
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNorbornene-tetrazine ligation chemistry for controlling RNA-guided CRISPR systems†
Qianqian
Qi‡
,
Yutong
Zhang‡
 ,
Wei
Xiong‡
,
Xingyu
Liu
,
Shuangyu
Cui
,
Xiaofang
Ye
,
Kaisong
Zhang
,
Tian
Tian
* and
Xiang Zhou
,
Wei
Xiong‡
,
Xingyu
Liu
,
Shuangyu
Cui
,
Xiaofang
Ye
,
Kaisong
Zhang
,
Tian
Tian
* and
Xiang Zhou
Key Laboratory of Biomedical Polymers of Ministry of Education, College of Chemistry and Molecular Sciences, Hubei Province Key Laboratory of Allergy and Immunology, Wuhan University, Wuhan 430072, Hubei, China. E-mail: ttian@whu.edu.cn
First published on 20th October 2022
Abstract
In the present study, norbornene-tetrazine ligation chemistry is harnessed for developing clickable RNA switches in biological contexts. This RNA control strategy is explored with a variety of applications. We further demonstrate the application of RNA-based norbornene-tetrazine ligation chemistry for controlling CRISPR systems. Moreover, the manipulation of gene editing in human cells is accomplished.
Introduction
RNAs are unique macromolecules, which play many roles in various biological processes. Loss of function tools offers a particularly attractive ‘negative’ approach to study RNAs of interest (ROIs). Covalent approaches, such as alkylating and cross-linking reactions, can be used to deactivate the function of macromolecules.1–3 However, most of these compounds may react with a number of endogenous biomolecules. The introduction of bioorthogonal chemistries into a suitably functionalized RNA is appealing. We prefer to use chemically inert molecules to avoid cross-reaction with common functional groups present in biological systems. In light of this, the inverse-electron-demand Diels–Alder (inv-DA) reaction between norbornene (NB) and tetrazine (TZ) derivatives stands out for its freedom from catalysts, nontoxic byproducts and substantial chemical optimizability.4–6 From the perspective of chemistry, chemical modifications of RNA can be made to the 5′- and 3′-ends of RNA oligomers, the carbon 2 (C2′) position of ribose, phosphate linkage, and nucleobase moieties. We targeted the ribose 2′-OH group because the abundance of this group is ideal for efficient reaction.7Here, NB-TZ ligation chemistry is harnessed for developing clickable RNA switches. The NB derivative of the formylized imidazole (NBFI) is used to equip the target of interest with reactive handles. The appended NB moieties in turn react with exogenously added complementary TZ partners (Fig. 1A). We characterize the formation of interstrand cross-links derived from the reaction of NB-bearing RNAs with multimeric TZ derivatives (Fig. 1B and S1†). This RNA control strategy is explored with a variety of applications. We further demonstrate the application of our strategy for controlling CRISPR (clustered regularly interspaced short palindromic repeat) systems. The NB-bearing guide RNAs (gRNAs) form cleavage–competent complexes almost as efficiently as native gRNAs. Importantly, selective and robust control of CRISPR is achieved with clickable RNA switches. Moreover, conditional control of gene editing in human cells is achieved.
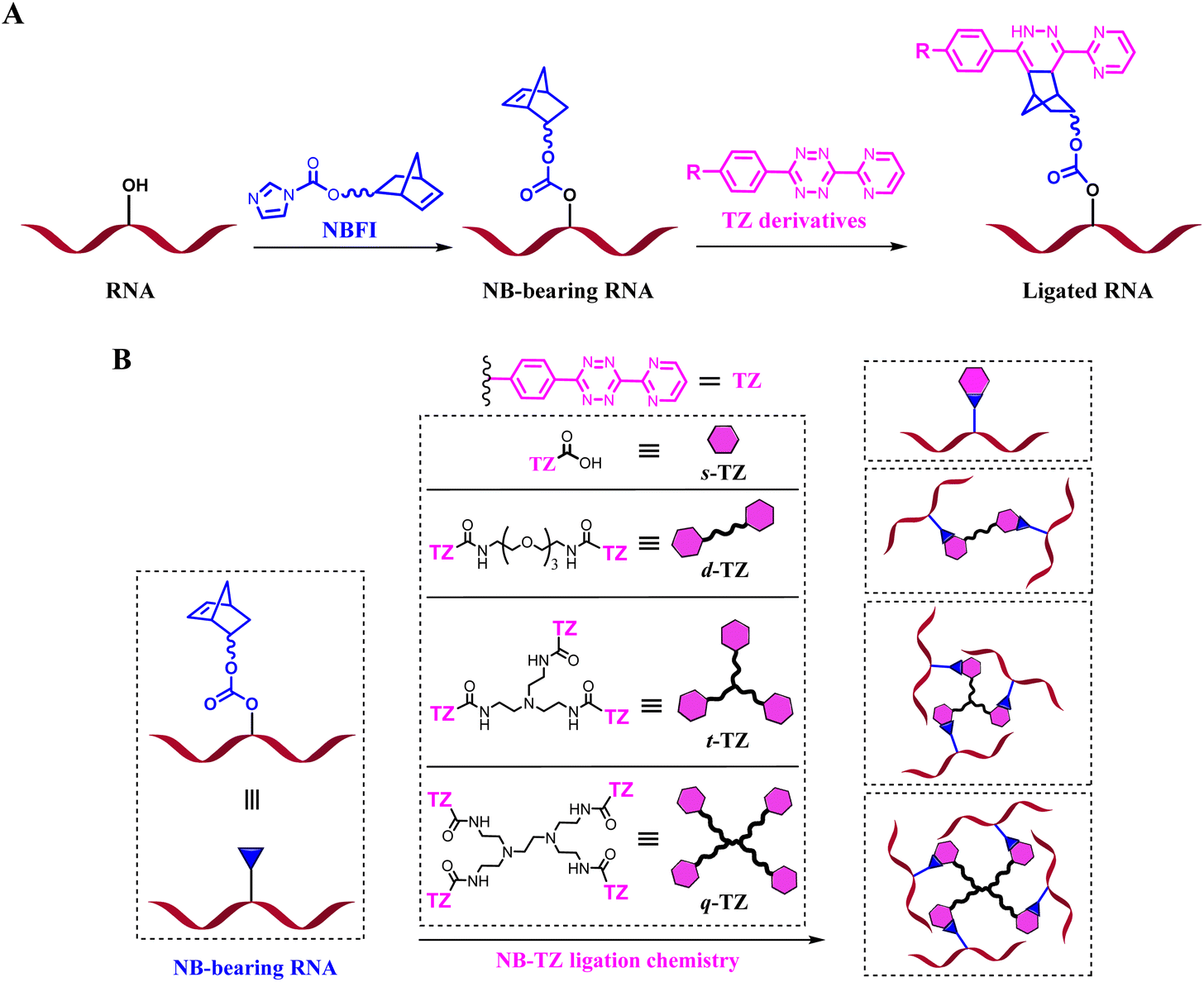 | ||
| Fig. 1 Schematic illustration of this study. (A) Workflow of RNA-based NB-TZ ligation chemistry. (B) The reaction between NB-bearing RNAs and each TZ. This demonstration gives just one possible product for each reaction. | ||
Results
Strategic design of this study
We strive to develop chemical tools for manipulating the functionality of RNAs in their native environments. Recent findings have established a set of bioorthogonal reactions to address the need for highly specific reactions in biological contexts.8–12 We have recognized that the NB-TZ ligation chemistry has fast reaction rate, high chemoselectivity, catalyst-free nature, and relatively simple synthetic procedures.13,14 Multimeric TZ molecules are further designed to introduce more constraints (Fig. S1†). The larger size of multimeric TZ derivatives also suggests their potential to improve the efficiency for disabling ROI functions.Management of RNA-based NB-TZ ligation chemistry
To perform our strategy, the first step is to obtain NB-functionalized RNAs. The NBFI molecule is prepared by a convenient one-step procedure (ESI†). A model RNA (R-32nt in Table S1†) was incubated with a specified concentration of NBFI (20 mM) for varied periods at room temperature. After reaction, the products were purified and the degree of NB modification was determined with denaturing polyacrylamide gel electrophoresis (PAGE). We demonstrate that the NB-bearing RNAs have reduced mobility compared to that of the control (Fig. 2A).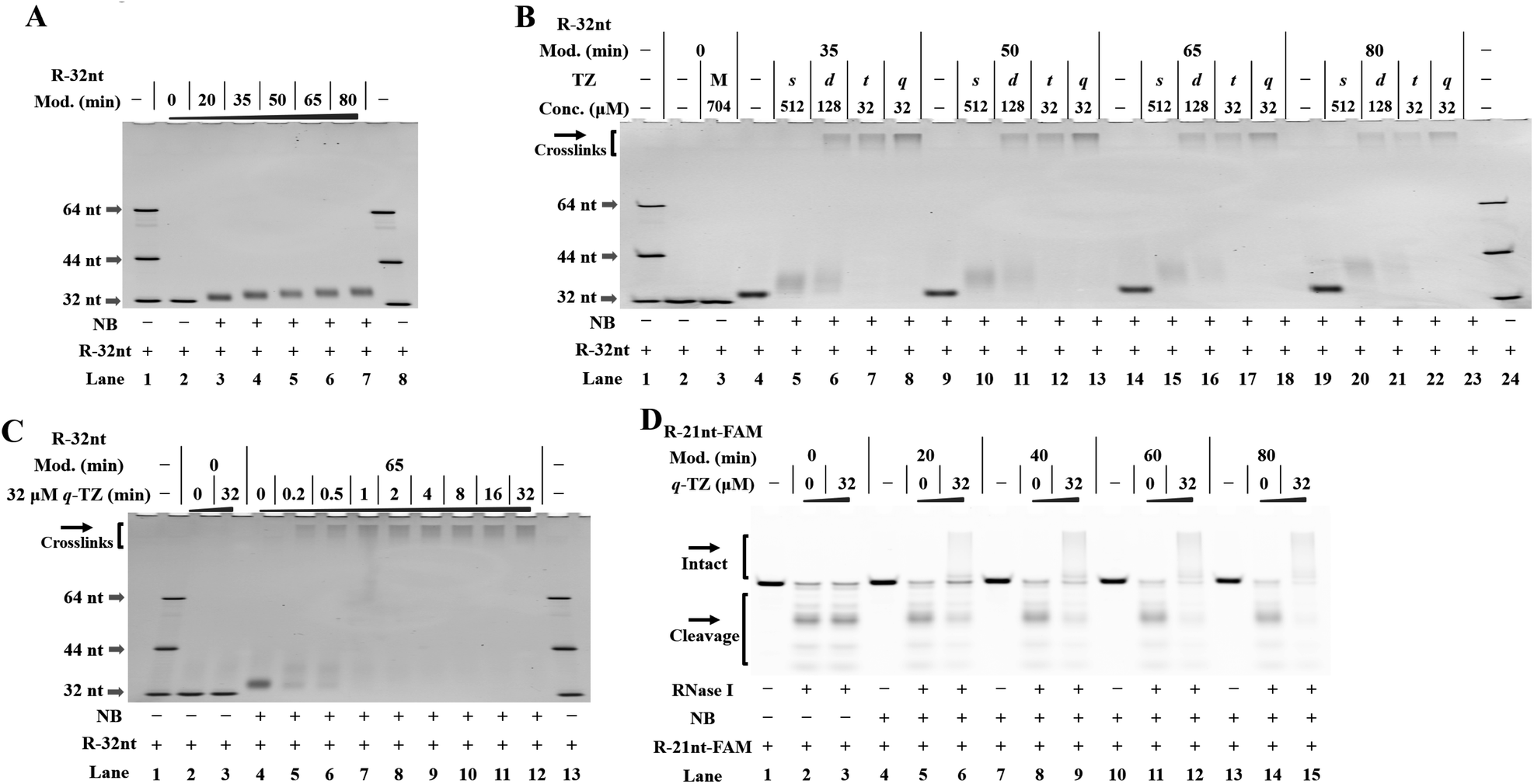 | ||
| Fig. 2 Management of RNA-based NB-TZ ligation chemistry (reactions were performed as described in the Experimental section). All samples were tested in three biological replicates. Image of representative data is shown here. (A) NB-functionalization of RNAs. Lanes 1, 8: RNA marker (R-32nt, R-44nt, R-64nt in Table S1†); lane 2 contains native R-32nt; lanes 3–7 contain NB-bearing R-32nt with increasing modification levels. (B) RNA-based NB-TZ ligation chemistry. For the TZ treatment, the letters s, d, t and q represent s-TZ, d-TZ, t-TZ and q-TZ, respectively. The capital letter M indicates the cotreatment with s-TZ (512 μM), d-TZ (128 μM), t-TZ (32 μM) and q-TZ (32 μM). Lanes 1, 24: RNA marker; lanes 2 and 3 contain native R-32nt; lanes 4–8, 9–13, 14–18, 19–23 contain NB-bearing R-32nt with increasing modification levels. (C) Time course experiment with q-TZ. Lanes 1, 13: RNA marker; lanes 2 and 3 contain native R-32nt; lanes 4–12 contain NB-bearing R-32nt (a 65 min modification with 20 mM NBFI). (D) Modification-dependent changes in RNA protection. Lanes 1–3 contain native R-21nt-FAM; lanes 4–6, 7–9, 10–12, 13–15 contain NB-bearing R-21nt-FAM with increasing modification levels. | ||
Next, we proceeded to test RNA-based NB-TZ ligation chemistry. In order to obtain multimeric TZ molecules (d-TZ, t-TZ and q-TZ), we first synthesized monomeric TZ with the carboxylic acid functionality (s-TZ). In the first study, each TZ was incubated with NB-bearing RNAs with different modification levels. The reaction products under each condition were assessed by measurement of the characteristic slowly migrating band on gels (Fig. 2B). Importantly, substantial yields of cross-linked RNAs were generated by incubating NB-bearing RNAs with d-TZ (lanes 6, 11, 16, 21 in Fig. 2B). Moreover, reactions carried out in the presence of t-TZ and q-TZ produced dramatically increased yields of the cross-linked RNAs. We also demonstrate that each TZ reacts with NB-bearing RNAs in a concentration-dependent manner (Fig. S2†). We further performed time course experiments to study the dynamic behavior of RNA-based NB-TZ ligation chemistry (Fig. S3† and 2C). Notably, the NB-TZ ligation reaction with q-TZ reached its final, maximum yield within 1 min under our standard conditions (lane 7 in Fig. 2C).
An RNase protection assay is performed to characterize and examine the reaction-introduced constraints. We demonstrate that the band of native and NB-bearing RNAs gradually degraded and a majority of RNA degradation was observed at an RNase I concentration of 0.1 U mL−1 in 20% denaturing PAGE gels. We further demonstrate that only low protection of RNA was provided by the ligation of s-TZ (lanes 9, 15, 21 in Fig. S4A and B†). Moreover, the NB-bearing RNAs showed a dramatic increase in stability after reaction with multimeric TZ molecules (Fig. S4A and B†), and no obvious degradation of cross-linked RNAs in q-TZ reactions was observed. This effect was found to be amplified upon increasing the level of NB modification of RNAs (Fig. 2D). These results demonstrate that the NB-TZ ligation reaction offers a large potential for tailoring the RNA properties.
Clickable RNA switches with NB-TZ ligation chemistry in vitro and in living cells
Encouraged by the above results, we sought to address whether NB-TZ ligation chemistry can be used to control RNA functions. In our studies, we used different methods to examine the efficiency of NB-TZ ligation chemistry to control RNA hybridization. We first performed native PAGE to show mobility shifts of a fluorescently labelled RNA upon hybridization with complementary RNAs. We confirm that RNA-based NB-TZ ligation reactions proceed well when using R-21nt (Fig. S5†). It is demonstrated that the NB modification had little impact on the formation of duplexes (lanes 5, 10, 15, 20 in Fig. 3A). We show that treating NB-bearing RNAs with s-TZ only mildly abolished the duplex formation (lanes 6, 11, 16, 21 in Fig. 3A). We further demonstrate that multimeric TZ derivatives are more inhibitory for the hybridization of NB-bearing RNAs to complementary strands (Fig. 3A). Further results demonstrated that NB-TZ ligation reactions disable RNA hybridization in a concentration-dependent manner (Fig. S6†).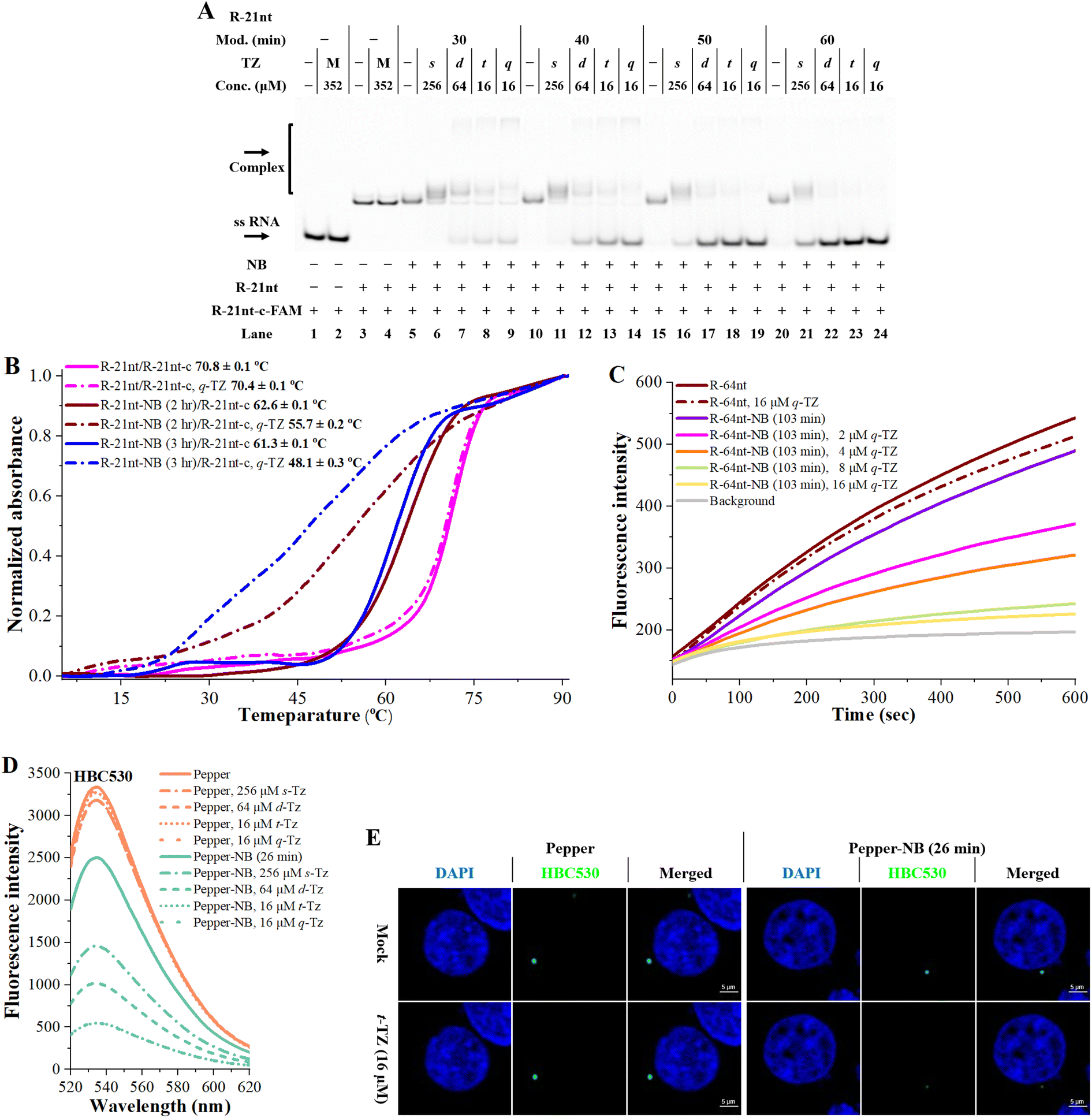 | ||
Fig. 3 Clickable RNA switches with NB-TZ ligation chemistry in vitro and in living cells (reactions were performed as described in the Experimental section). All samples were tested in three biological replicates. Image of representative data is shown here. (A) Mobility shifts of a fluorescently labelled RNA upon hybridization with a complementary sequence with different treatments. Fluorescent bands of the oligomers were visualized on gels. For the TZ treatment, the letters s, d, t and q represent s-TZ, d-TZ, t-TZ and q-TZ, respectively. The capital letter M indicates the cotreatment with s-TZ (256 μM), d-TZ (64 μM), t-TZ (16 μM) and q-TZ (16 μM). Lanes 1 and 2: control without R-21nt; lanes 3 and 4 contain native R-21nt; lanes 5–9, 10–14, 15–19, 20–24 contain NB-bearing R-21nt with increasing modification levels. (B) Melting curves from solutions of complementary RNAs hybridized with native, NB-bearing or NB-TZ-treated RNAs. The absorbance of each sample at 260 nm was monitored in a Tris-HCl buffer (10 mM, pH 7.4) containing 50 mM NaCl. (C) Fluorescence intensities from solutions of MB hybridized with native, NB-bearing or NB-TZ-treated RNAs. (D) Fluorescence response analysis of NB-TZ ligation chemistry on Pepper complexed with HBC530. (E) Effect of NB-TZ ligation chemistry on Pepper’s intracellular fluorescence when stained with HBC530. HEK293T cells transfected with Pepper were labeled with 2 μM HBC530 and imaged. The nucleus was stained with Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 342 (blue). The fields of view were randomly selected for each cell slide. Scale bars: 10 μm. 342 (blue). The fields of view were randomly selected for each cell slide. Scale bars: 10 μm. | ||
The UV melting assay was performed to corroborate the results found in gels. It was observed that TZ derivatives do not inhibit the hybridization of native RNAs to complementary sequences (magenta lines in Fig. 3B). Although the NB-bearing RNAs exhibit a lower melting temperature than native RNAs with complementary sequences, they are still capable of strong hybridization to their complements (solid red and blue lines in Fig. 3B). Significant changes in the thermal stability of RNA duplexes were observed after treating NB-bearing RNAs with q-TZ (dash-dotted red and blue lines in Fig. 3B). Being consistent with the gel analysis, multimeric TZ derivatives showed a higher inhibition of RNA hybridization, compared to s-TZ (Fig. S7†).
We further performed a fluorescence-based assay to assess the effects of NB-TZ ligation chemistry on RNA hybridization. Molecular beacons (MBs) are dual-labeled oligonucleotides with switch-like fluorescence properties.15 We demonstrate that MBs fluoresce well in the presence of the NB-bearing RNAs (violet line in Fig. 3C). Moreover, treating the NB-bearing RNAs with q-TZ significantly inhibited the MB fluorescence (Fig. 3C). Multimeric TZ derivatives show more pronounced inhibition of MB fluorescence compared to the s-TZ (Fig. S8†). These results together demonstrate that NB-TZ ligation chemistry provides a high degree of control over RNA hybridization.
It is important to examine whether NB-TZ ligation chemistry can be used to control RNA functionality in living cells. We recognized that fluorescently tracking RNAs (FRs) have good ability to activate fluorescent dyes.16 Particularly, Pepper RNAs can bind to (4-((2-hydroxyethyl)(methyl)amino)-benzylidene)-cyanophenylacetonitrile (HBC) and greatly improve its fluorescence in vitro and in cells.17 We tend to examine the manipulation of this process, since it can be readily measured. In our study, Pepper RNAs were synthesized with in vitro transcription (Pepper-F and Pepper-R in Table S1†). We tested a variety of HBC ligands with different maximum emission wavelengths (HBC525, HBC530, HBC555 and HBC620 in Fig. S9A†). Our in vitro assay demonstrated that the folding and function of Pepper RNAs can be regulated using NB-TZ ligation chemistry (Fig. 3D and S9B–D†).
We proceeded to demonstrate the effects of NB-TZ ligation chemistry on Pepper’s intracellular fluorescence in living cells. The in vitro results demonstrate that t-TZ and q-TZ have very similar efficiency for switching the fluorescence of NB-Pepper (Fig. 3D and S9B–D†). Next, NB-Pepper was transfected into HEK293T cells and then treated with HBC and t-TZ. The results demonstrate that the HBC fluorescence with native Pepper was not affected by TZ treatment (the left half of Fig. 3E). Not surprisingly, HEK293T cells with transfected NB-Pepper show bright fluorescence when stained with HBC530. Further results demonstrate that the intracellular fluorescence of NB-Pepper was significantly weakened by treating cells with 16 μM t-TZ, consistent with in vitro studies (the right half of Fig. 3E). The results from these experiments were replicated using HBC555 (Fig. S9E†).
Clickable RNA switches for controlling CRISPR systems
The need for safe gene editing has created the demand for developing CRISPR inhibitors.18 We tend to use NB-TZ ligation chemistry to switch off CRISPR/Cas9 (Fig. 4A). There are different types of formats to provide the RNA guides for CRISPR/Cas9.19 To avoid confusion, this study uses gRNA to describe all formats of RNA guides, and sgRNA refers to the simpler alternative that combines both the CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA) elements.20 The polymerase chain reaction (PCR) is used for amplifying target sequences from human genomic DNA (heparin binding EGF like growth factor, HBEGF sequence; hypoxanthine phosphoribosyltransferase 1, HPRT1 sequence; SLX4 interacting protein, SLX4IP sequence in Fig. S10†).21–23In vitro transcription is performed to synthesize sgRNAs with different target sequences (sg-HBEGF, sg-HPRT1, sg-SLX4IP in Table S1†).24 | ||
| Fig. 4 NB-TZ ligation chemistry for controlling CRISPR systems (reactions were performed as described in the Experimental section). All samples were tested in three biological replicates. Image of representative data is shown here. (A) Small molecule control of CRISPR systems with NB-TZ ligation chemistry. (B) Tolerance of CRISPR/Cas9 to NB-bearing sgRNAs (sg-HBEGF). Lane 1: no Cas9 control; lane 2 contains native sg-HBEGF; lanes 3–7 contain NB-bearing sg-HBEGF with increasing modification levels; lane 8: DNA marker (GeneRuler 100-bp DNA ladder). (C) Tolerance of CRISPR/Cas9 to NB-modification in various parts of gRNAs. Lane 1: no Cas9 control; lanes 2, 8 contain native cr-HBEGF and native tracrRNA; lanes 3–7 contain native tracrRNA and NB-bearing cr-HBEGF with increasing modification levels; lanes 9–13 contain native cr-HBEGF and NB-bearing tracrRNA with increasing modification levels; lane 14: DNA marker. (D) Modification-dependent inhibition of CRISPR/Cas9 with q-TZ. Lane 1: no Cas9 control; lanes 2 and 3 contain native sg-HBEGF; lanes 4 and 5, 6 and 7, 8 and 9, 10 and 11 contain NB-bearing sg-HBEGF with increasing modification levels; lane 12, DNA marker. (E) Dose-dependent effect of q-TZ on controlling CRISPR/Cas9. Lane 1: no Cas9 control; lanes 2 and 3 contain native cr-HBEGF and native tracrRNA; lanes 4 and 5, 6 and 7, 8 and 9, 10 and 11 contain native tracrRNA and NB-bearing cr-HBEGF with increasing modification levels; lanes 12 and 13, 14 and 15, 16 and 17, 18 and 19 contain native cr-HBEGF and NB-bearing tracrRNA with increasing modification levels; lane 20: DNA marker. (F) Modification-dependent inhibition of CRISPR/Cas13a with q-TZ. (G) Dose-dependent effect of q-TZ on controlling CRISPR/Cas13a. For (F) and (G), the NB-TZ ligation reactions were performed in the presence of all components except target RNAs. | ||
A gel-based assay is performed to investigate the tolerance of Cas9 to NB-bearing sgRNAs. In the first case, the NB-bearing sgRNAs targeting the HBEGF gene were prepared using the general protocol (Fig. S11A†). On the basis of our results, a 102-nt sg-HBEGF can tolerate the attachment of eight NB moieties (lane 6 in Fig. S11A†), and almost fully retain its activity (lane 5 in Fig. 4B). We further explore the tolerance of CRISPR/Cas9 to NB in various parts of the sgRNA. Following studies were performed by individually modifying the crRNA and tracrRNA components and then matching them with native counterparts (Fig. S11B and C†). We found that CRISPR/Cas9 generally tolerated the NB modifications both in the guide sequence and in the scaffold region, while the guide sequence exhibited higher tolerance (lanes 3–7 in Fig. 4C). Since crRNA is smaller than tracRNA, it is easier to modify tracrRNA with NB moieties.
We tend to determine whether RNA-based NB-TZ ligation chemistry is able to inhibit the function of CRISPR/Cas9. The results from our experiments are consistent: multimeric TZ derivatives show much more efficiency in abolishing the Cas9-mediated DNA cleavage than the s-TZ does (Fig. S11D and E†). It can be observed that the s-TZ treatment only barely influenced the function of NB-bearing sg-HBEGF with a relatively low level of modification (a 20 min modification with 20 mM NBFI, lane 5 in Fig. S11E†). Moreover, q-TZ functions well for controlling CRISPR/Cas9 with NB-bearing sgRNAs with a wide range low to high modification level (Fig. 4D). These results were fully replicated using sgRNAs targeting the SLX4IP gene and HPRT1 gene (Fig. S12 and S13†).
Further studies were performed to investigate the use of NB-TZ ligation chemistry in disabling CRISPR/Cas9 with the 2-part gRNAs. We demonstrate that NB-TZ ligation reactions work very well on NB-bearing crRNAs targeting the HBEGF gene (Fig. S14A†). NB-TZ ligation reactions significantly reduce the potency of NB-bearing cr-HBEGF to support Cas9-mediated DNA cleavage (Fig. S14B and C†). We further demonstrate that q-TZ exhibits a great propensity to control CRISPR/Cas9 with NB-bearing crRNAs and native tracrRNAs (lanes 4–11 in Fig. 4E). These results were replicated using another two crRNAs with different target sites (Fig. S15 and S16†).
We next investigated the use of NB-TZ ligation chemistry in controlling the function of tracrRNAs. We demonstrate that multimeric TZ derivatives exhibit a great propensity to generate interstrand crosslinks with NB-bearing tracrRNAs (Fig. S17A†). In vitro DNA cleavage assays show that multimeric TZ derivatives are much more effective in disabling the function of NB-bearing tracrRNAs than s-TZ does (Fig. S17B†). We also demonstrate that each TZ can inhibit the activity of Cas9 in complexation with NB-bearing tracrRNAs and native crRNAs in a concentration-dependent manner (Fig. S17C†). It is noteworthy that q-TZ efficiently inhibits the activity of NB-bearing tracrRNAs with a wide range low to high modification level (lanes 12–19 in Fig. 4E). These results were replicated by using NB-bearing tracrRNAs in complexation with another two crRNAs (Fig. S18 and S19†).
The RNA-guided RNA cleavage behavior of Cas13a (formerly C2c2) provides a powerful platform for RNA studies.25,26 We are encouraged to use RNA-based NB-TZ ligation chemistry for controlling this system.27 We prepared NB-bearing gRNAs for Cas13a and demonstrated the high efficiency of RNA-based NB-TZ ligation chemistry (Fig. S20†). We further performed a kinetic fluorescence assay to determine the Cas13a activity. In this study, the Cas13a starts cutting a quenched RNA reporter once it finds a perfect complementary target to its gRNA. The fluorescence intensity can therefore be largely increased. Our results revealed the general tolerance of CRISPR/Cas13a toward NB-bearing gRNAs (solid lines in Fig. 4F). Further results demonstrate that each TZ is effective in disabling CRISPR/Cas13a (Fig. S21,†4F and G). We believe the success in controlling CRISPR/Cas13a further demonstrates the value of RNA-based NB-TZ ligation chemistry.
NB-TZ ligation chemistry for controlling gene editing in Cas9-stable cells
We were encouraged to investigate the application of RNA-based NB-TZ ligation chemistry for controlling gene editing in living cells. We first used a Cas9-stable cell line (HeLa-OC) to simplify the management of experiments.21 By acknowledging the fact that t-TZ exhibits excellent performance for switching intracellular FR fluorescence, this compound was used in the following studies. By utilizing the MTT assay, we demonstrate that t-TZ does not appear to have significant toxicity issues with concentrations lower than 32 μM (Fig. S22†). Next, HeLa-OC cells were transfected with native or NB-bearing sgRNAs for 4 h, and the transfected cells were further cultured for 24 h. Then, the cells were harvested to analyze the gene editing efficiencies using the T7 endonuclease 1 (T7E1) mismatch detection assay. Although NB-bearing sgRNAs showed moderately reduced efficiency compared to native groups, they are still capable of supporting the Cas9-mediated genome editing (lanes 6, 8, 10, 12 in Fig. 5A).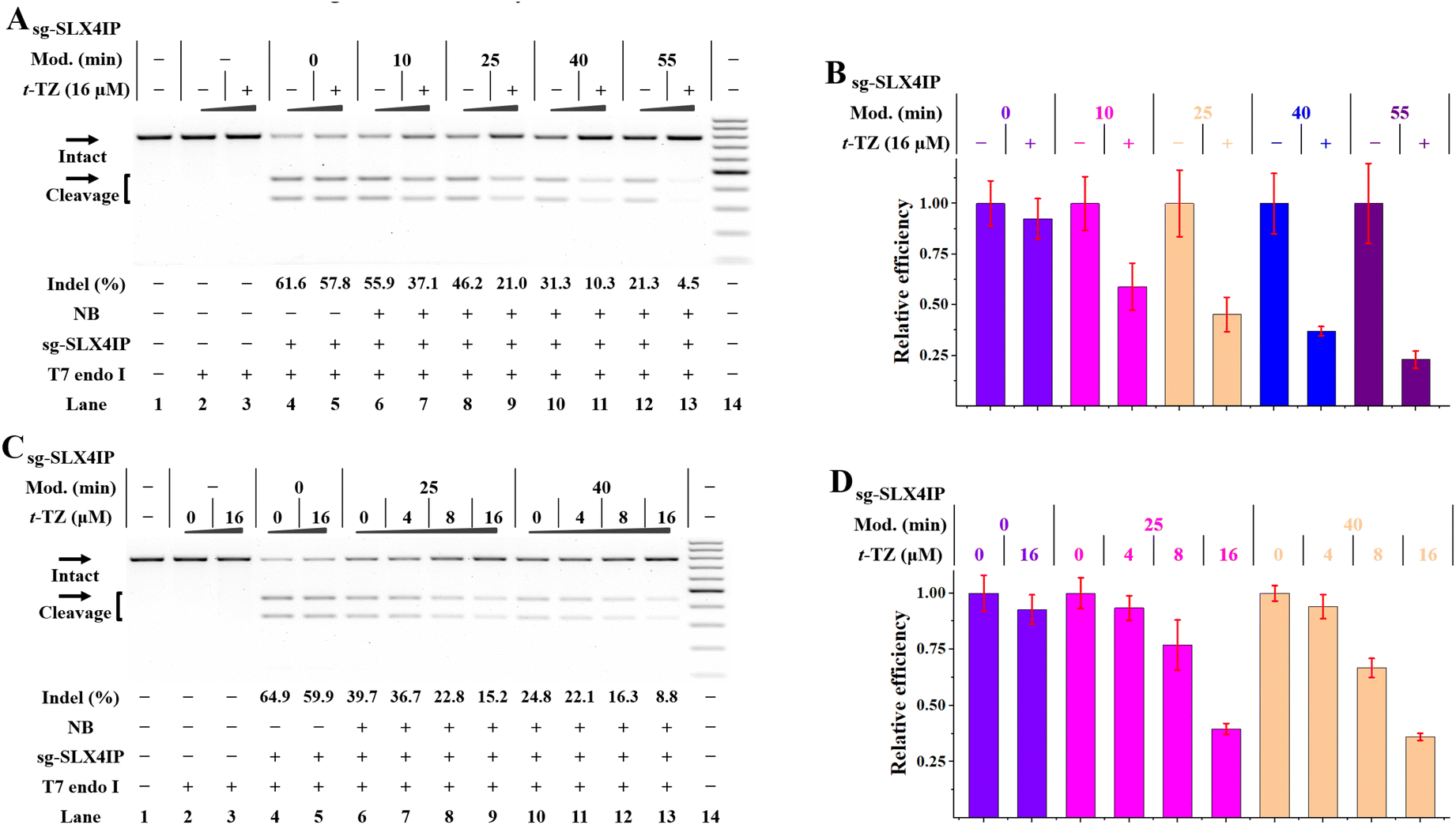 | ||
| Fig. 5 NB-TZ ligation chemistry for controlling genome editing in Cas9-stable cells (cellular studies were performed as described in the Experimental section). Different sgRNAs were delivered into HeLa-OC cells before t-TZ was added. The T7E1 nuclease assay was performed 24 h post-transfection. All samples were tested in three biological replicates. Image of representative data is shown here. (A) Modification-dependent inhibition of gene editing with t-TZ. Lane 1: target control; lanes 2 and 3: no sgRNA control; lanes 4 and 5 contain native sg-SLX4IP; lanes 6 and 7, 8 and 9, 10 and 11, 12 and 13 contain NB-bearing sg-SLX4IP with increasing modification levels; lane 14: DNA marker (GeneRuler 100-bp DNA ladder). (B) Modification–response bar graph plotted with error lines. (C) Dose-dependent effect of t-TZ on controlling gene editing. Lane 1: target control; lanes 2 and 3: no sgRNA control; lanes 4 and 5 contain native sg-SLX4IP; lanes 6–9, 10–13 contain NB-bearing sg-SLX4IP with increasing modification levels; lane 14: DNA marker. (D) Concentration–response bar graph plotted with error lines. For (A) and (C), uncleaved SLX4IP DNAs (773 bp) cut to shorter cleavage fragments (441 bp and 332 bp) are demonstrated. For (B) and (D), the indel formation of t-TZ-treated cells was normalized to that of mock-treated cells. The data are presented as the means ± SEM from three independent experiments. | ||
We next tested the efficiency of in vivo NB-TZ ligation chemistry. We demonstrate that NB-TZ ligation chemistry with t-TZ is substantially effective in suppressing the genome-editing activity of sgRNAs in HeLa-OC cells (Fig. 5A and B). These results were replicated using another two sgRNAs (Fig. S23A and B†). To optimize the concentration of t-TZ for inhibiting genome editing in cells, varying concentrations of t-TZ were investigated. We demonstrate that the treatment with t-TZ decreases the function of NB-bearing sgRNAs in a concentration-dependent manner (Fig.5C, D, S23C and D†). The control cellular assay revealed little or no change of the indel frequencies of native sgRNA groups that were treated with t-TZ, ruling out the possibility that the observed inhibitory effects of genome editing are the results of nonspecific inhibition. These results demonstrate that under these conditions (simply add t-TZ), the Cas9-mediated genome editing could be inhibited across different genes. Considering the fact that the transfected sgRNAs may trigger the Cas9-mediated genome editing before the t-TZ addition during the transfection process, the real-efficiency of RNA-based NB-TZ ligation chemistry is likely to be greater than the demonstrated results.
NB-TZ ligation chemistry for controlling plasmid-based gene editing
The above studies demonstrate the control of gene editing in a stable Cas9-expressing cell line. For the CRISPR/Cas9 study, both Cas9 and sgRNA can be introduced into cells in various ways. Acknowledging this fact, we tend to use NB-bearing sgRNAs while expressing Cas9 separately from plasmid DNA in cells. We therefore designed a validation system to use IVT sgRNAs and a Cas9-only plasmid (PX165).28,29 To investigate whether genomic loci can be edited using this hybrid system, three genes (SLX4IP, HPRT1 and HBEGF) were targeted in HeLa cells respectively. We delivered the HPRT1 targeting sgRNAs 4 h after the PX165 transfection and indels were quantified 24 h later by T7E1 analysis. We demonstrated that cells with both PX165 and IVT sgRNA exhibited evident indel formation (lane 4 in Fig. 6A).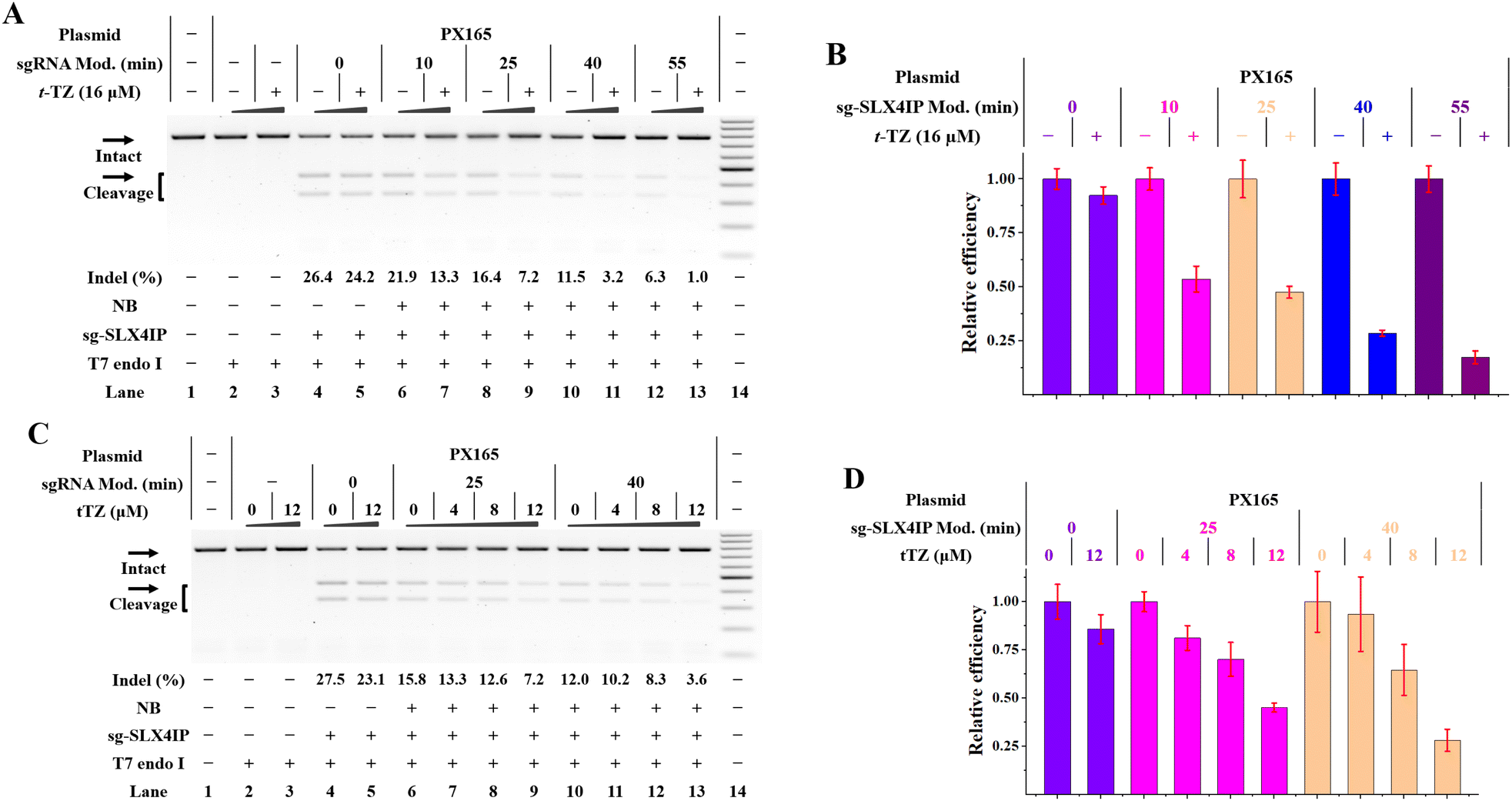 | ||
| Fig. 6 NB-TZ ligation chemistry for controlling plasmid-based gene editing (cellular studies were performed as described in the Experimental section). The plasmids and sgRNAs were delivered into HeLa cells before the treatment with t-TZ. All samples were tested in three biological replicates. Image of representative data is shown here. (A) Modification–response analysis of t-TZ on plasmid-based gene editing. Lane 1: target control; lanes 2 and 3: no sgRNA control; lanes 4 and 5 contain PX165 and sg-SLX4IP; lanes 6 and 7, 8 and 9, 10 and 11, 12 and 13 contain PX165 and NB-bearing sg-SLX4IP with increasing modification levels; lane 14: DNA marker (GeneRuler 100-bp DNA ladder). (B) Modification–response bar graph plotted with error lines. (C) Concentration–response analysis of t-TZ on plasmid-based gene editing. Lane 1: target control; lanes 2 and 3: no sgRNA control; lanes 4 and 5 contain PX165 and sg-SLX4IP; lanes 6–9, 10–13 contain PX165 and NB-bearing sg-SLX4IP with increasing modification levels; lane 14: DNA marker. (D) Concentration–response bar graph plotted with error lines. For (A) and (C), uncleaved SLX4IP DNAs (773 bp) cut to shorter cleavage fragments (441 bp and 332 bp) are demonstrated. For (B) and (D), the indel formation of t-TZ-treated cells was normalized to that of mock-treated cells. The data are presented as the means ± SEM from three independent experiments. | ||
We further investigated the effect of RNA-based NB-TZ ligation chemistry on the function of this hybrid system. The Cas9-expression plasmids and IVT sgRNAs were delivered into cells as above before increasing amounts of t-TZ were added. The amount of indel formation was determined 24 h after t-TZ treatment. We confirmed the potency of NB-TZ ligation chemistry in controlling the function of NB-bearing sg-SLX4IP and Cas9-only plasmids for gene editing (Fig. 6). These results were replicated using the Cas9-only plasmids and NB-bearing sgRNAs targeting another two genes (Fig. S24†). Hence, RNA-based NB-TZ ligation chemistry functions well in controlling the plasmid-based gene editing in human cells.
Discussion
The current work demonstrates that NB-TZ chemistry could work on exogenous RNA through NB modification. In this study, a series of multimeric TZ molecules are designed and synthesized to develop new tools for the loss of RNA function study. These compounds consist of different number of TZ units and have different MW and sizes. TZ units are hydrophobic moieties, and the hydrophobicity of whole TZ molecules differs depending on the number of TZ groups present in the molecules. We compared these compounds with different numbers of TZ arms for success of crosslinking. The order of the crosslinking capacities is as follows: d-TZ < t-TZ ≈ q-TZ. The crosslinking capacity first increases with increase in functionality from 2 to 3. However, the difference between the crosslinking capacities of t-TZ and q-TZ is not much pronounced.There is an increasing need for a universal strategy for suppression of unwanted CRISPR activities. Although gRNA is a biomolecule central to the function of CRISPR, it is an often overlooked target for CRISPR control. To date, relatively few strategies exist for CRISPR deactivation under biocompatible conditions. We demonstrate that both CRISPR/Cas9 and CRISPR/Cas13a are rendered inactive by RNA-based NB-TZ ligation chemistry. Although some strategies have been previously used for controlling crRNA in CRISPR systems to control gene editing, these systems were engineered for CRISPR activation rather than inhibition.30,31 Our strategy will be promising for use in an era now hungry for safe gene editing.
Conclusions
This work demonstrates that clickable RNA switches can be used broadly to target any RNA, thus creating an easy avenue for RNA control.Experimental procedures
Chemical synthesis
The synthetic data of NBFI and each TZ are demonstrated in the ESI.†Management of NB-TZ ligation on RNAs
The NB functionalization reaction was performed in 1 × modification buffer, which contained 100 mM HEPES, 6 mM MgCl2, 100 mM NaCl in 60% DMSO (pH 7.5 @ 25 °C). NBFI stock solutions (200 mM) were prepared by dissolving the powder in dimethyl sulfoxide (DMSO), and further dilutions were made with water. The reaction mixture containing RNA, NBFI and DMAP was incubated at 37 °C for various durations with shaking (speed, 200 r min−1). The final concentration of each component for a typical 10 μL reaction is as follows: 20 μM RNA, 20 mM NBFI, 20 mM DMAP. After incubation, 2.0 μL NaOAc (3 M), 75 μL ethanol and 1.0 μL glycogen (5 g L−1) were added into the mixture to quench the reaction. After centrifugation to collect the RNA, the pellets were rinsed twice with 75% ethanol, and then dried and resuspended in nuclease-free water.NB-TZ ligation reactions were started by adding a 20-fold concentrated solution of each TZ (0.5 μL) to a total volume of 10 μL mixture with RNAs. The NB-TZ ligation reaction was performed at room temperature for a total reaction time of 20 min. The reactions were quenched by adding 4 × v:v of stop buffer (95% formamide, 200 mM of 5-norbornen-2-ol at pH 8.0).
The modification levels of RNA were analyzed by denaturing electrophoresis (20% polyacrylamide, 19:1 acrylamide/bis acrylamide, urea 7 M, 1 × TBE buffer). The PAGE analysis was performed in a temperature-controlled vertical electrophoretic apparatus (DYCZ-22A, Liuyi Instrument Factory, Beijing, China). Electrophoresis was run at 10 °C for 4.0 h at 400 V. After electrophoresis, the oligomers in the gel were visualized using a Pharos FX Molecular imager (Bio-Rad, USA) in the fluorescence mode.
RNase protection assay
The NB functionalization of fluorescently labeled RNAs was performed using the above general protocol. NB-bearing RNAs (100 ng) were treated with TZ at varied concentrations for 60 min, and were subjected to RNase I degradation (0.1 U mL−1) in a reaction volume of 10 μL at 0 °C for 30 min. This assay was performed in 1 × reaction buffer in PCR grade water, which contained 50 mM Tris-HCl and 2 mM EDTA at pH 7.5 @ 25 °C. The reaction was quenched by adding a 5.0-fold excess of quenching solution (0.1% SDS in formamide). The samples were then denatured by heating at 95 °C for 5 min, immediately chilled on ice for 5 min, and analyzed on a 20% denaturing polyacrylamide gel (350 V, 1.5 h).NB-TZ ligation chemistry for controlling RNA hybridization
The NB functionalization of ROIs was performed using the above general protocol. The complementary RNA with the fluorophore was used as a probe to study the hybridization properties of NB-bearing RNAs. For the RNA hybridization experiment, NB-bearing R-21nt was treated with each TZ at varied concentrations for 60 min, and was annealed with the R-21nt-c-FAM in 1 × reaction buffer at 37 °C for 2 h. Samples were then resolved by 20% native PAGE in 1 × TBE buffer (89 mM Tris-borate, 2 mM EDTA) at room temperature (300 V, 4 h).UV melting studies
UV melting experiments were performed using a Jasco-810 CD spectropolarimeter (Jasco, Easton, MD, USA) equipped with a water bath temperature-control accessory. The detection is performed with a quartz cell (optical path length at 1 mm). The ss RNA was annealed with the complementary one in 10 mM Tris-HCl buffer (pH 6.8) containing 50 mM NaCl. The UV melting profiles were recorded by using a heating rate of 0.2 °C min−1 and the absorbance values were recorded every 1 °C at a wavelength of 260 nm. The melting point (Tm) corresponds to the mid-transition temperature, which was determined by the maximum of the first derivative of the absorbance as a function of temperature.Fluorescence measurements of MBs
Fluorescence of hybridization solutions was used to quantify the hybridization of MBs with R-64nt. 120 nM R-64nt was incubated with 100 nM of MB in 1 × hybridization buffer (100 mM Tris-HCl, pH 8.0, 25 °C, 100 mM NaCl, 5 mM MgCl2). The NB functionalization of R-64nt was performed using the above general protocol. To determine the effects of NB-TZ ligation reactions on the fluorescence emissions of MB, TZ was added to samples with R-64nt and MB and incubated for 60 min. Each of the 400 μL hybridization mixtures was excited at 496 nm and emitted fluorescence was quantified at 520 nm with a LS55 fluorescence spectrometer (PerkinElmer Inc., USA).Clickable RNA switches in vitro and in cells
This study was performed by using Pepper RNAs. The NB-bearing Pepper was prepared using the above general protocol. For the aptamer folding and fluorescence study, the Pepper RNA at a final concentration of 0.5 μM was incubated with 2 μM HBC in 100 μL folding buffer (40 mM HEPES at pH 7.4, 100 mM KCl, 5 mM MgCl2). After 1 h dark incubation at room temperature, the emission spectrum was collected using a 1 cm path-length cell. Fluorescence detection was performed at room temperature using a F-4600 FL spectrophotometer (Hitachi). The excitation wavelength was set as follows: HBC525 (λex = 491 nm), HBC530 (λex = 485 nm), HBC555 (λex = 488 nm), and HBC620 (λex = 577 nm), respectively. Slit width: excitation = 5 nm; emission = 5 nm. PMT voltage = 700 V.HEK293T cells were seeded in 20 mm glass bottom dishes 12 h before transfection. The cells were washed twice with DPBS (Dulbecco’s Phosphate-Buffered Saline), and 300 μL of pre-warmed DMEM was added to each well. Pepper RNAs (1.25 μg of each RNA at a concentration of 0.5 μg μL−1) were mixed with 50 μL of DMEM. The Lipofectamine® 3000 transfection agent (5.0 μL, Thermo Fisher Scientific) in 50 μL of DMEM per well was added to the diluted RNAs. The mixture was incubated at room temperature for 15 min to form the Pepper:Lipofectamine complexes, which were transferred to the well containing cells and growth medium. After 3 h incubation, the transfected cells were washed and stained with 1.0 mL Hoechest 33342 solutions (1 μg mL−1 in DPBS) at 37 °C for 10 min. The nuclear stained cells were washed three times with DPBS and dyed with 0.5 mL of each HBC fluorophore (2 μM in DPBS). Images were taken with a Leica TCS SP8 DIVE microscope equipped with a HC PL APO CS2 63×/1.40 OIL objective and HyD and HyD-RLD detectors. 488 nm laser excitation was used to image Hoechst 33342; 488 nm laser excitation was used to image HBC530 and HBC555; 552 nm laser excitation was used to image HBC555.
To accurately demonstrate clickable RNA switches, we first recorded the HEK293T cells stained with Hoechst and HBC in a specified region. The NB-TZ ligation reaction is performed by adding a 0.5 mL DPBS buffer with 32 μM t-TZ and 2 μM HBC and incubating for 0.5 h. The exactly same region was investigated using a confocal laser scanning microscope. The images were analyzed using ImageJ.
NB-TZ ligation chemistry for controlling CRISPR/Cas9
NB-bearing gRNAs were prepared using the above general protocol. Target DNA fragments were PCR amplified from genomic DNA (HeLa cells) using the following PCR primers: t-HBEGF-F and t-HBEGF-R for t-HBEGF; t-SLX4IP-F and t-SLX4IP-R for t-SLX4IP; t-HPRT1-F and t-HPRT1-R for t-HPRT1. An in vitro Cas9 cleavage assay was performed in 1 × NEBuffer™ 3.1 buffer, which contained 100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2 and 100 μg mL−1 BSA at pH 7.9 @ 25 °C. Briefly, native or NB-bearing sgRNAs (0.3 μM) were incubated with each TZ at various concentrations in the presence of Cas9 (0.2 μM), in 1 × NEBuffer™ 3.1 buffer at room temperature for 30 min. The Cas9-mediated DNA cleavage assays were performed by incubating the reaction components described in their respective figures at 37 °C. Specifically, cleavage reactions were started by rapid mixing of equal volumes of the sgRNA/Cas9 preparation with a solution containing target DNA fragments (50 ng) in 1 × NEBuffer™ 3.1 buffer, and subjected to incubation at 37 °C for 2.0 h. Reactions were terminated by adding SDS containing loading dye and resolved on 2% agarose gels (100 V, 1.5 h). The gels were then visualized using super GelRed stains. The GeneRuler 100-bp DNA ladder was used as a DNA size marker for these experiments.For the 2-part gRNA study, each crRNA (native or NB-bearing) and tracrRNA (native or NB-bearing) were annealed at room temperature for 10 min to form the gRNA complexes. For the NB-TZ ligation step, similar reactions were set up using 15 ng of each crRNA and 25 ng of tracrRNA instead of 50 ng of each sgRNA. For Cas9-mediated DNA cleavage reactions, the conditions were the same as the above sgRNA protocol except that 11 h incubation at 37 °C was used.
NB-TZ ligation chemistry to control CRISPR/Cas13a
NB-bearing gRNAs were prepared using the above general protocol. An in vitro Cas13a cleavage assay was performed in 1 × Cas13a buffer in RNase free water, which contained 20 mM HEPES, 50 mM KCl, 5 mM MgCl2 and 5% glycerol at pH 6.8 @ 25 °C. The gRNAs were pre-folded by heating to 65 °C for 5 min and then slowly cooling to room temperature in 1 × Cas13a buffer. Briefly, the gRNA was complexed with a 2.0 molar excess of Cas13a protein in 1 × Cas13a buffer at 37 °C for 20 min, in the presence of each TZ at various concentrations. For the kinetic fluorescence study, the reactions were started by rapid mixing of equal volumes of the Cas13a/gRNA preparation with a solution containing target and reporter RNAs in 1 × Cas13a buffer. The final concentration of all components except TZ was as follows: 45 nM purified LbuCas13a, 22.5 nM gRNA, 22.5 nM target (Cas13a target in Table S1†), 125 nM quenched fluorescent RNA reporter (Cas13a reporter in Table S1†) and 0.5 U RiboLock RNase inhibitor. The curve describing fluorescence increase versus time is determined using the LS55 fluorescence spectrometer in the kinetics mode at room temperature. Reactions were allowed to proceed for 1 h and measured every 1 s. The detection is conducted at room temperature with a 1 cm path-length cell. The excitation and emission wavelengths are set to 496 nm and 520 nm. Slit width: excitation = 10 nm; emission = 10 nm.NB-TZ ligation chemistry for controlling gene editing in Cas9-stable cells
HeLa-OC cells were passaged when 70–90% confluency was reached. For the endogenous genome editing experiments, gRNAs were designed using an online tool. HeLa-OC cells (4 × 105 per well) were seeded into 6-well plates overnight before transfection and washed twice with DPBS, and 300 μL of pre-warmed DMEM was added to each well. 2.5 μg of each sgRNA at a concentration of 0.5 μg μL−1 was mixed in 120 μL of DMEM. 5 μL of Lipofectamine 3000 transfection agent in 120 μL of DMEM per well was added to the diluted sgRNAs, followed by 15 min incubation. The complex was added to the cells, and the medium was changed to complete medium supplemented with t-TZ (16 μM) after 4 h incubation at 37 °C in 5% CO2.The transfected cells were further incubated at 37 °C with 5% CO2 for 24 h, after which the cells were detached from the plates and washed twice with DPBS. The genomic DNA of collected cells was extracted for mutation detection using a Qiagen DNeasy Blood and Tissue Kit. Subsequently, genomic PCR reaction was performed using 100 ng genomic DNA, corresponding primers, and PrimeSTAR HS DNA polymerase. The PCR products were purified via a Zymo Research DNA Clean and Concentrator Kit. The PCR product carrying the target loci (100 ng) is then denatured and reannealed to produce heteroduplex mismatches where double-strand breaks have occurred, resulting in indel introduction. These mismatches were recognized and cleaved by T7 endonuclease I (T7E1). The products were resolved on 2% agarose gel (100 V, 1.5 h) and then analyzed by densitometry measurements. Three biological replicates were performed for each condition.
NB-TZ ligation chemistry for controlling plasmid-based gene editing
HeLa cells were seeded at 4 × 105 cells per well in a 6-well plate and transfected with PX165/Lipofectamine 3000 for 4 h. The dose of PX165 was 2.5 μg per well. The cells were then treated with Lipofectamine 3000 loaded with endogenous gene targeting sgRNAs for 4 h. The sgRNA dose was 2.5 μg per well for each sgRNA. HeLa cells were then treated with various concentrations of t-TZ. At 24 h post-treatment, the target editing efficiency and inhibition thereof were quantified by the T7E1 assay. Three biological replicates were performed for each condition. Data were normalized on samples without TZ treatment.Data availability
All data are available within the article and its ESI.†Author contributions
T.T. conceived the original idea, designed the studies and led the project. Q.Q.Q., Y.T.Z. and X.Y.L. performed all biological studies. W.X. performed chemical synthesis. S.Y.C., X.F.Y. and K.S.Z. performed gel experiments. X.Z. provided consistent support during the running of this project. T.T. wrote the manuscript. All the authors provided feedback on the study and on the manuscript. The authors declare no competing financial interests.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (no. 22177089, 91853119, 21721005, 91753201, 21877086, and 22177088), the Hubei Natural Science Foundation for Distinguished Young Scholars (2019CFA064), and the Fundamental Research Funds for the Central Universities (2042019kf0189). The authors also thank Prof. Ping Yin (Huazhong Agricultural University) and Prof. Shaoru Wang (Wuhan University) for their valuable help.References
- A. Sosic, R. Gottlich, D. Fabris and B. Gatto, Nucleic Acids Res., 2021, 49, 6660–6672 CrossRef CAS PubMed.
- C. Huang, Y. Liu and S. E. Rokita, Signal Transduct Target Ther., 2016, 1, 16009 CrossRef PubMed.
- F. Fakhari and S. E. Rokita, Nat. Commun., 2014, 5, 5591 CrossRef CAS PubMed.
- C. F. Hansell, P. Espeel, M. M. Stamenovic, I. A. Barker, A. P. Dove, F. E. Du Prez and R. K. O'Reilly, J. Am. Chem. Soc., 2011, 133, 13828–13831 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed.
- W. A. Velema and E. T. Kool, Nat. Rev. Chem., 2020, 4, 22–37 CrossRef CAS PubMed.
- C. G. Gordon, J. L. Mackey, J. C. Jewett, E. M. Sletten, K. N. Houk and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 9199–9208 CrossRef CAS PubMed.
- J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem. Int., Ed. Engl., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654–9655 CrossRef CAS PubMed.
- H. Liu, D. Audisio, L. Plougastel, E. Decuypere, D. A. Buisson, O. Koniev, S. Kolodych, A. Wagner, M. Elhabiri, A. Krzyczmonik, S. Forsback, O. Solin, V. Gouverneur and F. Taran, Angew. Chem. Int., Ed. Engl., 2016, 55, 12073–12077 CrossRef CAS PubMed.
- K. Gutsmiedl, C. T. Wirges, V. Ehmke and T. Carell, Org. Lett., 2009, 11, 2405–2408 CrossRef CAS PubMed.
- R. D. Row and J. A. Prescher, ACS Cent. Sci., 2016, 2, 493–494 CrossRef PubMed.
- L. Wang, C. J. Yang, C. D. Medley, S. A. Benner and W. Tan, J. Am. Chem. Soc., 2005, 127, 15664–15665 CrossRef CAS PubMed.
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 16299–16308 CrossRef CAS PubMed.
- X. Chen, D. Zhang, N. Su, B. Bao, X. Xie, F. Zuo, L. Yang, H. Wang, L. Jiang, Q. Lin, M. Fang, N. Li, X. Hua, Z. Chen, C. Bao, J. Xu, W. Du, L. Zhang, Y. Zhao, L. Zhu, J. Loscalzo and Y. Yang, Nat. Biotechnol., 2019, 37, 1287–1293 CrossRef CAS PubMed.
- B. Maji, S. A. Gangopadhyay, M. Lee, M. Shi, P. Wu, R. Heler, B. Mok, D. Lim, S. U. Siriwardena, B. Paul, V. Dancik, A. Vetere, M. F. Mesleh, L. A. Marraffini, D. R. Liu, P. A. Clemons, B. K. Wagner and A. Choudhary, Cell, 2019, 177, 1067–1079 e1019 CrossRef CAS PubMed.
- A. Mir, J. F. Alterman, M. R. Hassler, A. J. Debacker, E. Hudgens, D. Echeverria, M. H. Brodsky, A. Khvorova, J. K. Watts and E. J. Sontheimer, Nat. Commun., 2018, 9, 2641 CrossRef PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- Y. Zhou, S. Zhu, C. Cai, P. Yuan, C. Li, Y. Huang and W. Wei, Nature, 2014, 509, 487–491 CrossRef CAS PubMed.
- M. Gasperini, G. M. Findlay, A. McKenna, J. H. Milbank, C. Lee, M. D. Zhang, D. A. Cusanovich and J. Shendure, Am. J. Hum. Genet., 2017, 101, 192–205 CrossRef CAS PubMed.
- S. Panier, M. Maric, G. Hewitt, E. Mason-Osann, H. Gali, A. Dai, A. Labadorf, J. H. Guervilly, P. Ruis, S. Segura-Bayona, O. Belan, P. Marzec, P. L. Gaillard, R. L. Flynn and S. J. Boulton, Mol. Cell, 2019, 76, 27–43.e11 CrossRef CAS PubMed.
- P. K. Jain, V. Ramanan, A. G. Schepers, N. S. Dalvie, A. Panda, H. E. Fleming and S. N. Bhatia, Angew. Chem. Int., Ed. Engl., 2016, 55, 12440–12444 CrossRef CAS PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed.
- A. Tambe, A. East-Seletsky, G. J. Knott, J. A. Doudna and M. R. O'Connell, Cell Rep., 2018, 24, 1025–1036 CrossRef CAS PubMed.
- A. East-Seletsky, M. R. O'Connell, S. C. Knight, D. Burstein, J. H. Cate, R. Tjian and J. A. Doudna, Nature, 2016, 538, 270–273 CrossRef CAS PubMed.
- F. A. Ran, P. D. Hsu, J. Wright, V. Agarwala, D. A. Scott and F. Zhang, Nat. Protoc., 2013, 8, 2281–2308 CrossRef CAS PubMed.
- H. X. Wang, Z. Song, Y. H. Lao, X. Xu, J. Gong, D. Cheng, S. Chakraborty, J. S. Park, M. Li, D. Huang, L. Yin, J. Cheng and K. W. Leong, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4903–4908 CrossRef CAS PubMed.
- Y. Zhang, X. Ling, X. Su, S. Zhang, J. Wang, P. Zhang, W. Feng, Y. Y. Zhu, T. Liu and X. Tang, Angew. Chem. Int. Ed., Engl., 2020, 59, 20895–20899 CrossRef CAS PubMed.
- M. Habibian, C. McKinlay, T. R. Blake, A. M. Kietrys, R. M. Waymouth, P. A. Wender and E. T. Kool, Chem. Sci., 2019, 11, 1011–1016 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Extended experimental procedures and figures. See DOI: https://doi.org/10.1039/d2sc02635j |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2022 |