
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular and hierarchical self-assembly of siRNAs into supramolecular nanomaterials for soft and homogeneous siRNA loading and precise and visualized intracellular delivery†
Xiaowen
Guan
,
Fanqi
Meng
,
Hongwei
Tan
,
Xiaoni
Wang
,
Jingjing
Li
,
Juanjuan
Wei
,
Jin
Ouyang
 and
Na
Na
*
and
Na
Na
*
Key Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: nana@bnu.edu.cn
First published on 5th July 2022
Abstract
siRNA therapeutics are challenged by homogeneous and efficient loading, maintenance of biological activities, and precise, dynamic and monitorable site-release. Herein, supramolecular nanomaterials of WP5⊃G–siRNA were constructed by modular and hierarchical self-assembly of siRNA with guest (3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione derivative, G) and host (pillar[5]arene, WP5) molecules in the same system. Demonstrated by experiments and theoretical calculations, WP5⊃G–siRNA was constructed via comprehensive weak interactions including electrostatic, hydrophobic–hydrophilic, host–guest and π–π interactions. Therefore, siRNAs were efficiently loaded, maintaining good stability, bioactivities and biocompatibilities. At pH 6.8, G was protonated to give weak acidic-responsive “turn-on” fluorescent signals, which realized the precise location of cancer sites. This triggered a subsequent delivery and a dynamic release of siRNA in cancer cells under acidic conditions for the entire collapse of WP5⊃G–siRNA by the protonation of both WP5 and G. By both in vitro and in vivo experiments, precise and visualized delivery to cancer sites was achieved to exhibit effective tumour inhibition. This provided an efficient and soft strategy of siRNA therapies and expanded the application of supramolecular nanomaterials in diagnosis and treatment.
Introduction
Small interference RNAs (siRNAs) have exhibited potential for efficient anticancer therapeutics to overcome tumour heterogeneity, drug resistance and systemic toxicities.1–3 As typical short/small double-stranded RNA molecules, siRNAs are capable of mobilizing RNA interference to modulate or block the defining hallmarks of cancers (such as drug resistance genes).4,5 To obtain therapeutic advancements of siRNAs in gene silencing, the selective and efficient delivery of siRNAs into cancer cells is a crucial premise. In contrast to small molecules and ions, siRNAs are densely negatively charged macromolecules and have difficulty crossing the cell membrane by passive diffusion.6 In addition, their poor resistance to enzymes, short biological half-life and inefficient cellular internalization are significant barriers to intercellular delivery and efficient therapy.7 Therefore, to develop clinical siRNA therapeutics, a “soft” (maintaining the biological activity of siRNAs) and efficient intercellular delivery remains a key challenge.For “soft” and efficient intercellular delivery of siRNAs, nanocarriers have become promising delivery tools for tumour diagnosis and therapy.8 Combined with significant advantages of the optical and physicochemical properties of nanoparticles, the visualized delivery strategy is continuously improving. For example, block copolymers,9 nano-gels,10 carbon nanomaterials11 and liposomes assemblies12,13 were devoted to deliveries14 with enhanced permeability and retention (EPR) effects. However, there are still challenges for the delivery of biological macromolecules, especially for gene silencing reagent of siRNAs with biological activities:
(1) The physicochemical properties and biological configurations of siRNAs are normally restricted by nanocarriers with rigid structures and modified species. This restriction affects the biological activity and stability of siRNAs in the subsequent intracellular therapy. (2) siRNAs were normally loaded onto nanocarriers via electrostatic adsorption, which led to low loading rates, poor stability and unsatisfactory pharmacokinetics. (3) The precise and simple release of siRNAs in the slightly acidic microenvironment (pH 6.8 to 7.0) of cancer sites is challenging. pH-dependent release is a simple release strategy without complicated stimuli-responsive units. The existing methods normally respond to relatively high acidic conditions (approximately pH 4–6)15 and hardly obtain the releasing gate within a 0.2 pH difference from neutral tissues. (4) The visualized monitoring of siRNA delivery is always pursued, while the chemical modification with fluorescent (FL) labels would alter the physicochemical properties of siRNAs. This can decrease the therapeutic effectiveness of siRNAs. Therefore, a new strategy for soft and homogeneous loading, slightly acidic-responsive delivery and visualized release with less effect on siRNA effectiveness is strongly worthy of pursuing.
In recent years, supramolecular assembled nanomaterials have attracted widespread attention as nanocarriers for cancer therapies.16,17 Unlike conventional nanomaterials, the driving forces for the assembly are noncovalent with diverse interactions, including hydrogen bonding,18 hydrophobic–hydrophilic interactions,19 coordinative bonds,20 π–π (or CH⋯π),21 host–guest interactions22 and electrostatic interactions.23 With this strategy, proteins were recently encapsulated by supramolecular coordination complexes via hydrophilic hydrophobic interactions24 in the form of hierarchical self-assembly. Since hierarchical self-assembly is a natural strategy to create high-order nanostructures,25 the assembly of biomolecule-encapsulated supramolecular nanoparticles will not sacrifice their activities. Initiating siRNA delivery by supramolecular assembly nanomaterials is an ideal pathway to maintain their biological activity and stability for intercellular therapy. However, reported hierarchical self-assemblies were normally employed in a single self-assembly manner, which exhibited limited functions and large-sized particles hard to be disassembled for biological applications. Therefore, further development is encouraged to meet biological applications. Simultaneously, more efforts to homogeneously guarantee high loading rates with dynamic pharmacokinetics and precise and visualized weak acidic site release are worth pursuing for ideal therapies.
Herein, a modular and hierarchical self-assembly of siRNAs with guest and host molecules in the same aqueous system was developed to obtain core–shell supramolecular nanovesicles (WP5⊃G–siRNA). The inner core module (G–siRNA) was prepared via self-assembly of siRNAs with a 3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione derivative of amphiphilic G, which shows weak FL signals in neutral and alkaline environments. Under electrostatic interactions, hierarchical self-assembly homogeneously endowed the modular core of G–siRNA with high loading rates and well-maintained bioactivities. Subsequently, the shell module of water-soluble pillar[5]arene (WP5) was self-assembled with the inner module of G–siRNA to obtain stable host–guest complexation of WP5⊃G–siRNA. Via FL changes of G protonation, visualized discrimination of cancer sites was obtained. Simultaneously, the dynamic pharmacokinetics for the release of siRNA under acidic conditions of cancer cells was visualized with less effects on the physicochemical properties of siRNA. Therefore, supramolecular core–shell nanovesicles were constructed to maintain the biological activity and stability of siRNAs for efficient and precise intracellular deliveries and subsequent effective therapies.
Results and discussion
Design of supramolecular WP5⊃G–siRNA nanovesicles
Core–shell supramolecular WP5⊃G–siRNA nanovesicles were prepared by modular and hierarchical self-assembly of siRNAs with host and guest molecules in the same aqueous system. The water-soluble macrocyclic molecule of WP5 was synthesized according to a previous report, which decorated many carboxylate groups on both sides (Scheme S1†).26–28 G was composed of a π-conjugated core and two alkyl chains terminated with quaternary ammonium, which promoted self-assembly through π–π interactions.29 In addition, G exhibited switchable off-on fluorescence between pH 6.8 and 7.0, which showed potential for cancer site discrimination in weakly acidic environments.30 In this work, G was prepared by a much simpler two-step synthesis and innovated into supramolecular hierarchical self-assembly. The detailed synthesis procedure is presented in Scheme S2,† which includes a nucleophilic substitution reaction, followed by a quaternization reaction. Their structures were confirmed by NMR and mass spectrometry analysis (Fig. S1–S11†).Generally, WP5 and G can self-assemble into supramolecular amphiphile WP5⊃G nanoparticles via the host–guest interaction between WP5 and G and π–π stacking between G (Scheme 1a). Interestingly, under slightly acidic conditions (pH 6.8), the supramolecular WP5⊃G nanoparticles collapsed under the protonation of G. In detail, the lactam N atoms on G were protonated to generate GH, which decreased π–π interactions to cause the nanostructure collapse. Significantly, under this collapse, the protonated GH emitted “turn-on” FL signals, which could be used to monitor nanostructure collapses.
 | ||
| Scheme 1 (a) Illustration of the formation of WP5⊃G nanoparticles by self-assembly of WP5 with G and the pH responsiveness of WP5⊃G. (b) Illustration of hierarchical self-assemblies to obtain WP5⊃G–siRNA supramolecular nanovesicles and the pH release of siRNA for intracellular therapy. | ||
Nevertheless, when three molecules of siRNA, G and WP5 were fed into the same aqueous solution, two hierarchical self-assemblies sequentially occurred (Scheme 1b). Unlike traditional hierarchical self-assemblies in a single manner,31–33 this process included two manners of self-assembly to construct the core–shell structure. The first self-assembly formed the G–siRNA modular core, which was followed by the subsequent self-assembly of the WP5 covering. In more detail, the G–siRNA modular core was fabricated by the first hierarchical self-assembly of siRNAs with the guest molecule of G under both π–π and electrostatic interactions. The G–siRNA modular core was nonfluorescent because the FL emission of G was ‘turn-off’ in a neutral environment. Furthermore, a large number of quaternary ammonium-terminated alkyl chains of G were exposed on the surface (Scheme 1b), which was convenient for binding with WP5. Subsequently, under the host–guest interaction and CH⋯π interaction between WP5 and G, the second hierarchical self-assembly occurred to obtain the final core–shell nanovesicles of WP5⊃G–siRNA. Therefore, supramolecular nanomaterials of modular core–shell nanovesicles were prepared via two sequential hierarchical self-assemblies in the same aqueous system. It should be noted that the obtained WP5⊃G–siRNA nanovesicles were negatively charged, being more biocompatible for siRNA delivery than traditional positively charged delivery systems. Via weak interactions, this not only maintained the activity and stability of siRNA, but also endowed the high efficiently loading with dynamic pharmacokinetics for siRNA release.
During the drug delivery, the nanovesicles of WP5⊃G–siRNA first concentrated at cancer sites through EPR effects. Given the weak acidic conditions of cancer cells, “turn on” FL signals were emitted under G protonation, which was convenient for indicating cancer sites. It should be noted that at pH 6.8, WP5 and G were still bound together. Then, WP5⊃G–siRNA was internalized by tumour cells with high endosome/lysosome activities.34,35 Subsequently, the WP5⊃G–siRNA nanovesicles completely collapsed, facilitated by the protonation of both WP5 (pKa 4–5) and G (pH 6.8) in the acidic environment of the lysosome (pH 4–6). In detail, the carboxylate groups on WP5 were protonated into carboxyl groups to form WP5S, which dramatically decreased the water solubility to break the supramolecular structures. Therefore, accompanying with breaking from endosomes, siRNAs were released for intracellular treatments via weakening or disappearance of π–π interactions and host–guest interactions (Scheme 1b). Therefore, supramolecular nanovesicles of WP5⊃G–siRNA can be internalized into cancer cells to release siRNAs for treatments, which can be monitored by FL signals of G.
Characterization of supramolecular WP5⊃G–siRNA nanovesicles
To confirm the successful preparation of supramolecular nanovesicles, WP5⊃G–siRNA was characterized. For comparison, WP5⊃G nanoparticles obtained by single self-assembly without siRNA were prepared in parallel. As characterized by both transmission electron microscopy (TEM) and dynamic light scattering (DLS), self-assembly between WP5 and G was successfully employed to obtain spherical WP5⊃G with a diameter of 110 nm (Fig. 1a). When siRNA participated, the single self-assembly was altered into modular and sequential hierarchical self-assembly, which realized siRNA loading. The new manner of self-assembly resulted in larger spherical nanovesicles of WP5⊃G–siRNA with a diameter of approximately 180 nm (Fig. 1b). Therefore, Tyndall effects were exhibited for supramolecular nanomaterials obtained by different self-assemblies (Fig. 1c). | ||
Fig. 1 TEM image and DLS data of (a) WP5⊃G and (b) WP5⊃G–siRNA nanovesicles. (c) Tyndall effects of WP5, WP5⊃G and WP5⊃G–siRNA. (d and e) HAADFS-TEM images and area elemental mappings of G–siRNA and WP5⊃G–siRNA. (Representative elements: N and O for WP5; G for siRNA; P for siRNA). (f) The zeta potential of G, WP5⊃G and WP5⊃G–siRNA in water (n = 3). (g(i)) Absorption spectra of the complex WP5⊃G with different molar ratios in water ([WP5] + [G] = 20 μM, 298 K, and wavelength range 220–650 nm); (ii) Job plot of WP5⊃G showing a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry. (h) The critical aggregation concentration of the WP5⊃G complex. :1 stoichiometry. (h) The critical aggregation concentration of the WP5⊃G complex. | ||
In addition, this hierarchical self-assembly in a modular and sequential manner can homogeneously load siRNA with high loading capacity. This homogeneous loading was conducted in two sequential hierarchical self-assemblies within the same system. Considering that the assembly time of G and siRNA has effect on the final size of WP5⊃G–siRNA (Fig. S12†), the first hierarchical self-assembly of siRNA with G was employed for 15 min to form the G–siRNA modular core. As confirmed by both TEM (Fig. S13†) and DLS (Fig. S14†) measurements, the final size of WP5⊃G–siRNA was ∼180 nm, which was feasible for intracellular applications. The high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) images of the elemental distributions in G–siRNA demonstrated homogeneous loading by the first self-assembly (Fig. 1d). Sequentially, G–siRNA hierarchically assembled with WP5 under host–guest interaction of WP5 with quaternary ammonium-terminated alkyl chains of G exposed on the core. This was confirmed by the homogeneous element distributions of supramolecular nanovesicles of WP5⊃G–siRNA (Fig. 1e). Furthermore, the element of P was homogeneously distributed inside the nanovesicles, which confirmed the successful loading of siRNA in the nanovesicles. This homogeneous loading contributed to maintaining the biological activity and facilitated the stable and efficient pharmacokinetic release of siRNA for intracellular delivery.
Moreover, ζ-potential was measured to characterize the hierarchical self-assembly and stability in aqueous solution.36 As shown in Fig. 1f, the guest molecule of G showed a positive ζ-potential at 17.6 mV. Without siRNA loading, WP5⊃G showed a significantly negative ζ-potential (−31.2 mV) due to the presence of a large number of carboxylate anionic groups on WP5. With siRNA presented for hierarchical self-assembly, the ζ-potential of WP5⊃G–siRNA increased to −14.6 mV, which indicated the good stability of the nanovesicles. This negatively charged property also endowed WP5⊃G–siRNA with low toxicity for biological systems during the delivery. This result further confirms the successful preparation of WP5⊃G–siRNA under sequential hierarchical self-assembly. In addition, the better stability of WP5⊃G–siRNA was demonstrated by the time-dependent DLS variations of G–siRNA and WP5⊃G–siRNA over 6 days (in PBS, pH = 7.0) (Fig. S15†). The good water stability guarantees the subsequent in vivo applications.
With siRNA presented, the binding stoichiometry of host–guest was significant for modular and sequential hierarchical self-assembly. As shown in Fig. 1g(i), different UV-vis absorptions were recorded for the hierarchical self-assembly systems with different molar ratios of host–guest (ranging from 9:1 to 1:9, WP5:G). By plotting the absorption at 350 nm versus different molar ratios, the optimized host–guest binding stoichiometry of WP5 to G was 2:1 (Fig. 1g(ii)). In addition, the critical aggregation concentration (CAC) of WP5 to G during self-assembly was calculated to be 31.6 μM by the water surface tension method (Fig. 1h). Therefore, a molar ratio of 2:1 (WP5 to G) was adopted in the subsequent experiment.
Mechanistic studies on modular and hierarchical self-assembly
The mechanism of the modular and hierarchical self-assembly was examined by both experiments and theoretical calculations. For better examination and comparison, the nanoparticles of WP5⊃G obtained by single hierarchical self-assembly without siRNA were also studied. First, isothermal titration calorimetry (ITC)37 experiments were employed to examine the hierarchical self-assembly. Fig. S16† shows the ITC thermograms of the raw signals for the self-assembly between WP5 and G (at pH 7.0), resulting in a binding constant (Ka) of (6.94 ± 0.30) × 105 M−1. This was generated from the cooperativity of multiple electrostatic interactions between the quaternary ammonium cationic units of G and the carboxylate anionic units on WP5. Thus, the supramolecular nanostructure of WP5⊃G was formed with amphiphilic properties. In addition, the hydrophobic–hydrophilic interactions in aqueous solution endowed this host–guest complex with high binding affinity, which was confirmed by the blueshift of UV-vis absorption (from 295 to 286 nm, Fig. S17†). Nevertheless, with siRNA in the self-assembly system to obtain WP5⊃G–siRNA, the self-assembly behaviour changed. Interestingly, the binding constant between siRNA and G was calculated according to the ITC data (Fig. 2a and S27†). The binding constant between siRNA and G was dramatically higher than that of WP5⊃G, which guaranteed sequential hierarchical self-assembly to obtain WP5⊃G–siRNA. As demonstrated by ITC experiments, Ka for the complex between G–siRNA and WP5 was calculated to be (1.21 ± 0.29) × 105 M−1 (Fig. S18†). Therefore, although siRNA, G and WP5 molecules were simultaneously present in the same aqueous system, their different binding abilities can definitely trigger the modular and sequential self-assembly.Furthermore, to investigate the binding sites to obtain WP5⊃G–siRNA by modular and sequential self-assembly, 1H NMR titration experiments were employed. Meanwhile, the self-assembly of G (Fig. S19†) and the interaction between WP5 and G were examined for comparison. Fig. S20† shows the 1H NMR titration experiment with WP5 gradually added into the D2O solution of G. For the G signal, quaternary ammonium salt protons (Ha and Hb) and alkyl chain protons (Hc) showed significant upfield shifts. The more significant upfield shift of Hc–f than Ha–f or Hb–f could be generated from the more stronger shielding effect of electron-rich cavities of WP5 toward G.22 These upfield shifts were generated from the enclosing of the positively charged trimethylammonium head of G in the cavity of WP5, shielding by the rings of WP5. For WP5, the signals of methylene (H2 and H3) and phenyl ring proton (H1) of WP5 were in the deshielding area of G and exhibited a slight downfield shift due to the encapsulation of G by WP5. In addition, the correlation signal between H1–3 on WP5 and Ha–c of the quaternary ammonium cations on G was determined by a 2D NOESY NMR study (Fig. S21†). This result confirms the spatial positions of related protons in the host–guest complex, which is in accordance with the abovementioned conformations. Therefore, a pseudorotaxane structure was confirmed to be formed by threading the quaternary ammonium head of G into the cavity of host WP5 (Scheme 1a).
In the presence of siRNA, the shift trends were unlike the single self-assembly between WP5 and G. 1H NMR titrations were employed by the titration of G into siRNA solution, followed by the titration of WP5. With the titration of G into siRNA (50 μM in D2O), upfield shifts in the quaternary ammonium salt proton (Hb) and thiophene protons (Hh and Hi) were observed (Fig. 2b). This was due to the self-assembly between siRNA and G under electrostatic, hydrophobic–hydrophilic and π–π interactions. Subsequently, with the addition of WP5 into the aforementioned solution, Hb significantly shifted upfield because Hb was shielded by the rings of WP5. This further indicates the enclosure of the quaternary ammonium salt in the WP5 cavity. In addition, the split peaks of protons became smoother and widened, which was generated from the formation of supramolecular polymer nanostructures. The self-assembly between siRNA and G was confirmed by Fourier transform infrared (FT-IR) spectroscopy analysis (Fig. 2c). The stretching vibrations of the PO43− backbone of siRNA (at 1253 cm−1)38 shifted to a lower frequency (at 1242 cm−1) upon the formation of G–siRNA. This result indicates the involvement of PO43− groups in the self-assembly via electrostatic interactions. Therefore, the final supramolecular WP5⊃G–siRNA nanovesicles were obtained under the subsequent sequential self-assembly of G–siRNA with WP5.
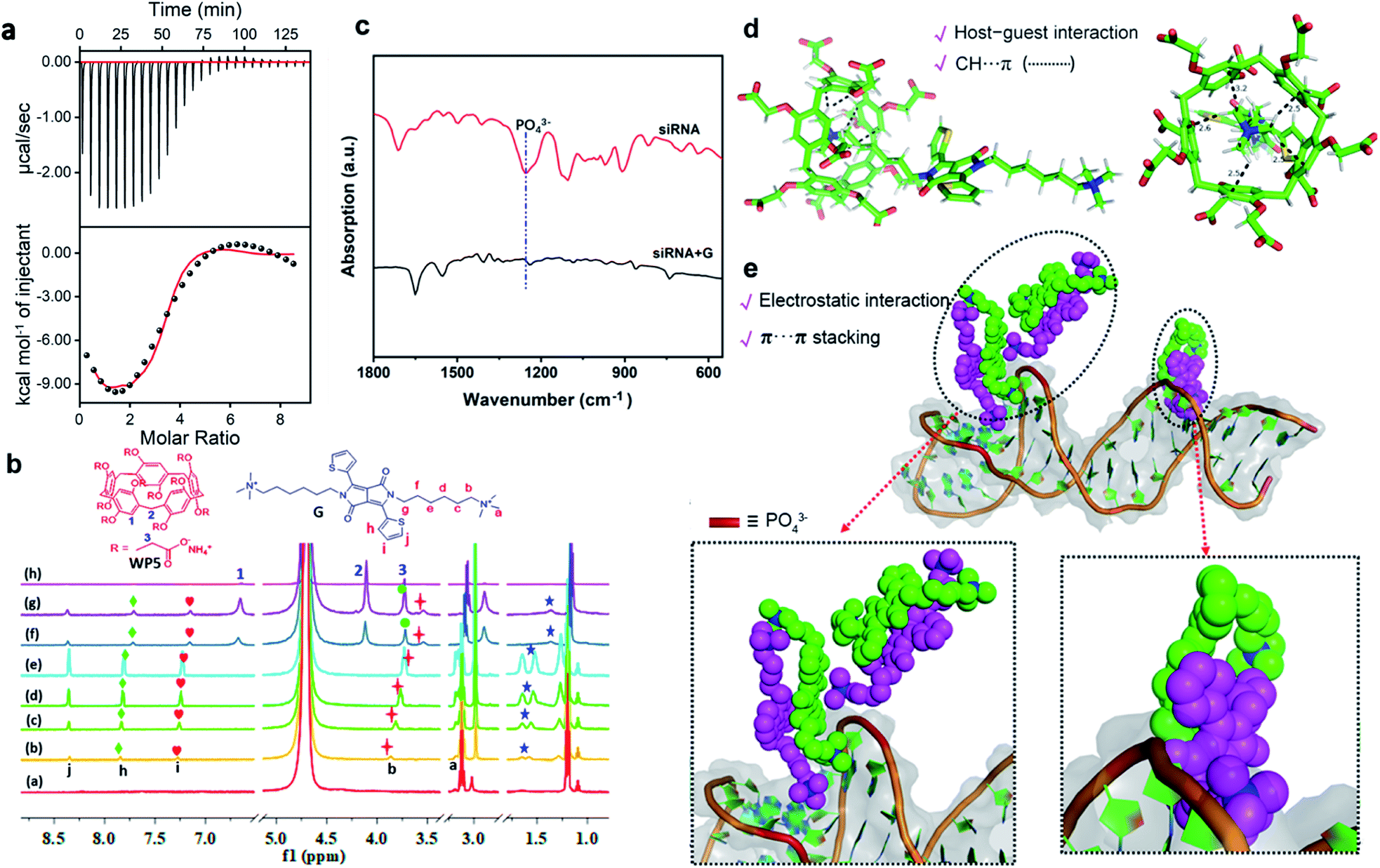 | ||
| Fig. 2 (a) ITC measurement of siRNA with G in PBS (pH = 7.0) at 298 K. Raw ITC data were obtained by sequential injections of a G solution (0.2 mM) into a siRNA solution (0.01 mM). The corresponding values of K, ΔH and ΔS are shown in Fig. S27.† (b) 1H NMR titration spectra of siRNA, G, and WP5. ((a) siRNA (50 μM). (b–e) Changes in resonance signals in the presence of 0.1 to 10.0 equiv. of G. (g and h) Changes in resonance signals in the presence of 5.0 to 15.0 equiv. of WP5 (600 MHz, D2O, 298 K)). (c) FT-IR spectra of free siRNA and siRNA + G. (d) Computer simulation of the interaction between WP5 and G. (e) Computer simulation of the interaction between siRNA and G. The siRNA is shown in greyish white as a cartoon representation. The phosphate groups are shown as a red columnar fragment, and quaternary ammonium groups are shown as blue spheres. | ||
To further examine the structure of the supramolecular WP5⊃G–siRNA, theoretical calculations of molecular dynamics simulations were employed. First, the PM6 semi-empirical quantum chemistry method was employed to obtain the host–guest self-assembly between WP5 and G. The energy minimized structure of the WP5–G complex is shown in Fig. 2d. This indicated the forming of a host–guest complex by enclosing a quaternary ammonium salt in the negative charged WP5 cavity and CH⋯π interactions between quaternary ammonium –CH3 and WP5.
To investigate the interaction between siRNA and G (generates the modular core), energy minimization and a 100 ns molecular dynamics (MD) simulation were performed on the single siRNA simulation system by using the sander module in the Amber20 program. The amber ff99SB force field and GAFF force field were applied for siRNA and G, respectively. After 1000 steps of energy minimization for the entire system (500 steepest descent steps and 500 conjugate gradient steps), MD simulation was performed at 1 atm and 300 K. Interestingly, the MD simulation indicated that the molecule G self-assembled on the surface of siRNA. As designed, G contains two hydrophobic alkyl chains that were terminated by a hydrophilic positively charged quaternary ammonium salt group. This symmetrical amphiphilic structure enables G to interact with phosphate anions both in the spiral groove and on the surface of siRNA (Fig. 2e). Thereafter, the aggregation of self-assembled G–siRNA was induced under electrostatic interactions between cation quaternary ammonium salt groups and siRNA phosphate anions. Moreover, the intermolecular interaction via π–π interactions of G could further contribute to G–siRNA aggregation, as exhibited in the MD simulation (Fig. 2e). Therefore, a modular core of G–siRNA was constructed under multiple interactions, which was consistent with the experimental characterization. Finally, G–siRNA was devoted to subsequent sequential self-assembly with WP5 to form supramolecular WP5⊃G–siRNA.
Performance of the siRNA release initiated by pH responses
Under weakly acidic (pH = 6.8) conditions, the molecule G can be protonated to initiate “turn on” FL signals from protonated G, which is convenient for the location of cancer sites.30 To investigate the feasibility of monitoring siRNA release by weak acidic responses, the FL responses of WP5⊃G at different pH values were first examined. As shown in Fig. S22,† when pH decreased from pH 9.0 to 7.0, the FL signals slightly increased. Significantly, when the pH further decreased to 6.8, dramatically increased FL signals were generated from GH and recorded. In fact, the slightly acidic FL responses of G (pH 6.8–7.0) were also valid when G was present in the form of supermolecules by assembly with WP5 and siRNA (Fig. 3a). Therefore, the supramolecular nanoparticles were weakly acidic-responsive with “turn-on” FL signals in the manner of protonated GH. | ||
| Fig. 3 (a) Fluorescence spectra of WP5⊃G–siRNA under different pH conditions. λex = 540 nm. (b) Time-dependent siRNA release efficiency of WP5⊃G–siRNA vesicles at different pH values. (c) TEM image of WP5⊃G–siRNA vesicles after incubating for 2 hours under different pH conditions. | ||
It should be noted that under weak acidic conditions (pH 6.8), WP5 and G are still bound together, independent of FL emission from GH. Demonstrated by ITC study of WP5 with G at pH 6.8 (Fig. S23†), Ka was calculated to be (1.65 ± 0.15) × 105 M−1 with an “N” value of 0.534 by curve fitting. A binding ratio of about 2:1 (WP5:G) was also exhibited, in accordance with Fig. 1a and a recent report.37b Furthermore, the binding behaviour at pH 6.8 was also demonstrated by the significant high-field shift of proton signals (Ha–f) of G in the NMR titration experiment (Fig. S24†). Therefore, under weak acidic conditions, WP5 still binds with G (at 2:1) to form a WP5⊃GH fluorescence complex (not the vesicle, indicated in Scheme 1a).
In addition, to study the siRNA release behaviours of the pH-responsive nanovesicles, FAM-siRNA, a green fluorescence dye labelled siRNA (FAM-siRNA) was prepared as the model for release examinations. As calculated by a reported method,39 the siRNA-loading efficiency and loading capacity were 96% and 7.7 wt%, respectively (Fig. S25†). To examine the release performance initiated at acidic responses,39 the dynamic changes in release rates at pH 7.0, 6.8 and 4.5 were examined. As shown in Fig. 3b, the slow release of the encapsulated siRNAs was recorded at pH 7.0, while a dramatically enhanced release profile was exhibited at pH 6.8. More obviously, the release at pH 4.5 was even faster than at pH 6.8. Moreover, up to 90% of siRNA was released at pH 4.5 after 4 h, which was mainly attributed to the decreased π–π stacking and host–guest interactions between G and WP5 under acidic conditions. This result was also consistent with the much serious collapse of supramolecular WP5⊃G–siRNA at lower pH values below 6.8, after incubating for 2 h (Fig. 3c) or 30 min (Fig. S26†).
Therefore, the release of siRNA from WP5⊃G–siRNA can be controllably initiated and monitored in acidic microenvironments, demonstrating the feasibility of siRNA release for in vivo therapy. In fact, the weak interactions for the present sequential self-assembly of siRNA with the guest and host molecules enable the efficient loading of siRNAs and dynamic pharmacokinetics for the therapy. In addition, this assembled supramolecular system also exhibits better biocompatibility for maintaining siRNA activities and in vivo applications. Thus, WP5⊃G–siRNA can act as a stable therapeutic agent to facilitate the efficient and selective release of siRNAs at tumour sites in acidic microenvironments.
Application of supramolecular WP5⊃G–siRNA nanovesicles to cancer cell imaging
For cell imaging, cell lines including normal 293T cells and SKOV-3 cancer cells were selected as models. First, the matrix cytotoxicity of the present supramolecular system was examined by cell viability experiments of WP5⊃G (without siRNA loaded). Fig. 4a demonstrates the low cytotoxicity of WP5⊃G, which resulted in greater than 85% cell viability in both lines of cells (0–80 μM). Subsequently, the cells were incubated with WP5⊃G–siRNA nanovesicles for cell imaging by confocal laser scanning microscopy (CLSM), where the nuclei were stained with Hoechst 33258 (in blue). As shown in Fig. 4b, no obvious red signal of GH was observed in the neutral microenvironment of 293T normal cells. For SKOV-3 cancer cells (acidic microenvironment), significant red signals of GH were recorded, which was generated from the internalization of WP5⊃G–siRNA by cancer cells with a significant increase in the lysosome number and endocytosis activity over normal cells.30 Therefore, based on red FL signals of protonated GH, the cancer cells were well discriminated from normal cells, which simultaneously monitored the intracellular drug release. | ||
| Fig. 4 (a) Relative viabilities of normal 293T (left) and SKOV-3 (right) tumour cells after incubation for 24 h at different WP5⊃G concentrations. (b) CLSM images of 293T and SKOV-3 cells after being treated with WP5⊃G–siRNA for 30 min. (c) CLSM images of SKOV-3 cells after being treated with WP5⊃G-FAM–siRNA. | ||
To further examine siRNA delivery by WP5⊃G–siRNA, a green FL probe of FAM was labelled onto siRNA to construct WP5⊃G-(FAM-siRNA) for cell co-imaging. As shown in Fig. 4c, after incubation with WP5⊃G-(FAM-siRNA), SKOV-3 cells exhibited significant green FAM signals and red signals of GH. This result confirms the successful internalization of WP5⊃G-(FAM-siRNA) by SKOV-3 cancer cells. Therefore, supramolecular WP5⊃G–siRNA nanovesicles were demonstrated to be effective for siRNA delivery and release for cancer therapies. Significantly, this release can also be monitored by the “turn on” signals of protonated GH in the weakly acidic environment of cancer sites (in red).
In vivo imaging and inhibition of tumours by supramolecular WP5⊃G–siRNA nanovesicles
The feasibility of in vivo gene therapy using WP5⊃G–siRNA nanovesicles was evaluated. The in vivo anti-tumour assessment of siRNA in SKOV-3 tumour-bearing xenograft nude mice was performed via intravenous injections of WP5⊃G-(FAM-siRNA) nanovesicles. During the examinations, the green signals of FAM-siRNAs were used to evaluate the distribution of siRNA in vivo. First, the biodistribution of WP5⊃G–siRNA was evaluated through ex-vivo imaging of major organs and tumours from tumour-bearing mice (4 h after tail vein injection). Two channels to image GH (in red) and FAM (in green) were adopted to examine the distribution of collapsed nanovesicles and siRNAs, respectively. Fig. 5a shows significant signals of both GH and FAM in tumour tissues, which were generated from WP5⊃G–siRNA accumulation in tumour tissue via the EPR40–43 effect. Therefore, the WP5⊃G–siRNA nanovesicles can be well used for the discrimination of cancer sites. | ||
| Fig. 5 (a) Ex vivo FL images of organs and tumour tissues from tumour-bearing mice (the images were obtained after 4 h of intravenous WP5⊃G-FAM-siRNA injection. (a) λex = 540 nm for WP5⊃G-FAM-siRNA detection; (b) λex = 480 nm for FAM-siRNA detection. (b) Photos of the tumour-bearing xenograft mice before and after 14 days of treatment. (c) Tumour volume and (d) body weight growth curves after different treatments. (e) H&E staining of organs and tumour tissues after different treatments for 14 days. | ||
Subsequently, the in vivo therapeutic effect of WP5⊃G–siRNA nanovesicles was evaluated in SKOV-3 tumour-bearing xenograft nude mice. After the tumours reached ∼100 mm3, the nude mice were randomly divided into 3 groups (n = 5 for each group) and treated with PBS, WP5⊃G without siRNA or WP5⊃G–siRNA. The tumour volume was monitored for 14 days to evaluate the anticancer effects of different groups. As expected, siRNA-loaded nanovesicles show the best capacity to suppress tumours among various treatments. As shown in Fig. 5b, the significant inhibition of tumour growth was only observed when treated with WP5⊃G–siRNA, which showed the smallest tumour. Fig. 5c shows the monitored tumour sizes over these 14 days for the three groups and confirms the good tumour inhibition by WP5⊃G–siRNA. Meanwhile, no significant changes in mouse weights were recorded during the treatment of the three groups (Fig. 5d), which verified the good biocompatibility of the developed nanovesicles. The treated mice in each group were sacrificed, and major organs (including the heart, liver, spleen, lung, and kidney) and tumours were sliced and stained for histological and immunohistochemical analyses. By haematoxylin–eosin (H&E) staining, the sliced major organs showed a normal morphology with no damage (Fig. 5e), which indicated the nanovesicles' excellent biocompatibility. Only significant apoptosis was recorded in the tumour sections after treatment with WP5⊃G–siRNA, which confirmed the treatment effects. Therefore, with good biocompatibility, the developed supramolecular WP5⊃G–siRNA nanovesicles can successfully deliver and release siRNAs for effective gene-silencing treatments of cancers.
Conclusion
In summary, a modular and hierarchical strategy for the sequential self-assembly of siRNA with guest and host molecules was developed in the same system. With comprehensive weak interactions, assembled supramolecular nanovesicles were obtained to achieve efficient and homogeneous loading of siRNAs. Without rigid structures and modified species like traditional nanocarriers, WP5⊃G–siRNA maintained the biological activity and stability of siRNAs. This is convenient for the dynamic pharmacokinetics for therapy in vivo. Moreover, the negatively charged WP5⊃G–siRNA exhibits better biocompatibility for delivery than other positively charged nanocarriers. Unlike the reported single hierarchical self-assembly manner, this modular and sequential self-assembly has initiated the multiple functions of supramolecular nanomaterials for in vivo biological applications. Furthermore, precise weak acidic “turn-on” FL responses at cancer sites were simply achieved by guest molecule protonation at pH 6.8, within a 0.2 pH difference from normal sites. Without other FL labels, visualized cancer site discrimination and monitoring of nanomaterial collapse for siRNA release were simply achieved by the “turn on” FL signals. The developed strategy of supramolecular nanomaterials maximizes the therapeutic efficacies of siRNA and accelerate the development and applications of supramolecular nanomaterials.Ethical statement
All animal experiments were conducted in accordance with the Principles of Laboratory Animal Care (People's Republic of China) and the Guidelines of the Animal Ethics Committee of Beijing Normal University.Author contributions
X. Guan and N. Na conceived and designed the project. X. Guan performed the experiments. F. Meng and H. Tan contributed to the calculations. X. Wang, J. Li and J. Wei provide some helps in biological experiments and figure preparations. J. Ouyang help to revise the manuscript. N. Na directed the whole research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by the National Natural Science Foundation of China (NNSFC, 21874012) and the National Key Research and Development Program of China (No. 2019YFC1805600). J. Ouyang is grateful for the financial support provided by NNSFC (21974010).Notes and references
- P. D. Zamore, T. Tuschl, P. A. Sharp and D. P. Bartel, Cell, 2000, 101, 25–33 CrossRef CAS PubMed.
- D. Luo, Adv. Drug Delivery Rev., 2010, 62, 591 CrossRef CAS PubMed.
- I. A. Sawyer, J. Bartek and M. Dundr, Semin. Cell Dev. Biol., 2019, 90, 94–103 CrossRef CAS PubMed.
- D. Castanotto and J. J. Rossi, Nature, 2009, 457, 426–433 CrossRef CAS PubMed.
- Y. Dong, T. Yu, L. Ding, E. Laurini, Y. Huang, M. Zhang, Y. Weng, S. Lin, P. Chen, D. Marson, Y. Jiang, S. Giorgio, S. Pricl, X. Liu, P. Rocchi and L. Peng, J. Am. Chem. Soc., 2018, 140, 16264–16274 CrossRef CAS PubMed.
- J. Shen, W. Zhang, R. Qi, Z.-W. Mao and H. Shen, Chem. Soc. Rev., 2018, 47, 1969–1995 RSC.
- M. H. Teplensky, M. Fantham, C. Poudel, C. Hockings, M. Lu, A. Guna, M. Aragones-Anglada, P. Z. Moghadam, P. Li, O. K. Farha, S. B. d. Q. Fernández, F. M. Richards, D. I. Jodrell, G. K. Schierle, C. F. Kaminski and D. Fairen-Jimenez, Chem, 2019, 5, 2926–2941 CAS.
- Y. Singh, P. Murat and E. Defrancq, Chem. Soc. Rev., 2010, 39, 2054–2070 RSC.
- J. M. Priegue, D. N. Crisan, J. Martínez-Costas, J. R. Granja, F. Fernandez-Trillo and J. Montenegro, Angew. Chem., Int. Ed., 2016, 55, 7492–7495 CrossRef CAS PubMed.
- J. Li, Y. Zhang, J. Sun, J. Ouyang and N. Na, Chem. Sci., 2021, 12, 15353–15361 RSC.
- J. M. Yoo, J. H. Kang and B. H. Hong, Chem. Soc. Rev., 2015, 44, 4835–4852 RSC.
- S. Lacerda, A. Delalande, S. V. Eliseeva, A. Pallier, C. S. Bonnet, F. Szeremeta, S. Même, C. Pichon, S. Petoud and É. Tóth, Angew. Chem., Int. Ed., 2021, 60, 23574–23577 CrossRef CAS PubMed.
- (a) B. Kim, J.-H. Park and M. J. Sailor, Adv. Mater., 2019, 31, 1903637 CrossRef CAS PubMed; (b) D.-Y. Wang, G. Yang, H. C. van der Mei, Y. Ren, H. J. Busscher and L. Shi, Angew. Chem., Int. Ed., 2021, 60, 17714–17719 CrossRef CAS PubMed.
- W. Tai and X. Gao, Adv. Drug Delivery Rev., 2017, 110, 157–168 CrossRef PubMed.
- B. Liu, S. Liang, Z. Wang, Q. Sun, F. He, S. Gai, P. Yang, Z. Cheng and J. Lin, Adv. Mater., 2021, 33, 2101223 CrossRef CAS PubMed.
- C. Sun, Z. Wang, K. Yang, L. Yue, Q. Cheng, Y.-L. Ma, S. Lu, G. Chen and R. Wang, Small, 2021, 17, 2101139 CrossRef CAS PubMed.
- (a) M.-X. Wu, J. Gao, F. Wang, J. Yang, N. Song, X. Jin, P. Mi, J. Tian, J. Luo, F. Liang and Y.-W. Yang, Small, 2018, 14, 1704440 CrossRef PubMed; (b) J. Xu, J. Wang, J. Ye, J. Jiao, Z. Liu, C. Zhao, B. Li and Y. Fu, Adv. Sci., 2021, 8, 2101101–2101141 CrossRef CAS PubMed.
- (a) Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Chem. Rev., 2021, 121, 13936–13995 CrossRef CAS PubMed; (b) C. Gao, Q. Cheng, J. Li, J. Chen, Q. Wang, J. Wei, Q. Huang, S. M. Y. Lee, D. Gu and R. Wang, Adv. Funct. Mater., 2021, 31, 2102440 CrossRef CAS.
- Y. Chang, K. Yang, P. Wei, S. Huang, Y. Pei, W. Zhao and Z. Pei, Angew. Chem., Int. Ed., 2014, 53, 13126–13130 CrossRef CAS PubMed.
- N. Song, Z. Zhang, P. Liu, D. Dai, C. Chen, Y. Li, L. Wang, T. Han, Y.-W. Yang, D. Wang and B. Z. Tang, Adv. Funct. Mater., 2021, 31, 2009924 CrossRef CAS.
- X. Ji, M. Ahmed, L. Long, N. M. Khashab, F. Huang and J. L. Sessler, Chem. Soc. Rev., 2019, 48, 2682–2697 RSC.
- J. Yao, W. Wu, C. Xiao, D. Su, Z. Zhong, T. Mori and C. Yang, Nat. Commun., 2021, 12, 2600 CrossRef CAS PubMed; M. A. Beatty and F. Hof, Chem. Soc. Rev., 2021, 50, 4812–4832 RSC.
- J. Chen, Y. Zhang, Z. Meng, L. Guo, X. Yuan, Y. Zhang, Y. Chai, J. L. Sessler, Q. Meng and C. Li, Chem. Sci., 2020, 11, 6275–6282 RSC.
- D. Fujita, R. Suzuki, Y. Fujii, M. Yamada, T. Nakama, A. Matsugami, F. Hayashi, J.-K. Weng, M. Yagi-Utsumi and M. Fujita, Chem, 2021, 7, 2672–2683 CAS.
- Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889–3919 RSC.
- J. Zhou, L. Rao, G. Yu, T. R. Cook, X. Chen and F. Huang, Chem. Soc. Rev., 2021, 50, 2839–2891 RSC.
- (a) T. Ogoshi, T. Kakuta and T. Yamagishi, Angew. Chem., Int. Ed., 2019, 58, 2197–2206 CrossRef CAS PubMed; (b) L. Yue, K. Yang, X.-Y. Lou, Y.-W. Yang and R. Wang, Matter, 2020, 3, 1557–1588 CrossRef.
- B. Li, Z. Meng, Q. Li, X. Huang, Z. Kang, H. Dong, J. Chen, J. Sun, Y. Dong, J. Li, X. Jia, J. L. Sessler, Q. Meng and C. Li, Chem. Sci., 2017, 8, 4458–4464 RSC.
- Z. M. Hao and A. Iqbal, Chem. Soc. Rev., 1997, 26, 203–213 RSC.
- Y. Liu, Z. Qu, H. Cao, H. Sun, Y. Gao and X. Jiang, ACS Nano, 2017, 11, 12446–12452 CrossRef CAS PubMed.
- Y. He, T. Ye, M. Su, C. Zhang, A. E. Ribbe, W. Jiang and C. Mao, Nature, 2008, 452, 198–201 CrossRef CAS PubMed.
- J. Liu, T. Luo, Y. Xue, L. Mao, P. J. Stang and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 5429–5435 CrossRef CAS PubMed.
- G.-L. Li, Z. Zhuo, B. Wang, X.-L. Cao, H.-F. Su, W. Wang, Y.-G. Huang and M. Hong, J. Am. Chem. Soc., 2021, 143, 10920–10929 CrossRef CAS PubMed.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- P. R. Gil, M. Nazarenus, S. Ashraf and W. J. Parak, Small, 2012, 8, 943–948 CrossRef PubMed.
- Y. Cao, X.-Y. Hu, Y. Li, X. Zou, S. Xiong, C. Lin, Y.-Z. Shen and L. Wang, J. Am. Chem. Soc., 2014, 136, 10762–10769 CrossRef CAS PubMed.
- (a) W.-J. Wang, X. Mu, C.-P. Tan, Y.-J. Wang, Y. Zhang, G. Li and Z.-W. Mao, J. Am. Chem. Soc., 2021, 143, 11370–11381 CrossRef CAS PubMed; (b) D. Li, Z. Feng, Y. Han, C. Chen, Q.-W. Zhang and Y. Tian, Adv. Sci., 2022, 9, 2104790 CrossRef CAS PubMed.
- M. Li, C. Wang, Z. Di, H. Li, J. Zhang, W. Xue, M. Zhao, K. Zhang, Y. Zhao and L. Li, Angew. Chem., Int. Ed., 2019, 58, 1350–1354 CrossRef CAS PubMed.
- Z. Wang, J. Niu, C. Zhao, X. Wang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2021, 60, 12431–12437 CrossRef CAS PubMed.
- M. Izci, C. Maksoudian, B. B. Manshian and S. J. Soenen, Chem. Rev., 2021, 121, 1746–1803 CrossRef CAS PubMed.
- Y. Deng, Y. Wang, F. Jia, W. Liu, D. Zhou, Q. Jin and J. Ji, ACS Nano, 2021, 15, 8663–8675 CrossRef CAS PubMed.
- S. Shen, X. Xu, S. Lin, Y. Zhang, H. Liu, C. Zhang and R. Mo, Nat. Nanotechnol., 2021, 16, 104–113 CrossRef CAS PubMed.
- X. Xue, Y. Huang, R. Bo, B. Jia, H. Wu, Y. Yuan, Z. Wang, Z. Ma, D. Jing, X. Xu, W. Yu, T. Lin and Y. Li, Nat. Commun., 2018, 9, 3653 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02488h |
| This journal is © The Royal Society of Chemistry 2022 |