
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn atomically precise silver nanocluster for artificial light-harvesting system through supramolecular functionalization†
Anish Kumar
Das‡
a,
Sourav
Biswas‡
a,
Surya Sekhar
Manna
 b,
Biswarup
Pathak
b and
Sukhendu
Mandal
*a
b,
Biswarup
Pathak
b and
Sukhendu
Mandal
*a
aSchool of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Kerala 69551, India. E-mail: sukhendu@iisertvm.ac.in
bDepartment of Chemistry, Indian Institute of Technology, Indore, Madhya Pradesh 453552, India
First published on 20th June 2022
Abstract
Designing an artificial light-harvesting system (LHS) with high energy transfer efficiency has been a challenging task. Herein, we report an atom-precise silver nanocluster (Ag NC) as a unique platform to fabricate the artificial LHS. A facile one-pot synthesis of [Cl@Ag16S(S-Adm)8(CF3COO)5(DMF)3(H2O)2]·DMF (Ag16) NC by using a bulky adamantanethiolate ligand is portrayed here which, in turn, alleviates the issues related to the smaller NC core designed from a highly steric environment. The surface molecular motion of this NC extends the non-radiative relaxation rate which is strategically restricted by a recognition site-specific supramolecular adduct with β-cyclodextrin (β-CD) that results in the generation of a blue emission. This emission property is further controlled by the number of attached β-CD which eventually imposes more rigidity. The higher emission quantum yield and the larger emission lifetime relative to the lesser numbered β-CD conjugation signify Ag16 ∩ β-CD2 as a good LHS donor component. In the presence of an organic dye (β-carotene) as an energy acceptor, an LHS is fabricated here via the Förster resonance energy transfer pathway. The opposite charges on the surfaces and the matched electronic energy distribution result in a 93% energy transfer efficiency with a great antenna effect from the UV-to-visible region. Finally, the harvested energy is utilized successfully for efficient photocurrent generation with much-enhanced yields compared to the individual components. This fundamental investigation into highly-efficient energy transfer through atom-precise NC-based systems will inspire additional opportunities for designing new LHSs in the near future.
Introduction
Light-harvesting systems (LHSs) have a crucial role in natural photosynthesis for transforming photonic energy into chemical energy, that covers an outlook for the utilization of renewable energy sources.1,2 In natural photosynthesis, the system-specific organization of chlorophylls advances strong photon absorptivity and constituent emission energy; simultaneously conquering energy-dissipating excited-states decay processes. These unique features permit excited-states energy transfer to the next neighbour of LHS and, ultimately, to the reaction centre with impressive efficiency.3–6 For the fabrication of artificial LHSs, two important prerequisites are crucial for efficient energy transfer between the donor–acceptor through the Förster Resonance Energy Transfer (FRET) pathway. Firstly, the packing of donors should not be dense and intramolecular self-quenching should be avoided.7–11 Secondly, the multiple donors should correspond to one acceptor. In a natural LHS, proteins and chlorophylls interacted through non-covalent interaction. Several artificial LHSs were evolved to mimic the outstanding structure and functionality of the natural photosynthesis system. However, the efficient charge transfer property of such a system is the main concern for practical applicability.12,13 To solve the issues of traditional chromophores, an encouraging strategy depends on host–guest interaction to keep up miniature rift chromophores during the suppression of exciting energy quenching.1,2 The use of large hosts like cucurbit[n]uril and pillar[n]arene have the advantage of boosting the fluorescence intensity through encapsulation of the guest group of the chromophores while impeding chromophores from stacking.14–19 So, it is clear that the design of the donor molecules is the most essential factor for determining the activity of the artificial LHS. In this current circumstance, to continue the development of the artificial LHSs with the efficient FRET process, atom-precise metal NCs would be the appropriate alternative candidate.Ligand-stabilized silver nanoclusters (Ag NCs) have gained tremendous attention due to their fascinating structural architectures with molecular precision and exotic photophysical properties.20–28 Many recent studies have identified that the geometry, size, surface ligands, inorganic–organic interfaces, and chemical functionalization implement an important role in controlling their distinct physical and chemical properties.20,21,29–31 Although the high-nuclear Ag NCs related to the core–shell arrangement are well studied, the anion templated mono-layered core containing the d10 configuration of Ag(I) ions is relatively less researched. It has been reported that templating species were utilized to control the formation of the Ag(I) core based on electrostatic interactions that regulate the size and geometry of the core.32–34 However, most of the reported anion template silver NCs rarely exhibit photoluminescence (PL) properties at room temperature and it is the most perplexing issue which needs to be resolved.35–38 Progressing research activities suggest that imposing bulkiness on the surface protecting groups can restrict the intramolecular motion of the NC in solution that can directly decrease the non-radiative relaxation pathway and enhance the emission properties.39–48 Therefore, it will be very interesting to encapsulate the ligand-protected Ag NC with a bulky host to build the host–guest adduct at the recognition site of the NC to improve the emission properties by introducing surface rigidity through bulkiness to counteract the thermally activated non-radiative relaxation which will be beneficial for the artificial LHS fabrication.
Herein, we have designed an atom-precise Ag NC-based novel artificial LHS with a very high energy transfer efficiency. We have strategically synthesized bulky adamantanethiolate ligand-protected, [Cl@Ag16S(S-Adm)8(CF3COO)5(DMF)3(H2O)2]·DMF, Ag16 NC. Although a bulky ligand system is utilized to restrict the surface molecular vibrations in solution, it is not sufficient for generating the emission properties of this as-synthesized NC at room temperature. To alleviate this issue, we have applied a host–guest approach to rigidify the outer surface of the cluster by anchoring a specific hydrophobic guest molecule β-cyclodextrin (β-CD) to alter the relaxation mechanism. The successful entrapment of the adamantane moieties inside the cavity of the β-CD is fully characterized by various spectroscopic tools and confirms the number of the supramolecular adduct, Ag16 ∩ β-CDn (n = 1, 2), while the facile position is identified by the binding energy calculation. We have then utilized this system for generating blue emission energy to construct an LHS via the FRET process with an organic dye β-carotene. A 93% energy transfer efficiency and good antenna effect from the UV-to-visible region are detected, which is further utilized for efficient photocurrent generation.
Results and discussion
To begin our investigation, a facile one-pot chemical reaction pathway was followed to synthesize Ag16 NC. At first, the reaction took place between the [AgS-Adm]n complex and CF3COOAg in dimethylformamide (DMF) solvent (Scheme 1). Afterward, PPh4Cl was added into the mixture as a source of a templating agent of chloride ion. The final mixture was kept in dark for 3 days and transparent colorless block-shaped crystals were acquired, yielding 58% (Ag-based). In general, synthesizing smaller NCs by using the bulky thiolate groups is a challenging task as the bulkiness of the ligand provides a higher oxidation resistance to larger Ag NCs.49 In this report, we successfully achieve the smaller core-sized Ag NC utilizing the bulky adamantanethiolate ligand which will surely throw light on the synthesis procedure with the controlled size of the NCs.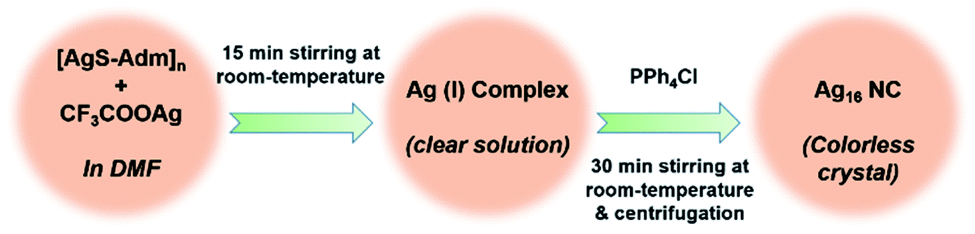 | ||
| Scheme 1 Synthetic outline of the adamantanethiolate protected Ag16 NC. | ||
The obtained high-quality colorless crystals are suitable to elucidate the structural information exclusively via the single-crystal X-ray diffraction (SCXRD) technique. The block-shaped crystals are crystallized in a triclinic crystal system with space group ![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) 1 (no. 2) (Table S1†). The crystallographic data implies that the overall molecular formula of the as-synthesized NC is [Cl@Ag16S(S-Adm)8(CF3COO)5(DMF)3(H2O)2]·DMF, (Ag16), depicted in Fig. 1 as top and side views. The chemical formula also reveals the neutral state of the Ag16 NC where all the Ag atoms are in the +1 state.
1 (no. 2) (Table S1†). The crystallographic data implies that the overall molecular formula of the as-synthesized NC is [Cl@Ag16S(S-Adm)8(CF3COO)5(DMF)3(H2O)2]·DMF, (Ag16), depicted in Fig. 1 as top and side views. The chemical formula also reveals the neutral state of the Ag16 NC where all the Ag atoms are in the +1 state.
 | ||
| Fig. 1 Structural illustration of the overall Ag16 NC from (a) top and (b) side views. | ||
In the view of the structural aspects, the skeleton of the Ag16 core is constructed through metallophilic interactions among Ag(I) ions and adopts a geometry of square gyrobicupola (J29; Johnson solid) with a slight distortion (Fig. 2a). The crystallographic data reveals that the bond distances of AgI⋯AgI in the Ag16 core fall in the range of 2.95–3.39 Å, which are lower compared to twice the van der Waals radius of Ag (3.44 Å).50,51 The average distance indicates that some sort of long-range non-covalent interaction (NCI) is present between the two Ag atoms which must be an argentophilic interaction.52 We have calculated the weak metallophilic interaction such as bond critical point (BCP), bond order, and NCI of two adjacent Ag atoms of the Ag16 core. The electron density [∇2ρ(rc) = 0.055] between two Ag atoms with a positive Laplacian indicates the existence of bond critical point (BCP) (Fig. S1†). A low Mayer bond order (0.17) and an attractive NCI have been noticed as well between two Ag atoms of the Ag16 core (Fig. S2†) which indicates the reality of the nonbonding argentophilic interaction.53,54 The geometry of the distorted Ag16 core comprises eight triangular facets and ten square facets (Fig. S3†). The Ag16 core can also be divided into three layers; one square (top, made by four Ag atoms), one octagon (middle, made by eight Ag atoms), and another square (bottom, made by four Ag atoms) (Fig. S4†). The middle octagon layer is connected with the two square layers from the opposite sides via eight square and eight triangular facets. In addition, one square layer is related to another square layer by 45° of rotation, and that classifies the gyro configuration of the core geometry instead of the ortho (Fig. S5†). The chloride atom is enclosed at the center of the octagon layer, resulting in a Cl− templated Ag16 core (Fig. 2b). We have observed that one S2− ion is attached to the octagon layer by μ2 coordination mode (Fig. 2c) which could be produced from the breakage of the C–S bond in DMF medium at room temperature.35 The outer surface of the skeleton is protected by eight Adm-S− ligands (Fig. 2d). Each of the thiolate ligands occupies each of the square facets by μ4 ligation mode with Ag–S bond lengths ranging from 2.40 Å to 2.56 Å. The skeleton of the Ag16 is further capped by the five CF3COO− auxiliary ligands (Fig. 2e). Among the five CF3COO−, four are bonded with the Ag present in the octagon layer by μ2 bridging mode with Ag–O bond lengths in the range of 2.28–2.38 Å, whereas the remaining one is attached with one of the square layers by μ1 bridging with an Ag–O bond length of 2.49 Å. Additionally, three DMF molecules and two water molecules are coordinated with the octagon layer of the skeleton (Fig. 2f). Also, one non-coordinated DMF molecule is detected in the lattice by careful crystallographic measurement (Fig. S6†).
 | ||
| Fig. 2 Structural investigation of the Ag16 NC. (a) Square gyrobicupola geometry of the Ag16 skeleton, (b) chloride ion template, (c) attachment of S2− with the middle octagon layer of the skeleton, (d) connection of eight S atoms of the thiolate groups with the square facets of the skeleton, and (e) and (f) linking of CF3COO− as auxiliary ligands and DMF and water as solvents with Ag16 skeleton, respectively. | ||
Further, to verify the chemical composition and identify the charge of the as-synthesized NC as well as its associated fragments, electrospray ionization mass spectrometry (ESI-MS) is carried out (Fig. 3). In the positive mode ESI-MS spectrum, three prominent peaks are identified. The peak corresponding to the highest m/z value at 4026.30 is associated with [{Cl@Ag16S(S-Adm)8(CF3COO)5(DMF)3(H2O)2·(DMF)} + H]+, which further justifies that the cluster itself is neutral. The experimental m/z value is well-matched with the simulated isotropic patterns, demonstrating the consistency of the assigned molecular formula of the Ag16 NC with the single-crystal data. The presence of the other two predominant peaks in the ESI-MS spectrum corroborates the associated fragments of [{Cl@Ag15S(S-Adm)8(CF3COO)4(DMF)3(H2O)2·(DMF)} + H]+ and [{Cl@Ag15S(S-Adm)8(CF3COO)4(DMF)2(H2O)·(DMF)} + H]+ (Fig. S7†). Further, to verify the stability of the as-synthesized NC, 1H, and 19F nuclear magnetic resonance (NMR) studies are performed in CDCl3. The shift of the adamantane proton in the NC compared to the free [Ag-Adm]n complex confirms the presence of bonded Ag-Adm moieties in the structure of the Ag16 NC (Fig. S8†). Moreover, the presence of F atoms in the structure is also proved by 19F NMR (Fig. S9†).
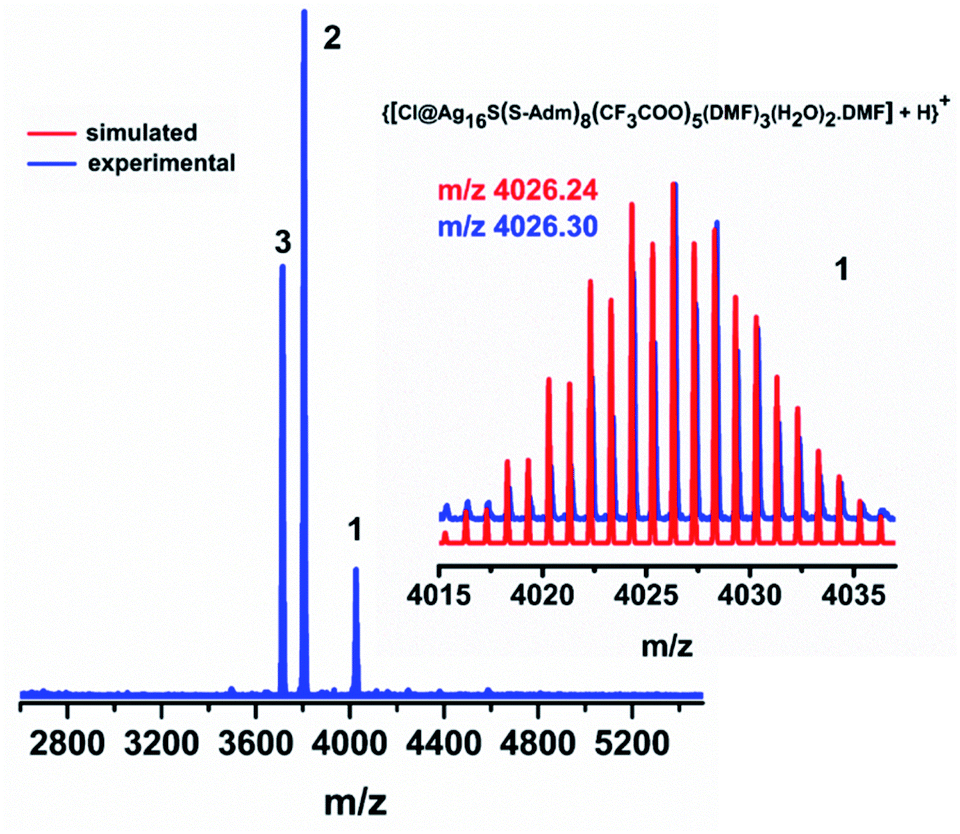 | ||
| Fig. 3 Positive mode ESI-MS spectrum of the as-synthesized Ag16 NC over the m/z range from 2600 to 5600. Inset: experimental and simulated patterns of the parent Ag16 NC. | ||
The good agreement of the experimental powder X-ray diffraction patterns with the simulated data further dictates the high purity of the crystals of Ag16 NC (Fig. S10†). The high-resolution transmission electron microscopic (HR-TEM) image confirms that the diameter of the individual NC is ∼0.8 nm (Fig. 4a). The corresponding scanning electron microscopy (SEM) image and optical microscopic image of the bulk crystals corroborate the block shape of this NC (Fig. 4b). The X-ray photoelectron spectroscopy (XPS) survey spectrum illustrates the existence of the respective elements in this NC (Fig. 4c), that is matched with the energy dispersive X-ray analysis (Fig. S11†). The high-resolution binding-energy spectra of each element reveal their corresponding oxidation states. The binding energy spectrum of Ag 3d demonstrates the presence of Ag(I) by showing two peaks of 3d5/2 and 3d3/2 at 368.76 eV and 374.76 eV, respectively (Fig. 4d). The existence of S, Cl, F, O, N, and C is also confirmed (Fig. S12†).
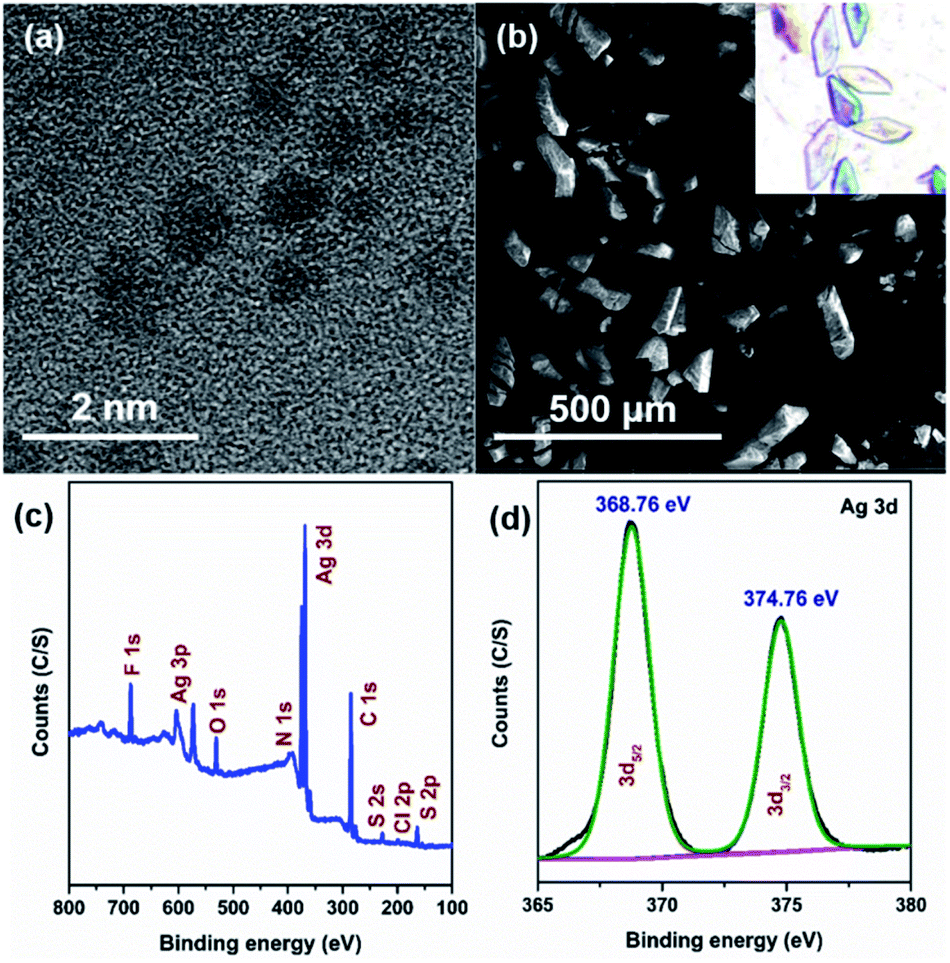 | ||
| Fig. 4 (a) HR-TEM and (b) SEM images of the Ag16 NC, (c) XPS survey spectrum, and (d) binding energy spectrum of Ag of the Ag16 NC. | ||
To elucidate the photophysical properties of this newly synthesized adamantanethiol-protected Ag16 NC, UV-vis absorbance is measured in DCM solvent. A monotonic decrease in the absorbance spectrum with a small shoulder peak at 374 nm is observed (Fig. 5a). From the time-dependent density functional theory (TD-DFT) analysis, the calculated Kohn–Sham orbital population suggests that the occupied states have the major contribution of s- and p-like states of the ligand, whereas the unoccupied orbitals are mainly driven through the s-, p-, and d-like states of the core (Fig. 5b). Hence, the ligand to core transition is clearly understood by the Kohn–Sham orbital population analysis. Further, the computational study reveals that the shoulder peak at 369 nm originates due to the optical transition of HOMO−4 → LUMO (Fig. 5c). The frontier molecular orbitals arrangement describes that the low intense peak of 369 nm is mainly constituted by the transition occurring from the p-like (2p) bonding state of adamantane carbon (ligand) to the antibonding states of Ag (4d) and S (3p). This transition is related to the small shoulder peak at 374 nm in the experimental absorbance spectrum. Thus, the observation of low intense peak is mainly derived due to the transition from the ligand to the core which justifies the simulated Kohn–Sham orbital population analysis of the Ag16 NC dissolved in DCM.
 | ||
| Fig. 5 (a) UV-vis absorbance spectrum of the Ag16 NC dissolved in DCM, inset: simulated absorbance spectrum of the Ag16 NC, (b) Kohn–Sham molecular energy diagram of the NC, and (c) corresponding transitions and related frontier-molecular orbitals diagram. | ||
This as-synthesized NC is not emissive at room temperature. However, the strategical modification of the cluster surface bulkiness using β-CD through host–guest interaction is able to generate the luminescence properties of this Ag16 NC at room temperature upon excitation at 380 nm (Fig. S13†). Owing to the specific size of the inner hydrophobic cavity, the guest β-CD molecule can easily encapsulate the hydrophobic surface of the host NC i.e., the adamantane group which can act as a recognition site for constructing the β-CD-functionalized stable host–guest hybrid assemblies.44,55 To verify the intermolecular host–guest interaction between the adamantane moiety and the β-CD molecule, the 1H NMR study shows more splitting at the adamantane region which confirms the insertion of the ligand in the β-CD cavity (Fig. S14†). However, no certain change is witnessed from the UV-vis absorbance spectra of Ag16 NC after the supramolecular adduct formation with β-CD, suggesting the unaffected ground-state electronic structure (Fig. S15†). Further, the electrochemical properties of the parent Ag16 NC vs. β-CD functionalized Ag16 NC dissolved in DCM are examined. In cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements, the redox peaks disappear in the case of β-CD functionalized Ag16 NC, which suggests that the host–guest complex is resistant to the redox reaction (Fig. S16†). Finally, the number of interacted β-CD molecules with the surface of the NC via non-covalent interactions is identified in the solution medium by ESI-MS measurements. The positive mode spectrum of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Ag16 NC with β-CD reveals that the predominate m/z peak at 2565.15 corresponds to Ag16 ∩ β-CD12+ (∩ symbolizes the supramolecular adducts) along with a small amount of Ag162+ species and two other associated fragments represented in Fig. S17.† The +2 charge originates after the removal of S2− from the parent NC which is further validated by the characteristic isotropic distribution with the peak separation of m/z ∼ 0.50. The well-matched experimental and simulated mass spectrum data indicates that only one of the adamantane moieties is entrapped in the β-CD cavity. Interestingly, the mass data of 1:2 or excess (1:3, 1:4, 1:5, and 1:8) mixture of Ag16 NC with β-CD reveals that a maximum of 2 molecules of β-CD can be attached with the NC yielding Ag16 ∩ β-CD22+ at an m/z value of 3132.65 along with a lesser amount of Ag16 ∩ β-CD12+ and Ag162+ species (Fig. S18†). The packing fashion of the two β-CD molecules anchored on the cluster surface induces steric hindrance in such a way that the approach of any further CD molecules towards the cluster surface is restricted. However, the peak maximum of the adduct is shifted toward the higher m/z value from the one β-CD attachment to the two β-CD attachments in solution signifying the complexation of β-CD on the NC.
:1 mixture of Ag16 NC with β-CD reveals that the predominate m/z peak at 2565.15 corresponds to Ag16 ∩ β-CD12+ (∩ symbolizes the supramolecular adducts) along with a small amount of Ag162+ species and two other associated fragments represented in Fig. S17.† The +2 charge originates after the removal of S2− from the parent NC which is further validated by the characteristic isotropic distribution with the peak separation of m/z ∼ 0.50. The well-matched experimental and simulated mass spectrum data indicates that only one of the adamantane moieties is entrapped in the β-CD cavity. Interestingly, the mass data of 1:2 or excess (1:3, 1:4, 1:5, and 1:8) mixture of Ag16 NC with β-CD reveals that a maximum of 2 molecules of β-CD can be attached with the NC yielding Ag16 ∩ β-CD22+ at an m/z value of 3132.65 along with a lesser amount of Ag16 ∩ β-CD12+ and Ag162+ species (Fig. S18†). The packing fashion of the two β-CD molecules anchored on the cluster surface induces steric hindrance in such a way that the approach of any further CD molecules towards the cluster surface is restricted. However, the peak maximum of the adduct is shifted toward the higher m/z value from the one β-CD attachment to the two β-CD attachments in solution signifying the complexation of β-CD on the NC.
To understand the stability of Ag16 ∩ β-CD2, we have modelled the stepwise encapsulation process of β-CD over the adamantane group. Initially, we have considered the three different possible attachment positions of β-CD on the Ag16 NC (S2, S4, S8) depending on the surrounding chemical environment of each adamantane group (Fig. S19†). Here, we have considered both the head and tail conformation of β-CD. Our calculated binding energy suggested that the β-CD encapsulation in its head conformation on the S4 position is more favorable compared to other positions due to the lowest binding energy of −3.3 eV (Fig. S20, Table S2 and eqn (S1)†). We have then identified the preferred position of the next attachment of β-CD on the Ag16 NC. For this, seven possible attachment positions (S4–S1, S4–S2, S4–S3, S4–S5, S4–S6, S4–S7, and S4–S8) are considered with respect to the encapsulated β-CD on the S4 position of Ag16 ∩ β-CD1. Among these, the S4–S6 attachment position is found to be more stable because of the lowest binding energy of −2.66 eV compared to other positions which are experiencing high steric repulsion (Table S3†). So, the experimental observation and computational findings suggest that β-CD molecules would always prefer the oppositely-placed adamantane groups over the adjacent for host–guest adduct formation, yielding Ag16 ∩ β-CD2 (Fig. 6a).
 | ||
| Fig. 6 (a) Reaction scheme of the formation of supramolecular adducts, Ag16 ∩ β-CD1, and Ag16 ∩ β-CD2 from the parent NC, (b) emission spectra of the Ag16 NC, Ag16 ∩ β-CD1, and Ag16 ∩ β-CD2 dissolved in DCM upon excitation at 380 nm, (c) corresponding 2D-contour plot for visualizing the difference in PL intensities with the number of the attached β-CD molecules, and (d) emission lifetimes data upon excitation at 375 nm. | ||
So, we have strategically designed the adamantanethiolate-protected Ag16 NC with inspiration for using their molecular recognition properties to originate the photoluminescence through host–guest chemistry. The precise supramolecular functionalization of the NC with the β-CD restricts the intramolecular motion of this NC in the solution phase. Interestingly, we have observed that the luminescence intensity and emission lifetimes (0.48 μs to 1.03 μs) increase with the number of attached β-CD molecules (Fig. 6b–d). The physicochemical interactions arising at the ligand surface may alter the efficacy of the relaxation process, which may eventually result in the improvement of their emission properties. So, the origin of the pronounced enhancement in the PL emission QY (relative QY from 0.12 to 0.26) lies in the formation of the host–guest supramolecular adduct which certainly suppresses the surface ligand motions. The theoretical investigation revealed that at this excitation, charge transition is majorly contributed by the p-like (2p) bonding state of the adamantane moiety. However, a similar UV-vis absorbance study excludes the possibilities of any ground-state overlapping, so the rigidification will clearly affect the relaxation pathway. After calculating the radiative and nonradiative relaxation rates, we have identified that the suppression of surface ligand vibrations is closely correlated with the nonradiative relaxation pathway (eqn (S2), (S3) and Table S4†). So, the enhancement in the luminescence behaviour is observed here due to the effective suppression of the nonradiative relaxation rate (from 1.83 × 106 to 7.18 × 105), while the radiative relaxation rate is almost intact after the supramolecular adduct formation. Hence, interfacing individual NCs with functional supramolecular arrangements is an intriguing outlook that delivers new potential.
For utilizing the enhanced emission properties of the β-CD functionalized supramolecular adduct, i.e., Ag16 ∩ β-CD2 in an artificial light-harvesting application through efficient energy transfer by FRET, an acceptor dye molecule is required whose absorption considerably overlaps with the emission of Ag16 ∩ β-CD2.7 Here, we have strategically chosen β-carotene as a dye that has a broad absorptivity with an absorbance maximum at 462 nm in DCM (Fig. 7a). The energy transfer process is examined by the fluorescence titration of β-carotene (2 × 10−6 M) with the Ag16 ∩ β-CD2 (10−5 M) in DCM upon excitation at 380 nm (Fig. 7b). With the gradual addition of 25 μL of β-carotene in the Ag16 ∩ β-CD2 containing solution, a drop in the PL intensity is detected at 444 nm, corresponding to the emission maximum of Ag16 ∩ β-CD2 (donor), while a secondary emission is generated at the ∼540 nm region which corresponds to the emission of β-carotene (Fig. 7c, d and S21†). So, this observation clearly suggests the energy transfer from donor Ag16 ∩ β-CD2 to acceptor β-carotene is facilitated. The CIE chromaticity diagram further verifies the FRET process between acceptor and donor molecules by a change in fluorescence color from blue (co-ordinate of CIE (0.15, 0.09)) to green (co-ordinate of CIE (0.36, 0.53)) via an intermediate color of the donor–acceptor system of light blue (co-ordinate of CIE (0.24, 0.32)) (Fig. 7e). The considerable overlap between the emission and absorption band of the donor and acceptor molecules is the primary factor for the energy transfer process, while the two opposite surface zeta potential values (ζ of Ag16 ∩ β-CD2 = 60 mV, and ζ of β-carotene = −29 mV) support the formation of the adducts for charge transfer feasibility (Fig. S22† and Scheme 2). However, the unchanged absorbance spectra with continuous addition of the similar amount of β-carotene on the Ag16 ∩ β-CD2 solution eliminates the possibility of the ground state interaction (Fig. S23†). This observation is consistent with the 1H NMR spectrum of the Ag16 ∩ β-CD2 and β-carotene mixture which displays the essentially non-shifted proton peaks compared to the individual components (Fig. S24†). So, the energy transfer exclusively originates from the excited state of the Ag16 ∩ β-CD2 by forming an effective sphere with the acceptor that leads to a fast quenching of the excited donor before they diffuse apart. The continuous enhancement in the emission intensity at a higher wavelength region gets saturated after the addition of 200 μL of the β-carotene solution to 2 mL of Ag16 ∩ β-CD2, suggesting the detectable maximum energy transfer limit when the maximum number of acceptor molecules is present in the effective sphere. The theoretically optimized structure of this mixture suggests that both molecules are maintaining an average ∼2 nm gap at the ground state (Fig. S25†). However, the Förster radius of the effective sphere (∼3.7 nm) is calculated experimentally by eqn (S4)† from the average emission lifetime and the energy transfer rate per donor–acceptor pair.56–60 As per the size of each donor molecule, it is obvious that during the excitation, both the molecules will be in a favorable position for the FRET. The calculated radiative rate of the donor (2.52 × 105 s−1) is much slower than the radiative energy transfer rate between the donor and acceptor (2.6 × 107 s−1). Thus, we can assume that during the excitation of a donor, acceptor molecules are present in a favorable position and the excited donor molecule can promptly transfer the energy to the adjacent acceptor molecules which results in a quenching of the excited state of the donor (Scheme 2).8 The energy transfer efficiency between the donor and acceptor is calculated from the emission intensities to be 93% by following eqn (S5).† The additional evidence for the energy transfer is envisioned by obtaining a 17 times shorter average emission lifetime of the conjugated materials than the parent Ag16 ∩ β-CD2 (Fig. S26†). In a controlled experiment, the conjugated material and β-carotene were excited at the original excitation of the β-carotene, showing an impressive antenna effect of 34 (Fig. S27 and eqn (S6)†).
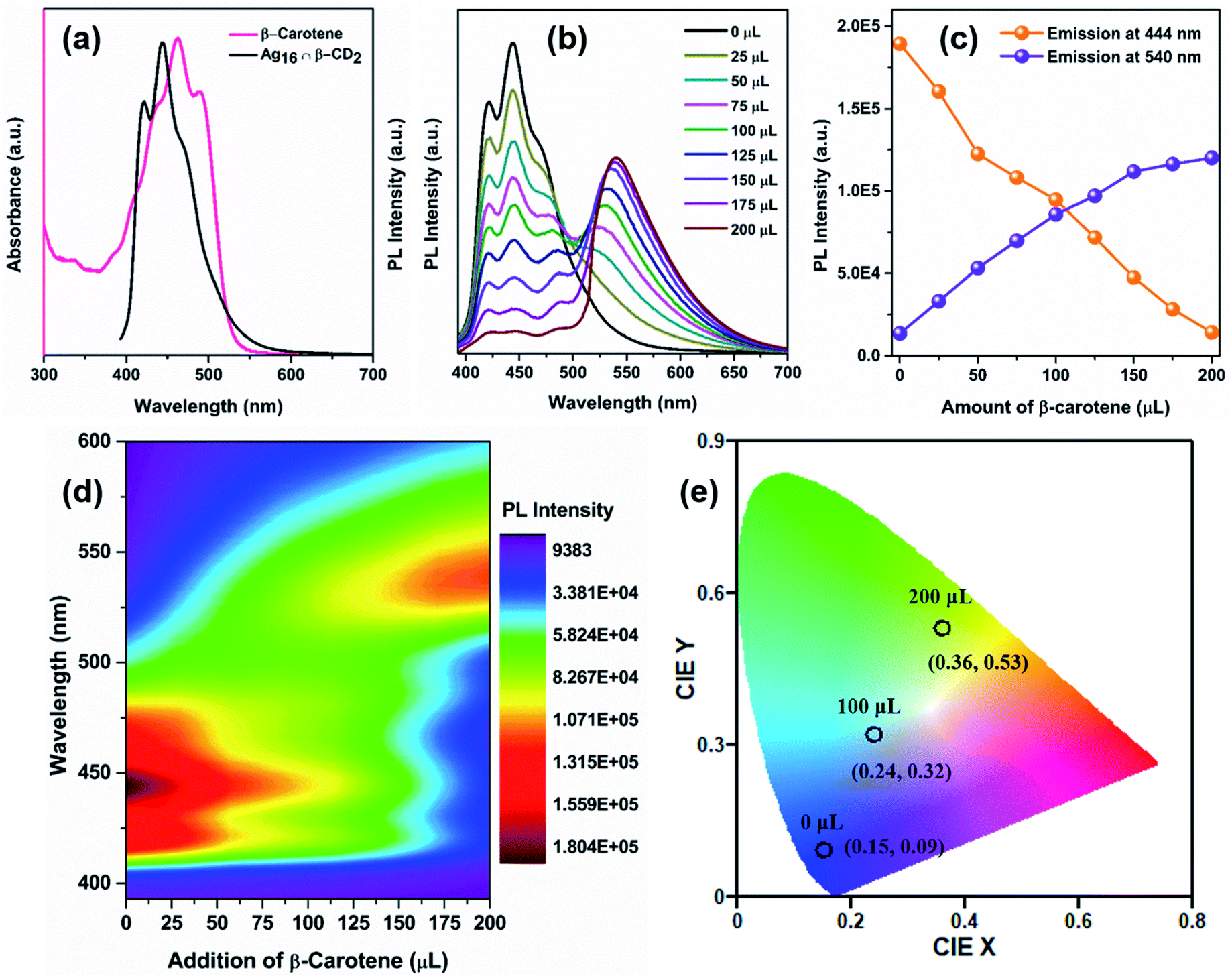 | ||
| Fig. 7 (a) Absorbance spectrum of acceptor β-carotene and emission spectrum of donor Ag16 ∩ β-CD2, (b) emission spectra of the donor molecule with the addition of the measured amount of β-carotene (λex = 380 nm) in DCM, (c) variation in emission intensity for Ag16 ∩ β-CD2 during the addition of the same amount of β-carotene at 444 nm and 540 nm, (d) associated 2D-contour plot of the FRET process for the visualization of the PL intensity transferring process, and (e) change of 1931 CIE chromaticity coordinates of Ag16 ∩ β-CD2 upon titration with β-carotene. | ||
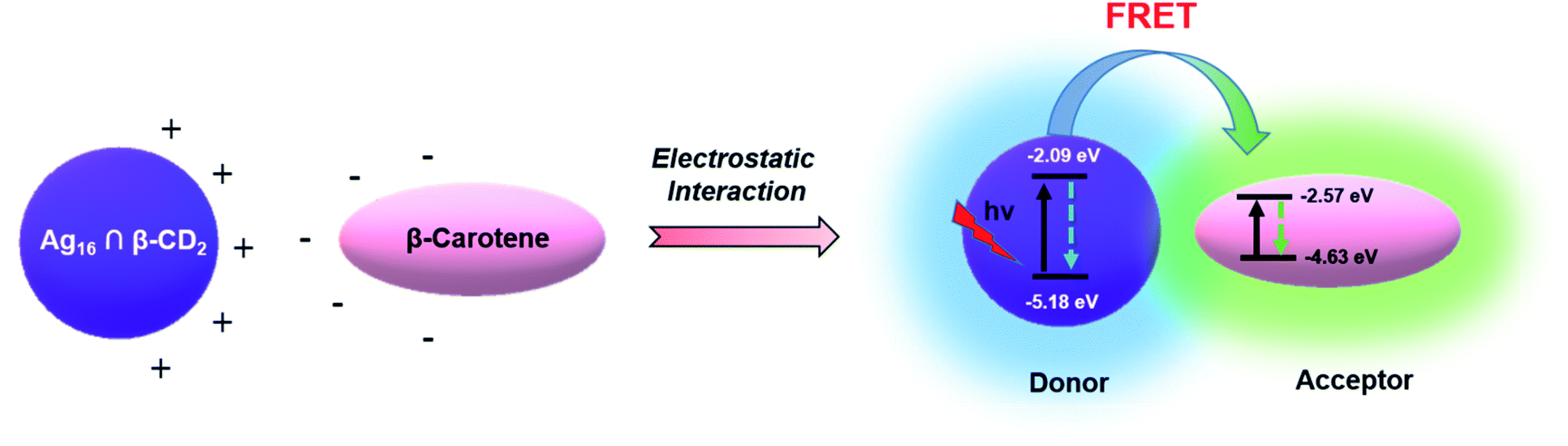 | ||
| Scheme 2 Proposed mechanism of the FRET process between Ag16 ∩ β-CD2 (donor) and β-carotene (acceptor). | ||
To explore the practical usability of this fabricated LHS, we explore its photocurrent generation ability under irradiation of sunlight at a 10 V bias on a patterned gold electrode. As sunlight is a combination of UV and visible light, the β-carotene has low absorption in the UV region whereas the synthesized Ag16 ∩ β-CD2 exhibits strong absorption in the UV region. So, as a combination, the conjugation serves as an antenna source to efficiently transfer the energy from the UV-to-visible region to generate the maximum photocurrent (472 nA) on the gold electrode (Fig. S28†), whereas the individual components show a much lesser photocurrent generation ability which is depicted in a 2D contour plot (Fig. S29†).
Conclusions
In summary, a unique artificial LHS has been strategically designed with an outstanding energy transfer efficiency based on atom-precise Ag(I) NC. Initially, we successfully synthesized bulky ligand-protected Ag16 NC with the small core via controlling the reaction condition. Owing to the hydrophobic nature of the cluster surface, we have implemented a versatile approach of specific host–guest supramolecular adduct formation between β-CD and the adamantane ligand anchored on the cluster. Due to this host–guest interaction, we have effectively generated luminescence properties in the Ag16 NC at room temperature by altering the relaxation mechanism. This adduct is further utilized to construct the artificial LHS through an efficient energy transfer process through FRET occurring from the donor (Ag16 ∩ β-CD2) to the acceptor (β-carotene) with an energy transfer efficiency of 93% and a good antenna effect. The energy transfer process is well justified by the decrease in emission lifetime and emission color change from blue to green which is depicted in the CIE chromaticity diagram. Finally, the designed LHS has been successfully utilized as a source of unconventional energy generation with much-enhanced yields compared to the individual components. So, the unique design strategy of the atom-precise nanocluster based-LHS may surely open up a new direction for nanocluster research in sustainable energy generation.Experimental section
Synthesis of Ag16 NC
An equimolar (0.1 mmol) mixture of [AgS-Adm]n complex and CF3COOAg was treated in 3 mL DMF at room temperature for 15 min. Then, 18 mg PPh4Cl was added to the clear mixture and continued stirring for a further 30 min. A white color precipitate was formed which was separated by centrifugation. The clear solution mixture was kept in a dark environment for crystallization. After 3 days, colorless crystals were obtained at room temperature.The [AgS-Adm]n complex was prepared by a reported method where an equivalent amount of AgNO3 was treated with AdmSH and Et3N in methanol.
Synthesis of Ag16 ∩ β-CDn (where n = 1, 2)
After dissolving Ag16 NC (3 mg) in 4 mL DCM, a 2 mL aqueous solution of β-CD was added in such a way that the desired Ag16/β-CD ratio was preserved in the mixture (1:1 for Ag16 ∩ β-CD1 and 1:2, 1:3, 1:4, 1:5, and 1:8 for Ag16 ∩ β-CD2). Then, the above solution mixture was carefully kept under sonication for 20 min. Further, the solution mixture was stirred for 40 min with intermittent sonication for 3 min at 10 min intervals. After separating the aqueous phase, the organic phase was dried by a rotatory evaporator.
Data availability
The ESI† contains experimental details, crystal structure refinement parameters, binding energies and emission properties of the host–guest inclusion complexes, QTAIM molecular plot of the Ag16 NC, RDG isosurface of argentophilic interaction, different facets of the Ag16 skeleton, different layers of the Ag16 skeleton, relation between the top and bottom square facets, presence of the lattice DMF molecule in the lattice of the Ag16 NC, positive mode ESI-MS data of the Ag16 NC, 1H NMR spectrum in CDCl3 of Ag16 NC, 19F NMR spectrum in CDCl3 of the Ag16 NC, PXRD, EDS analysis of the Ag16 NC, deconvoluted XPS spectra of each element of the Ag16 NC, UV-vis spectrum of the Ag16 NC, 1H NMR spectra in CDCl3 of the β-CD attached Ag16 NC and pure β-CD, UV-vis absorbance spectra of the Ag16 NC and the β-CD functionalized Ag16 NC, CV, DPV of the Ag16 NC and the β-CD functionalized Ag16 NC, partial positive mode ESI-MS data of the Ag16 ∩ β-CD12+, partial positive mode ESI-MS data of the Ag16 ∩ β-CD22+, numbering of the possible β-CD attachment sites on the Ag16 NC, binding energy calculation of the tail and head configuration of β-CD attachment on the stable S4 position, PL emission of the organic dye β-carotene, zeta potential data of the donor and acceptor, UV-visible spectra of Ag16 ∩ β-CD2 with the gradual addition of β-carotene, 1H NMR spectra in CDCl3 of the β-CD attached Ag16 NC with β-carotene and pure β-carotene, optimized distance between the donor and acceptor, emission lifetime data of Ag16 ∩ β-CD2 and Ag16 ∩ β-CD2 + β-carotene, fluorescence intensity of the donor–acceptor system to calculate the antenna effect, photocurrent from the donor–acceptor light-harvesting system, 2-D contour plot for differentiating the photocurrent generation, and references.Author contributions
A. K. D. and S. B. performed the synthesis, characterization and data interpretation of the nanocluster and light harvesting system. S. S. M. and B. P. performed the theoretical calculation. S. K. M. was involved in manuscript preparation. All authors discussed the results and were involved in manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. B. acknowledges the NPDF fund from SERB (PDF/2020/001085).References
- G. McDermott, S. Prince, A. Freer, A. Hawthornthwaite-Lawless, M. Papiz, R. Cogdell and N. Isaacs, Nature, 1995, 374, 517–521 CrossRef CAS.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. Van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- H.-Q. Peng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Rev., 2015, 115, 7502–7542 CrossRef CAS PubMed.
- A. H. Proppe, Y. C. Li, A. Aspuru-Guzik, C. P. Berlinguette, C. J. Chang, R. Cogdell, A. G. Doyle, J. Flick, N. M. Gabor and R. van Grondelle, Nat. Rev. Mater., 2020, 5, 828–846 CrossRef CAS.
- A. Kumar, R. Saha and P. S. Mukherjee, Chem. Sci., 2021, 12, 5319–5329 RSC.
- Y. Li, S. S. Rajasree, G. Y. Lee, J. Yu, J.-H. Tang, R. Ni, G. Li, K. N. Houk, P. Deria and P. J. Stang, J. Am. Chem. Soc., 2021, 143, 2908–2919 CrossRef CAS PubMed.
- M. Hao, G. Sun, M. Zuo, Z. Xu, Y. Chen, X. Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 10095–10100 CrossRef CAS PubMed.
- J. J. Li, Y. Chen, J. Yu, N. Cheng and Y. Liu, Adv. Mater., 2017, 29, 1701905 CrossRef PubMed.
- D. Zhang, W. Yu, S. Li, Y. Xia, X. Li, Y. Li and T. Yi, J. Am. Chem. Soc., 2021, 143, 1313–1317 CrossRef CAS PubMed.
- S. Kundu and A. Patra, Chem. Rev., 2017, 117, 712–757 CrossRef CAS PubMed.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- Y. Zeng, Y. Li, M. Li, G. Yang and Y. Li, J. Am. Chem. Soc., 2009, 131, 9100–9106 CrossRef CAS PubMed.
- F. Biedermann, E. Elmalem, I. Ghosh, W. M. Nau and O. A. Scherman, Angew. Chem., 2012, 124, 7859–7863 CrossRef.
- K. Liu, Y. Liu, Y. Yao, H. Yuan, S. Wang, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8285–8289 CrossRef CAS PubMed.
- Y. Yao, X. Chi, Y. Zhou and F. Huang, Chem. Sci., 2014, 5, 2778–2782 RSC.
- Z. Xu, S. Peng, Y. Y. Wang, J. K. Zhang, A. I. Lazar and D. S. Guo, Adv. Mater., 2016, 28, 7666–7671 CrossRef CAS PubMed.
- C.-L. Sun, H.-Q. Peng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Commun., 2018, 54, 1117–1120 RSC.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- J. Yang and R. Jin, ACS Mater. Lett., 2019, 1, 482–489 CrossRef CAS.
- Y. Jin, C. Zhang, X.-Y. Dong, S.-Q. Zang and T. C. Mak, Chem. Soc. Rev., 2021, 50, 2297–2319 RSC.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019, 120, 526–622 CrossRef PubMed.
- K. D. M. Weerawardene, H. Häkkinen and C. M. Aikens, Annu. Rev. Phys. Chem., 2018, 69, 205–229 CrossRef CAS PubMed.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 17, 2005328 CrossRef CAS PubMed.
- H. Hirai, S. Ito, S. Takano, K. Koyasu and T. Tsukuda, Chem. Sci., 2020, 11, 12233–12248 RSC.
- Q. Yao, Z. Wu, Z. Liu, Y. Lin, X. Yuan and J. Xie, Chem. Sci., 2021, 12, 99–127 RSC.
- I. Chakraborty, W. Kurashige, K. Kanehira, L. Gell, H. Häkkinen, Y. Negishi and T. Pradeep, J. Phys. Chem. Lett., 2013, 4, 3351–3355 CrossRef CAS PubMed.
- M. Ganguly, A. Pal, Y. Negishi and T. Pal, Langmuir, 2013, 29, 2033–2043 CrossRef CAS PubMed.
- Q.-M. Wang, Y.-M. Lin and K.-G. Liu, Acc. Chem. Res., 2015, 48, 1570–1579 CrossRef CAS PubMed.
- Z. Wang, R. K. Gupta, G. G. Luo and D. Sun, Chem. Rec., 2020, 20, 389–402 CrossRef CAS PubMed.
- S. Biswas, A. K. Das, A. C. Reber, S. Biswas, S. Bhandary, V. B. Kamble, S. N. Khanna and S. Mandal, Nano Lett., 2022, 22, 3721–3727 CrossRef CAS PubMed.
- D. Sun, H. Wang, H.-F. Lu, S.-Y. Feng, Z.-W. Zhang, G.-X. Sun and D.-F. Sun, Dalton Trans., 2013, 42, 6281–6284 RSC.
- S. Yuan, Y.-K. Deng, X.-P. Wang and D. Sun, New J. Chem., 2013, 37, 2973–2977 RSC.
- K. Zhou, C. Qin, X.-L. Wang, K.-Z. Shao, L.-K. Yan and Z.-M. Su, CrystEngComm, 2014, 16, 7860–7864 RSC.
- M. J. Alhilaly, R.-W. Huang, R. Naphade, B. Alamer, M. N. Hedhili, A.-H. Emwas, P. Maity, J. Yin, A. Shkurenko, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2019, 141, 9585–9592 CrossRef CAS PubMed.
- E. Khatun, A. Ghosh, P. Chakraborty, P. Singh, M. Bodiuzzaman, P. Ganesan, G. Nataranjan, J. Ghosh, S. K. Pal and T. Pradeep, Nanoscale, 2018, 10, 20033–20042 RSC.
- X. Kang and M. Zhu, Coord. Chem. Rev., 2019, 394, 1–38 CrossRef CAS.
- A. Nag and T. Pradeep, ACS Nanosci. Au, 2022, 2, 160–178 CrossRef CAS.
- M. A. Ganayee, C. Manju, W. A. Dar, B. Mondal and T. Pradeep, Ind. Eng. Chem. Res., 2020, 59, 12737–12744 CrossRef CAS.
- T. Jiang, G. Qu, J. Wang, X. Ma and H. Tian, Chem. Sci., 2020, 11, 3531–3537 RSC.
- A. Mathew, G. Natarajan, L. Lehtovaara, H. Hakkinen, R. M. Kumar, V. Subramanian, A. Jaleel and T. Pradeep, ACS Nano, 2014, 8, 139–152 CrossRef CAS PubMed.
- A. Nag, P. Chakraborty, A. Thacharon, G. Paramasivam, B. Mondal, M. Bodiuzzaman and T. Pradeep, J. Phys. Chem. C, 2020, 124, 22298–22303 CrossRef CAS.
- C. Yan, C. Liu, H. Abroshan, Z. Li, R. Qiu and G. Li, Phys. Chem. Chem. Phys., 2016, 18, 23358–23364 RSC.
- M. A. H. Muhammed, L. K. Cruz, A. H. Emwas, A. M. El-Zohry, B. Moosa, O. F. Mohammed and N. M. Khashab, Angew. Chem., 2019, 131, 15812–15817 CrossRef.
- H.-H. Deng, X.-Q. Shi, F.-F. Wang, H.-P. Peng, A.-L. Liu, X.-H. Xia and W. Chen, Chem. Mater., 2017, 29, 1362–1369 CrossRef CAS.
- L. Ren, P. Yuan, H. Su, S. Malola, S. Lin, Z. Tang, B. K. Teo, H. Häkkinen, L. Zheng and N. Zheng, J. Am. Chem. Soc., 2017, 139, 13288–13291 CrossRef CAS PubMed.
- A. K. Das, S. Biswas, A. Thomas, S. Paul, A. S. Nair, B. Pathak, M. S. Singh and S. Mandal, Mater. Chem. Front., 2021, 5, 8380–8386 RSC.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. K. Das, S. Biswas, S. S. Manna, B. Pathak and S. Mandal, Inorg. Chem., 2021, 60, 18234–18241 CrossRef CAS PubMed.
- A. K. Das, S. Maity, T. Sengupta, D. Bista, A. C. Reber, A. Patra, S. N. Khanna and S. Mandal, J. Phys. Chem. Lett., 2021, 12, 2154–2159 CrossRef CAS PubMed.
- M. Mohamadhoseini and Z. Mohamadnia, Coord. Chem. Rev., 2021, 432, 213711 CrossRef CAS.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- R. C. Powell and Z. G. Soos, J. Lumin., 1975, 11, 1–45 CrossRef CAS.
- S. Crooker, J. Hollingsworth, S. Tretiak and V. I. Klimov, Phys. Rev. Lett., 2002, 89, 186802 CrossRef CAS PubMed.
- S. Halivni, A. Sitt, I. Hadar and U. Banin, ACS Nano, 2012, 6, 2758–2765 CrossRef CAS PubMed.
- A. J. Mork, M. C. Weidman, F. Prins and W. A. Tisdale, J. Phys. Chem. C, 2014, 118, 13920–13928 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2169435. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02786k |
| ‡ Shows the equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |