
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitchable photocatalysis for the chemodivergent benzylation of 4-cyanopyridines†
Eleni
Georgiou‡
ab,
Davide
Spinnato‡
 ab,
Kang
Chen
a,
Paolo
Melchiorre
*ac and
Kilian
Muñiz§
ab,
Kang
Chen
a,
Paolo
Melchiorre
*ac and
Kilian
Muñiz§
aICIQ – Institute of Chemical Research of Catalonia, The Barcelona Institute of Science and Technology, Avenida Països Catalans 16 – 43007, Tarragona, Spain. E-mail: pmelchiorre@iciq.es; Web: https://www.iciq.org/research/research_group/prof-paolo-melchiorre
bDepartment of Analytical Chemistry and Organic Chemistry, University Rovira i Virgili (URV), 43007, Tarragona, Spain
cICREA, Passeig Lluís Companys 23 – 08010, Barcelona, Spain
First published on 7th June 2022
Abstract
We report a photocatalytic strategy for the chemodivergent radical benzylation of 4-cyanopyridines. The chemistry uses a single photoredox catalyst to generate benzyl radicals upon N–F bond activation of 2-alkyl N-fluorobenzamides. The judicious choice of different photocatalyst quenchers allowed us to select at will between mechanistically divergent processes. The two reaction manifolds, an ipso-substitution path proceeding via radical coupling and a Minisci-type addition, enabled selective access to regioisomeric C4 or C2 benzylated pyridines, respectively. Mechanistic investigations shed light on the origin of the chemoselectivity switch.
Introduction
Pyridines and related nitrogen heterocycles are prevalent in pharmaceuticals and agrochemicals,1 so methods are needed for their regioselective functionalization. The recent resurgence of photoredox catalysis2 and electrochemistry3 has triggered the development of new pyridine functionalization strategies based on radical pathways. One such approach exploits the unique reactivity of 4-cyanopyridines 1 (Fig. 1a), which are prone to single-electron transfer (SET) reduction to form kinetically stable dearomatized radical anions I (path i). These open-shell intermediates I can then couple with other radicals offering, upon cyanide extrusion, a selective ipso-substitution manifold to access C4-functionalized pyridines.4 The venerable Minisci reaction5 is another effective approach for the functionalization of pyridines, which is based on the addition of nucleophilic carbon radicals onto protonated heteroaromatic compounds. Due to their decreased π-electron density, 4-cyanopyridines generally participate in Minisci-type functionalization with C2 regioselectivity (path ii in Fig. 1a).6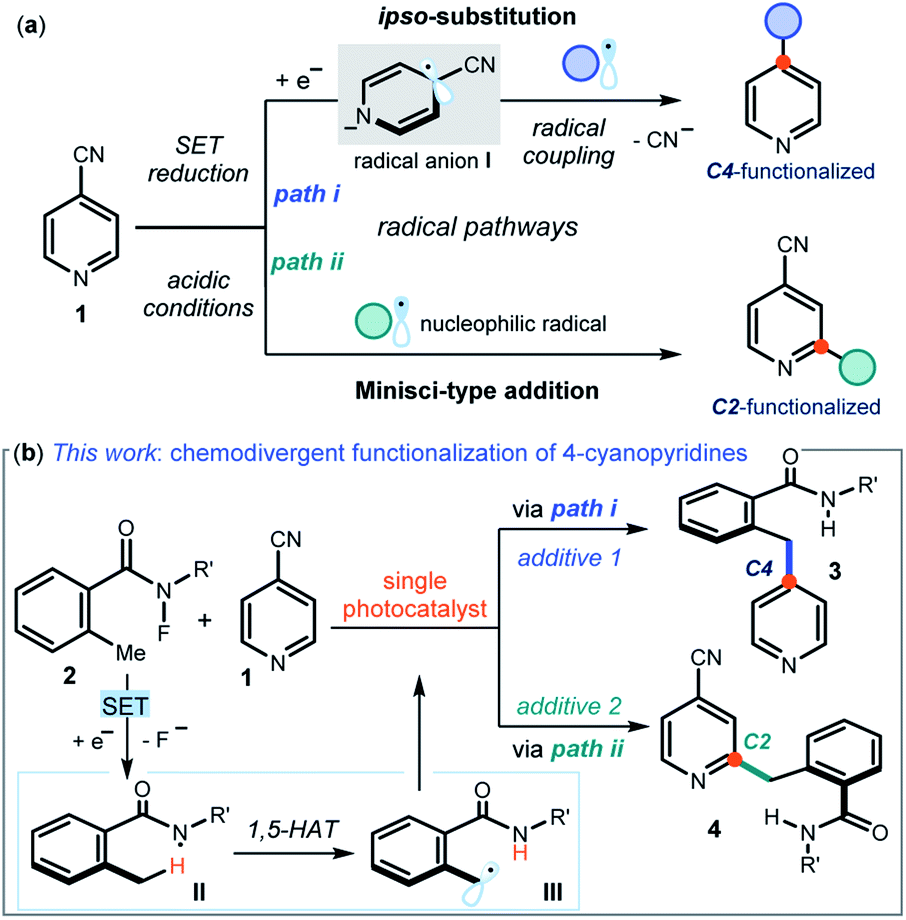 | ||
| Fig. 1 (a) Radical pathways available for the functionalization of 4-cyanopyridines. (b) A photoredox system that uses different additives to switch between different mechanisms to afford C4 or C2 benzylated products 3 or 4 at will. | ||
In this manuscript, we describe the development of switchable photocatalytic methods7 that allowed us to select at will between the functionalization of 4-cyanopyridines at the C4 or C2 position, leading to products 3 and 4 respectively (Fig. 1b).
Specifically, we used a single photoredox catalyst to activate, via an SET reduction, the N–F bond within 2-alkyl N-fluorobenzamides 2.8 A 1,5-hydrogen atom transfer (1,5-HAT), mastered by the resulting amidyl radical II, then afforded the benzylic radical III. We serendipitously observed that different photocatalyst quenchers enabled mechanistically divergent processes, triggering either an ipso-substitution (path i) or a Minisci manifold (path ii). This additive-controlled mechanistic switch afforded benzylated pyridines 3 and 4 with different regioselectivity. Considering the paucity of methods available for the benzylation of pyridines,9 and the ability of controlling the chemoselectivity to dictate the regio-isomeric outcome of the process by simply changing additives, we decided that a detailed investigation was worth pursuing.
Design plan
This study was prompted by the interest of the late Kilian Muñiz in the activation of N-halogenated compounds to form the corresponding N-centered radicals, and their use in 1,5-HAT to initiate position-selective C(sp3)–H functionalization reactions.10 The original plan (Fig. 2a) was to use a photoredox catalyst (PC) to activate, via an SET reduction, the N–F bond within N-(tert-butyl)-N-fluoro-2-methylbenzamide (2a, Ered = −0.84 V vs. SCE).8a By triggering the sequence detailed in Fig. 1b, this SET would afford the benzyl radical III. Engaging III in an ipso-substitution manifold would also require the SET reduction of 4-cyanopyridine 1a (Ered = −1.60 V vs. SCE)11 to form the radical anion I. We surmised that the activation of 1a could also be mastered by PC. Crucial for the sequential activation of substrates 1 and 2 would be therefore a donor additive capable of efficiently turning over the photocatalyst PC by facilitating SET reduction of the oxidized PC+·.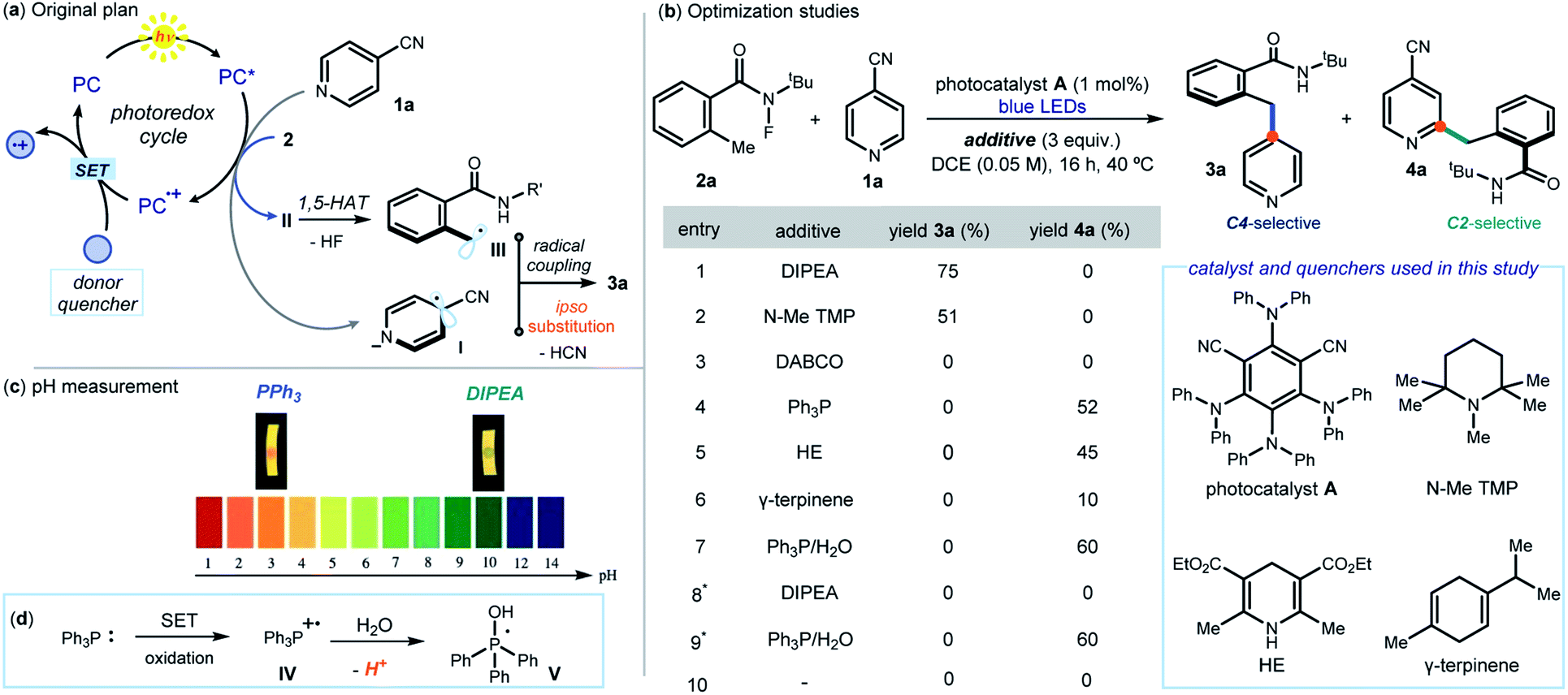 | ||
| Fig. 2 Initial explorations. (a) Design plan. (b) Optimization studies: reactions performed under argon on a 0.1 mmol scale at 40 °C for 16 h under illumination by a blue LED strip (λmax = 465 nm, 14 W) using 3 equiv. of 1a and additive. Yield determined by 1H NMR analysis. (c) Measuring the pH of the reaction media. (d) Rationale for the acidity increase in the PPh3-based protocol. *Performed under air. | ||
Results and discussion
The feasibility of our plan was tested by selecting 1,3-dicyano-2,4,5,6-tetrakis(diphenylamino)-benzene A as photocatalyst (see Section E1 in the ESI† for the screening of other photoredox catalysts). This choice was informed by the redox potential of A (E1/2(A+/A*) = −1.52 vs. SCE),12 which makes it potentially suitable for activation of both 1a and 2avia SET reduction.13 Initial experiments were conducted in 1,2-dichloroethane (DCE) using a blue LED emitting at 465 nm (Fig. 2b). The choice of the donor additives (3 equiv.), which served as reductive quenchers of A, was critical for efficiency. Tertiary amines, including DIPEA (N,N-diisopropylethylamine) and pentamethyl piperidine (N–Me TMP), smoothly afforded the ipso-benzylated product 3a (entries 1–2), with DIPEA offering the best yield (75%, entry 1). A strained bicyclic amine (DABCO) was not suitable for this process (entry 3). Surprisingly, non-basic quenchers completely switched the reaction manifold towards a Minisci pathway, leading exclusively to the formation of the C2 benzylated pyridine 4a. Specifically, triphenylphosphine (PPh3), Hantzsch ester (HE), and γ-terpinene all channeled the radical process towards a C2-selective pattern (entries 4–6). Further optimization using PPh3 as the quencher established that H2O (5 equiv., entry 7) improved the yield of 4a to 60%. Performing the process under air completely suppressed the ipso-substitution manifold, while the Minisci-type reaction remained unaffected (entries 8–9). Control experiments indicated that the reductive quencher (entry 10), the photocatalyst, and light irradiation were necessary for reactivity.Investigations were then performed to gain insight into the origin of the mechanistic switch. Since Minisci reactions require acidic conditions5 for effective heteroarene activation, we measured the pH of the two reaction systems by means of a pH meter (see Fig. 2c and Section F in the ESI† for details). The C2 selective system, mastered by PPh3, had a pH of 3.1, which is suitable for a Minisci pathway. In contrast, we measured a pH of 9.8 for the DIPEA-based protocol, leading to the ipso-substitution product 3a. This observation hints to the acidity of the reaction medium as responsible for channeling the process towards different mechanistic paths. A basic quencher (DIPEA) ensures photocatalyst A turnover while keeping a basic pH by neutralizing the HCN and HF generated upon SET activation of substrates 1a and 2a, respectively. Switching to neutral quenchers (PPh3, but also HE and γ-terpinene, entries 4–6 in Fig. 2b) avoids the possibility of buffering the build-up of acidity. In addition, the possible SET oxidation of PPh3 (E1/2 = +0.98 V vs. SCE) from the photocatalyst would afford the radical cation IV, which is known to deliver the phosphoranyl radical V liberating protons upon addition of water (Fig. 2d).14 Overall, the PPh3-based protocol ensures the acidic conditions needed for an effective Minisci pathway, which overrides the ipso-substitution manifold. This scenario is congruent with kinetic profile analyses of the two systems, detailed in Section I of the ESI,† that established the Minisci C2-selective path to proceed faster than the C4-selective process (after 75 minutes, the Minisci product is formed in 60% yield, while the ipso-substitution adduct in about 5% yield). The Minisci pathway also showed an induction period, which was suppressed upon addition of TFA (5 mol%). This reactivity profile is coherent with the need for the reaction medium's pH to be low enough to switch on the Minisci pathway.13 Importantly, the Minisci reaction cannot be promoted by simple addition of stoichiometric amounts of TFA and removal of PPh3 (see Section E2, Table S3, entry 12–13 in the ESI†). This indicates the unique ability of the neutral quencher to switch on an effective photocatalytic pathway that enables the C2-selective process.
After delineating a rationale for the observed chemodivergency, we explored the synthetic applicability of our photocatalytic systems by adopting the optimized conditions in entries 1 and 7 of Fig. 2b. Both protocols required the same reaction conditions (substrates, photocatalyst A, and solvent), but differed in the additive used (DIPEA or PPh3). We first evaluated the 2-alkyl N-fluorobenzamides 2 that could participate in the ipso-substitution and the Minisci processes with 4-cyanopyridine 1a leading to C4 or C2 benzylated pyridines 3 and 4, respectively (Fig. 3a).
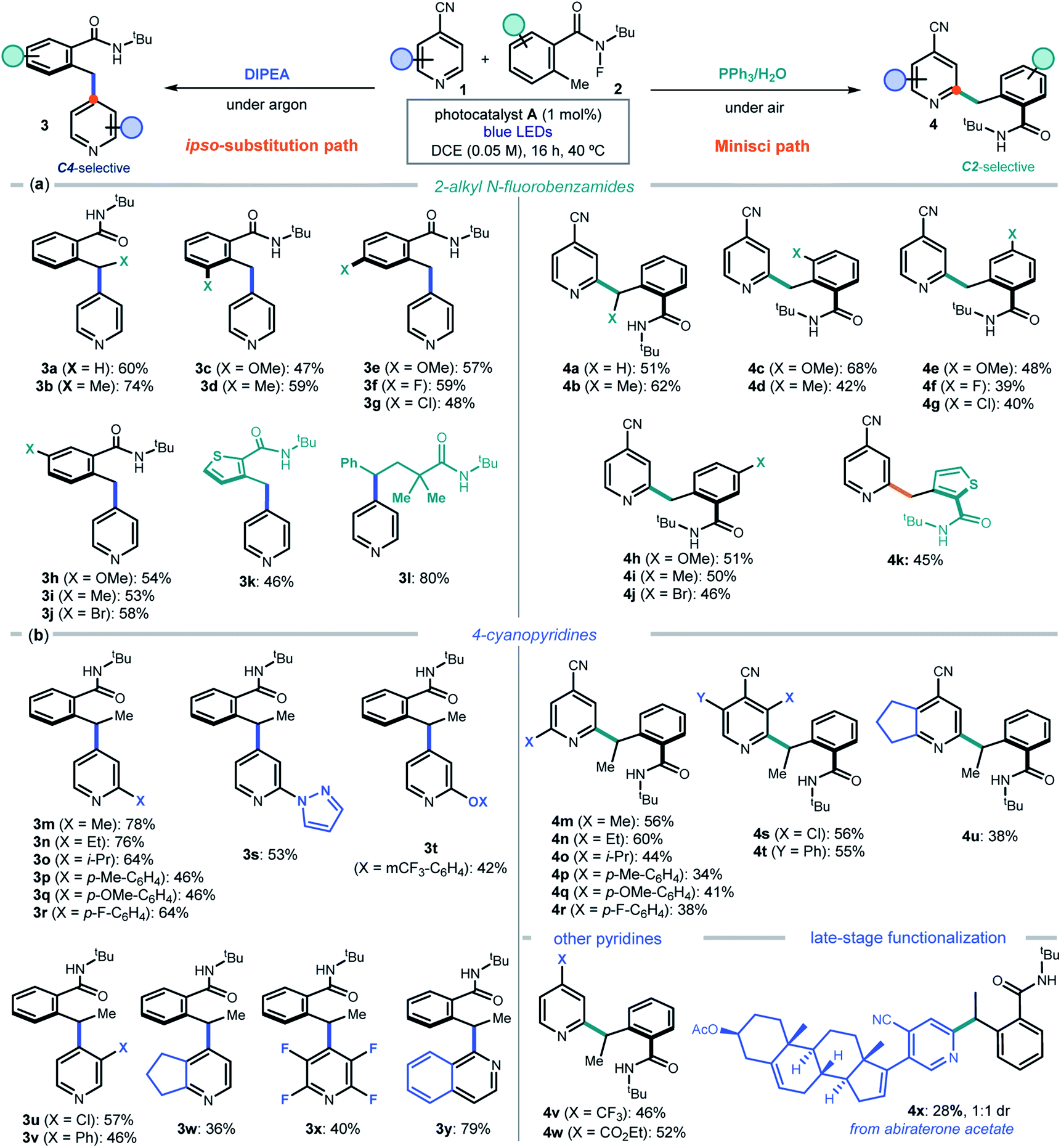 | ||
| Fig. 3 Switchable photocatalytic strategy for the chemodivergent C4 and C2 radical benzylation of 4-cyanopyridines. Survey of the (a) N-fluoroamides 2 and (b) 4-cyanopyridines 1 that can participate in the processes. Reactions performed using 3 equiv. of 1 and 3 equiv. of additive; yields refer to isolated products 3 and 4 after purification. | ||
A methyl substituent at the benzylic position of 2 was well-tolerated (products 3b and 4b), as was the presence of both electron-donating groups and halides at various positions of the aryl ring (adducts c–j in both protocols). A heteroaryl moiety, such as a thiophenyl, could be installed in products 3k and 4k. The ipso-substitution protocol could be extended to include a linear N-fluoramide that, upon N–F activation and 1,5-HAT, formed a benzylic radical eventually leading to product 3l. The linear substrate offered poor reactivity (<20%) in the Minisci protocol. Linear N-fluoramides that do not lead to benzylic radicals proved unreactive (a list of unsuccessful and moderately reactive substrates is reported in Fig. S4 of the ESI†).
We then explored the cyanopyridines 1 that could react with 2-ethyl N-fluorobenzamide 2b, applying both C4 and C2-selective protocols (Fig. 3b). Various substituents were tolerated at the pyridine 2-position, leading to products 3m–r and 4m–r. A fused cyclopentane substrate provided products 3w and 4u in moderate yield. Focusing on the ipso-substitution pathway, we found that a pyrazole and a phenol ether substituent could be successfully installed in pyridines 3s and 3t. Moreover, 3-substituted cyanopyridines and 2,3,5,6-tetrafluoro-4-cyanopyrine also reacted smoothly, leading to products 3u–v and 3x, respectively. A different heteroarene scaffold, such as 1-cyanoquinoline, afforded the ipso-substitution product 3y in high yield. As for the C2-selective Minisci protocol, substitution at the 3-position of the cyanopyridines was tolerated, with a chloro substituent affording the more crowded regioisomer 4s (halogens at the C3 position act as ortho-activators)5d while the sterically hindered phenyl group leading to adduct 4t. 4-Substituted pyridines bearing electron-withdrawing groups other than nitrile, including a trifluoromethyl and an ester, afforded 4v and 4w in good yields. The Minisci-type addition was also suitable for the direct functionalization of a biorelevant compound, since abiraterone acetate (a medication used to treat prostate cancer) was successfully benzylated, leading to adduct 4x in satisfactory yield.
We then considered the development of a sequential protocol for the difunctionalization of pyridines (Scheme 1a). Capitalizing on the pH-regulated switchable methods, we first conducted the Ph3P-enabled Minisci process to couple 4-cyanopyridine 1a and 2-ethyl N-fluorobenzamide 2b with C2-selectivity. The resulting product 4b, upon isolation, was reacted with the linear N-fluoroamide 2c using DIPEA to select the C4-selective ipso-substitution manifold. The overall sequence smoothly delivered the complex pyrdine 5.
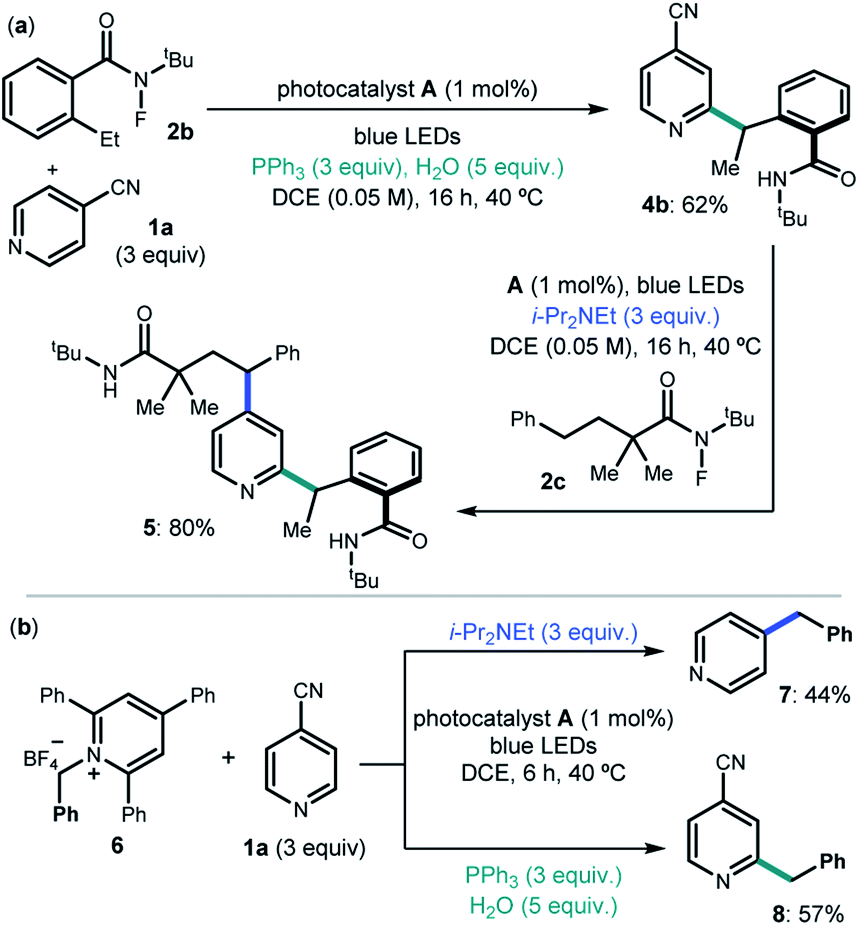 | ||
| Scheme 1 (a) Sequential procedure for the difunctionalization of 4-cyanopyridine 1a. (b) Ipso-substitution (C4 selective) and Minisci-type reaction (C2 selective) for the benzylation of 1a with N-benzyl pyridinium 6. | ||
We also investigated if the generality of our chemodivergent system could be expanded to include different radical precursors (Scheme 1b). Specifically, the pyridinium salt 6, which is prone to facile SET reduction (Ered = −1.01 V vs. Ag/AgCl),15 was successfully activated towards benzyl radical formation in both systems, delivering C2 and C4 benzylated pyridines 7 and 8 in satisfactory yields.
Conclusions
In summary, we have developed a photoredox strategy that allows switchable control of the chemoselectivity in the radical benzylation of 4-cyanopyridines. An ipso-substitution path leads to C4 benzylated pyridines, while a Minisci-type addition affords products with C2 selectivity. The two reactivity manifolds, which require the same photoredox catalyst, reaction conditions, and substrates, are selected at will by the choice of the photocatalyst quencher. The switchable photocatalytic system was successfully applied with two different benzylic radical precursors. Mechanistic studies established that a change in the reaction medium's pH is responsible for channeling the process towards chemodivergent pathways.Data availability
Electronic supplementary information (ESI†) available: details of experimental procedures, full characterization data, and copies of NMR spectra (PDF).Author contributions
E. G., D. S., and K. C. performed the experiments. K. M. was involved in the conceptualization of the process. P. M. directed the project. P. M. wrote the manuscript with assistance from all authors. E. G. and D. S. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by Agencia Estatal de Investigación (PID2019-106278GB-I00 to PM and CTQ2017-88496-R to KM) and the MCIN/AEI/10.13039/501100011033 (CEX2019-000925-S). E.G. thanks MINECO (CTQ2017-88158-R) for a predoctoral fellowship. The authors thank Dr Will C. Hartley (ICIQ) for insightful discussions.Notes and references
- E. Vitaku, D. T. Smith and J. T. J. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) P. Melchiorre, Chem. Rev., 2022, 122, 1483–1484 CrossRef CAS PubMed , and reviews cited therein.; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- For a review: (a) S. Tong, K. Li, X. Ouyang, R. Song and J. Li, Green Synth. Catal., 2021, 2, 145–155 CrossRefFor seminal examples: (b) T. Caronna, A. Clerici, D. Coggiola and S. Morrocchi, Tetrahedron Lett., 1981, 22, 2115–2118 CrossRef CAS; (c) R. Bernardi, T. Caronna, S. Morrocchi and M. Ursini, J. Heterocyclic Chem., 1996, 33, 1137–1142 CrossRef CASFor selected recent examples: (d) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed; (e) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed; (f) T. Hoshikawa and M. Inoue, Chem. Sci., 2013, 4, 3118–3123 RSC.
- For a review: (a) R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed and references therein.For seminal reports: (b) F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3580 CrossRef CAS; (c) F. Minisci, F. Fontana and E. Vismara, J. Heterocycl. Chem., 1990, 27, 79 CrossRef CASFor recent selected examples: (d) F. O'Hara, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2013, 135, 12122–12134 CrossRef PubMed; (e) A. Gutiérrez-Bonet, C. Remeur, J. K. Matsui and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 12251–12258 CrossRef PubMed; (f) J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565–1569 CrossRef CAS PubMed.
- (a) A. Nikolaev, C. Y. Legault, M. Zhang and A. Orellana, Org. Lett., 2018, 20, 796–799 CrossRef CAS PubMed; (b) X. Li, H.-Y. Wang and Z.-J. Shi, New J. Chem., 2013, 37, 1704–1706 RSC.
- Y. Sakakibara and K. Murakami, ACS Catal., 2022, 12, 1857–1878 CrossRef CAS and references therein..
- (a) Q. Guo, Q. Peng, H. Chai, Y. Huo, S. Wang and Z. Xu, Nat. Commun., 2020, 11, 1463–1472 CrossRef CAS PubMedFor metal-catalyzed N-F bond activation protocols, see: (b) B. J. Groendyke, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 12771–12774 CrossRef CAS PubMed; (c) Z. Li, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 13288–13292 CrossRef CAS PubMed; (d) Z. Zhang, L. H. Stateman and D. A. Nagib, Chem. Sci., 2019, 10, 1207–1211 RSC; (e) Q.-Q. Min, W.-Y. Shi, Z. Zhang and Y.-M. Liang, Org. Lett., 2021, 23, 2693–2698 CrossRef PubMed; (f) D. Bafaluy, J. M. Muñoz-Molina, I. Funes-Ardoiz, S. Herold, A. J. de Aguirre, G. Zhang, F. Maseras, T. R. Belderrain, P. J. Pérez and K. Muñiz, Angew. Chem., Int. Ed., 2019, 58, 8912–8916 CrossRef CAS PubMed.
- For ipso-benzylation: (a) B. M. Vittimberga, F. Minisci and S. Morrocchi, J. Am. Chem. Soc., 1975, 97, 4397–4398 CrossRef CASFor Minisci-type processes: (b) M. Wan, H. Lou and L. Liu, Chem. Commun., 2015, 51, 13953–13956 RSC.
- (a) C. Martinez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287–8291 CrossRef CAS PubMed; (b) A. E. Bosnidou, T. Duhamel and K. Muñiz, Eur. J. Org. Chem., 2020, 6361–6365 CrossRef CAS and references therein. See also Ref. 8f..
- M. C. Nicastri, D. Lehnherr, Y.-H. Lam, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2020, 142, 987–998 CrossRef CAS PubMed.
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182–8186 CrossRef CAS PubMed.
- The simplistic mechanistic picture detailed in Fig. 2a, based on the SET reduction of 1a and 2a by photocatalyst A, served as our initial design plan. Detailed investigations, including quantum yield determination, Stern–Volmer quenching studies, and cyclic voltammetry, suggested a more complex mechanistic scenario for both protocols based on a reductive quenching cycle, which is discussed in Section G of the ESI†.
- (a) G. Pandey, D. Pooranchand and U. T. Bhalerao, Tetrahedron, 1991, 47, 1745–1752 CrossRef CAS; (b) Y. Masuda, H. Tsuda and M. Murakami, Angew. Chem., Int. Ed., 2021, 60, 3551–3555 CrossRef CAS PubMed.
- M. Ociepa, J. Turkowska and D. Gryko, ACS Catal., 2018, 8, 11362–11367 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, full characterization data, and copies of NMR spectra (PDF). See https://doi.org/10.1039/d2sc02698h |
| ‡ These authors contributed equally to this work. |
| § Prof. Dr Kilian Muñiz: deceased, March 16, 2020. |
| This journal is © The Royal Society of Chemistry 2022 |