
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical ring-opening metathesis polymerization: development, scope, and mechano-exclusive polymer synthesis†
Gue Seon
Lee
a,
Hyo Won
Lee
a,
Hyun Sub
Lee
a,
Taeyang
Do
a,
Jean-Louis
Do
 b,
Jeewoo
Lim
c,
Gregory I.
Peterson
*d,
Tomislav
Friščić
*b and
Jeung Gon
Kim
*ae
b,
Jeewoo
Lim
c,
Gregory I.
Peterson
*d,
Tomislav
Friščić
*b and
Jeung Gon
Kim
*ae
aDepartment of Chemistry and Research Institute of Physics and Chemistry, Jeonbuk National University, Jeonju, 54896, Republic of Korea. E-mail: jeunggonkim@jbnu.ac.kr
bDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, H3A0B8 Montreal, Canada. E-mail: tomislav.friscic@mcgill.ca
cDepartment of Chemistry and Research Institute for Basic Science, Kyung Hee University, Seoul 02447, Republic of Korea
dDepartment of Chemistry, Inchon National University, Incheon, 22012, Republic of Korea. E-mail: gpeterson@inu.ac.kr
eInstitute of Advanced Composite Materials, Korea Institute of Science and Technology (KIST), Jeonbuk, 55324, Republic of Korea
First published on 7th September 2022
Abstract
Ruthenium-alkylidene initiated ring-opening metathesis polymerization has been realized under solid-state conditions by employing a mechanochemical ball milling method. This method promotes greenness and broadens the scope to include mechano-exclusive products. The carbene- and pyridine-based Grubbs 3rd-generation complex outperformed other catalysts and maintained similar mechanistic features of solution-phase reactions. High-speed ball milling provides sufficient mixing and energy to the solid reaction mixture, which is composed of an initiator and monomers, to minimize or eliminate the use of solvents. Therefore, the solubility and miscibility of monomers and Ru-initiators are not limiting factors in solid-state ball milling. A wide variety of solid monomers, including ionomers, fluorous monomers, and macromonomers, were successfully polymerized under ball milling conditions. Importantly, direct copolymerization of immiscible (ionic/hydrophobic) monomers exemplifies the synthesis of mechano-exclusive polymers that are difficult to make using traditional solution procedures. Finally, the addition of a small amount of a liquid additive (i.e., liquid-assisted grinding) minimized chain-degradation, enabling high-molecular-weight polymer synthesis.
Introduction
Ring-opening metathesis polymerization (ROMP) has deeply impacted polymer chemistry by providing a robust synthetic technique for synthesizing functional polymers with wide-ranging applications.1 Studies on initiator structures, reaction kinetics, new monomers, and reaction conditions have led to dramatic advances in efficiency, selectivity, scope, and applicability of ROMP. Conventionally, most ROMPs have been conducted in the liquid state, with the initiator and monomer reacting in the same phase.2 When a common solvent is not available, ROMP does not proceed smoothly. Thus, many monomers and the resulting polymers remain unexplored. A new synthetic system not limited by solubility could substantially expand the realm of ROMP.Mechanochemical synthesis employs chemical transformations induced by mechanical force.3 Efficient mixing and energy delivery using mechanical methods, such as ball milling, have many merits including solvent-free conditions, less energy input, enhanced reactivity, and unique products.4 Over the years, mechanochemical syntheses have been successfully established in many areas of chemical synthesis.5 Mechanochemical polymerization has a long history as well.6 The first report was the free radical polymerization of vinyl monomers with ball milling by Kargin in 1959.7 Other critical studies include those by Oprea in the 1970s and by Kuzuya in the 1990s.8 In the 21st century, green chemistry has become an important topic, and sustainable mechanochemical polymerization has recently received increasing attention.6 Direct ball milling of reactive monomers has enabled step-growth polymerizations that have produced poly(phenylene vinylene)s, polyphenylenes, polyazomethines, polyurethanes, and polyimides.9 Mechanochemical chain-growth polymerizations have also been developed, for example, ring-opening polymerizations of lactide and trimethylene carbonate and anion-initiated vinyl polymerization and atom transfer radical polymerization of 2-vinyl naphthalene.10 These examples have demonstrated that the general merits of mechanochemical synthesis remain valid when applied to polymerizations. However, research on mechanochemical polymer synthesis is still in its infancy compared to solution-phase polymerizations. The scope of the reported examples is limited, and the feasibility of many common polymerization techniques under mechanochemical conditions is obscure. Thus, the synthesis of polymers only obtainable by mechanochemical means (i.e., mechano-exclusive polymers) is rarely explored.
Recently, Friščić and coworkers reported that Ru-alkylidene catalysts are active under ball milling conditions for ring-closing and cross-metathesis reactions.11 Our team envisioned that these catalysts might also be active for the ring-opening reactions required in ROMP. The combination of ROMP and mechanochemistry would improve the greenness of the polymerization and unlock the polymerizations of immiscible monomers, leading to new classes of polymers. Herein, we describe the development and scope of mechanochemical ROMP (Mech-ROMP) (Fig. 1).
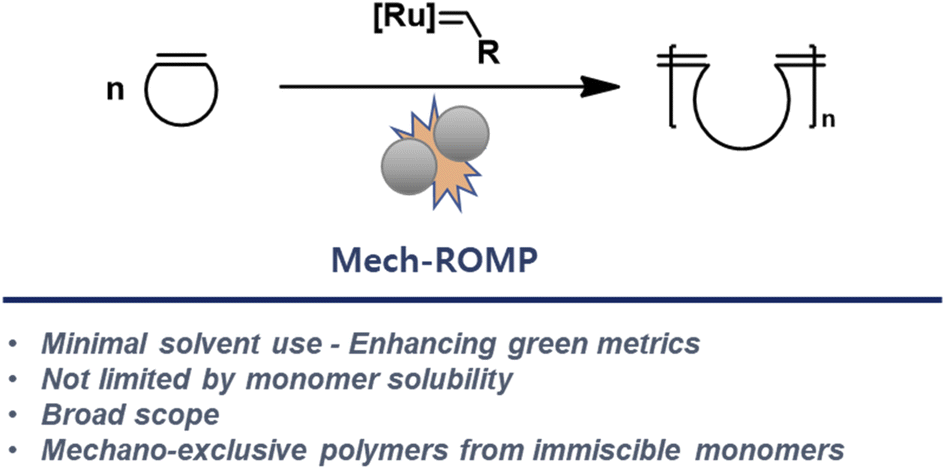 | ||
| Fig. 1 Mech-ROMP using ball mill grinding. | ||
Results & discussion
Ru-initiator & ball milling parameter study
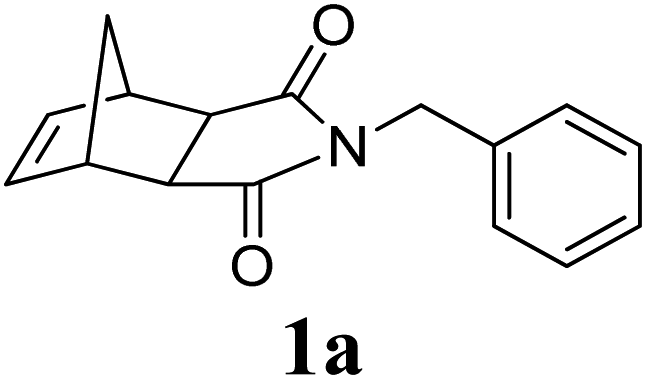
A series of representative ruthenium-alkylidene initiators were chosen to conduct Mech-ROMP, including Ru-phosphine (G1) and highly reactive Ru–N-heterocyclic carbene (NHC) complexes (G2, G3, and HG2) (Scheme 1). Each initiator (1 mol%) polymerized the model norbornene monomer (1a) under solvent-free ball milling conditions (Table 1). Monomer 1a (50 mg, 100 equiv.) and Ru-alkylidene (1 equiv.) were added into a 10 mL zirconia milling jar with three zirconia balls with a diameter of 8 mm. The tightly closed vessel was placed in a vibratory ball-milling equipment (Retsch MM400). After 30 min at 30 Hz, all reactions were quenched using a few drops of ethyl vinyl ether and milled for 5 min.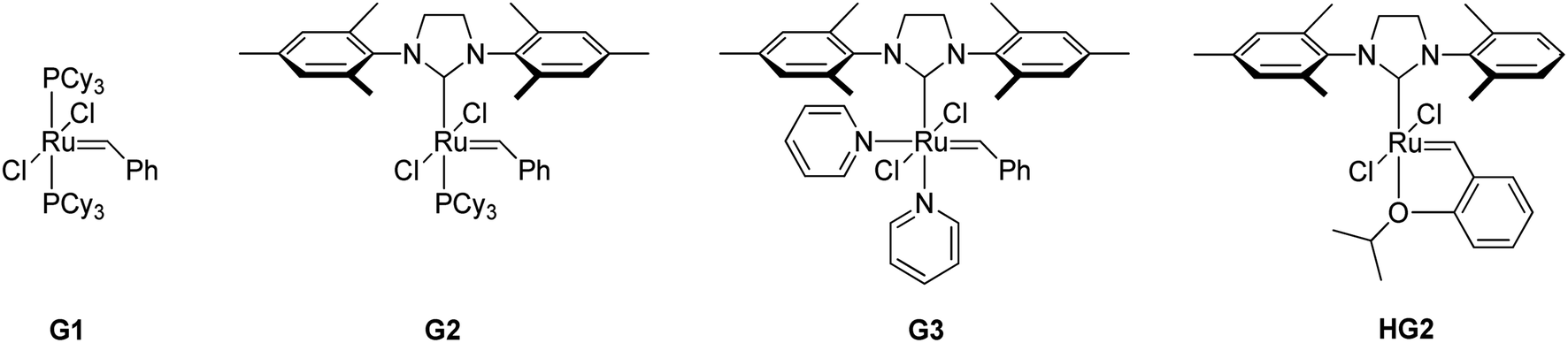 | ||
| Scheme 1 Structures of Ru-initiators used in this study. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Ru] | Conv.c (%) | M n (kg mol−1) | Đ | E/Zc |
| a Average values from two experiments. b Reaction condition: 1a (50 mg) and [Ru] = 1 mol% in a 10 mL zirconia jar containing three zirconia balls (8 mm diameter), followed by 30 Hz vibration for 30 min. c Determined using 1H NMR spectroscopy. Conv. (%) = a portion of polymeric alkenes of total alkenes. d Determined using size exclusion chromatography (SEC) with polystyrene (PS) standards in tetrahydrofuran (THF) at 40 °C. | |||||
| 1 | G1 | 23 | 6.4 | 1.29 | 76/24 |
| 2 | G2 | 98 | 14.1 | 1.69 | 57/43 |
| 3 | G3 | 97 | 14.5 | 1.53 | 57/43 |
| 4 | HG | 98 | 21.3 | 2.12 | 57/43 |
The general reactivity trends observed in the solution-phase polymerization reaction were maintained in the solid-state ball milling ROMP.12 Phosphine-based initiator G1 exhibited the slowest rate (23% monomer conversion) after 30 min (entry 1), producing the corresponding polymer with a number-average molecular weight (Mn) of 6.4 kg mol−1. G1 was inactive in the cross-metathesis reaction in Friščić’s experiments, which is not the case for ROMP.11 Highly reactive Ru–NHC species (G2, G3, and HG2) exhibit >90% conversion and high molecular weight (entries 2–4). G3 with fast-dissociating pyridine ligands showed the narrowest dispersity (Đ = 1.53, entry 2) compared to G2 (Đ = 1.69, entry 3) and HG2 (Đ = 2.12, entry 4). HG2 exhibited a higher Mn than G2 and G3 due to slow initiation and fast propagation. The trans and cis ratios of the product polymers were comparable to those observed in their corresponding solution-phase polymerization reactions.13 Ru-phosphine G1 preferably produces the (E)-isomer (E/Z = 76/24, entry 1), and the Ru–NHC initiators (G2, G3, and HG2) exhibited near-equivalent E/Z selectivity (E/Z = 57/43, entries 2–4). These results support that mech-ROMP proceeds via a mechanism similar to that observed in the solution-phase ROMP.
Next, ball milling parameters were investigated (Table 2). Considering that long polymer chains would undergo chain degradation under mechanochemical ball milling conditions,14 [M]/[G3] = 200 conditions were used to evaluate both the polymerization and competing chain degradation. Changing the number of milling balls was not a regulating factor (entries 1–3). Ball milling with one 8 mm ball converted 87% of monomer 1a to the polymer (entry 1). Three and five 8 mm balls resulted in 90 and 93% conversion, respectively (entries 2 and 3). Similar molecular weights were obtained regardless of the number of milling balls used (Mn = 24.1 kg mol−1, 22.6 kg mol−1, and 23.0 kg mol−1, respectively). However, variations in the ball size presented a pronounced effect on the polymerization. Changing to a heavier 10 mm ball resulted in the full consumption of the monomer (entry 4), whereas the use of 5 mm balls (×12) only gave a 16% turnover (entry 5), and 3 mm balls (×20) showed almost no conversion (entry 6). As seen in ring-opening lactide polymerization,10a the importance of the collision energy on the reaction efficiency was again confirmed in Ru-ROMP. However, high-energy ball milling enhanced the degree of chain degradation. Specifically, the experiment using a 10 mm ball produced a polymer product with a lower Mn, despite high monomer conversion, than that obtained under 8 mm ball conditions (16.5 vs. 24.1 kg mol−1, respectively). This observation is consistent with Choi and Peterson's results of decreasing degradation kinetics as the ball size decreases.14a The effect of the vibration frequency was evaluated (entry 7). A slower vibration (20 Hz) provided poor mixing and low-energy delivery, resulting in a low conversion (16%). These results also rule out the possibility of significant background polymerization not induced by ball milling.
|
|
||||
|---|---|---|---|---|
| Entry | Balls | Conv.c (%) | M n (kg mol−1) | Đ |
| a Average values from two experiments. b Reaction conditions: 1a (50 mg) and G3 in a 10 mL zirconia jar, followed by 30 Hz vibration for 30 min. c Determined using 1H NMR spectroscopy. Conv. (%) = a portion of polymeric alkenes of total alkenes. d Determined using SEC with PS standards in THF at 40 °C. | ||||
| 1 | 8 mm × 1 | 87 | 24.1 | 1.64 |
| 2 | 8 mm × 3 | 90 | 22.6 | 1.67 |
| 3 | 8 mm × 5 | 93 | 23.0 | 1.65 |
| 4 | 10 mm × 1 | 98 | 16.5 | 1.65 |
| 5 | 5 mm × 12 | 16 | N/A | N/A |
| 6 | 3 mm × 20 | <5 | N/A | N/A |
| 7 | 8 mm × 3 (20 Hz) | 20 | N/A | N/A |
The temperature variation in each reaction was monitored to determine the reaction phase. The ball milling equipment was placed in an isotherm container at 30 °C, which is the initial reaction temperature. At the end of the reaction, the temperature of the reaction mixture was measured using an IR thermometer and was found to have increased to 45–50 °C; these temperature were much lower than the melting point of monomer 1a (104 °C) and the glass transition temperature (Tg) of its resulting polymer (116 °C). The reaction mixtures at low, medium and high conversions did not exhibit any eutectic state. These observations indicate that the polymerization proceeds in the solid-state.
Scope of monomers
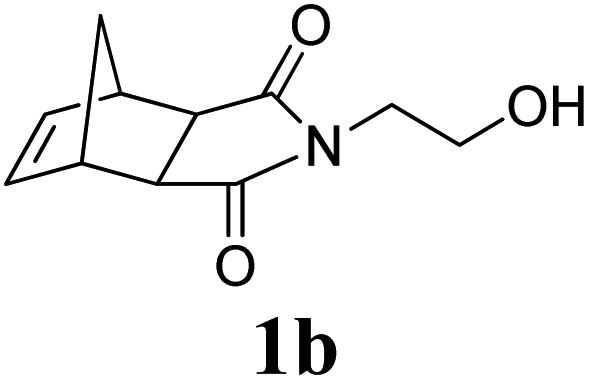
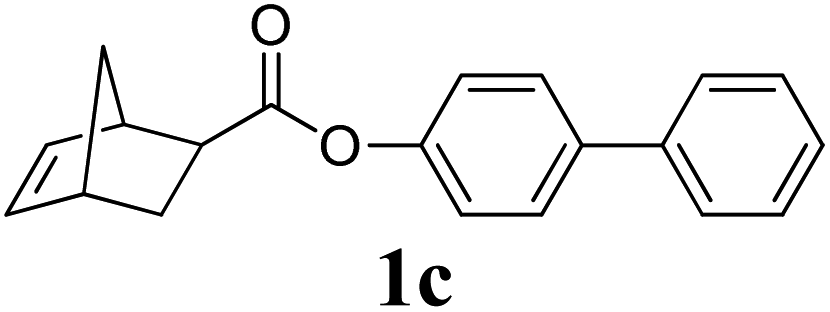
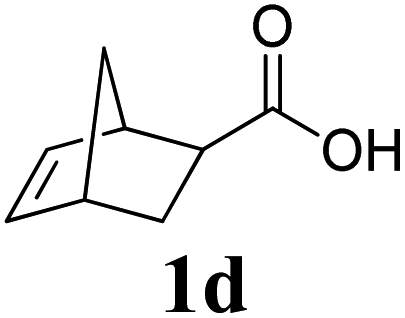
A variety of solid monomers were evaluated under the Mech-ROMP conditions (Table 3). The high functional group compatibility of the Ru-alkylidene initiator was also observed in the solid-state ROMP (entries 1–4). Initiator G3 maintained its metathesis reactivity in the presence of highly concentrated hydroxy (1b), ester (1c), and carboxylic acid (1d) groups (entries 2–4). 1H NMR and SEC confirmed high conversion and high molecular weight product formation.| Entry | Monomer | [M]/[G3] | LAG | Conv.c (%) | M n (kg mol−1) | Đ |
|---|---|---|---|---|---|---|
| a Average values from two experiments. b Reaction conditions: monomer 1a (50 mg), G3, and liquid (20 μL) in a 10 mL zirconia jar containing three zirconia balls (8 mm diameter), followed by 30 Hz vibration for 30 min. c Determined using 1H NMR spectroscopy. Conv. (%) = a portion of polymeric alkenes of total alkenes. d Determined using SEC with PS standards in THF at 40 °C. e Determined using SEC with PS standards in DMF at 40 °C. f Products did not elude the column. g Determined using SEC with poly(ethylene oxide) (PEO) standards in H2O at 40 °C. h Milling time = 60 min. i 1j (57 μL) and G3 in a 10 mL Teflon jar containing three zirconia balls (8 mm diameter). | ||||||
| 1 |
|
100 | None | 97 | 14.5d | 1.53d |
| 2 |
|
100 | None | 98 | 69.9e | 1.73e |
| 3 |
|
100 | None | 91 | 18.2d | 1.54d |
| 4 |
|
100 | None | 98 | N/Af | N/Af |
| 5 |
|
100 | None | 86 | 5.4g | 1.13g |
| H2O (η = 0.4) | 99 | 24.7g | 1.36g | |||
| 6h |
|
100 | None | 9 | 4.7g | 1.03g |
| H2O (η = 0.4) | 54 | 44.7g | 1.41g | |||
| DMF (η = 0.4) | 99 | 57.5g | 1.32g | |||
| 7 |
|
50 | THF/HFEd (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (η = 0.4) :1) (η = 0.4) |
99 | N/Af | N/Af |
| 8h |

|
100 | THF (η = 0.4) | 97d | 97.3d | 1.29d |
| 9h |
|
50 | THF (η = 0.4) | 93e | 72.4e | 1.25e |
| 10i |
|
100 | None | 99 | 26.3d | 2.48d |
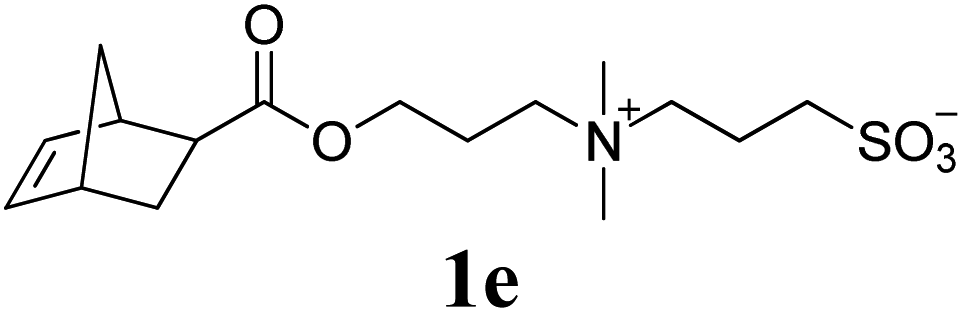
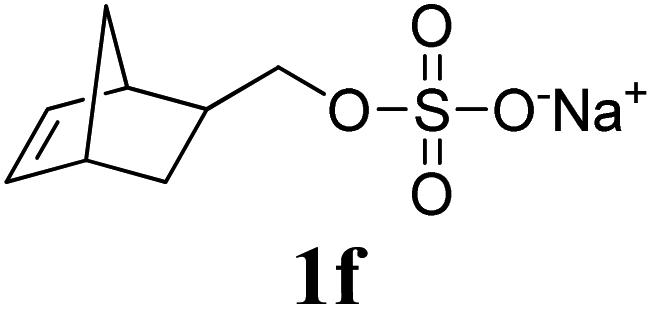
Next, the polymerizations of ionic monomers were investigated (entries 5 and 6). Several ROMP examples of ionic monomers in aqueous media have been previously reported.15 However, modification of the Ru initiator with highly polar pendants was required to make it soluble in water. Mech-ROMP of ionic monomers 1e and 1f without a liquid additive resulted in diminished conversions (86 and 9%, respectively) and low molecular weight products (Mn = 5.4 and 4.7 kg mol−1, respectively). In our previous lactide polymerization study, liquid-assisted grinding (LAG, in which a small amount of liquid is added) facilitated the polymerization and retarded polymer chain scission under ball-milling.10,16 For the Mech-ROMP with the liquid additives of 1e and 1f, LAG exhibited positive effects. The unmodified G3 initiator gave excellent efficiency (99% monomer conversion) in polymerizing ionic monomers 1e and 1f with high molecular weights (Mn = 24.7 and 57.5 kg mol−1, respectively) and narrow dispersity values (Đ = 1.36 and 1.32, respectively) with water or dimethylformamide (DMF) assisted grinding (η = 0.4 μL mg−1).16
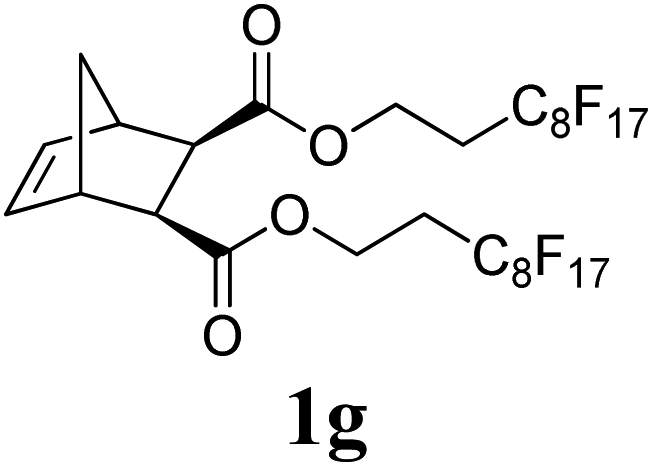
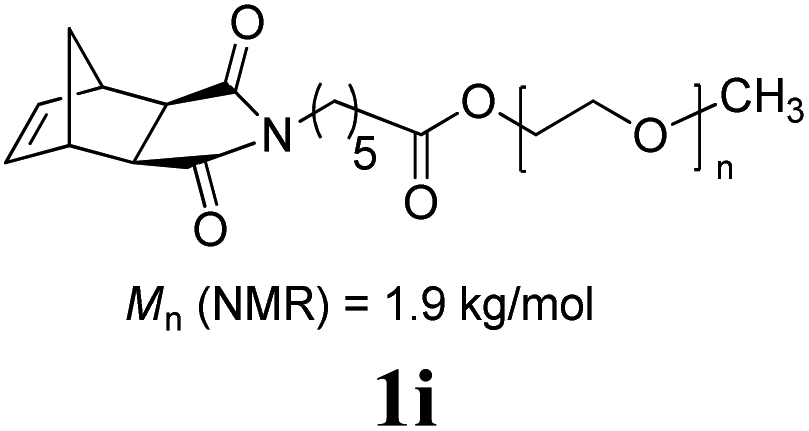
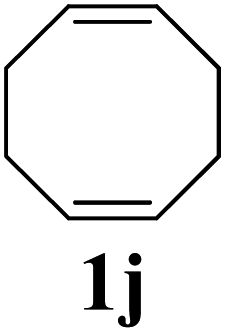
The production of a fluorous polymer was also achieved (entry 7).17 The LAG of 1g and G3 with a 1:1 mixture of THF and C4F7OCH3 (HFE) (η = 0.4 μL mg−1) provided sufficient mixing, and the corresponding fluorous-polymer was obtained quantitatively. Another interesting class of monomers, norbornenyl-terminated macromolecules (NB-MM), could also be polymerized (entries 8 and 9) with Mech-ROMP, giving bottlebrush polymers.18 The excellent catalytic efficiency of G3 in the grafting-through polymerization of NB-MM was sustained under solid-state conditions. Representative norbornene-terminated poly(lactic acid) (1h) and poly(ethylene glycol) (1i) macromonomers were efficiently polymerized using THF-LAG (η = 0.4 μL mg−1). Cyclooctadienen (1j) is a class of ROMP monomer for highly regulated polybutadiene synthesis. Mech-ROMP of 1j was also successful (entry 10). The fast second metathesis reaction under highly concentrated conditions resulted in predominantly trans poly(cyclooctadiene) (Mn = 26.3 kg mol−1, 92% trans) and a broad dispersity (Đ = 2.48).
Copolymers composed of neutral and ionic monomers are found in many applications.19 However, the orthogonal solubility of monomers leads to a lack of common solvents, which has challenged their polymerization in a one-pot process. Related polymers have been prepared using multi-step polymerizations or post-polymerization modifications.20 For the case of Ru-based ROMP, the compatibility of the Ru-initiator with monomers is also a concern. As previously stated, polymerization in a hydrophilic system necessitated Ru-initiator modification, such as adding a polar unit to a ligand.15 However, these initiators are not compatible with hydrophobic monomers and vice versa. Several recent studies have shown that immiscibility in solution is not a problem under ball milling settings.21 For example, solvent-free direct ball milling was used for post-polymerization modification of hydrophobic polymers and ionic reagents21a or ionic polymers and hydrophobic reagents.21b Bielawski also showcased the atom transfer radical polymerization of incompatible monomers in the solid-state.10f Thus, we envisioned that mech-ROMP would copolymerize monomers of orthogonal solubility with one initiator. Table 3 shows that regardless of the polarity of the monomers, mechanochemical ball milling with G3 resulted in high conversion. Subsequently, their copolymerizations were investigated. In ball milling, the hydrophobic monomer 1a and the ionic monomer 1e were polymerized, resulting in copolymers with varying compositions (Table 4). After each reaction, an aliquot was transferred to quantify the amount of unreacted monomers. All reactions exhibited good conversion of both 1a and 1e, and LAG was necessary for highly efficient copolymerizations.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry |
n:m (mol ratio) |
LAG | Time (min) | Conv.c (1a/1e) | M n (kg mol−1) | Đ | T g (°C) |
| a Average values from two experiments. b Reaction conditions: 1a + 1e (50 mg), G3 (1.0 mol%), and LAG liquid (20 μL) in a 10 mL zirconia jar containing three zirconia balls (8 mm diameter), followed by 30 Hz vibration. c Determined using 1H NMR spectroscopy. Conv. (%) = a portion of polymeric alkenes of total alkenes. d Determined using SEC with PS standards in THF at 40 °C. e Determined using SEC with PEO standards in H2O at 40 °C. | |||||||
| Poly-1a | 1:0 |
116 | |||||
| 1 | 10:1 |
THF (18.2 μL) + H2O (1.8 μL) | 30 | 96%/99% | 14.2d | 1.38d | 137 |
| 2 | 4:1 |
THF (16 μL) + H2O (4 μL) | 30 | 99%/99% | 14.0d | 1.36d | 143 |
| 3 | 2:1 |
THF (13.3 μL) + H2O (6.7 μL) | 30 | 99%/99% | 15.5d | 1.30d | 154 |
| 4 | 1:2 |
THF (6.7 μL) + H2O (13.3 μL) | 30 | 99%/99% | 14.2e | 1.39e | 153 |
| 5 | 1:4 |
THF (4 μL) + H2O (16 μL) | 30 | 99%/99% | 17.4e | 1.40e | 156 |
| 6 | 1:10 |
THF (1.8 μL) + H2O (18.2 μL) | 30 | 99%/99% | 18.1e | 1.40e | 161 |
| Poly-1e | 0:1 |
170 | |||||
The solubility of the resulting copolymers was dictated by the major component. Hydrophobic polymers were produced in entries 1–3, and hydrophilic polymers in entries 4–6. Only signals from the major components were observed in the 1H-NMR spectra (Fig. S4 and S5†). However, the coexistence of two repeat units was established by IR spectroscopy (Fig. S3†). The peak corresponding to S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1034 cm−1) in 1e and the peaks corresponding to C–N (1165 cm−1) and CO (1697 cm−1) in 1a were observed for all copolymers. The measured Tg values of each copolymer also supported copolymer formation. Differential scanning calorimeter (DSC) traces exhibited a single glass transition. The Tg values changed gradually from 116 °C (poly-1a) to 170 °C (poly-1e) with increasing 1e composition (Fig. S6†). These observations excluded the formation of mixtures of poly-1a and poly-1e or block-like structures, which would give rise to two separate glass transitions. Finally, we attempted to dissolve both monomers at various molar ratios in a large volume of THF/water mixtures (targeting [M] = 0.5 M) to pursue the possibility of solution copolymerizations (Fig. S15†). However, full monomer solubilization was not observed in any case, implying that statistical copolymerization in the solution state was not possible.
O (1034 cm−1) in 1e and the peaks corresponding to C–N (1165 cm−1) and CO (1697 cm−1) in 1a were observed for all copolymers. The measured Tg values of each copolymer also supported copolymer formation. Differential scanning calorimeter (DSC) traces exhibited a single glass transition. The Tg values changed gradually from 116 °C (poly-1a) to 170 °C (poly-1e) with increasing 1e composition (Fig. S6†). These observations excluded the formation of mixtures of poly-1a and poly-1e or block-like structures, which would give rise to two separate glass transitions. Finally, we attempted to dissolve both monomers at various molar ratios in a large volume of THF/water mixtures (targeting [M] = 0.5 M) to pursue the possibility of solution copolymerizations (Fig. S15†). However, full monomer solubilization was not observed in any case, implying that statistical copolymerization in the solution state was not possible.
LAG: retarding mechanically induced chain-degradation
Mechanical force induces both polymerization and chain degradation.6 To evaluate its effect on mechanochemical ROMP, the polymerization reaction of neat monomer 1a and G3 was monitored with increasing ball milling time (Fig. 2). The conversion and Mn were plotted versus the reaction time for two different initiator-to-monomer 1a ratios ([1a]/[G3] = 100 and 200). The conversion reached >90% completion after 30 min under both polymerization reaction conditions. However, the molecular weight growth did not follow the monomer conversion. In the case of [1a]/[G3] = 100, the highest Mn (16.7 kg mol−1) was obtained at 30 min (90% conversion); further increasing the ball milling time gave a diminished molecular weight (60 min, 96% conversion, 14.2 kg mol−1). When using [1a]/[G3] = 200, the final product did not exhibit a two-fold increase in the Mn when compared to the product obtained using [1a]/[G3] = 100; however, the product exhibited a similar Mn (16.2 kg mol−1). Reaction monitoring revealed that the maximum Mn (24.3 kg mol−1) was reached at an early stage of the polymerization (10 min, 50% conversion). Gradual degradation was observed upon the further reaction of the monomer. These results imply that chain propagation and degradation occur at the same time. The chain degradation process was considerable after Mn ∼ 15 kg mol−1. Similar observations have been repeatedly reported in the ball milling synthesis of poly(phenylene vinylene),9a poly(lactic acid),10a poly(trimethylene carbonate),10c polyphenylene,9c and poly(2-vinyl naphthalene).10e To elucidate the nature of chain-degradation in Mech-ROMP, separately prepared poly-1a (Mn = 33.3 kg mol−1) was ground in otherwise identical milling conditions. Fast chain-degradation to 18.2 kg mol−1 in 30 min suggested that mechanical force can solely cause the chain-degradation process (Table S7†). Another possibility is that the decreased molecular weight originates from the chain backbiting reaction of the reactive Ru-chain end.22 However, the backbiting process is dominant when the monomer concentration is low. In this case, the decrease in the molecular weight begins at a low monomer conversion. Thus, mechanical action was proposed to be the dominant cause of the chain scission process.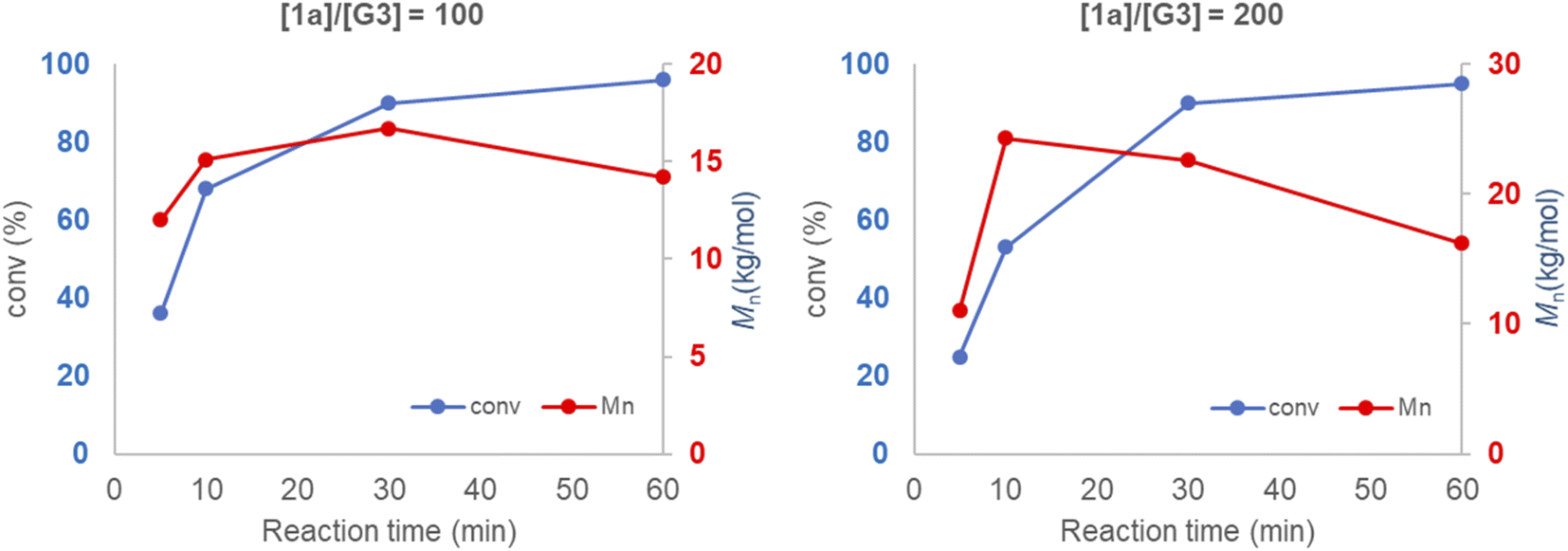 | ||
| Fig. 2 Conversion and Mnvs. ball milling time of neat 1a and G3. | ||
LAG was examined to alleviate the chain degradation process (Table 5 and Fig. 3). Previously, we observed that the addition of a very small portion of liquid could reduce the chain degradation process.10a,b The uniform distribution of small molecules in the polymer matrix was expected to lubricate the polymer chains or dissipate the impact energy. Therefore, 20 μL of the selected liquid (η = 0.4 μLmg−1) was added to the mixture consisting of monomer 1a (50 mg) and G3 (0.5 mol%). We chose a group of solvents conventionally used in Ru-ROMP for our LAG experiments (entries 2–4) and compared them with their corresponding neat grinding (entry 1) and solution-phase reactions (in DCE, entry 5). Toluene did not effectively improve the Mn (entry 2). THF resulted in a marginal increase of 3.1 kg mol−1 in the Mn and an improvement of 6.9 kg mol−1 in the Mw (entry 3). Interestingly, 1,2-dichloroethane (DCE) exhibited a substantial effect (entry 4) with an ∼10 kg mol−1 increase in the Mn and a significant narrowing of the dispersity (Đ = 1.35). To gain more details on the LAG effect, we compared the shape of the SEC traces obtained for selected polymer products (Fig. 3). The addition of toluene gave similar Mn and Mw to the neat grinding reaction (entry 1, blue line in Fig. 3). However, the peak molecular weight (Mp) of the toluene LAG polymerization product (46.6 kg mol−1, red line in Fig. 3) was 12.9 kg mol−1 higher than that obtained using neat grinding (33.7 kg mol−1, blue line in Fig. 3) and close to the Mp of the solution-phase reaction (51.4 kg mol−1, green line in Fig. 3). Thus, LAG with toluene efficiently protects the polymer chains from mechanical forces. The increased small Mw portion, which was probably due to the slow initiation, accounts for the low Mn. In the case of THF (entry 3), the Mp shifts to a higher value when compared to the solution-phase reactions. Chain protection and rapid initiation were simultaneously achieved when DCE was added (dashed line in Fig. 3). It is feasible that a small amount of a liquid could promote a highly concentrated solution polymerization, with no need for milling.23 To rule out this possibility and deduce the apparent LAG additive effect, the reaction mixture of entry 4 was placed in a V-shape vial, and magnetic stirring was applied (Fig. S17†). After 30 min, no polymer was detected by 1H-NMR. Therefore, mechanochemical ball-milling was necessary to promote the polymerization, and LAG helped both material dispersion and chain protection. The use of LAG also enabled the synthesis of high-molecular-weight polymers. A gradual increase in the molecular weight of poly-1a was observed with increasing [1a]/[G3] using G3 and DCE (20 μL) (Table S9† and Fig. 4). At [1a]/[G3] = 100, 200 and 300, polymers with Mn = 15.2 ± 0.9 kg mol−1 (Đ = 1.54 ± 0.01), Mn = 31.7 ± 1.8 kg mol−1 (Đ = 1.43 ± 0.11) and Mn = 53.1 ± 2.1 kg mol−1 (Đ = 1.13 ± 0.04) were obtained.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Liquid | Conv.c (%) | M n (kg mol−1) | M w (kg mol−1) | M p (kg mol−1) | Đ |
| a Average values from two experiments. b Reaction conditions: 1a (50 mg), G3, and liquid (20 μL) in a 10 mL zirconia jar containing three zirconia balls (8 mm diameter), followed by 30 Hz vibration for 30 min. c Determined using 1H NMR spectroscopy. Conv. (%) = a portion of polymeric alkenes of total alkenes. d Determined using SEC with PS standards in THF at 40 °C. e Reactions were performed using 0.4 mL of solvent ([M] = 0.5 M) for 30 min. f Reaction was proceeded in 20 μL DCE solvent in a V-shaped vial with magnetic stirring. | ||||||
| 1 | None | 90 | 22.6 | 37.7 | 33.7 | 1.67 |
| 2 | Toluene | 99 | 23.1 | 38.9 | 46.7 | 1.71 |
| 3 | THF | 96 | 25.7 | 44.6 | 55.7 | 1.74 |
| 4 | DCE | 99 | 32.3 | 43.4 | 46.1 | 1.35 |
| 5e | DCE (solution) | 99 | 40.4 | 48.1 | 51.4 | 1.19 |
| 6f | DCE (20 μL) mechanical stirring | < 1 | — | — | — | — |
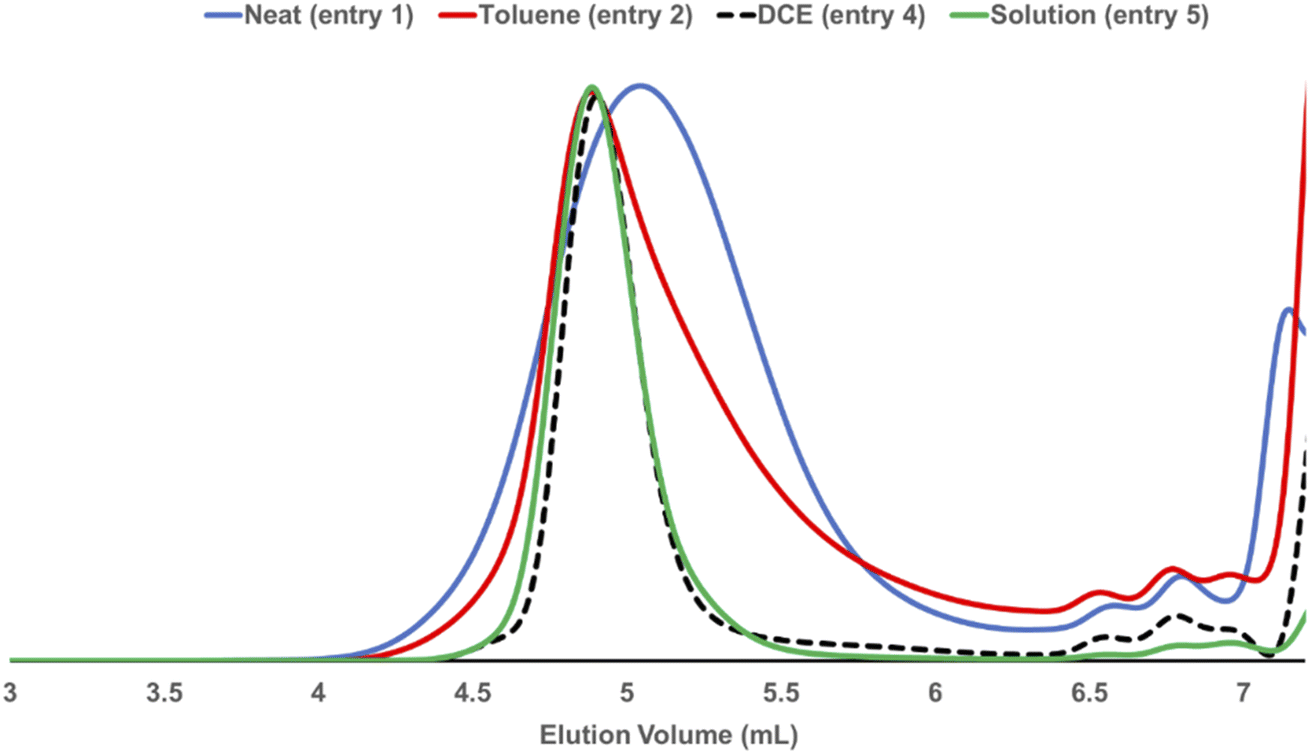 | ||
| Fig. 3 SEC traces obtained for selected polymers from Table 5. | ||
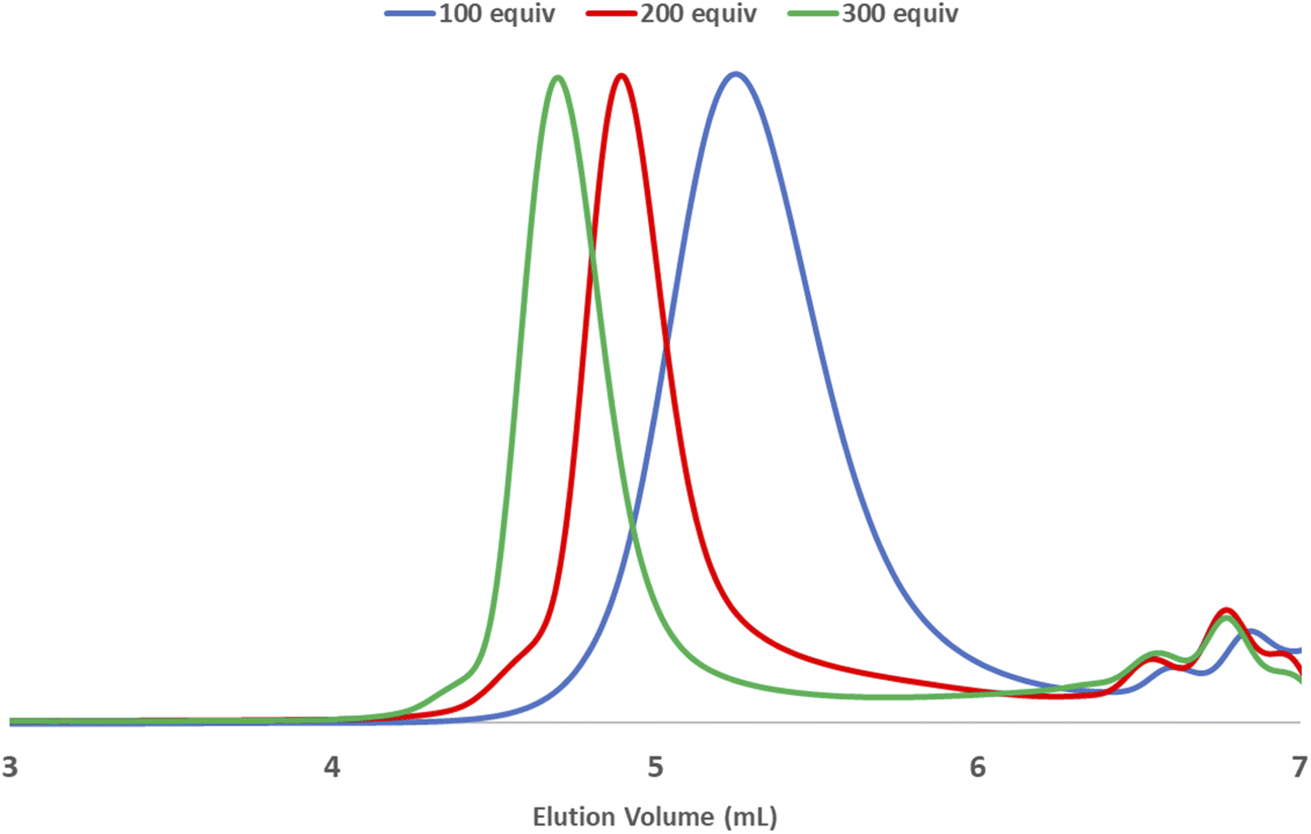 | ||
| Fig. 4 SEC traces of [1a]/[G3] = 100, 200, and 300. [1a (50 mg), G3, and DCE (20 μL) in a 10 mL zirconia jar, three 8 mm zirconia balls, and 30 Hz vibration for 30 min]. | ||
Conclusion
We have shown that ROMP, one of the most versatile methods used for functional polymer synthesis, is possible under mechanochemical ball milling conditions. Unmodified Ru-alkylidene G3 maintained its reactivity and versatility in the solid state, and the mechanical energy clearly regulated the mech-ROMP process, likely following a similar mechanism to the solution-based ROMP. The mech-ROMP examples showcased enhanced green chemistry and broadening of the field. Efficient mixing and energy supply by ball milling significantly lowered the solvent portion. The polymerization of immiscible reagents such as hydrophilic and hydrophobic monomers produced mechano-exclusive polymers, which will lead the expedition to new properties and applications.Experimental
General experimental procedure for mech-ROMP (Table 5, entry 4)
All chemical transfers and vessel assemblies were conducted in a nitrogen-purged dry-box. Monomer 1a (50 mg) and DCE (20 μL) were added to a zirconia milling container (10 mL) having 8 mm zirconia balls (3 ea.). A solution of G3 in DCE was added to the top closure. This part was left for a minute to allow the DCE to evaporate, leaving the designated amount of G3 (0.50 mol%). The main vessel and top closure were assembled. The vessel was placed in a vibrational ball milling machine and milled for 30 min in a thermostat at 30 °C. The milling vial was opened. A few drops of ethyl vinyl ether were added to quench the polymerization, followed by an additional 5 min of ball milling. A portion of the solid mixture was subjected to 1H NMR spectroscopy and SEC analysis to determine the conversion and molecular weights. The average of the two experiments was reported: 99% conversion (1H NMR, CDCl3); Mn = 32.3 kg mol−1, and Đ = 1.29 (SEC in THF, PS-standard).Data availability
All experimental data and detailed experimental procedures are available in the ESI.†Author contributions
J. G. K. and T. F. conceived, designed, and initiated this project. D. T. conducted the initial experiments. G. S. L, H. W. L, H. S. L, and J.-L. D. performed the experiments, obtained all data and analysed the results. J. G. K. and G. I. P. validated all data. J. L. provided the fluorine monomer and confirmed the data. J. G. K. wrote the original draft, and G. I. P. reviewed and edited the manuscript. All authors read and confirmed the manuscript and ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Samsung Science & Technology Foundation (SRFC-MA1902-05). This paper is dedicated to Prof. Sukbuk Chang (IBS/KAIST) on the occasion of his 60th birthday.References
- (a) C. W. Bielawski and R. H. Grubbs, Living ring-opening metathesis polymerization, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS; (b) S. Sutthasupa, M. Shiotsuki and F. Sanda, Recent advances in ring-opening metathesis polymerization, and application to synthesis of functional materials, Polym. J., 2010, 42, 905–915 CrossRef CAS; (c) O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Recent advances in ruthenium-based olefin metathesis, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- (a) K. Skowerski, J. Biatecki, A. Tracz and T. K. Olszewski, An attempt to provide an environmentally friendly solvent selection guide for olefin metathesis, Green Chem., 2014, 16, 1125–1130 RSC; (b) A. Fürstner, L. Ackermann, K. Beck, H. Hori, D. Koch, K. Langmann, M. Liebl, C. Six and W. Leitner, Olefin metathesis in supercritical carbon dioxide, J. Am. Chem. Soc., 2001, 123, 9000–9006 CrossRef; (c) H. G. Shin, H. S. Lee, E. J. Hong and J. G. Kim, Study of green solvents for ruthenium alkylidene mediated ring-opening Metathesis Polymerization, Bull. Korean Chem. Soc., 2021, 42, 502–505 CrossRef CAS; (d) M. Shetty, V. A. Kothapalli and C. E. Hobbs, Toward the (nearly) complete elimination of solvent waste in ring opening metathesis polymerization (ROMP) reactions, Polymer, 2015, 80, 64–66 CrossRef CAS.
- (a) S. L. James and T. Friščić, Mechanochemistry, Chem. Soc. Rev., 2013, 42, 7494–7496 RSC; (b) S. L. James and T. Friščić, Mechanochemistry: A web themed issue, Chem. Commun., 2013, 49, 5349–5350 RSC; (c) J. J. Gilman, Mechanochemistry, Science, 1996, 274, 65 CrossRef CAS.
- (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Mechanochemistry: opportunities for new and cleaner synthesis, Chem. Soc. Rev., 2012, 41, 413–447 RSC; (b) J. G. Hernández, Mechanochemistry, Beilstein J. Org. Chem., 2017, 13, 2372–2373 CrossRef; (c) J.-L. Do and T. Friščić, Mechanochemistry: A force of synthesis, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed; (d) T. Friščić, C. Mottillo and H. M. Titi, Mechanochemistry for synthesis, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed; (e) K. J. Ardila-Fierro and J. G. Hernández, Sustainability assessment of mechanochemistry by using the twelve principles of green chemistry, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- (a) G.-W. Wang, Mechanochemical organic synthesis, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC; (b) A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, Metal-mediated and metal-catalyzed reactions under mechanochemical conditions, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS; (c) A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Ball milling in organic synthesis: solutions and challenges, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC; (d) D. Tan and T. Friščić, Mechanochemistry for organic chemists: An update, Eur. J. Org. Chem., 2018, 2018, 18–33 CrossRef CAS; (e) N. R. Rightmire and T. P. Hanusa, Advances in organometallic synthesis with mechanochemical methods, Dalton Trans., 2016, 45, 2352–2362 RSC; (f) A. Moores, Bottom up, solid-phase syntheses of inorganic nanomaterials by mechanochemistry and aging, Curr. Opin. Green Sustainable Chem., 2018, 12, 33–37 CrossRef; (g) E. Boldyreva, Mechanochemistry of inorganic and organic systems: what is similar, what is different?, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC; (h) D. Tan and F. García, Main group mechanochemistry: from curiosity to established protocols, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- A. Krusenbaum, S. Gratz, G. T. Tigineh, L. Borchardt and J. G. Kim, The mechanochemical synthesis of polymers, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC.
- (a) K. A. Plate and V. A. Kargin, Chemical grafting on crystal surfaces, Vysokomol. Soedin., 1959, 1, 330 Search PubMed; (b) K. A. Plate and V. A. Kargin, Mechanochemical reactions of polymerization and degradation at low temperatures, J. Polym. Sci., Part C: Polym. Symp., 1963, 4, 1027–1041 CrossRef.
- (a) C. V. Oprea and M. Popa, Mechanochemisch ausgelöste polymerisationsreaktionen I. mechanochemische homopolymerisation durch schwingmahlung von styrol und acrylnitril, Angew. Makromol. Chem., 1978, 68, 1–15 CrossRef CAS; (b) C. V. Oprea and M. Popa, Mechanochemically synthesized polymers with special properties, Polym.-Plast. Technol. Eng., 1989, 28, 1025–1058 CrossRef CAS; (c) C. Simionescu, C. V. Oprea and J. Nicoleanu, Mechanochemically initiated polymerizations—5. polymerization by vibratory milling of acrylamide and methacrylamide, Eur. Polym. J., 1983, 19, 525–528 CrossRef CAS; (d) M. Kuzuya, S. Kondo and A. Noguchi, A new development of mechanochemical solid-state polymerization of vinyl monomers: prodrug syntheses and its detailed mechanistic study, Macromolecules, 1991, 24, 4047–4053 CrossRef CAS; (e) M. Kuzuya, S.-I. Kondo, A. Noguchi and N. Noda, Mechanistic study on mechanochemical polymerization of acrylamide, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 489–494 CrossRef CAS.
- (a) J. B. Ravnsbæk and T. M. Swager, Mechanochemical synthesis of poly(phenylene vinylene), ACS Macro Lett., 2014, 3, 305–309 CrossRef; (b) S. Grätz, B. Wolfrum and L. Borchardt, Mechanochemical Suzuki polycondensation – from linear to hyperbranched polyphenylenes, Green Chem., 2017, 19, 2973–2979 RSC; (c) C. G. Vogt, S. Grätz, S. Lukin, I. Halasz, M. Etter, J. D. Evans and L. Borchardt, Direct mechanocatalysis: Palladium as milling media and catalyst in the mechanochemical Suzuki polymerization, Angew. Chem., Int. Ed., 2019, 58, 18942–18947 CrossRef CAS PubMed; (d) S. Grätz and L. Borchardt, Mechanochemical polymerization – controlling a polycondensation reaction between a diamine and a dialdehyde in a ball mill, RSC Adv., 2016, 6, 64799–64802 RSC; (e) C. Oh, E. H. Choi, E. J. Choi, T. Premkumar and C. Song, Facile solid-state mechanochemical synthesis of eco-friendly thermoplastic polyurethanes and copolymers using a biomass-derived furan diol, ACS Sustainable Chem. Eng., 2020, 8, 4400–4406 CrossRef CAS; (f) T. Rensch, S. Fabig, S. Grätz and L. Borchardt, Mechanochemically-assisted Synthesis of Polyimides, ChemSusChem, 2022, 15, e202101975 CAS.
- (a) N. Ohn, J. Shin, S. S. Kim and J. G. Kim, ChemSusChem, 2017, 10, 3529–3533 CrossRef CAS; (b) G. S. Lee, B. R. Moon, J. Shin and J. G. Kim, Mechanochemical synthesis of poly(lactic acid) block copolymers: overcoming the miscibility of the macroinitiator, monomer and catalyst under solvent-free conditions, Polym. Chem., 2019, 10, 539–545 RSC; (c) S. Park and J. G. Kim, Mechanochemical synthesis of poly (trimethylene carbonate)s: an example of rate acceleration, Beilstein J. Org. Chem., 2019, 15, 963–970 CrossRef CAS; (d) T. F. Burton, J. Pinaud, N. Pétry, F. Lamaty and O. Giani, Simple and rapid mechanochemical synthesis of lactide and 3S-(isobutyl)morpholine-2,5-dione-based random copolymers using DBU and thiourea, ACS Macro Lett., 2021, 10, 1454–1459 CrossRef CAS PubMed; (e) K. Yoo, G. S. Lee, H. W. Lee, B.-S. Kim and J. G. Kim, Mechanochemical solid-state polymerization with anionic initiator, Faraday Discuss., 2022 10.1039/D2FD00080F; (f) H. Y. Cho and C. W. Bielawski, Atom Transfer Radical Polymerization in the Solid-State, Angew. Chem., Int. Ed., 2020, 59, 13929–13935 CrossRef CAS PubMed.
- J.-L. Do, C. Mottillo, D. Tan, V. Štrukil and T. Friščić, Mechanochemical ruthenium-catalyzed olefin metathesis, J. Am. Chem. Soc., 2015, 137, 2476–2479 CrossRef CAS.
- (a) T.-L. Choi and R. H. Grubbs, Controlled living ring-opening-metathesis polymerization by a fast-initiating ruthenium catalyst, Angew. Chem., Int. Ed., 2003, 42, 1743–1746 CrossRef CAS PubMed; (b) Y. Vidavsky, A. Anaby and N. G. Lemcoff, Chelating alkylidene ligands as pacifiers for ruthenium catalysed olefin metathesis, Dalton Trans., 2012, 41, 32–43 RSC; (c) K. M. Engle, G. Lu, S.-X. Luo, L. M. Henling, M. K. Takase, P. Liu, K. N. Houk and R. H. Grubbs, Origins of initiation rate differences in ruthenium olefin metathesis catalysts containing chelating benzylidenes, J. Am. Chem. Soc., 2015, 137, 5782–5792 CrossRef CAS; (d) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1, 3-dimesityl-4, 5-dihydroimidazol-2-ylidene ligands, Org. Lett., 1999, 1, 953–956 CrossRef CAS; (e) J. Huang, E. D. Steven, S. P. Nolan and J. L. Peterson, Olefin metathesis-active ruthenium complexes bearing a nucleophilic carbene ligand, J. Am. Chem. Soc., 1999, 121, 2674–2678 CrossRef CAS; (f) S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, Efficient and recyclable monomeric and dendritic Ru-based metathesis catalysts, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS; (g) R. Liu, Z. Dong and A. F. M. Kilbinger, Mono-telechelic polymers by catalytic living ring-opening metathesis polymerization with second-generation Hoveyda–Grubbs catalyst, Mater. Chem. Front., 2020, 4, 2791–2796 RSC.
- T. Ritter, A. Hejl, A. G. Wenzel, T. W. Funk and R. H. Grubbs, A standard system of characterization for olefin metathesis catalysts, Organometallics, 2006, 25, 5740–5745 CrossRef CAS.
- (a) G. I. Peterson, W. Ko, Y.-J. Hwang and T.-L. Choi, Mechanochemical degradation of amorphous polymers with ball mill grinding: Influence of the glass transition temperature, Macromolecules, 2020, 53, 7795–7802 CrossRef CAS; (b) T. Q. Nguyen and H.-H. Kausch, GPC data interpretation in mechanochemical polymer degradation, Int. J. Polym. Anal. Charact., 1998, 4, 447–470 CrossRef CAS; (c) V. P. Balema, I. Z. Hlova, S. L. Carnahan, M. Seyedi, O. Dolotko, A. J. Rossini and I. Luzinov, Depolymerization of polystyrene under ambient conditions, New J. Chem., 2021, 45, 2935–2938 RSC.
- (a) S. H. Hong and R. H. Grubbs, Highly active water-soluble olefin metathesis catalyst, J. Am. Chem. Soc., 2006, 128, 3508–3509 CrossRef CAS PubMed; (b) S. A. Isarov and J. K. Pokorski, Protein ROMP: Aqueous graft-from ring-opening metathesis polymerization, ACS Macro Lett., 2015, 4, 969–973 CrossRef CAS; (c) J. C. Foster, S. Varlas, L. D. Blackman, L. A. Arkinstall and R. K. O'Reilly, Ring-opening metathesis polymerization in aqueous media using a macroinitiator approach, Angew. Chem., Int. Ed., 2018, 57, 10672–10676 CrossRef CAS PubMed; (d) D. C. Chruch, L. Takiguchi and J. K. L. Pokorski, Optimization of ring-opening metathesis polymerization (ROMP) under physiologically relevant conditions, Polym. Chem., 2020, 11, 4492–4499 RSC; (e) C. Kim and H. Chung, Oligo(ethylene glycol) length effect of water-soluble Ru-based olefin metathesis catalysts on reactivity and removability, J. Org. Chem., 2018, 83, 9787–9794 CrossRef CAS PubMed.
- LAG parameter η is the ratio between the amount of liquid (μL) and the total mass of solid components (mg) and usually lies between 0 and 1 (a) L. Chen, M. Regan and J. Mack, The choice is yours: using liquid-assisted grinding to choose between products in the palladium-catalyzed dimerization of terminal alkynes, ACS Catal., 2016, 6, 868–872 CrossRef CAS; (b) J. L. Howard, Y. Sagatov, L. Repusseau, C. Schotten and D. L. Browne, Controlling reactivity through liquid assisted grinding: the curious case of mechanochemical fluorination, Green Chem., 2017, 19, 2798–2802 RSC.
- S. Song, Y. Chang, S. H. Oh, S. Kim, S. Choi, S. Kim, J. K. Lee, S. H. Choi and J. Lim, Fluorous dispersion ring-opening metathesis polymerization, Macromolecules, 2022, 55, 1515–1523 CrossRef CAS.
- (a) Y. Xia, J. A. Kornfield and R. H. Grubbs, Efficient synthesis of narrowly dispersed brush polymers via living ring-opening metathesis polymerization of macromonomers, Macromolecules, 2009, 42, 3761–3766 CrossRef CAS; (b) Y. Xia, B. D. Olsen, J. A. Kornfield and R. H. Grubbs, Efficient synthesis of narrowly dispersed brush copolymers and study of their assemblies: The importance of side chain arrangement, J. Am. Chem. Soc., 2009, 131, 18525–18532 CrossRef CAS PubMed.
- (a) P. Raffa, D. A. Z. Wever, F. Picchioni and A. A. Broekhuis, Polymeric surfactants: synthesis, properties, and links to applications, Chem. Rev., 2015, 115, 8504–8563 CrossRef CAS PubMed; (b) P. Banerjee, S. Jana and T. K. Mandal, Coulomb interaction-driven UCST in poly(ionic liquid) random copolymers, Eur. Polym. J., 2020, 133, 109747 CrossRef CAS; (c) M. V. Ramos-Garcés, K. Li, Q. Lei, D. Bhattacharya, S. Kole, Q. Zhang, J. Strzalke, P. P. Angelopoulou, G. Sakellariou, R. Kumar and C. G. Arges, Understanding the ionic activity and conductivity value differences between random copolymer electrolytes and block copolymer electrolytes of the same chemistry, RSC Adv., 2021, 11, 15078–15084 RSC.
- (a) H. Okamura, Y. Takarori, M. Tsunooka and M. Shirai, Synthesis of random and block copolymers of styrene and styrenesulfonic acid with low polydispersity using nitroxide-mediated living radical polymerization technique, Polymer, 2002, 43, 3155–3162 CrossRef CAS; (b) H. Hu, W. Yuan, Z. Jia and G. L. Baker, Ionic liquid-based random copolymers: a new type of polymer electrolyte with low glass transition temperature, RSC Adv., 2015, 5, 3135–3140 RSC.
- (a) J. W. Lee, J. Park, J. Lee, S. Park, J. G. Kim and B.-S. Kim, Solvent-free mechanochemical post-polymerization modification of ionic polymers, ChemSusChem, 2021, 14, 3801–3805 CrossRef CAS PubMed; (b) N. Ohn and J. G. Kim, Mechanochemical post-polymerization modification: Solvent-free solid-State synthesis of functional polymers, ACS Macro Lett., 2018, 7, 561–565 CrossRef CAS PubMed; (c) T. Seo, N. Toyoshima, K. Kubota and H. Ito, Tackling solubility issues in organic synthesis: Solid-state cross-coupling of insoluble aryl halides, J. Am. Chem. Soc., 2021, 143, 6165–6175 CrossRef CAS PubMed; (d) S. Grätz, D. Beyer, V. Tkachova, S. Hellmann, R. Berger, X. Feng and L. Borchardt, The mechanochemical Scholl reaction – a solvent-free and versatile graphitization tool, Chem. Commun., 2018, 54, 5307–5310 RSC.
- S. Sutthasupa, M. Shiotsuki and F. Sanda, Recent advances in ring-opening metathesis polymerization, and application to synthesis of functional materials, Polym. J., 2010, 42, 905–915 CrossRef CAS.
- A. J. Wooten, J. G. Kim and P. J. Walsh, Highly concentrated catalytic asymmetric allylation of ketones, Org. Lett., 2007, 9, 381–384 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02536a |
| This journal is © The Royal Society of Chemistry 2022 |