
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of Cu(II)-specific peptide shuttles capable of preventing Cu–amyloid beta toxicity and importing bioavailable Cu into cells†
Michael
Okafor
 ab,
Paulina
Gonzalez
a,
Pascale
Ronot
c,
Islah
El Masoudi
c,
Anne
Boos
c,
Stéphane
Ory
b,
Sylvette
Chasserot-Golaz
b,
Stéphane
Gasman
b,
Laurent
Raibaut
a,
Christelle
Hureau
*d,
Nicolas
Vitale‡
*b and
Peter
Faller‡
*a
ab,
Paulina
Gonzalez
a,
Pascale
Ronot
c,
Islah
El Masoudi
c,
Anne
Boos
c,
Stéphane
Ory
b,
Sylvette
Chasserot-Golaz
b,
Stéphane
Gasman
b,
Laurent
Raibaut
a,
Christelle
Hureau
*d,
Nicolas
Vitale‡
*b and
Peter
Faller‡
*a
aLaboratory of Biometals and Biological Chemistry, Institut de Chimie (UMR 7177), Université de Strasbourg-CNRS, 4 Rue Blaise Pascal, 67000 Strasbourg, France. E-mail: pfaller@unistra.fr
bCentre National de la Recherche Scientifique, Université de Strasbourg, Institut des Neurosciences Cellulaires et Intégratives, F-67000 Strasbourg, France. E-mail: vitalen@unistra.fr
cUniversité de Strasbourg, CNRS, IPHC, UMR 7178, F-67000 Strasbourg, France
dLCC-CNRS, Université de Toulouse, CNRS, Toulouse, France. E-mail: Christelle.hureau@lcc-toulouse.fr
First published on 21st September 2022
Abstract
Copper (Cu) in its ionic forms is an essential element for mammals and its homeostasis is tightly controlled. Accordingly, Cu-dyshomeostasis can be lethal as is the case in the well-established genetic Wilson's and Menkes diseases. In Alzheimer's disease (AD), Cu-accumulation occurs in amyloid plaques, where it is bound to the amyloid-beta peptide (Aβ). In vitro, Cu–Aβ is competent to catalyze the production of reactive oxygen species (ROS) in the presence of ascorbate under aerobic conditions, and hence Cu–Aβ is believed to contribute to the oxidative stress in AD. Several molecules that can recover extracellular Cu from Aβ and transport it back into cells with beneficial effects in cell culture and transgenic AD models were identified. However, all the Cu-shuttles currently available are not satisfactory due to various potential limitations including ion selectivity and toxicity. Hence, we designed a novel peptide-based Cu shuttle with the following properties: (i) it contains a Cu(II)-binding motif that is very selective to Cu(II) over all other essential metal ions; (ii) it is tagged with a fluorophore sensitive to Cu(II)-binding and release; (iii) it is made of a peptide platform, which is very versatile to add new functions. The work presented here reports on the characterization of AKH-αR5W4NBD, which is able to transport Cu ions selectively into PC12 cells and the imported Cu appeared bioavailable, likely via reductive release induced by glutathione. Moreover, AKH-αR5W4NBD was able to withdraw Cu from the Aβ1–16 peptide and consequently inhibited the Cu-Aβ based reactive oxygen species production and related cell toxicity. Hence, AKH-αR5W4NBD could be a valuable new tool for Cu-transport into cells and suitable for mechanistic studies in cell culture, with potential applications in restoring Cu-homeostasis in Cu-related diseases such as AD.
Introduction
Copper (Cu), being an essential transition metal in biology, is very important for an array of biological functions in the body and to a large extent in the brain. Cu is present as ions (Cu(I) and Cu(II)) and is absorbed by cells of the choroid plexus at the blood–brain barrier into the cerebrospinal fluid (CSF),1 from where it is transported into brain cells by the transmembrane Cu-transporter, CTR1, in its reduced Cu(I) form.2 The entering Cu(I) is transferred to Cu-chaperons like CCS (copper chaperone for superoxide dismutase), responsible for the delivery of Cu(I) to superoxide dismutase (SOD), an antioxidant enzyme.3,4 Another Cu-chaperon, COX17, transports Cu(I) to cytochrome C oxidase in the mitochondria,5 whereas ATOX1 (ref. 6 and 7) delivers Cu(I) to the Trans Golgi Network (TGN), where Cu is incorporated into cupro-proteins mainly destined to the extracellular space. These cupro-proteins play important roles in maintaining physiological activities. For example, the cupro-protein ceruloplasmin is primordial for Fe homeostasis and transport, while another cupro-protein, dopamine-β-hydroxylase, is needed for the conversion of dopamine to norepinephrine.7 Despite the importance of Cu in biology, excess Cu can provoke an array of dysfunctions, and therefore systems are in place in the advent of excess intracellular Cu. The Cu-transporting P-type ATPase, ATP7A, which is normally located to the Trans-Golgi Network (TGN), relocates to vesicles in proximity to the plasma membrane during excessive intracellular Cu levels and pumps Cu into these vesicles which are then excreted from the cell.8,9 This pathway of Cu excretion is also used to regulate synaptic signals, as Cu has been shown to plays a role in regulating NMDA receptor activity by binding to the post-synaptic NMDA receptors.10 Cu has also been shown to bind to other cell surface proteins such as PrP, AMPA, and GABA receptors amongst others.1Given the crucial role of Cu in biological systems, the deregulation of Cu homeostasis by mutations in the ATP7 transporters provoke Menkes and Wilson's diseases, both leading to neurodegeneration in patients.11–13 Moreover, Cu dyshomeostasis has also been linked to other neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), Parkinson's, Huntington's and Alzheimer's diseases (AD).13 The role of Cu dyshomeostasis in AD is one of the most studied in the literature with the main hallmark for AD being the appearance of neurofibrillary tangles (NFTs) and amyloid plaques in brain regions affected.14 Recent models actually suggest that these alterations may be cumulative and have demonstrated a connection between the amyloid hypothesis (amyloid plaques) and tau pathology (NFTs).14–16 Additionally, there has also been an increasing number of studies on other causative factors including mitochondrial dysfunction and increased reactive oxygen species (ROS) levels.17–20 Equally, emerging studies have also demonstrated the important role of amyloid species in the production of ROS.21 These studies show that amyloid-beta (Aβ) the main component of amyloid plaques is capable of binding metal ions at a relatively high affinity. Interestingly, high concentrations of Fe(III) ≈ 1 mM, Zn(II) ≈ 1 mM and Cu(II) ≈ 0.4 mM have been found in amyloid plaques, with Aβ having the highest affinity for Cu(II) with a conditional affinity value at pH 7.4 of 1010 M−1.22 In the reducing intracellular environment of cells, Cu is mainly found in its reduced form, whereas in the less reductive extracellular environment, Cu is found mainly in its oxidized form. However, Cu is never free, as it is bound to chaperones or cupro-proteins or other molecular ligands such as Aβ, where Cu can switch between its oxidized and reduced forms as a result of reductants such as ascorbate found in abundance in living organisms.23,24 These reductants reduce Cu(II) to Cu(I), which can be oxidized back to Cu(II) by oxidizing agents. The reaction of Cu(II) with dioxygen produce ROS, which can cause damage to proteins, lipids (including cell membranes), and DNA.21,25 Under physiological conditions, brain cells are protected from overproduction and accumulation of ROS by the production of antioxidants such as glutathione and SOD and by the cleavage of Aβ1–40/42 to Aβ4–40/42.26,27 In the Aβ4–40/42 form, Aβ possesses an Amino Terminal Cu(II)- and Ni(II)-Binding (ATCUN) motif, which is composed of three amino acids with histidine (His) at position 3 (Xxx–Zzz–His). ATCUN motifs are highly selective for Cu(II) and possess a high affinity ranging from 1012 to 1015 M−1.28 The first two positions in ATCUN motifs can be composed of any amino acid (but Pro in position 2) with the motif in Aβ4–40/42 being Phe–Arg–His.27,29 They have been shown to bind Cu(II) in a coordination unsuitable for Cu(I), thereby preventing its transition from the oxidized to the reduced form of Cu with a physiological reductant.30 These ATCUN motifs are found in numerous endogenous proteins, with the most common being Asp–Ala–His (DAH) found in Human Serum Albumin (HSA).31
Cu binding to Aβ has been reported to increase toxicity to cells, with proposed mechanisms such as Cu-catalyzed ROS production and/or its ability to change the Aβ conformation and hence the promotion of more toxic aggregates of Aβ. Both mechanisms could also occur together in the case of ROS induced tyrosine bridges between two Aβ, which reduces clearance efficiency by degrading the Aβ oligomers.32 Additionally, the binding of Cu to amyloid plaques increases the dyshomeostasis between intracellular and extracellular Cu. There have been studies and clinical trials with the main objective to correct this Cu dyshomeostasis found in AD, which led to studies on Clioquinol, PBT2 and GSTM.33–35 These molecules, termed ionophores or shuttles, are synthetic chemical molecules capable of binding Cu extracellularly such as retrieving Cu from extracellular plaques, entering the cell and releasing the Cu intracellularly.33,34 These compounds are not specific to Cu(II) as they have been shown to bind to other metals such as Zn. However, in AD there is also accumulation of Zn, Fe as well as Cu. Hence from a therapeutic point of view it is not clear which is the best strategy, either targeting all metal ions at the same time and place or to use selective ionophores targeting one defined metal ion. The latter would allow the targeting of each type of metal ion in a time and may be space specific manner. However, as a tool to better understand the role of Cu in AD, a selective shuttle would be very useful. Another potential problem of ionophores such as PBT-2 is linked to their toxicity and recently the term cuproptosis was coined. Elesclomol, a Cu ionophore, has been shown to induce the so-called cuproptosis at low nanomolar concentrations, which was attributed the Cu induced aggregation of lipoylated mitochondrial proteins in the TCA cycle.36 This may partially explain why Cu ionophores in clinical trials, such as Clioquinol, were unsuccessful.33 On the other hand, the same compound Cu-elesclomol could alleviate the Menkes pathology, a genetic disease of Cu-deficiency.37 This indicates that the amount, kinetics, and route of Cu-supply to cells are very critical for the restoration of Cu-homeostasis. Hence, new shuttles that are specific to Cu(II) and presenting less toxicity are attractive alternatives.
To this end, we have developed Cu(II) selective shuttles capable of importing Cu into cells thanks to a cell penetrating peptide (CPP) domain. CPPs are composed of amino acids forming usually an aliphatic or cationic chain.38,39 These peptides are capable of forming secondary structures when in contact with the cell membrane and driving cellular entry usually through direct penetration and/or endocytosis.38,40 Among the most studied CPPs are TAT,41–43 penetratin,44–46 and pVEC.47–49 For this study, we identified CPPs capable of penetrating PC12 cells used as a model of neuronal cells. These CPPs were coupled with the high Cu(II) affinity ATCUN motif AKH and a NBD (4-chloro-7-nitrobenzofurazan) molecule to permit tracking of the molecule in cells. The novel Cu(II) specific shuttles were tested for their ability to retrieve Cu(II) from Aβ1–16, to transport Cu into PC12 cells and release the Cu intracellularly.
Results and discussion
Characterization of a Cu(II)-binding module grafted to CPP (AKH–CPPNBD)
| Peptide | ATCUN motif | Fluorophore (AA position) | Linker + CPP sequence | C-ter |
|---|---|---|---|---|
| AKH-αR5W4NBD | AKH- | NBD (4) | -KGRRWWRRWWR | Amide |
| AKH-LAH4NBD | AKH- | NBD (2) | -KKALLALALHHLAHLALHLALALKKA | Amide |
| AKHKNBD | AKH- | NBD (4) | -K | Amide |
| DAHK | DAH- | NBD (4) | -K | Amide |
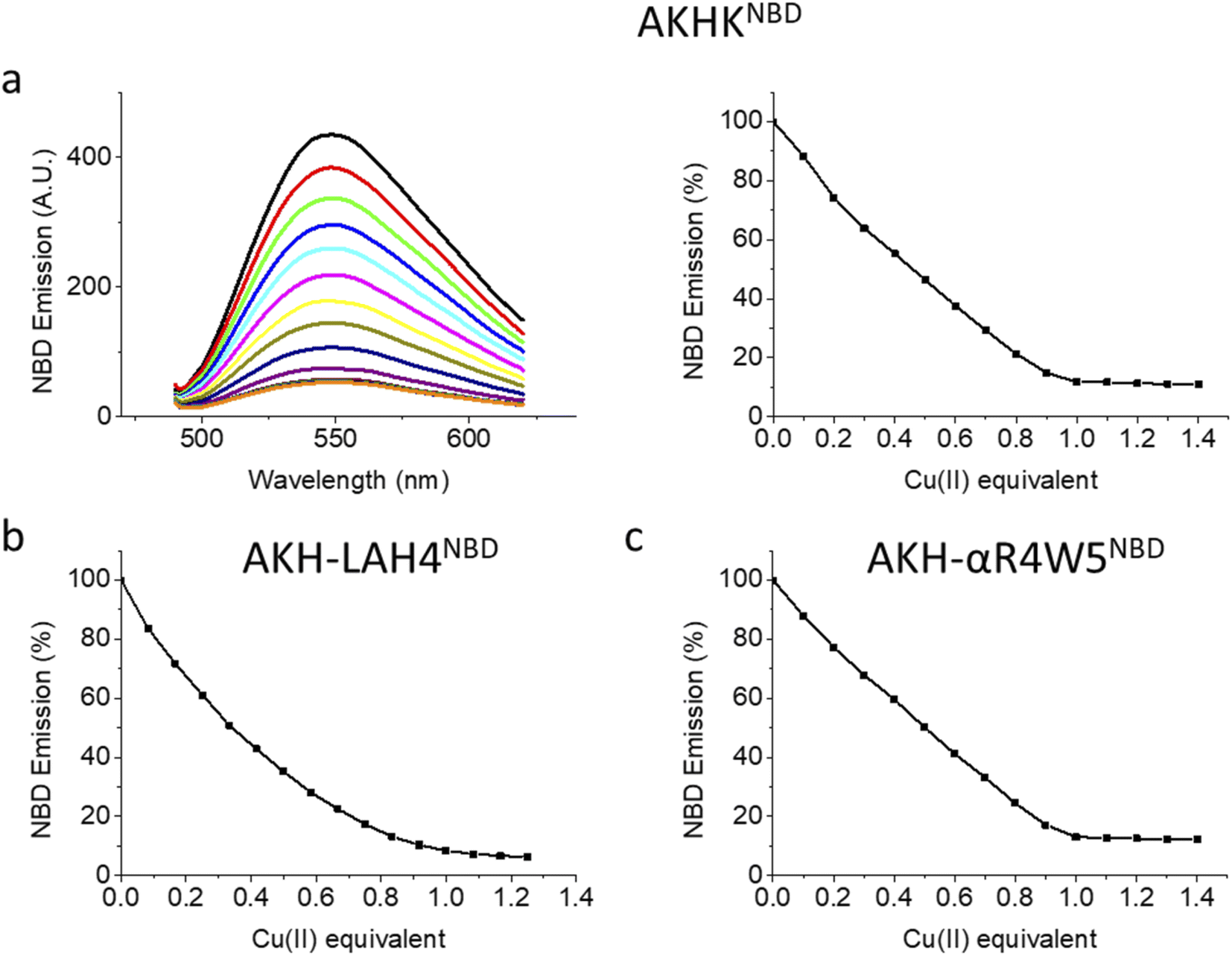 | ||
| Fig. 1 Fluorescence spectra and Cu(II) dependent quenching: (a) AKH-αR5W4NBD: left: spectra taken after each addition of 0.1 equivalents of Cu(II), and titration from black to brown; right: fluorescence intensity at 545 nm vs. added Cu(II); (b) AKH-LAH4NBD and (c) AKHKNBD. Conditions: 4 μM peptides, addition of Cu(II); 0.1 equivalent = 0.4 μM; HEPES buffer: 100 mM, pH 7.4. Excitation wavelength: 478 nm; emission wavelength: 545 nm. | ||
Upon the addition of Cu(II), all three peptides showed a steady decrease in fluorescence emission. AKHKNBD and AKH-αR5W4NBD had a clear break point at 1 equivalent with an ensuing plateau. The total quenching was about 90% in HEPES solution. This is in line with the strong affinity of the Xxx–Zzz–His motif for Cu(II) and the well-known quenching effect of Cu(II).54 For AKH-LAH4NBD, a similar behavior was observed, but the break point was less marked (absence of a sharp break point). This could in principle be due to a lower affinity, but further experiments suggest rather a time-dependent fluorescence change of NBD emission due to peptide aggregation. Indeed, AKH-LAH4NBD fluorescence evolved with time at pH 7.4, but not in the presence of 6 M GndHCl, pH 7.4 (or at pH 2) (Fig. S3†). This observation shows the importance of prudence in the interpretation of fluorescence data relating to AKH-LAH4NBD (Fig. S3†). For further tests, only data with AKH-αR5W4NBD will be shown, but the results of AKH-LAH4NBD are available in the ESI (Fig. S1–S11†).
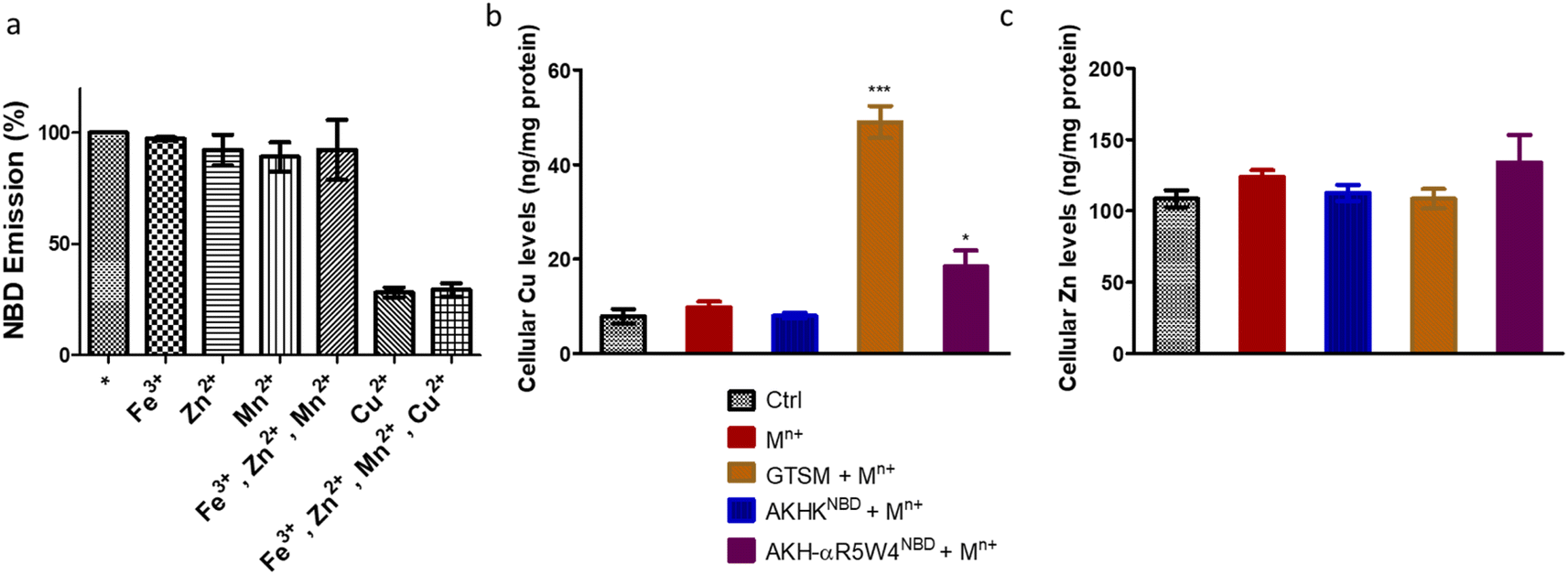 | ||
| Fig. 2 Specific binding of AKH–CPPNBD to Cu(II) in the presence of Fe(III), Zn(II) and Mn(II). (a) The emission spectrum of AKH-αR5W4 NBD was recorded before (*) and after the addition of Cu(II), Zn(II), Fe(III), Mn(II) or all of the above. Conditions: AKH-αR5W4NBD: 7 μM; Cu(II), Zn(II), Fe(III), and Mn(II): 5 μM; HEPES 100 mM, pH 7.4. (b) Import of Cu ions or (c) Zn ions in PC12 by AKH-αR5W4NBD. The peptides were pre-complexed with an equimolar concentration of Mn+(Cu(II), Zn(II), Fe(III), and Mn(II)) in 100 mM HEPES, and then cells were treated for 1 h at 37 °C in DMEM. Conditions: AKH-αR5W4NBD: 5 μM; Cu(II), Zn(II), Fe(III), and Mn(II). Each condition was done in duplicate; n = 3 independent experiments with 4 × 106 PC12 cells. A parametric ANOVA test was carried out with Dunnett's multiple comparison test, *p < 0.01 and ***p < 0.0001. | ||
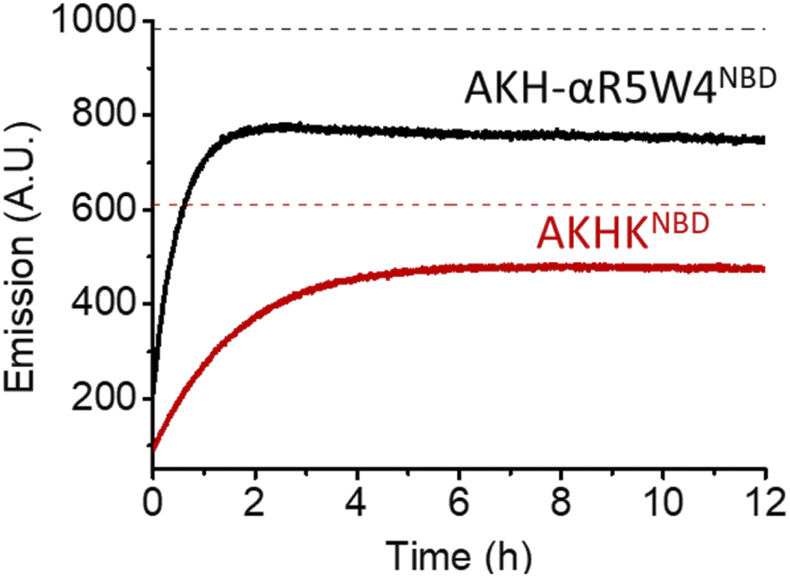 | ||
| Fig. 3 Reduction of Cu(II) bound to AKH-αR5W4NBD and AKHKNBD by glutathione monitored by the increase in NBD fluorescence at 545 nm over time. Black and red dashes mark the expected emission endpoint (emission in the absence of Cu(II)) of AKH-αR5W4NBD and AKHKNBD, respectively. Conditions: 4 μM Cu(II)-AKH–CPPNBD, 5 mM GSH, HEPES 100 mM, pH 7.4 and 37 °C; representative traces of n = 2 independent experiments are shown. | ||
As expected, there was a recovery in fluorescence emission levels after incubation with GSH. Unexpectedly the release of Cu from AKH-αR5W4NBD was 3 times faster than that from AKHKNBD, indicating that the additional amino acids in AKH-αR5W4NBD favor the reduction of Cu(II) and its removal by GSH. These data support a model in which most of the Cu(II) can be released from our shuttle intracellularly when triggered by physiological millimolar GSH concentrations.
AKH–CPPNBD peptide as a bioavailable Cu importer
After showing in vitro that the two AKH–CPPNBD peptides can bind Cu(II) and that 5 mM GSH can reduce Cu(II) and that this release can be followed by fluorescence, we aimed to measure their Cu-importer activity on cultured cells. We used three different approaches for this evaluation, including intracellular Cu-concentration measurements via ICP-MS, confocal microscopy to follow expected fluorescence increase upon Cu(II)-release from AKH–CPPNBD in the cell and the translocation of the Cu(I) extruder, ATP7A, used as a sensor for bioavailable intracellular Cu.Basal Cu concentrations in PC12 cells are similar to what was obtained by Ogra et al., 2016, i.e. about 10 ng mg−1 protein.55 For the positive control, PC12 cells were treated with 1 μM GTSM, after treatment for 1 h, and we detected a 3–4-fold increase in intracellular Cu levels, lower than the increase previously reported in NGF-treated PC12 cells.56 It is of note that NGF differentiated PC12 cells have an increased Cu(II) intake capacity, also verified by a higher basal Cu(II) content in these cells.55
After confirming ICP-MS as a reliable tool for detecting cellular Cu uptake, 1 μM Cu(II)GTSM, 5 μM Cu(II)-AKH-αR5W4NBD and 1 μM Cu(II)-AKH-LAH4NBD were tested. These concentrations did not induce significant toxicity on PC12 cells, as determined by a MTT assay (Fig. S5†). The addition of Cu(II)-AKH-αR5W4NBD and Cu(II)-AKH-LAH4NBD yielded an increase in Cu concentration in PC12 cells (Fig. 4 and S7†). 5 μM Cu(II)-AKH-αR5W4NBD was about as efficient as 1 μM GTSM (3–4 times increase), whereas 1 μM Cu(II)-AKH-LAH4NBD was less efficient (2–3 times increase). From these experiments, the AKH-αR5W4NBD peptide proved to be a reliable candidate for a Cu(II) shuttle. Subsequently we analyzed its capacity to specifically bind Cu(II) in the presence of other bioavailable transition metals Mn+ (Zn(II), Fe(III) and Mn(II)). For this, we mixed the AKH-αR5W4NBD peptide with Mn+ and Cu(II) in HEPES before addition to PC12 cells in DMEM (Dulbecco's modified Eagle's medium) media for 1 h. ICP-MS data (Fig. 2b) showed that the AKH-αR5W4NBD peptide is still capable of importing Cu(II) ions but did not import Zn(II) (Fig. 2c), the most competitive ion.
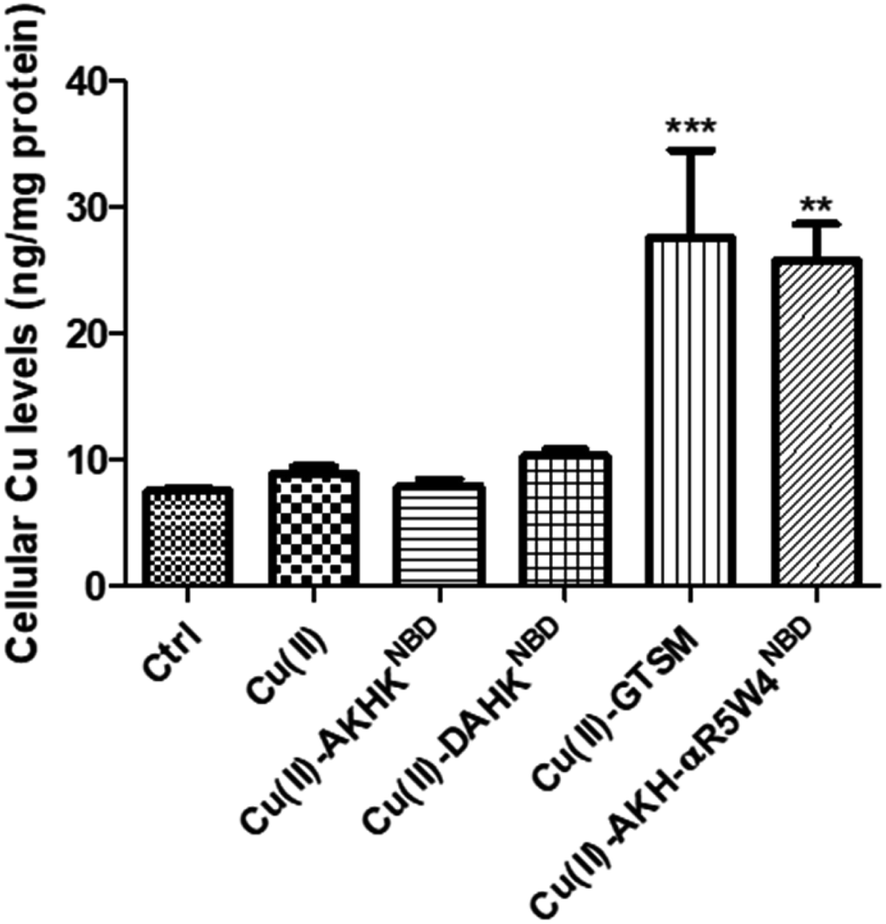 | ||
| Fig. 4 AKH-αR5W4NBD imports Cu into PC12 cells after 1 h incubation at 37 °C, measured by ICP-MS. Each condition was done in duplicate; n = 3 independent experiments with 2 × 106 PC12 cells. A parametric ANOVA test was carried out with a Dunnett's multiple comparison test, **p < 0.001 and ***p < 0.0001. | ||
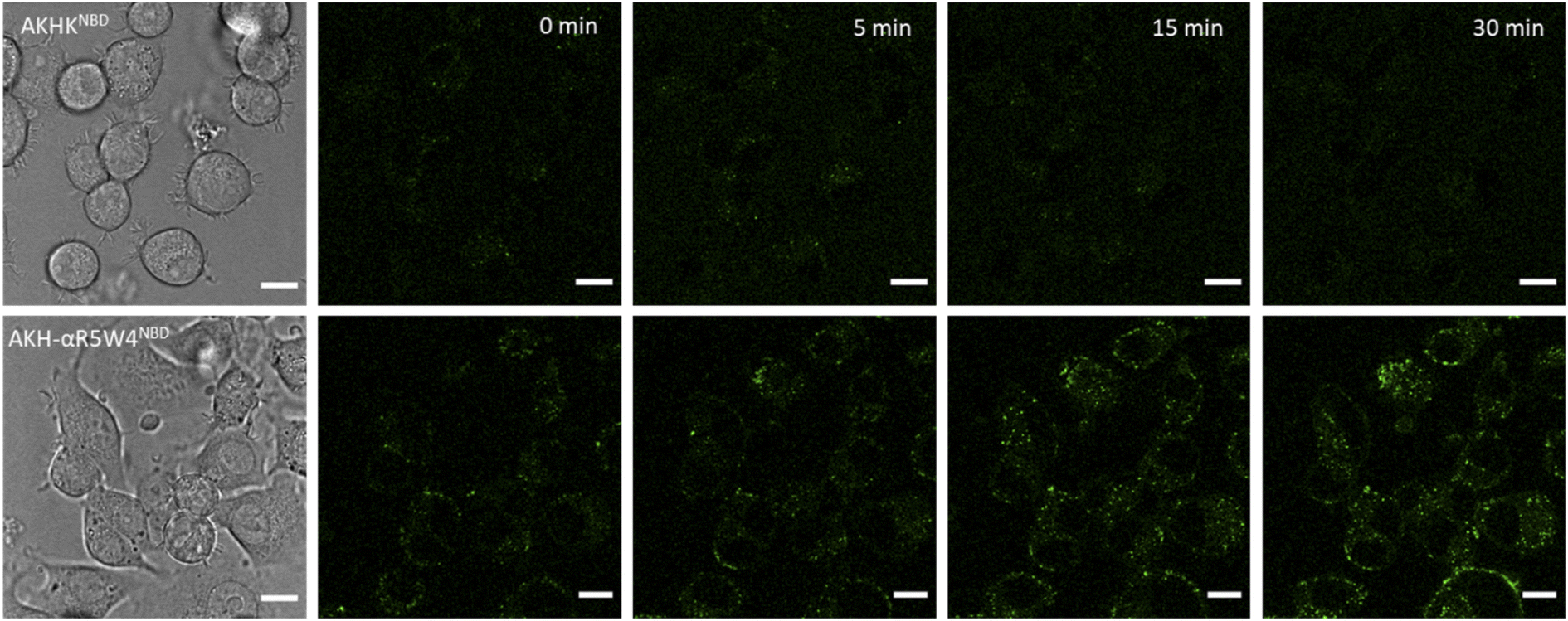 | ||
| Fig. 5 Penetration of Cu(II)-AKH-αR5W4NBD into PC12 cells. Representative images at the indicated time obtained from live imaging performed on 5 × 10 4 PC12 cells incubated with Cu(II)-AHKHNBD or Cu(II)-AKH-αR5W4NBD = 5 μM, at 37 °C using a Leica TCS SP5 II confocal microscope, with an excitation wavelength at 478 nm for 30 min. Time point zero is about 1 minute after incubation with the peptides. Bars = 10 μm. Similar observations were obtained with 5 different cell cultures. | ||
A clear entry of Cu(II)-AKH-αR5W4NBD (Fig. 5) and Cu(II)-AKH-LAH4NBD (Fig. S8†) into PC12 cells was seen as early as t0 (0 min) after the cells were incubated with the peptide complexes. Both Cu(II)-AKH-αR5W4NBD and Cu(II)-AKH-LAH4NBD seem to accumulate in vesicular compartments with their fluorescence increasing with time, which could suggest the reduction and release of Cu in these compartments (Fig. 3). For the case of Cu(II)-AKHKNBD (absence of CPP), no detectable cell penetration is detected within an incubation period of 30 minutes. In conclusion, AKH-αR5W4NBD and AKH-LAH4NBD proved to be good Cu(II) shuttles as observed using fluorescence microscopy as they seem to import Cu and possibly release Cu in PC12 cells.
Under basal conditions, a good colocalization is observed in the PC12 cell between ATP7A and giantin, a marker of the TGN. However, upon increase in Cu(I) levels, ATP7A is known to relocate from the TGN to vesicles located in proximity to the plasma membrane.57,58 Accordingly, upon treatment with Cu(II)-AKH-αR5W4NBD and Cu(II)GTSM, the staining of ATP7A delocalized from the TGN to vesicles in proximity to the plasma membrane (Fig. 6), suggesting an increase in Cu levels in the cells. As negative controls, Cu(II)-AKHKNBD, Cu(II)EDTA and free Cu(II) were used. Given the negative charge of EDTA, it is unexpected to penetrate the cell membrane and accordingly, no delocalization of ATP7A was observed under these conditions after treatment. The peptide, AKHKNBD, not having a CPP did not provoke a significant delocalization of ATP7A, neither did free Cu(II) which could enter through the Cu transporter, CTR1.
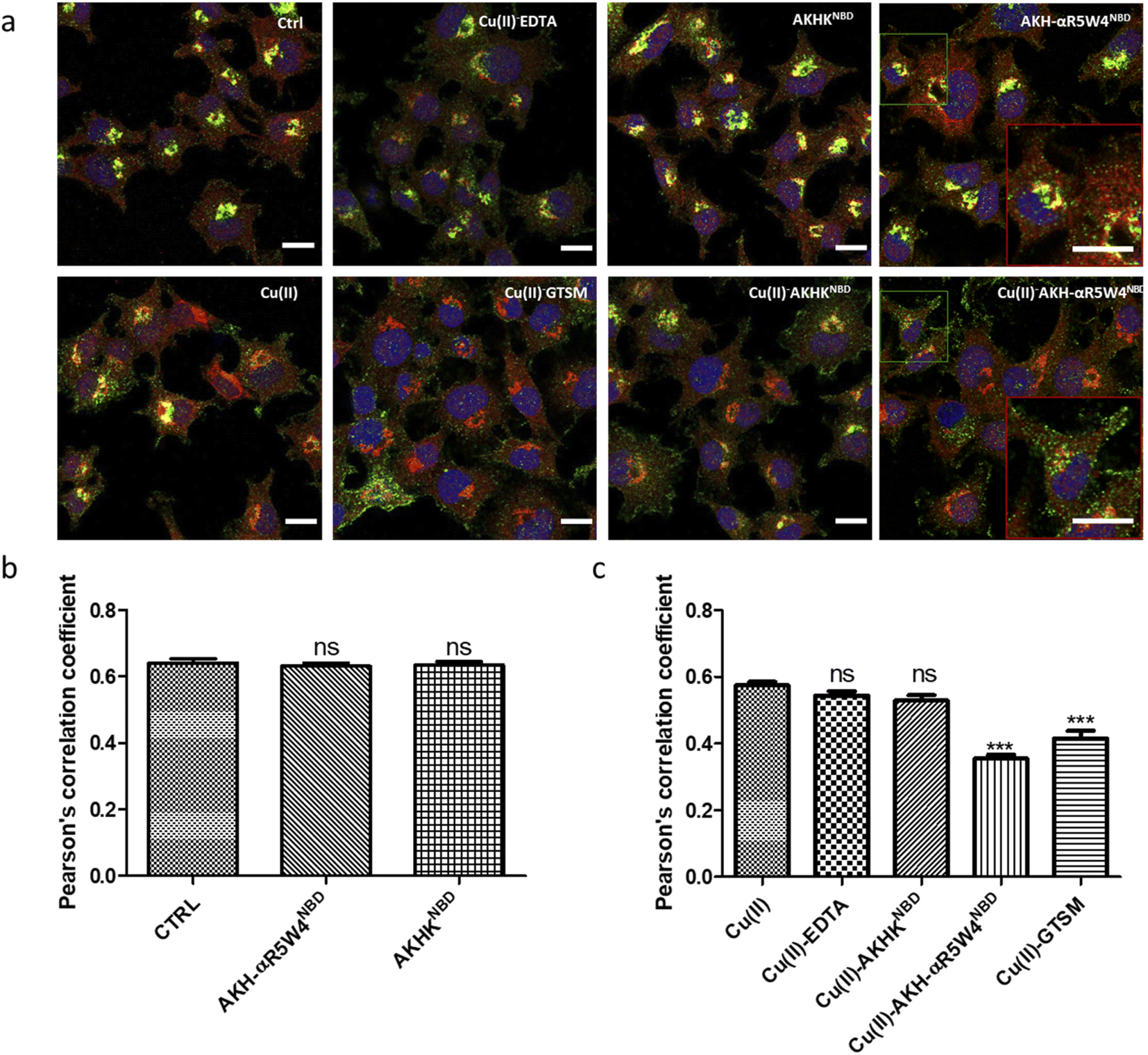 | ||
| Fig. 6 Cu import by AKH-αR5W4NBD induced delocalization of ATP7A from the TGN to vesicular structures. (a) PC12 cells were incubated for 1 h in DMEM media alone (control = Ctrl) or 5 μM Cu(II), 5 μM Cu(II)-EDTA, 5 μM AKHK4NBD, 5 μM Cu(II)-AKHK4NBD, 5 μM AKH-αR5W4NBD, 5 μM Cu(II)-AKH-αR5W4NBD or 1 μM Cu(II)-GTSM, before fixation and processing for immunostaining; blue: Hoechst (nucleus marker); green: ATP7A; red: Giantin (Golgi marker). Representative images are shown. Bars = 10 μm. n = 3 independent experiments. A zoomed inset is displayed for AKH-αR5W4NBD and Cu(II)-AKH-αR5W4NBD to illustrate the vesicular subplasmalemmal redistribution of ATP7A staining. (b and c) Quantification of the colocalization between ATP7A and Giantin staining measured as Pearson's correlation coefficient. Number of cells analyzed ≥ 27 for each condition. A parametric ANOVA test was carried out with Dunnett's multiple comparison test ***p < 0.0001. | ||
In summary, all three methods support the notion that Cu(II)-AKH-αR5W4NBD is able to import Cu into cells, which becomes bioavailable as revealed by the ATP7A delocalization from the TGN. Bioavailable intracellular Cu is likely to be Cu(I), in line with the reduction of Cu(II) peptide complexes by intracellular reductants leading to the subsequent release of Cu(I).
Inhibition of ROS production in vitro and in vivo by the AKH–CPPNBD peptide
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was incubated with 0.5 equivalents (with respect to Aβ1–16) of AKH-αR5W4NBD or AKHKNBD and the transfer of Cu(II) from Aβ1–16 to our Cu(II) shuttles was followed using the emission quenching of NBD when Cu(II) is coordinated to the ATCUN motif (AKH) in AKH-αR5W4NBD or AKHKNBD.
:1 was incubated with 0.5 equivalents (with respect to Aβ1–16) of AKH-αR5W4NBD or AKHKNBD and the transfer of Cu(II) from Aβ1–16 to our Cu(II) shuttles was followed using the emission quenching of NBD when Cu(II) is coordinated to the ATCUN motif (AKH) in AKH-αR5W4NBD or AKHKNBD.
In 10% DMEM buffer, the addition of AKH-αR5W4NBD and AKHKNBD to Cu(II)–Aβ1–16 resulted in fluorescence quenching in line with their high Cu(II)-binding affinity. The intensity of the quenching after 90 min is similar to the quenching of their respective pre-formed complexes (dashed lines), indicating that most of the Cu(II) was transferred. Given the aggregative propensity of AKH-LAH4NBD (Fig. S3†), the transfer of Cu from Aβ1–16 was not studied. However, it is expected to be similar to AKH-αR5W4NBD and AKHKNBD, since all peptides have the same ATCUN motifs.
 | ||
| Scheme 1 Cycling of Cu between its oxidized and reduced forms on Aβ1–16 in the presence of ascorbate. | ||
In agreement with the literature, Cu(II)-Aβ1–16 induced a fast decrease in the absorption of ascorbate at 265 nm (Fig. 8, black) due to the oxidation of ascorbate and the concomitant ROS production.61,62 Then Cu(II)–Aβ1–16 was added at the same time without pre-incubation (co-addition) with AKH-αR5W4NBD or AKHKNBD. In this case, AKH-LAH4NBD (Fig. S9†), AKH-αR5W4NBD or AKHKNBD (Fig. 8a and b) was able to decrease ascorbate oxidation, albeit not being immediate. At the beginning of the recordings, most of the Cu is still bound to Aβ1–16. Nevertheless, with time ascorbate oxidation slowed down and a plateau is reached, higher than for Cu(II)–Aβ1–16 alone, indicating that more and more Cu is transferred to AKH-LAH4NBD, AKH-αR5W4NBD or AKHKNBD. This is indeed in accordance with the time dependent transfer of Cu from Aβ1–16 to AKH-LAH4NBD, AKH-αR5W4NBD or AKHKNBD as seen in Fig. 7.
 | ||
| Fig. 7 Retrieval of Cu(II) from Aβ1–16 by AKH-αR5W4NBD and AKHKNBD. Dashed lines indicate the expected emission endpoint of AKH-αR5W4NBD (black) and AKHKNBD (red), respectively, in the presence of Cu(II) in a 1:1 complex. Conditions: AKH-αR5W4NBD and AKHKNBD: 5 μM; Aβ1–16: 10 μM; Cu: 5 μM; 25 °C; n = 3 independent experiments. Experiments were carried out in DMEM diluted to 10% (10% DMEM) with salts: 0.2 g L−1 CaCl2, 0.0001 g L−1 Fe(NO3)3, 0.098 g L−1 MgSO4, 0.4 g L−1 KCl, 3.7 g L−1 NaHCO3, 6.4 g L−1 NaCl, 0.11g L−1 NaH2PO4, and 4.5 g L−1D-glucose. | ||
Next, we tested the reaction with a 1 h pre-incubation of AKH-LAH4NBD, AKH-αR5W4NBD or AKHKNBD with Cu(II)–Aβ1–16, to allow Cu(II) transfer from Aβ1–16 to the ATCUN motif. The ascorbate consumption was markedly insignificant (Fig. 8a and b). This is in line with the quasi-inexistent redox-activity of Cu(II) bound in the Xxx–Zzz–His motif.30 When Cu(II)–Aβ1–16 is pre-incubated with AKH–CPPNBD for 1 h, there is complete transfer of Cu(II) from Aβ1–16 to the AKH Cu(II)-binding site, and therefore there is no efficient redox cycling of Cu and thus no reduction in ascorbate absorption. Hence, once Cu(II) is bound to the AKH motif in AKH-LAH4NBD, AKH-αR5W4NBD or AKHKNBD, it is almost completely redox silenced and incapable of catalyzing deleterious amounts of ROS.
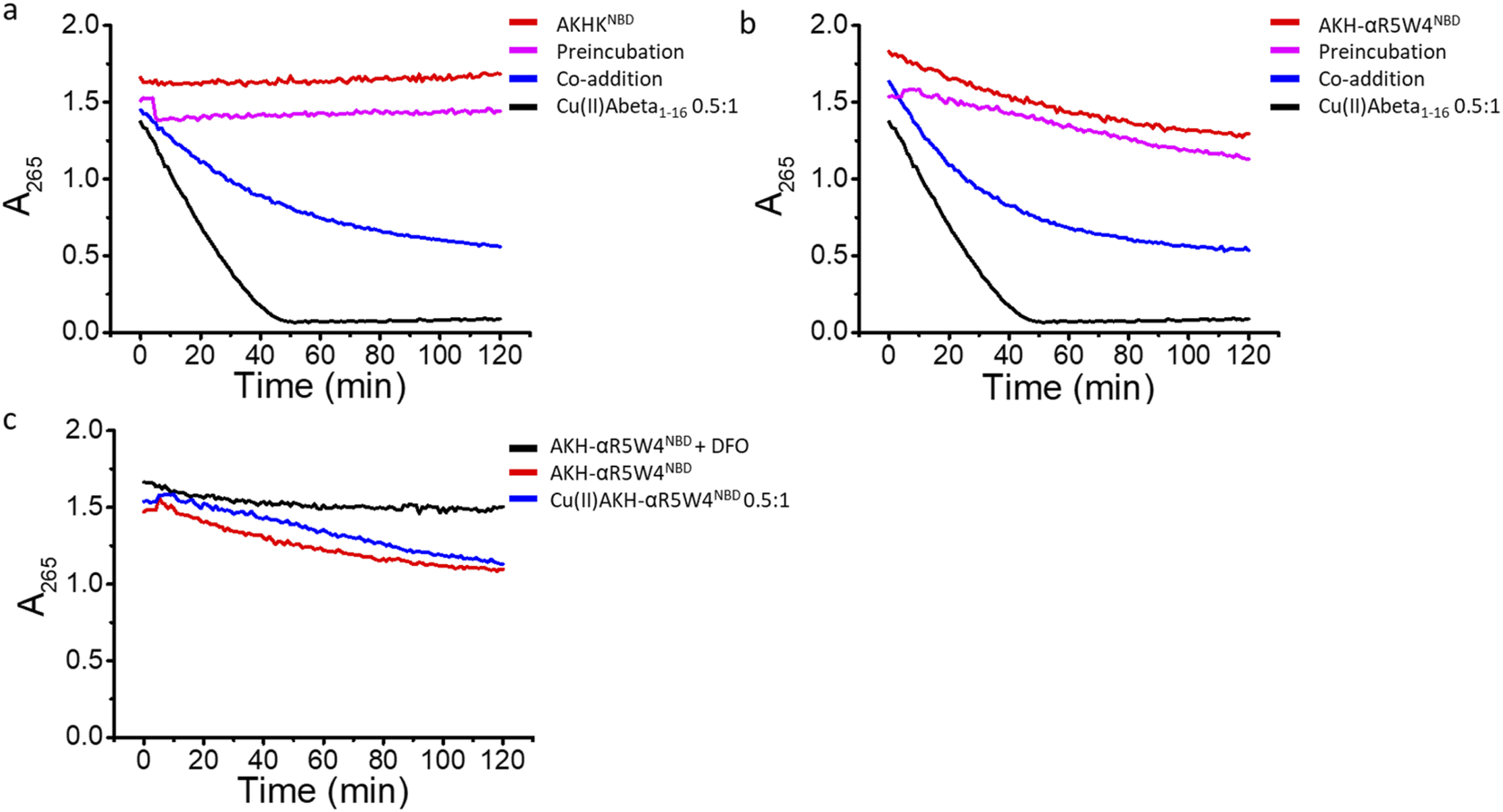 | ||
| Fig. 8 Retrieval of Cu(II) from Aβ1–16 by AKH-αR5W4NBD or AKHKNBD reduces ROS production. Inhibition of ROS production by (a) AKH-αR5W4NBD or AKHKNBD (b) monitored by the absorbance at 265 nm of ascorbate. Preincubation: 10 μM Cu(II)-Aß1–16 0.5:1 and 5 μM AKH-αR5W4NBD or AKHKNBD was incubated for 1 h before the addition of ascorbate followed by absorbance measurement; co-addition: 10 μM Cu(II)–Aβ1–16 0.5:1, 5 μM AKH-αR5W4NBD or AKHKNBD is added together with ascorbate followed by absorbance measurement. (c) Comparison of ascorbate absorption in the presence of 5 μM AKH-αR5W4NBD with or without 10 μM deferoxamine (DFO). Conditions: Asc: 100 μM, AKH-αR5W4NBD = AKHKNBD = Aβ1–16: 10 μM, Cu: 5 μM, DFO: 10 μM; HEPES 100 mM, pH 7.4. n = 2 independent experiments. | ||
Intriguingly, the higher ascorbate oxidation rate observed in the case of AKH-αR5W4NBD was unexpected, as it has the same Cu(II)-binding motif as AKHKNBD. This higher ROS production rate (Fig. S9d†) in the presence of AKH-αR5W4NBD was also seen to the same extent in the presence of pre-complexed Cu(II)-AKH-αR5W4NBD (Fig. 8c). Further experiments were able to attribute this higher ascorbate oxidation to Fe(III) contamination in the AKH-αR5W4NBD sample, which is markedly reduced by the addition of a strong Fe(III) chelator, deferoxamine (DFO) (Fig. 8c). Such Fe(III) contamination in the batch could contribute to the ROS-based toxicity to cells as studied below. However, it did not hamper the protecting effect of AKH-αR5W4NBD against Cu-Aß1–16 generated ROS, possible due to the much higher affinity to Cu(II) compared to Fe(III) of AKH-αR5W4NBD (Fig. 2).
Altogether, these results indicate that both in the absence or presence of pre-incubation, AKH-LAH4NBD, AKH-αR5W4NBD or AKHKNBD was able to retrieve Cu from Aβ1–16 and lower ROS production. As a control, we carried out the same experiment using Cu(II)Aβ1–42, which is the main toxic element of Aβ and showed that AKH-αR5W4NBD or AKHKNBD was equally able to lessen ROS production in vitro (Fig. S9b and c†).
:1 with 5 μM AKH–CPPNBD for 1 h, after which 300 μM ascorbate was added to the mixture prior to incubation with PC12 cells for 24 h, with cell toxicity being evaluated using a MTT assay.
Treatment with AKH-αR5W4NBD or AKHKNBD effectively prevented Cu(II)–Aβ1–16 toxicity, in line with Cu-binding and redox silencing. Of note AKH-LAH4NBD seemed toxic to PC12 cells at 5 μM when present in 10% DMEM (Fig. S11†), which was unexpected given that no toxicity was observed in 100% DMEM at that concentration (Fig. S6†). Recapitulating the in vitro data, there were almost no rescue effects from AKH-LAH4NBD, AKH-αR5W4NBD or AKHKNBD on Cu(II)–Aβ1–16-induced cell toxicity without 1 h preincubation (data not shown). Indeed in vitro experiments above showed that the transfer of Cu from Cu(II)-Aβ1–16 to AKH–CPPNBD (Fig. 7) takes time, thereby allowing the production of a significant amount of ROS catalyzed by Cu(II)–Aβ1–16 before complete transfer occurred.
We tested two different batches Aβ1–42 to evaluate their toxicity on PC12 cells (individual batch data in Fig. S11b†). The incubation of Aβ1–42 was toxic to PC12 cells at 10 μM and the complexation of Aβ1–42 with Cu(II) did not change the level of toxicity. However, upon addition of ascorbate, the latter showed a significant increase in toxicity on PC12 cells (p < 0.0001). An hour pre-incubation of Cu(II)–Aβ1–42 with either AKH-αR5W4NBD or AKHKNBD rescued the toxicity induced by the Cu, although the toxicity due to Aβ1–42 alone was not remediated as expected (Fig. 9c).
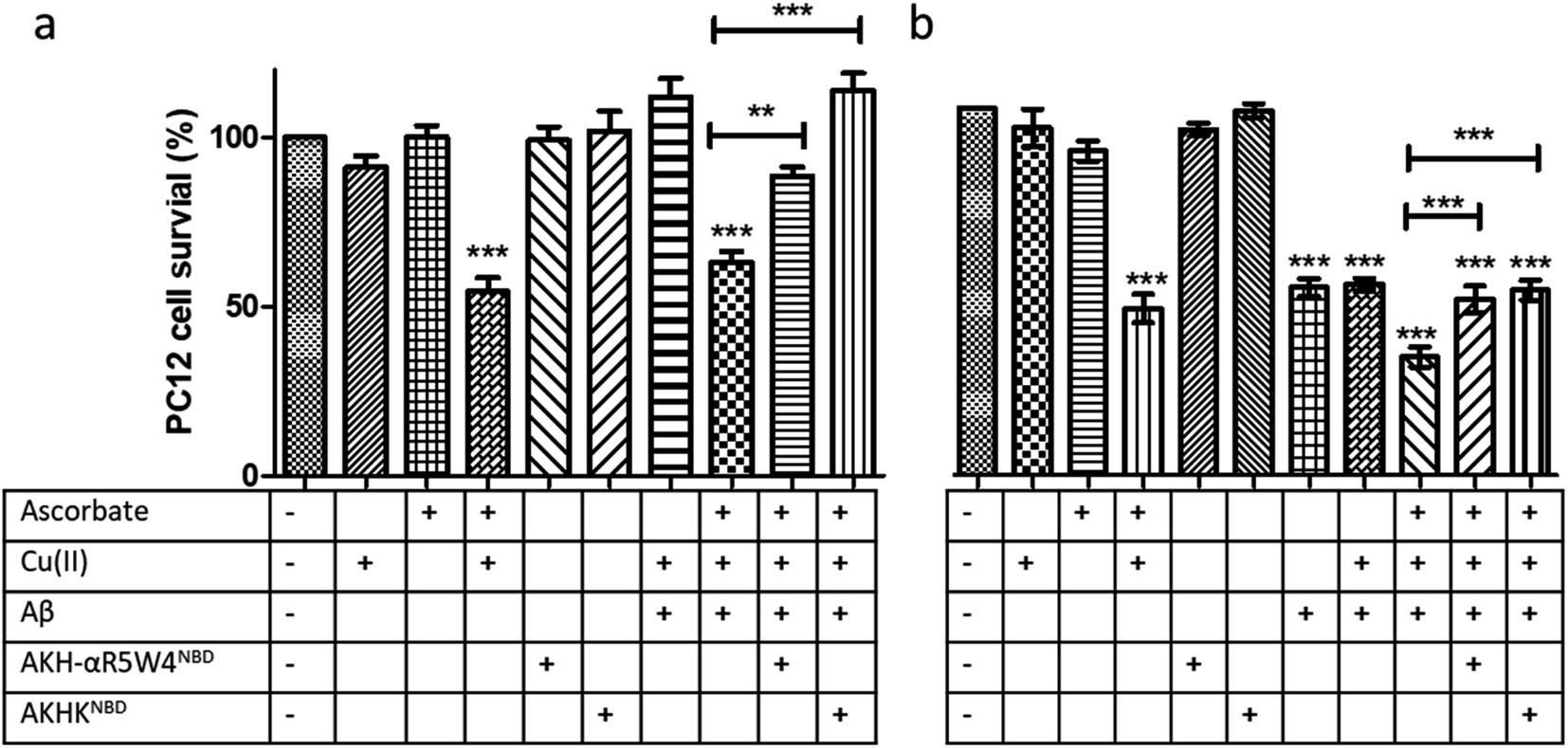 | ||
| Fig. 9 Transfer of Cu(II) from Aβ to the ATCUN motif prevents Cu-induced ROS production and toxicity to PC12 cells. (a) Aβ1–16 and (b) Aβ1–42 (compilation of two batches, see Fig. S11†). 5 μM AKH-αR5W4NBD or AKHKNBD was pre-incubated with 10 μM Cu(II)–Aβ1–16/42 0.5:1 in 10% DMEM in a test tube for 1 h before the addition of ascorbate and immediate administration on the PC12 cells. Experiments were done in triplicate, n = 3. A parametric ANOVA test was carried out with a Tukey post-test, **p < 0.001 and ***p < 0.0001. Experiments were carried out in DMEM dilutes to 10% (10% DMEM) with salts: 0.2 g L−1 CaCl2, 0.0001 g L−1 Fe(NO3)3, 0.098g L−1 MgSO4, 0.4 g L−1 KCl, 3.7 g L−1 NaHCO3, 6.4g L−1 NaCl, 0.11 g L−1 NaH2PO4, and 4.5 g L−1D-glucose. | ||
Conclusions
Re-equilibrating Cu homeostasis in neurodegenerative pathologies such as AD is a complex task, but it deserves to be addressed based on the potentially important outcomes, such as reducing oxidative stress and Aβ toxicity. This involves the design of a molecule that is highly selective to Cu(II) in comparison to other more concentrated bioavailable metals in the brain such as Zn(II) and Fe(III). Equally the molecule has to have a high enough affinity for Cu(II), to withdraw the latter from the Cu(II)-binding domain of Aβ (Aβ1–16) in amyloid plaques in the brain but not from other essential Cu(II)–proteins, such as ceruloplasmin. Finally, the molecule should be capable of penetrating the plasma membrane of neuronal cells as well as releasing imported Cu into neurons or glia cells.Probing here the potential of the AKH–CPPNBD shuttle, we have been able to synthesize, for the first time, a Cu(II) peptide shuttle capable of binding specifically Cu(II) in biological media in the presence of Zn(II), Mn(II) and Fe(III). This shuttle is selective for Cu(II) since it is based on the ATCUN motif that binds very weakly to other essential metal ions under extracellular relevant conditions. This differs from CQ and PBT2, used in clinical trials in the treatment of AD, which are able to shuttle Zn as well. The absence of selectivity of these shuttles for Cu makes it difficult to study the effect on Cu dyshomeostasis singularly in several pathologies including AD.
Cu(II) bound to the AKH–CPPNBD shuttle is also capable of being reduced to Cu(I) by a physiological concentration of GSH in vitro. This seems also to occur in cellulo, as suggested by the translocation of ATP7A from the TGN to vesicles in proximity to the plasma membrane. This is in line with a Cu(II) import by AKH-αR5W4NBD, reduction to Cu(I) by GSH and subsequent release in a bioavailable Cu(I) pool. Our construct AKH-αR5W4NBD has several important features needed to re-distribute Cu from extracellular space to an intracellular bioavailable Cu pool, and hence can be an interesting tool for studying dyshomeostasis in Cu-related pathologies. In addition, AKH-αR5W4NBD is capable of retrieving Cu(II) from Aβ1–16, the metal-binding domain responsible for Cu complexation by Aβ1–40/42 as well as from the full length Aβ1–42. In contrast to Aβ, AKH-αR5W4NBD binds Cu(II) and stabilizes it, such that ascorbate oxidation and concomitant ROS production are precluded. In line with this, AKH-αR5W4NBD is able to protect PC12 from cells from Cu(II)–Aβ induced damage in the presence of ascorbate. However, the transfer of Cu from Aβ to AKH-αR5W4NBD is relatively slow, and hence protection is not immediate. In conclusion, the shuttle described here might serve as a seminal scaffold to identify derived shuttles with faster Cu(II) complexation in future studies, as well as in studies with more elaborated cell models related to AD.
Materials and methods
Materials
All compounds used during this study were purchased from accredited merchants unless stated otherwise: Dulbecco's modified Eagle's medium–high glucose (Sigma, D5796), horse serum (Gibco, 26050070), Fetal calf serum (Gibco, 10270-160), penicillin/streptomycin (Sigma, P4458), Trypsin (Gibco, 25300-054), MTT (Fisher Scientific, 10133722), anti-giantin antibodies (Abcam, ab24586), monoclonal anti-ATP7A antibodies (Abcam, ab13995), secondary antibodies (Life sciences) and Hoechst (Thermo Fisher, 62249). Batch 1 Aβ1–42 (AnaSpec, >95% purity) and Batch 2 Aβ1–42 (Genecust, >95% purity).Peptide synthesis
Peptide synthesis was carried out according to a general SPPS protocol.63 The peptide synthesis was carried out using the Biotage® Initiator+ Alstra™. A Fmoc-Rink-Amide Tenta XV RAM resin (charge of 0.24 mmol g−1) was used for the synthesis of all peptides. 0.5 M 1:1 ethyl cyano(hydroxyimino)acetate (Oxyma)/diisopropylcarbodiimide (DIC) was used for activation, and 2 M DIEA dissolved in N-methyl-2-pyrrolidone (NMP) was used as the base. 5 M anhydride acetic acid was used to cap unreacted free amino groups.
The reaction time was 2 × 30 minutes coupling at ambient temperature and 2 × 5 minutes coupling when using microwaves (μWF) for compatible amino acids at 75 °C with the exception of cysteine, basic amino acids, and glycine after acidic amino acids. Fmoc deprotection was carried out using μWF at 75 °C. Lysine was added as the Nε-allyloxycarbonyl-protected (Fmoc-Lys (Alloc)–OH) form to allow specific sidechain deprotection in order to graft the fluorophore NBD. The Alloc protecting group of the lysine side chain is deprotected using tetrakis(triphenylphosphine)palladium(0), which is a palladium-based mechanism.64 The NBD fluorophore is added to the free lysine by nucleophilic substitution of 4 eq. NBD-Cl in the presence of DIEA. Finally, deprotection was carried out using TFA in the presence of scavengers (H2O and triisopropylsilane).
Preparation of stock solution
All peptide stock solutions were prepared in ultra-pure Milli Q water (ρ = 18.2 MΩ cm−1) and were stored at −20 °C. The pH of the solutions was maintained at ≈pH 2, except when complexed to Cu(II) in which case they were buffered at pH 7.4 using 100 mM HEPES.Fluorescence spectroscopy
The emission of NBD on AKH–CPP was taken in the presence and absence of Cu(II) using a Horiba Fluoromax plus fluorimeter using a 500 μL 1 cm path quartz cuvette. 4 μM of each peptide was titrated by adding 0.1 equivalent Cu into the cuvette (Fig. 1). AKH-αR5W4NBD; excitation slit: 4 mm; emission slit: 3 mm, AKH-LAH4NBD and AKHKNBD; excitation slit: 2 mm; emission slit: 2 mm.Ascorbate assay (UV/Vis spectroscopy)
Using a CLARIOstar (BMG LABTECH) plate reader, experiments were carried out in a UV-Star® 96 well microplate (Greiner), Half Area, clear. The absorbance of 100 μM ascorbate (DO ≈ 1.5) at 265 nm was followed under different conditions to indirectly follow ROS production. The final volume per well was 100 μL and was buffered at pH 7.4 using 100 mM HEPES.Cell culture
PC12 cells were maintained in culture in a DMEM high glucose medium (4500 mg mL−1) with 10% horse serum, 5% FBS and 1% penicillin/streptomycin as described previously.65 The cells were split once a week using trypsin for detachment and replaced by a fresh batch of cells after a maximum of 15 splits.Immunohistochemistry and live imaging
PC12 cells were cultured on poly-D-lysine-coated glass coverslips in NUNC 4-well plates at a density of 5 × 104 per well, 24 h before experiments. After the experiment, the medium was removed and the wells were washed with 1X PBS, and the cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 10 min at room temperature. The cells were permeabilised in 0.1% Triton X-100 (Thermo Fisher Scientific) for 10 min followed by 1 h saturation in 1X PBS containing 0.2% Triton X-100, 10% donkey serum (Sigma-Aldrich) and 3% Bovine Serum Albumin (BSA, Sigma – Aldrich) at 37 °C. The cells were then incubated overnight at 4 °C with primary antibodies, and after washing in PBS 1X were incubated with secondary antibodies and Hoechst for 45 min at 37 °C before being rinsed and mounted.For live imaging, PC12 was grown in glass bottom Ibidi chamber coverslip cells and maintained in Locke solution (140 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 11 mM glucose, 0.56 mM ascorbic acid, 0.01 mM EDTA and 15 mM HEPES, pH 7.5) for recording. Fluorescent peptides are added < 30 seconds before recording for a duration of 30 minutes at 10 frames/min.
MTT assay
A MTT stock solution at 5 mg mL−1 was prepared in PBS 1X. The cells were incubated with a final MTT concentration of 0.5 mg mL−1 in DMEM media for 4 h at 37 °C after which the medium was removed, and the formazan precipitate dissolved in DMSO. The absorption was recorded at 595 nm with a plate reader.ICP-MS
| ICP-MS | Agilent 7700 |
|---|---|
| RF power | 1550 W |
| Sampling and skimmer cones | Nickel |
| Nebulizer | MicroMist |
| Spray chamber | Scott double-pass, quartz |
| Plasma gas | 15.0 L min−1 |
| Nebulizer gas | 1.03 L min−1 |
| Makeup gas | 0.00 L min−1 |
| Auxiliary gas | 0.9 L min−1 |
| Spray chamber temp | 2 °C |
| Sampling depth | 8 mm |
| Dwell time | 0.003 s |
| Isotopes | 63Cu65Cu115In |
| ORS mode | He collision ORS3 |
| Nebulizer pump | 0.10 rps |
Statistical analysis
All analyses were carried out using a GraphPad prism. The data set underwent the Shapiro test for normality. For comparisons of the mean, a parametric test with Dunnett's multiple comparison post-test or non-parametric Kruskal–Wallis with Dunn's multiple comparisons test was performed.Data availability
We added as much as possible of the experimental data in the ESI.†Author contributions
Investigation: M. O., P. G., P. R., and I. E.; writing – original draft: M. O.; supervision: A. B., L. R., N. V., and P. F.; initial conceptualization: C. H.; funding acquisition: C. H., N. V., and P. F.; writing – review & editing: L. R., C. H., N. V., and P. F.; methodology: L. R., N. V., P. F., S. C.-G., S. O., and S.G.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge the support of the IdEx PhD program, University of Strasbourg for funding MO. Grants from the Agence Nationale pour la Recherche (ANR-19-CE44-0019) to N.V. and ERC StG aLzINK to C.H. INSERM to NV are acknowledged. We thank Dr Fabienne Burlina and Dr Sandrine Sagan for the synthesis protocol of RW9, Dr Silvie Ferlay for access to the fluorimeter, Dr Thomas Hermans and Dr George Formon for access to the atomic absorption spectroscope and Dr Charlène Esmieu for fruitful discussions.References
- E. Falcone, M. Okafor, N. Vitale, L. Raibaut, A. Sour and P. Faller, Coord. Chem. Rev., 2021, 433, 213727 CrossRef CAS.
- T. Galler, V. Lebrun, L. Raibaut, P. Faller and N. E. Wezynfeld, Chem. Commun., 2020, 56, 12194–12197 RSC.
- A. Skopp, S. D. Boyd, M. S. Ullrich, L. Liu and D. D. Winkler, BioMetals, 2019, 32, 695–705 CrossRef CAS PubMed.
- S. D. Boyd, M. S. Ullrich, A. Skopp and D. D. Winkler, Antioxidants, 2020, 9, E500 CrossRef PubMed.
- M. D. Harrison, C. E. Jones and C. T. Dameron, J. Biol. Inorg Chem., 1999, 4, 145–153 CrossRef CAS PubMed.
- Y. Hatori, S. Inouye and R. Akagi, IUBMB Life, 2017, 69, 246–254 CrossRef CAS PubMed.
- Y. Hatori and S. Lutsenko, Antioxidants, 2016, 5, E25 CrossRef PubMed.
- I. D. Goodyer, E. E. Jones, A. P. Monaco and M. J. Francis, Hum. Mol. Genet., 1999, 8, 1473–1478 CrossRef CAS PubMed.
- C. Lane, M. J. Petris, A. Benmerah, M. Greenough and J. Camakaris, BioMetals, 2004, 17, 87–98 CrossRef CAS PubMed.
- M. L. Schlief and J. D. Gitlin, Mol. Neurobiol., 2006, 33, 81–90 CrossRef CAS.
- Z. Tümer and L. B. Møller, Eur. J. Hum. Genet., 2010, 18, 511–518 Search PubMed.
- M. T. Lorincz, Handb. Clin. Neurol., 2018, 147, 279–292 Search PubMed.
- G. Gromadzka, B. Tarnacka, A. Flaga and A. Adamczyk, Int. J. Mol. Sci., 2020, 21, E9259 CrossRef PubMed.
- G. S. Bloom, JAMA Neurol., 2014, 71, 505–508 CrossRef PubMed.
- M. A. Busche and B. T. Hyman, Nat. Neurosci., 2020, 23, 1183–1193 CrossRef CAS PubMed.
- H. Zhang, W. Wei, M. Zhao, L. Ma, X. Jiang, H. Pei, Y. Cao and H. Li, Int. J. Biol. Sci., 2021, 17, 2181–2192 CrossRef CAS PubMed.
- X. Wang, W. Wang, L. Li, G. Perry, H. Lee and X. Zhu, Biochim. Biophys. Acta, 2014, 1842, 1240–1247 CrossRef CAS PubMed.
- J. M. Perez Ortiz and R. H. Swerdlow, Br. J. Pharmacol., 2019, 176, 3489–3507 CrossRef CAS PubMed.
- W. Ahmad, B. Ijaz, K. Shabbiri, F. Ahmed and S. Rehman, J. Biomed. Sci., 2017, 24, 76 CrossRef PubMed.
- C. Hureau, in Alzheimer's Disease, 2021, pp. 170–192 Search PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2017, 14, 450–464 CrossRef PubMed.
- E. Atrián-Blasco, P. Gonzalez, A. Santoro, B. Alies, P. Faller and C. Hureau, Coord. Chem. Rev., 2018, 375, 38–55 CrossRef PubMed.
- R. C. Broad, J. P. Bonneau, R. P. Hellens and A. A. T. Johnson, Int. J. Mol. Sci., 2020, 21, E1790 CrossRef.
- F. E. Harrison and J. M. May, Free Radical Biol. Med., 2009, 46, 719–730 CrossRef CAS PubMed.
- K. Jomova and M. Valko, Toxicology, 2011, 283, 65–87 CrossRef CAS PubMed.
- P. Poprac, K. Jomova, M. Simunkova, V. Kollar, C. J. Rhodes and M. Valko, Trends Pharmacol. Sci., 2017, 38, 592–607 CrossRef CAS PubMed.
- C. Esmieu, G. Ferrand, V. Borghesani and C. Hureau, Chem. - Eur. J., 2021, 27, 1777–1786 CrossRef CAS PubMed.
- E. Falcone, P. Gonzalez, L. Lorusso, O. Sénèque, P. Faller and L. Raibaut, Chem. Commun., 2020, 56, 4797–4800 RSC.
- E. Stefaniak, D. Płonka, P. Szczerba, N. E. Wezynfeld and W. Bal, Inorg. Chem., 2020, 59, 4186–4190 CrossRef CAS PubMed.
- A. Santoro, G. Walke, B. Vileno, P. P. Kulkarni, L. Raibaut and P. Faller, Chem. Commun., 2018, 54, 11945–11948 RSC.
- R. Sankararamakrishnan, S. Verma and S. Kumar, Proteins: Struct., Funct., Bioinf., 2005, 58, 211–221 CrossRef CAS.
- M. G. M. Weibull, S. Simonsen, C. R. Oksbjerg, M. K. Tiwari and L. Hemmingsen, J. Biol. Inorg Chem., 2019, 24, 1197–1215 CrossRef CAS PubMed.
- J. F. Quinn, S. Crane, C. Harris and T. L. Wadsworth, Expert Rev. Neurother., 2009, 9, 631–637 CrossRef CAS PubMed.
- P. J. Crouch, L. W. Hung, P. A. Adlard, M. Cortes, V. Lal, G. Filiz, K. A. Perez, M. Nurjono, A. Caragounis, T. Du, K. Laughton, I. Volitakis, A. I. Bush, Q.-X. Li, C. L. Masters, R. Cappai, R. A. Cherny, P. S. Donnelly, A. R. White and K. J. Barnham, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 381–386 CrossRef CAS.
- C. Esmieu, D. Guettas, A. Conte-Daban, L. Sabater, P. Faller and C. Hureau, Inorg. Chem., 2019, 58, 13509–13527 CrossRef CAS PubMed.
- P. Tsvetkov, S. Coy, B. Petrova, M. Dreishpoon, A. Verma, M. Abdusamad, J. Rossen, L. Joesch-Cohen, R. Humeidi, R. D. Spangler, J. K. Eaton, E. Frenkel, M. Kocak, S. M. Corsello, S. Lutsenko, N. Kanarek, S. Santagata and T. R. Golub, Science, 2022, 375, 1254–1261 CrossRef CAS.
- L. M. Guthrie, S. Soma, S. Yuan, A. Silva, M. Zulkifli, T. C. Snavely, H. F. Greene, E. Nunez, B. Lynch, C. De Ville, V. Shanbhag, F. R. Lopez, A. Acharya, M. J. Petris, B.-E. Kim, V. M. Gohil and J. C. Sacchettini, Science, 2020, 368, 620–625 CrossRef CAS PubMed.
- E. Böhmová, D. Machová, M. Pechar, R. Pola, K. Venclíková, O. Janoušková and T. Etrych, Physiol. Res., 2018, 67, S267–S279 Search PubMed.
- A. Walrant, A. Bauzá, C. Girardet, I. D. Alves, S. Lecomte, F. Illien, S. Cardon, N. Chaianantakul, M. Pallerla, F. Burlina, A. Frontera and S. Sagan, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183098 CrossRef CAS PubMed.
- K. Kardani, A. Milani, S. H Shabani and A. Bolhassani, Expert Opin. Drug Delivery, 2019, 16, 1227–1258 CrossRef CAS PubMed.
- M. Green and P. M. Loewenstein, Cell, 1988, 55, 1179–1188 CrossRef CAS PubMed.
- R. P. Viscidi, K. Mayur, H. M. Lederman and A. D. Frankel, Science, 1989, 246, 1606–1608 CrossRef CAS PubMed.
- E. Vivès, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef.
- A. H. A. Clayton, B. W. Atcliffe, G. J. Howlett and W. H. Sawyer, J. Pept. Sci., 2006, 12, 233–238 CrossRef CAS PubMed.
- E. Dupont, A. Prochiantz and A. Joliot, Methods Mol. Biol., 2015, 1324, 29–37 CrossRef PubMed.
- T. Keum, G. Noh, J.-E. Seo, S. Bashyal and S. Lee, Pharmaceuticals, 2020, 13, E408 CrossRef.
- A. Elmquist, M. Lindgren, T. Bartfai and U. Ülo Langel, Exp. Cell Res., 2001, 269, 237–244 CrossRef CAS PubMed.
- B. Alaybeyoglu, B. Sariyar Akbulut and E. Ozkirimli, J. Pept. Sci., 2018, 24, e3083 CrossRef PubMed.
- A. Elmquist and U. Langel, Biol. Chem., 2003, 384, 387–393 CAS.
- S. Gao, M. J. Simon, C. D. Hue, B. Morrison and S. Banta, ACS Chem. Biol., 2011, 6, 484–491 CrossRef CAS PubMed.
- B. Bechinger, J. Mol. Biol., 1996, 263, 768–775 CrossRef CAS.
- P. Gonzalez, K. Bossak, E. Stefaniak, C. Hureau, L. Raibaut, W. Bal and P. Faller, Chemistry, 2018, 24, 8029–8041 CrossRef CAS PubMed.
- R. H. Sigg, P. L. Luisi and A. A. Aboderin, J. Biol. Chem., 1977, 252, 2507–2514 CrossRef CAS PubMed.
- P. Eli and A. Chakrabartty, Protein Sci., 2006, 15, 2442–2447 CrossRef CAS.
- Y. Ogra, A. Tejima, N. Hatakeyama, M. Shiraiwa, S. Wu, T. Ishikawa, A. Yawata, Y. Anan and N. Suzuki, Sci. Rep., 2016, 6, 33007 CrossRef CAS PubMed.
- C. Duncan, L. Bica, P. J. Crouch, A. Caragounis, G. E. Lidgerwood, S. J. Parker, J. Meyerowitz, I. Volitakis, J. R. Liddell, R. Raghupathi, B. M. Paterson, M. D. Duffield, R. Cappai, P. S. Donnelly, A. Grubman, J. Camakaris, D. J. Keating and A. R. White, Metallomics, 2013, 5, 700–714 CrossRef CAS PubMed.
- R. Curnock and P. J. Cullen, J. Cell Sci., 2020, 133, jcs249201 CrossRef CAS PubMed.
- J.-F. Monty, R. M. Llanos, J. F. B. Mercer and D. R. Kramer, J. Nutr., 2005, 135, 2762–2766 CrossRef CAS PubMed.
- M. E. Rice, Trends Neurosci., 2000, 23, 209–216 CrossRef CAS.
- L. Guilloreau, S. Combalbert, A. Sournia-Saquet, H. Mazarguil and P. Faller, ChemBioChem, 2007, 8, 1317–1325 CrossRef CAS PubMed.
- B. Alies, I. Sasaki, O. Proux, S. Sayen, E. Guillon, P. Faller and C. Hureau, Chem. Commun., 2013, 49, 1214–1216 RSC.
- E. Atrián-Blasco, M. del Barrio, P. Faller and C. Hureau, Anal. Chem., 2018, 90, 5909–5915 CrossRef PubMed.
- E. Falcone, B. Vileno, M. Hoang, L. Raibaut and P. Faller, J. Inorg. Biochem., 2021, 221, 111478 CrossRef CAS PubMed.
- N. Thieriet, J. Alsina, E. Giralt, F. Guibé and F. Albericio, Tetrahedron Lett., 1997, 38, 7275–7278 CrossRef CAS.
- E. Tanguy, P. Costé de Bagneaux, N. Kassas, M.-R. Ammar, Q. Wang, A.-M. Haeberlé, J. Raherindratsara, L. Fouillen, P.-Y. Renard, M. Montero-Hadjadje, S. Chasserot-Golaz, S. Ory, S. Gasman, M.-F. Bader and N. Vitale, Cell Rep., 2020, 32, 108026 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02593k |
| ‡ Contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |