF-1/AB film experiences one full switching cycle within 15 s (ON/OFF), which is one of the fastest observed structural transitions for azobenzene-containing solid crystalline films56,64,65 and one of the first for structurally photo-switchable MOF films.25,26,30–33,66
Moreover, in relation to the bulk DMOF-1/AB system which was reported to require >300 min for its structural change,34,36 preparation of DMOF-1/AB in oriented films accelerated this process by 3 orders of magnitude (see Fig. S11† for bulk DMOF-1/AB experiments). These findings imply that the fabrication of crystalline and oriented film structures is the key for the successful and fast photo-switching in solids, which we hypothesize to arise from the improved light penetration in the MOF film system.3
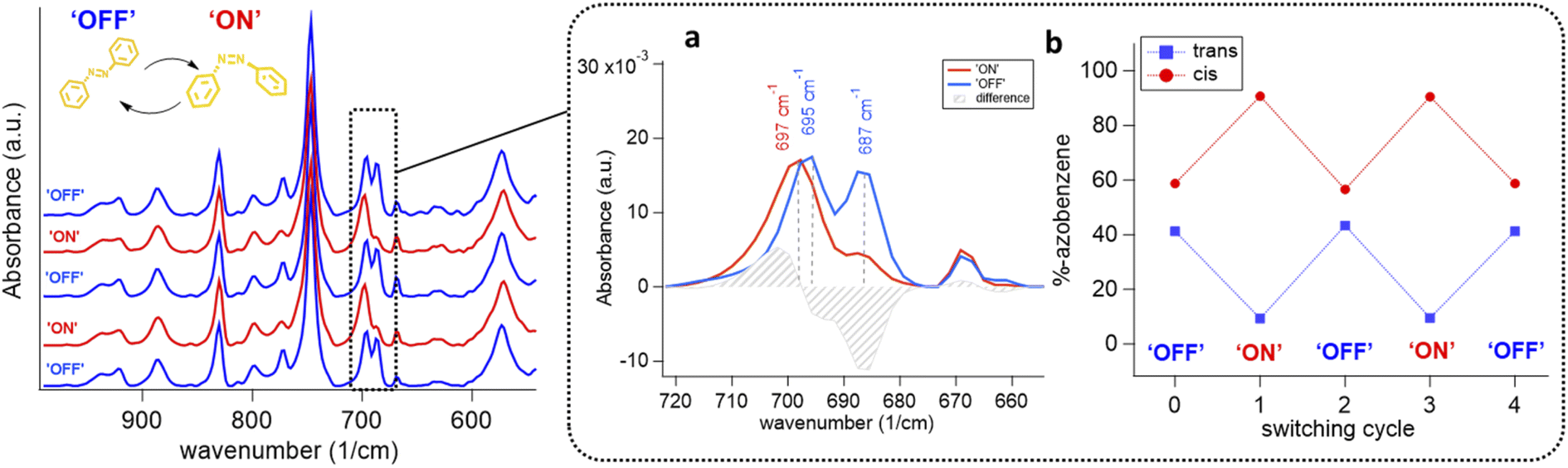 | ||
| Fig. 6 Infrared measurements on the photo-switched structures. Infrared spectra of the oriented DMOF-1/AB films switched repeatedly from OFF (blue trace) to ON (red trace). (a) Inset displays the infiltrated azobenzene when in trans (blue) and cis-configuration (red) with a shift of Δν = 2 cm−1. (b) The active photo-switchable azobenzene portion between the ON and the OFF state was determined from infrared measurements to account for 40% of the infiltrated azobenzene. The ON and OFF cycling as a function of the azobenzene percentage supports the reversible switching behavior. | ||
The freshly infiltrated DMOF-1/AB films comprise trans-azobenzene within the MOF pores (vide supra). However, the first photo-isomerization cycle of azobenzene within the DMOF-1/AB pores (ON/OFF) was found to prevent the azobenzene molecules from fully reversing to their initial trans-conformer (Fig. S12†). Repetition of the photo-cycling experiment revealed an irreversibly ‘trapped’ portion of cis-azobenzene molecules being present in the relaxed OFF state (Fig. 6(a)). In the OFF state, the DMOF-1/AB film structure reaches a trans/cis-azobenzene ratio of 0.70. Comparing the ON to the OFF state, the two cis-azobenzene absorption bands comprise a red-shift of Δν = 2 cm−1 (Fig. 6(b)).67 Similarly, the strong C–O vibration68 arising from the carboxylate in the DMOF-1 structure is shifted by Δν = 8 cm−1 (Fig. S9(d)†).69 This property points to a strong interaction between the DMOF-1 cage and the infiltrated azobenzene molecules.36 Interestingly, for poorly oriented DMOF-1/AB films almost no shift of the vibrational bands was found to arise between the ON and the OFF switch (Fig. S13†). This property indicates the importance of fabricating an oriented DMOF-1/AB film structure as the concomitant interactions seem to be crucial for the progress of the photo-switch.16
Based on this, we presume that the initial trans-to-cis azobenzene isomerization already leads to the full structural change of the oriented DMOF-1/AB film system. During the initial azobenzene isomerization the interactions between the DMOF-1 cage and the infiltrated azobenzene molecules are weakened, provoking adjacent azobenzene molecules to isomerize whilst the DMOF-1 structure has already switched to the ON state.36 The switching process can therefore be described by a ‘cooperative movement’ of the oriented DMOF-1/AB crystallites, which would correlate with the seconds-time scale that is required to switch the system.16,23 To account for the origin of the strained cis-azobenzene molecules, we increased the power density of the 343 nm light source from 1.2 to 95 mW cm−2 to understand if the cis/trans ratio would change (Fig. S14†). However, independent of the photon power density the portion of photo-active azobenzene molecules in the DMOF-1 pores remained unchanged, which accounts for ∼40% (Fig. 6(b)). This percentage is comparable to the cis-portion reported for bulk DMOF-1/AB systems28,34 and is eventually an intrinsic property of the system that can be attributed to the loading level of one azobenzene molecule per DMOF-1 pore, which is the estimated loading for the DMOF-1/AB films (see the ESI†).28,31,36
However, the presence of azobenzene molecules frozen in the cis-configuration is still unclear, since no correlation between the film preparation conditions or chemical parameters could be established.31 As a comparison, this behavior was not reported for the isostructural well-oriented yet non-flexible Cu2BDC2DABCO system when using Cu(OH)2 nanobelts.39 Based on this, we presume that these irreversibly ‘trapped’ molecules are both a consequence of the interaction of the azobenzene molecule with the DMOF-1 cage28,31,36 and the considerable lattice mismatch between the DMOF-1 and the Cu2BDC2-on-Cu(OH)2 structure (vide supra). This property eventually creates ‘strained’ DMOF-1 pores, which trap these in their cis-conformation upon azobenzene infiltration thus inhibiting the backward relaxation due to a better stabilization of the cis-isomer.28 Therefore, the ON-to-OFF switching characteristics of the DMOF-1/AB film are mainly related to the interactions arising between the MOF cage and the infiltrated azobenzene molecules,28,31,36 as well as to the degree of azobenzene loading.23,28,36 In addition, the influence of the total DMOF-1-Cu2BDC2-on-Cu(OH)2 film thickness is another crucial parameter to assess, which will be the scope of future research. Nevertheless, IR data (Fig. 6(b)) support the reversibility of the photo-switching process and verify that the crystalline DMOF-1 structure responds to the photo-switch of the azobenzene.
Conclusions
This paper demonstrates an experimental strategy to characterize and quantify the time-resolved structural transformations by time-resolved GIWAXS measurements, as outlined on the herein employed photo-switchable DMOF-1/AB-on-Cu2BDC2-on-Cu(OH)2 oriented film system. The described set-up involves two light sources of 343 and 450 nm wavelengths, which triggered the isomerization of azobenzene within the DMOF-1 pores that was found to provoke a strong structural response along the c axis of the DMOF-1 structure. As further evidenced by infrared spectroscopic measurements, strong interactions within the oriented film system promoted the DMOF-1 film structure to cooperatively switch within 15 s for one ON/OFF cycle. In comparison to the bulk system, the herein fabricated oriented DMOF-1/AB film was found to accelerate the photo-switching process from hours to the seconds-scale, being moreover reversible and free of fatigue for several switching repetitions. The photo-switching of the oriented DMOF-1/AB film was shown to be one of the fastest observed structural transitions for azobenzene-containing solid crystalline films, and one of the first for photo-switchable MOF films. With the herein selected approach to design photo-switchable MOF films together with the employed methodology we aim to introduce a general conceptual advance towards 4D-MOFs to assess the photo-responsive behavior of similar or even more advanced solid film systems.Data availability
Experimental data have been provided in the ESI.†Author contributions
H. A., P. F. and S. K. conceived and designed the project. S. K. carried out most of the experimental work including the optimization and fabrication of the system and wrote the manuscript. D. N. and B. M. assisted with the data analysis and completed the GIWAXS measurements with S. K., S. D.-Z. provided some starting materials and performed SEM imaging. G. B. completed the infrared measurements. F. C. and M. L.-M. completed the XRD measurements, whilst R. K. conducted the QCM measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Max Burian, Dr Barbara Sartori, PhD Marcello Solomon, Ing. Andrea Radeticchio, DI Florian Lackner and Tobias Steindorfer for experimental and technical support. The authors acknowledge support from the European Research Council under the European Union's Horizon 2020 Programme (FP/2014-2020)/ERC Grant Agreement No. 771834—POPCRYSTAL and TU Graz for the Lead Project LP-03: Porous Materials @ Work for Sustainability. The authors also acknowledge the CERIC-ERIC Consortium for access to the Austrian SAXS beamline and SISSI beamline at ELETTRA.Notes and references
- A. Gonzalez, E. S. Kengmana, M. V. Fonseca and G. Han, Mater. Today Adv., 2020, 6, 100058 CrossRef.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, e1905966 CrossRef.
- R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2020, 32, e1905227 CrossRef PubMed.
- E. Moulin, L. Faour, C. C. Carmona-Vargas and N. Giuseppone, Adv. Mater., 2020, 32, e1906036 CrossRef PubMed.
- A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600 CrossRef CAS PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed.
- Z. Chang, D.-H. Yang, J. Xu, T.-L. Hu and X.-H. Bu, Adv. Mater., 2015, 27, 5432 CrossRef CAS PubMed.
- F. Bigdeli, C. T. Lollar, A. Morsali and H.-C. Zhou, Angew. Chem., Int. Ed., 2020, 59, 4652 CrossRef CAS.
- W. Cai, J. Wang, C. Chu, W. Chen, C. Wu and G. Liu, Adv. Sci., 2019, 6, 1801526 CrossRef PubMed.
- L. Vanduyfhuys, S. M. J. Rogge, J. Wieme, S. Vandenbrande, G. Maurin, M. Waroquier and V. van Speybroeck, Nat. Commun., 2018, 9, 204 CrossRef CAS PubMed.
- S. Bureekaew, S. Amirjalayer and R. Schmid, J. Mater. Chem., 2012, 22, 10249 RSC.
- S. Henke, A. Schneemann, A. Wütscher and R. A. Fischer, JACS, 2012, 134, 9464 CrossRef CAS PubMed.
- S.-Q. Wang, S. Mukherjee and M. J. Zaworotko, Faraday Discuss., 2021, 231, 9 RSC.
- Z. Liu, L. Zhang and D. Sun, Chem. Commun., 2020, 56, 9416 RSC.
- S. Kitagawa and M. Kondo, Bull. Chem. Soc. Jpn., 1998, 71, 1739 CrossRef CAS.
- S. Ehrling, H. Miura, I. Senkovska and S. Kaskel, Trends in Chem., 2021, 3, 291 CrossRef CAS.
- A. E. Hoffman, J. Wieme, S. M. Rogge, L. Vanduyfhuys and V. van Speybroeck, Cryst. Mater., 2019, 234, 529 CAS.
- A. U. Ortiz, A. Boutin and F.-X. Coudert, Chem. Commun., 2014, 50, 5867 RSC.
- (a) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC; (b) J. H. Lee, S. Jeoung, Y. G. Chung and H. R. Moon, Coord. Chem. Rev., 2019, 389, 161 CrossRef CAS; (c) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (d) A. M. Rice, C. R. Martin, V. A. Galitskiy, A. A. Berseneva, G. A. Leith and N. B. Shustova, Chem. Rev., 2020, 120, 8790 CrossRef CAS.
- É. Whelan, F. W. Steuber, T. Gunnlaugsson and W. Schmitt, Coord. Chem. Rev., 2021, 437, 213757 CrossRef.
- N. D. Shepherd and D. M. D'Alessandro, Chem. Phys. Rev., 2021, 2, 11301 CrossRef.
- J. D. Evans, V. Bon, I. Senkovska, H.-C. Lee and S. Kaskel, Nat. Commun., 2020, 11, 2690 CrossRef CAS PubMed.
- J. Nishida, A. Tamimi, H. Fei, S. Pullen, S. Ott, S. M. Cohen and M. D. Fayer, PNAS, 2014, 111, 18442 CrossRef CAS PubMed.
- A. B. Kanj, K. Müller and L. Heinke, Macromol. Rapid Commun., 2018, 39, 1700239 CrossRef.
- X. Yu, Z. Wang, M. Buchholz, N. Füllgrabe, S. Grosjean, F. Bebensee, S. Bräse, C. Wöll and L. Heinke, PCCP, 2015, 17, 22721 RSC.
- D. Mutruc, A. Goulet-Hanssens, S. Fairman, S. Wahl, A. Zimathies, C. Knie and S. Hecht, Angew. Chem., Int. Ed., 2019, 58, 12862 CrossRef CAS.
- H. Huang, H. Sato and T. Aida, JACS, 2017, 139, 8784 CrossRef CAS PubMed.
- K. Griffiths, N. R. Halcovitch and J. M. Griffin, Chem. Mater., 2020, 32, 9925 CrossRef CAS.
- A. Modrow, D. Zargarani, R. Herges and N. Stock, Dalton Trans., 2011, 40, 4217 RSC.
- K. Müller, J. Wadhwa, J. Singh Malhi, L. Schöttner, A. Welle, H. Schwartz, D. Hermann, U. Ruschewitz and L. Heinke, Chem. Commun., 2017, 53, 8070 RSC.
- T. Koehler, I. Strauss, A. Mundstock, J. Caro and F. Marlow, J. Phys. Chem. Lett., 2021, 12(36), 8903–8908 CrossRef CAS.
- Z. Wang, K. Müller, M. Valášek, S. Grosjean, S. Bräse, C. Wöll, M. Mayor and L. Heinke, J. Phys. Chem. C, 2018, 122, 19044 CrossRef CAS.
- K. Müller, A. Knebel, F. Zhao, D. Bléger, J. Caro and L. Heinke, Chemistry, 2017, 23, 5434 CrossRef PubMed.
- N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda and S. Kitagawa, JACS, 2012, 134, 4501 CrossRef CAS PubMed.
- Y. Kim, R. Haldar, H. Kim, J. Koo and K. Kim, Dalton Trans., 2016, 45, 4187 RSC.
- M. Rödl, S. Kerschbaumer, H. Kopacka, L. Blaser, F. R. S. Purtscher, H. Huppertz, T. S. Hofer and H. A. Schwartz, RSC Adv., 2021, 11, 3917 RSC.
- K. Ikigaki, K. Okada and M. Takahashi, ACS Appl. Nano Mater., 2021, 4, 3467 CrossRef CAS.
- P. Falcaro, K. Okada, T. Hara, K. Ikigaki, Y. Tokudome, A. W. Thornton, A. J. Hill, T. Williams, C. Doonan and M. Takahashi, Nat. Mater., 2017, 16, 342 CrossRef CAS PubMed.
- K. Okada, M. Nakanishi, K. Ikigaki, Y. Tokudome, P. Falcaro, C. J. Doonan and M. Takahashi, Chem. Sci., 2020, 11, 8005 RSC.
- (a) K. Ikigaki, K. Okada, Y. Tokudome, T. Toyao, P. Falcaro, C. J. Doonan and M. Takahashi, Angew. Chem., Int. Ed., 2019, 58, 6886 CrossRef CAS; (b) K. Ikigaki, K. Okada and M. Takahashi, ACS Appl. Nano Mater., 2021, 4, 3467–3475 CrossRef CAS; (c) T. Stassin, S. Rodríguez-Hermida, B. Schrode, A. Cruz, F. Carraro, D. Kravchenko, V. Creemers, I. Stassen, T. Hauffman and D. De Vos, et al., Chem. Commun., 2019, 55, 10056–10059 RSC.
- M. Linares-Moreau, L. A. Brandner, T. Kamencek, S. Klokic, F. Carraro, K. Okada, M. Takahashi, E. Zojer, C. J. Doonan and P. Falcaro, Adv. Mater. Interfaces, 2021, 8, 2101039 CrossRef CAS.
- (a) D. Das and H. Agarkar, ACS Omega, 2018, 3, 7630 CrossRef CAS PubMed; (b) N. Prasetya and B. Ladewig, ACS Appl. Mater. Interfaces, 2018, 10, 34291–34301 CrossRef CAS PubMed.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033 CrossRef CAS PubMed.
- R. Klajn, Pure Appl. Chem., 2010, 82, 2247 Search PubMed.
- (a) K. Titov, Z. Zeng, M. R. Ryder, A. K. Chaudhari, B. Civalleri, C. S. Kelley, M. D. Frogley, G. Cinque and J.-C. Tan, J. Phys. Chem. Lett., 2017, 8, 5035 CrossRef CAS PubMed; (b) B. Pattengale, S. Ostresh, C. A. Schmuttenmaer and J. Neu, Chem. Rev., 2022, 122, 132 CrossRef CAS PubMed.
- C. L. Hobday, R. J. Marshall, C. F. Murphie, J. Sotelo, T. Richards, D. R. Allan, T. Düren, F.-X. Coudert, R. S. Forgan and C. A. Morrison, et al., Angew. Chem., Int. Ed., 2016, 55, 2401 CrossRef CAS.
- S. Devautour-Vinot, G. Maurin, F. Henn, C. Serre, T. Devic and G. Férey, Chem. Commun., 2009, 2733 RSC.
- (a) M. Burian, B. Marmiroli, A. Radeticchio, C. Morello, D. Naumenko, G. Biasiol and H. Amenitsch, J. Synchrotron Rad., 2020, 27, 51 CrossRef CAS PubMed; (b) H. Amenitsch, M. Rappolt, M. Kriechbaum, H. Mio, P. Laggner and S. Bernstorff, J. Synchrotron Rad., 1998, 5, 506 CrossRef CAS.
- (a) A. Hexemer and P. Müller-Buschbaum, IUCrJ, 2015, 2, 106 CrossRef CAS PubMed; (b) M. Pflüger, V. Soltwisch, J. Probst, F. Scholze and M. Krumrey, IUCrJ, 2017, 4, 431 CrossRef PubMed.
- N. Kavoosi, V. Bon, I. Senkovska, S. Krause, C. Atzori, F. Bonino, J. Pallmann, S. Paasch, E. Brunner and S. Kaskel, Dalton Trans., 2017, 46, 4685 RSC.
- (a) F. Ercole, T. P. Davis and R. A. Evans, Polym. Chem., 2010, 1, 37 RSC; (b) Z. G. Fthenakis, Z. Zhu, D. Teich, G. Seifert and D. Tománek, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 245402 CrossRef; (c) Y. Jiang and L. Heinke, Langmuir, 2021, 37, 2 CrossRef CAS.
- R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, C. J. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695 CrossRef CAS PubMed.
- S. Castellanos, F. Kapteijn and J. Gascon, CrystEngComm, 2016, 18, 4006 RSC.
- Z. Wang, J. Liu, B. Lukose, Z. Gu, P. G. Weidler, H. Gliemann, T. Heine and C. Wöll, Nano Lett., 2014, 14, 1526 CrossRef CAS PubMed.
- (a) L. Oesinghaus, J. Schlipf, N. Giesbrecht, L. Song, Y. Hu, T. Bein, P. Docampo and P. Müller-Buschbaum, Adv. Mater. Interfaces, 2016, 3, 1600403 CrossRef; (b) Z. Jiang, J. Appl. Crystallogr., 2015, 48, 917 CrossRef CAS.
- C. Weber, L. Pithan, A. Zykov, S. Bommel, F. Carla, R. Felici, C. Knie, D. Bléger and S. Kowarik, J. Phys. Condens. Mater., 2017, 29, 434001 CrossRef CAS PubMed.
- H. A. Schwartz, U. Ruschewitz and L. Heinke, Photochem. Photobiol. Sci., 2018, 17, 864 CrossRef CAS PubMed.
- Z.-F. Liu, B. H. Loo, R. Baba and A. Fujishima, Chem. Lett., 1990, 19, 1023 CrossRef.
- L. Pesce, C. Perego, A. B. Grommet, R. Klajn and G. M. Pavan, JACS, 2020, 142, 9792 CrossRef CAS PubMed.
- T. Fujino, S. Y. Arzhantsev and T. Tahara, J. Phys. Chem. A, 2001, 105, 8123 CrossRef CAS.
- I. K. Lednev, T.-Q. Ye, P. Matousek, M. Towrie, P. Foggi, F. Neuwahl, S. Umapathy, R. E. Hester and J. N. Moore, Chem. Phys. Lett., 1998, 290, 68 CrossRef CAS.
- M. Hada, D. Yamaguchi, T. Ishikawa, T. Sawa, K. Tsuruta, K. Ishikawa, S.-Y. Koshihara, Y. Hayashi and T. Kato, Nat. Commun., 2019, 10, 4159 CrossRef.
- M. L. Rahman, S. M. Sarkar, M. M. Yusoff, S. Kumar and C. Tschierske, RSC Adv., 2015, 5, 87019 RSC.
- U. Jung, C. Schütt, O. Filinova, J. Kubitschke, R. Herges and O. Magnussen, J. Phys. Chem. C, 2012, 116, 25943 CrossRef CAS.
- (a) I. V. Malyar, E. Titov, N. Lomadze, P. Saalfrank and S. Santer, J. Chem. Phys., 2017, 146, 104703 CrossRef PubMed; (b) A. Khayyami, A. Philip, J. Multia and M. Karppinen, Dalton Trans., 2020, 49, 11310 RSC; (c) E. Sundin, F. Johansson, V. Saavedra Becerril, J. Wallenstein, A. Gasslander, J. Mårtensson and M. Abrahamsson, Mater. Adv., 2021, 2, 2328 RSC.
- L. Heinke, M. Cakici, M. Dommaschk, S. Grosjean, R. Herges, S. Bräse and C. Wöll, ACS Nano, 2014, 8, 1463 CrossRef CAS PubMed.
- D. Hermann, H. A. Schwartz, M. Werker, D. Schaniel and U. Ruschewitz, Chem.–Eur. J., 2019, 25, 3606 CrossRef CAS PubMed.
- H. Nabipour, B. Soltani and N. Ahmadi Nasab, J. Inorg. Organomet. Polym., 2018, 28, 1206 CrossRef CAS.
- (a) J. Wieme, S. M. J. Rogge, P. G. Yot, L. Vanduyfhuys, S.-K. Lee, J.-S. Chang, M. Waroquier, G. Maurin and V. van Speybroeck, J. Mater. Chem. A, 2019, 7, 22663 RSC; (b) A. E. J. Hoffman, L. Vanduyfhuys, I. Nevjestić, J. Wieme, S. M. J. Rogge, H. Depauw, P. van der Voort, H. Vrielinck and V. van Speybroeck, J. Phys. Chem. C, 2018, 122, 2734 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02405e |
| This journal is © The Royal Society of Chemistry 2022 |