
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atom and step economical synthesis of acyclic quaternary centers via iridium-catalyzed hydroarylative cross-coupling of 1,1-disubstituted alkenes†
Phillippa
Cooper
a,
Andrew G.
Dalling
a,
Elliot H. E.
Farrar
 b,
Timothy P.
Aldhous
ad,
Simon
Grélaud
a,
Eleanor
Lester
a,
Lyman J.
Feron
c,
Paul D.
Kemmitt
c,
Matthew N.
Grayson
*b and
John F.
Bower
*d
b,
Timothy P.
Aldhous
ad,
Simon
Grélaud
a,
Eleanor
Lester
a,
Lyman J.
Feron
c,
Paul D.
Kemmitt
c,
Matthew N.
Grayson
*b and
John F.
Bower
*d
aSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: M.N.Grayson@bath.ac.uk
cMedicinal Chemistry, Oncology, IMED Biotech Unit, AstraZeneca, Cambridge, UK
dDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: john.bower@liverpool.ac.uk
First published on 13th September 2022
Abstract
Quaternary benzylic centers are accessed with high atom and step economy by Ir-catalyzed alkene hydroarylation. These studies provide unique examples of the use of non-polarized 1,1-disubstituted alkenes in branch selective Murai-type hydro(hetero)arylations. Detailed mechanistic studies have been undertaken, and these indicate that the first irreversible step is the demanding alkene carbometallation process. Structure-reactivity studies show that the efficiency of this is critically dependent on key structural features of the ligand. Computational studies have been undertaken to rationalize this experimental data, showing how more sterically demanding ligands reduce the reaction barrier via predistortion of the reacting intermediate. The key insight disclosed here will underpin the ongoing development of increasingly sophisticated branch selective Murai hydroarylations.
Introduction
It is well appreciated that the synthesis of acyclic quaternary centers is challenging. The development of step and atom economical methods, especially those that can be adapted to enantioselective settings, represents an enduring challenge.1 Aryl substituted quaternary centers are especially important as they are key motifs in a variety of bioactive compounds (Scheme 1A).2 A conceptually ideal framework for accessing these subunits involves Friedel–Crafts-type addition of an aryl C–H bond across a 1,1-disubstituted alkene; however, this approach is notoriously problematic, offers limited scope and is not well suited to enantioselective protocols (Scheme 1B, eqn (1)).3 These limitations have stimulated the development of other methods that require prefunctionalized reactants. For example, “formal” (non-enantioselective) alkene hydroarylations can be achieved through the use of a reductant and an aryl halide.4 Outside of allylic substitution5 and conjugate addition reactions,6 the most prominent asymmetric arylative protocols for accessing acyclic quaternary centers rely on the oxidative coupling of α-tertiary boronic esters with aryl lithium reagents (eqn (2)).7 Ni-catalyzed cross-couplings of α-tertiary benzylic acetates with aryl boronic esters offer a powerful alternative (eqn (3)).8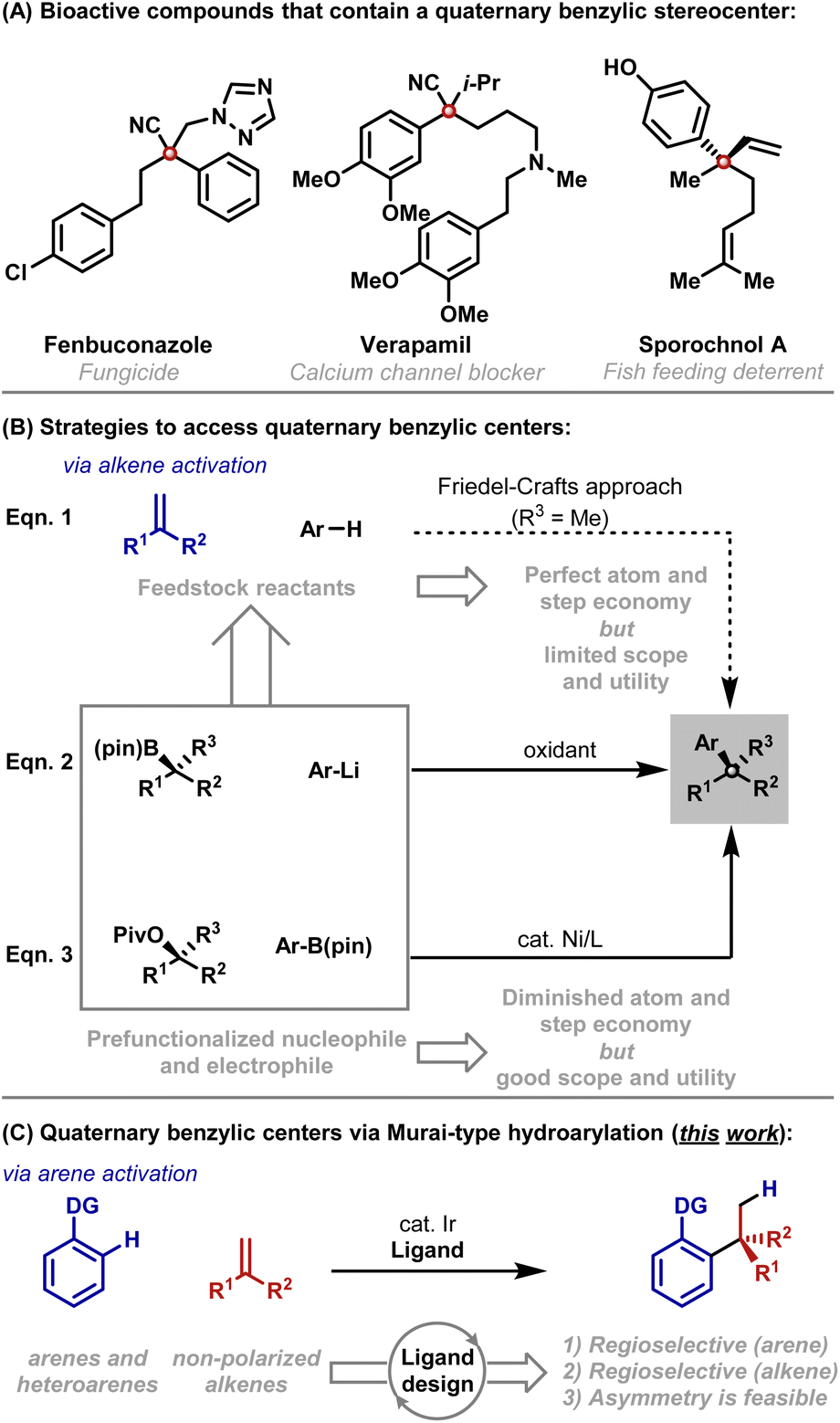 | ||
| Scheme 1 Introduction. | ||
As outlined above, state of the art cross-coupling methods require substantial levels of prefunctionalization, and the desired stereochemistry is established in an earlier step, thereby detracting from atom and step economy. Consequently, we considered whether the Friedel–Crafts approach might be re-engineered to offer broader scope (e.g. a wider electronic range) and greater control (e.g. predictable mono-alkylation and regiocontrol). This would then provide a viable method where feedstock reactants are used directly, and C–C bond formation and stereocenter creation are united.9,10 To achieve this, we envisaged initiating the reaction by metal-catalyzed C–H activation of the arene rather than by Friedel–Crafts-type activation of the alkene (Scheme 1C). Murai hydroarylations of this type are extremely rare,11 with Ellman's Rh-catalyzed method for the branch selective hydroheteroarylation of methyl methacrylate with specific classes of N-heteroarene being the most significant development so far.12 The paucity of methods reflects wider difficulties in developing intermolecular branch selective Murai-hydroarylations.11b–d Indeed, broad scope protocols involving non-polarized mono-substituted alkenes have only been developed recently,13 whereas processes involving non-polarized 1,1-disubstituted alkenes have remained elusive. The key impediment lies in designing catalysts that can tolerate sterically demanding alkenes, and, at the same time, exert threefold control of (1) C–H bond activation regioselectivity, (2) branched![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :linear regioselectivity11b and (3) enantioselectivity.11c In this report, we outline efforts to address these objectives, which have resulted in an efficient C–H activation protocol for the hydro(hetero)arylation of non-polarized 1,1-disubstituted alkenes. Our results make a significant contribution to the area of Murai-type hydroarylations,11c provide an enabling cross-coupling methodology and set the stage for the development of an enantioselective method.
:linear regioselectivity11b and (3) enantioselectivity.11c In this report, we outline efforts to address these objectives, which have resulted in an efficient C–H activation protocol for the hydro(hetero)arylation of non-polarized 1,1-disubstituted alkenes. Our results make a significant contribution to the area of Murai-type hydroarylations,11c provide an enabling cross-coupling methodology and set the stage for the development of an enantioselective method.
Results and discussion
Reaction development and scope
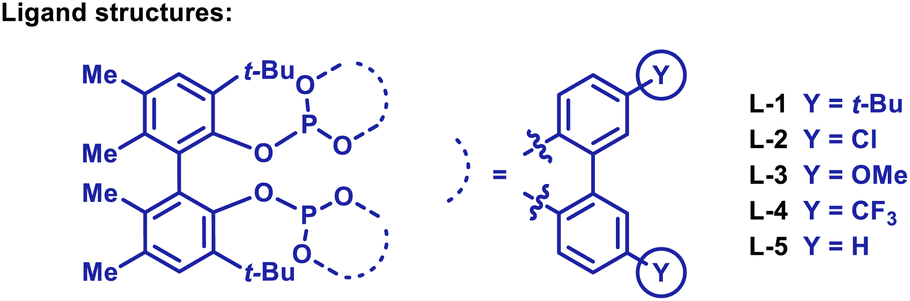
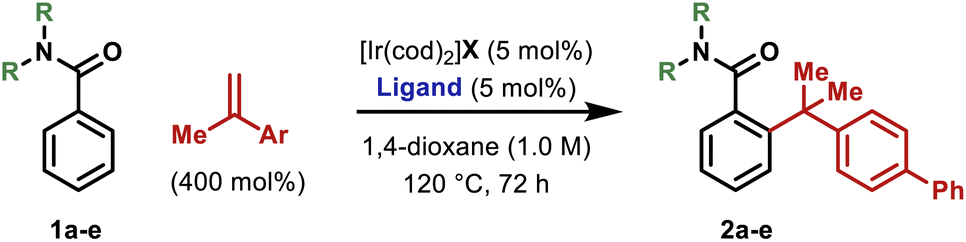
We have previously developed an Ir-system modified with bisphosphite ligand L-1 for the enantioselective hydroarylative cross-coupling of anilide ortho-C–H bonds with monosubstituted alkenes.13,14 These processes are highly efficient and this prompted us to explore hydroarylations of much more demanding 1,1-disubstituted alkenes. Initially, we focused on generating 2a by benzamide directed cross-coupling of 1a with p-phenyl-α-methylstyrene (Table 1). Remarkably, use of [Ir(cod)2]BARF/L-1 provided 2a in 68% yield and, importantly, with >20:1 branched:linear (B:L) selectivity (entry 1). As part of extensive optimization efforts, we varied the electronics of the flanking biaryl units of the ligand. Replacement of the t-Bu groups of L-1 with chloride, methoxy or trifluoromethyl groups (L2–4) resulted in lower yields (entries 2–4). Pleasingly, L-5 (Y = H) was effective, and this allowed 2a to be isolated in 73% yield (entry 5). Higher catalyst loadings led to diminished yields and 1,4-dioxane was found to be the optimal solvent.15 Ir-precatalysts possessing more strongly coordinating counterions (e.g. triflate) were less effective (entry 6). Replacement of the N,N-diethyl groups of 1a with other substituents gave targets 2b–e in lower yields (entries 7–10). The attempted use of weaker 6-membered anilide-based chelates or stronger 5-membered 2-pyridyl-based chelates was not successful. As discussed later, reaction efficiency is specifically dependent on the structural features of the central biaryl unit of L-5, such that we have been unable to identify commercially available ligands that offer comparable efficiencies. Additionally, dFppb, a ligand developed previously for branch selective hydroarylations of monosubstituted alkenes,16 is ineffective.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | X | Ligand | B:L selectivity |
Yield (%)a |
| a NMR yield. b Isolated yield. | |||||
| 1 | Et (1a) | BARF | L-1 | >20:1 |
68 |
| 2 | Et | BARF | L-2 | >20:1 |
55 |
| 3 | Et | BARF | L-3 | >20:1 |
64 |
| 4 | Et | BARF | L-4 | >20:1 |
37 |
| 5 | Et | BARF | L-5 |
>20:1 |
75 (73) |
| 6 | Et | OTf | L-5 | >20:1 |
44 |
| 7 | Me (1b) | BARF | L-5 | >20:1 |
48 |
| 8 | –C4H8– (1c) | BARF | L-5 | >20:1 |
51 |
| 9 | –C5H10– (1d) | BARF | L-5 | >20:1 |
31 |
| 10 | i-Pr (1e) | BARF | L-5 | >20:1 |
<10 |
|
|||||
As outlined in Table 2, the optimized conditions offer broad scope. Hydroarylation of electronically diverse α-substituted styrenes with benzamide 1a delivered 2f–i in high yields (Table 2A). In each case, complete branch selectivity was observed and only mono-ortho-alkylation of the arene occurred. The latter is consistent with initial monoalkylation causing the directing group to twist from the plane of the arene, such that alkylation of the remaining ortho-position cannot occur. Significantly, the process extends to aliphatic alkenes; for example, 2j was accessed in 57% yield. Substitution at the meta- and para-positions of the benzamide is tolerated (2k–m). For 2l, C–C bond formation was highly selective for the less hindered ortho-position, and this presumably reflects the steric demands of L-5. Very electron poor arenes (e.g. R1 = p-NO2) are not suitable, perhaps because these systems are less effective at stabilizing the cyclometallated Ir(III)-intermediate (vide infra).
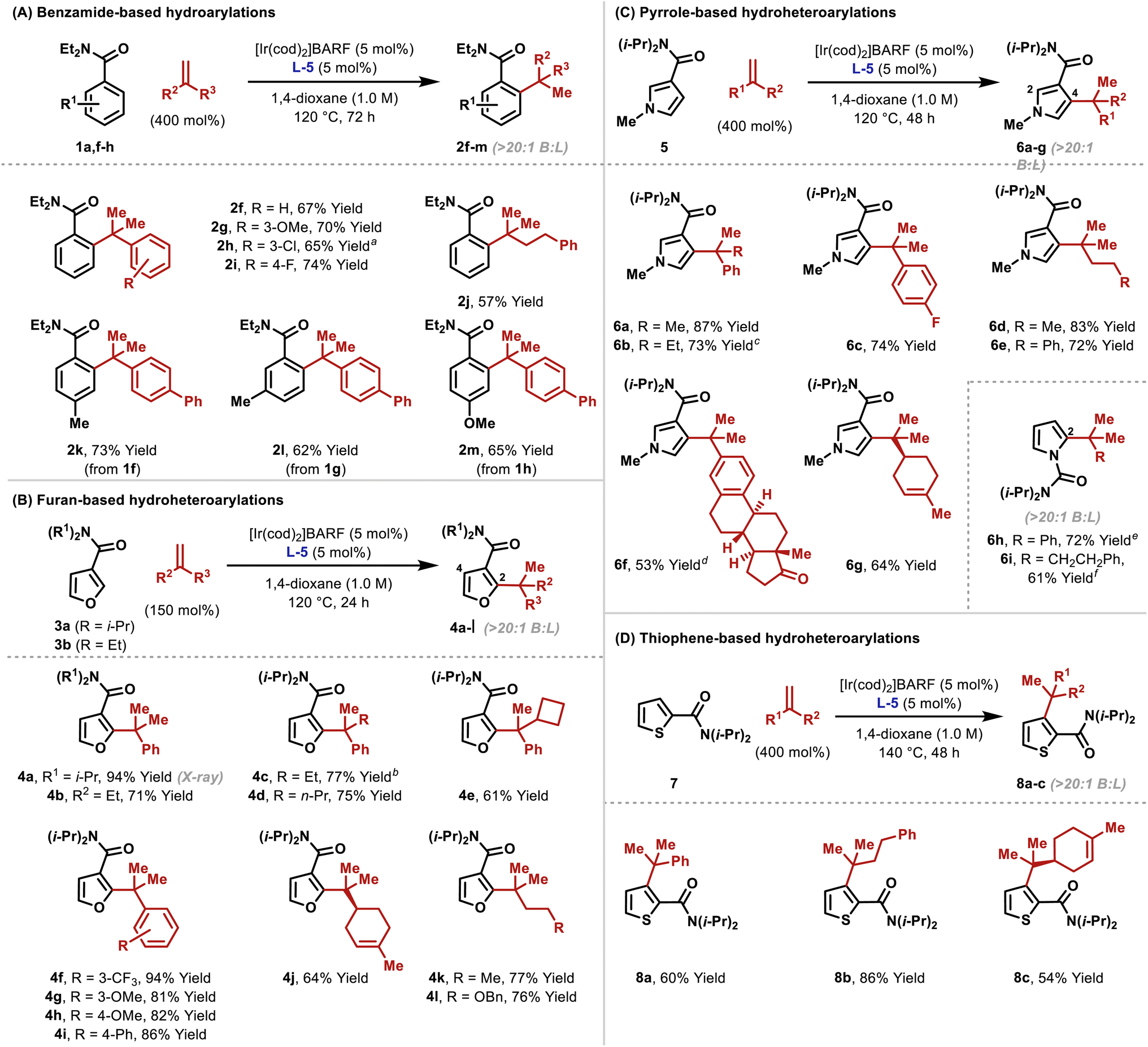
To highlight complementarity to Friedel–Crafts alkylations, we evaluated hydroheteroarylations using electron rich 5-membered heteroaromatics. Cross-coupling of furans 3a–b with α-methylstyrene revealed that an N,N-diisopropyl directing group is more efficient than an N,N-diethyl variant (Table 2B). In both cases, alkylation was observed at the furan C2 position only, which likely reflects an electronic preference. Quaternary benzylic stereocenters could be installed (4c–e), and electronically diverse styrenes underwent efficient hydroheteroarylation to afford furans 4f–i. The cross-coupling extended to alkenes where the R2 and R3 groups are both aliphatic, to give, for example, 4j–l. For the former, the generation of a quaternary center adjacent to a tertiary center is notable, especially as complete branch selectivity was maintained and competing hydroheteroarylation of the trisubstituted alkene was not observed. To validate the prospects of an enantioselective protocol, we prepared L-5 as a single enantiomer, and this provided 4c in 60:40 e.r. Further improvements are clearly required, and this aspect will be facilitated by the mechanistic studies outlined later.
To the best of our knowledge, examples of pyrroles participating in intermolecular branch selective Murai-type processes have not been reported. Consequently, we were pleased to find that the protocol extended to pyrroles possessing directing groups at C3. Hydroheteroarylation of α-methyl and α-ethyl styrene with pyrrole 5 afforded products 6a and 6b in 87% and 73% yield, respectively, and with high selectivity for the C4 position (Table 2C). Here, steric effects of the N-Me group likely disfavor bond formation at C2 (cf.Table 2B). As with furan-based processes, electronically distinct styrenes and non-activated aliphatic alkenes are viable reaction partners (6c–g). The directing group can also be placed on the pyrrole nitrogen, and this allowed the generation of C2-alkylated pyrroles 6h–i. These results validate the use of a distinct pyrrole unit and demonstrate that the method offers a true alternative to problematic Friedel–Crafts reactions of (acid sensitive) pyrroles.
To explore scope further, we examined hydroheteroarylations with thiophene 7, which possesses a C2 directing group (Table 2D). Although this substrate required a higher reaction temperature (140 °C vs. 120 °C for other processes), both styrenic and “all-aliphatic” 1,1-disubstituted alkenes participated to provide 8a–c in 54–86% yields. As with 6g, the trisubstituted alkene associated with 8c did not undergo hydroheteroarylation.
Mechanistic studies
A series of experiments support the working mechanistic hypothesis in Scheme 2D. Hydroheteroarylation of deuterio-9 with furan 3a resulted in deuterium transfer to C4–H of deuterio-4i, and scrambling in recovered deuterio-9′ (Scheme 2A). These results indicate (a) that ortho-C–H activation is reversible and non-selective for C4–H vs. C2–H, (b) that reversible alkene hydrometallation is operative (III to IV and V) and (c) that the alkene isomerizes under the reaction conditions. Based on these data, C–C bond formation could occur either via C–C reductive elimination from V or via carbometallation from III. To distinguish these options, natural abundance 13C KIE experiments were undertaken on alkene 9 using the Singleton method, which establishes which carbon centers are involved in the first irreversible step.17 This revealed significant KIEs at both C2 (1.013) and C1 (1.035), as well as a large inverse KIE at C3 (0.981) (Scheme 2B). The KIEs were determined by integration of 13C NMR data for recovered versus starting alkene. In this case, this provides an enhanced KIE at C1 and a diminished KIE at C3, because the starting alkene is depleted in carbon-13 at C1 relative to C3 owing to KIEs that are inherent to the method used for its synthesis.18 Under the hydroheteroarylation conditions, alkene isomerization between C1–C2 and C2–C3 occurs, such that a (more) even carbon-13 distribution is established. To confirm this, alkene 9 was exposed to the reaction conditions in the absence of arene 3a; integration of the 13C NMR spectra of recovered 9 revealed a relative enhancement at C1 (1.020) and a depletion at C3 (0.984), with the average approximating unity as expected (Scheme 2C). In Scheme 2B, the sum of the KIE enhancements at C1 and C3 is 0.016; if this is derived solely from the terminal alkene carbon during C–C bond formation, then a hypothetical “isomerization-free” KIE value for this center can be approximated as 1.016, which is similar to the value obtained for C2.19 Accordingly, C–C bond formation likely occurs via irreversible alkene carbometallation from III (both C-centers involved), and the large inverse KIE at C3 in Scheme 2B is an artefact of both the method used for alkene synthesis and alkene isomerization under the reaction conditions. The assertion that carbometallation is irreversible contrasts earlier work with monosubstituted alkenes,13 and likely reflects the increased steric demands of 1,1-disubstituted variants. Branched selectivity is presumably favored during carbometallation because the bulky Ir-center ends up at the less hindered position. The processes described here require 5-membered amide-based chelates, which are presumably sufficiently tractable to be intercepted by the hindered alkene. Weaker 6-membered anilide-based chelates are not suitable.13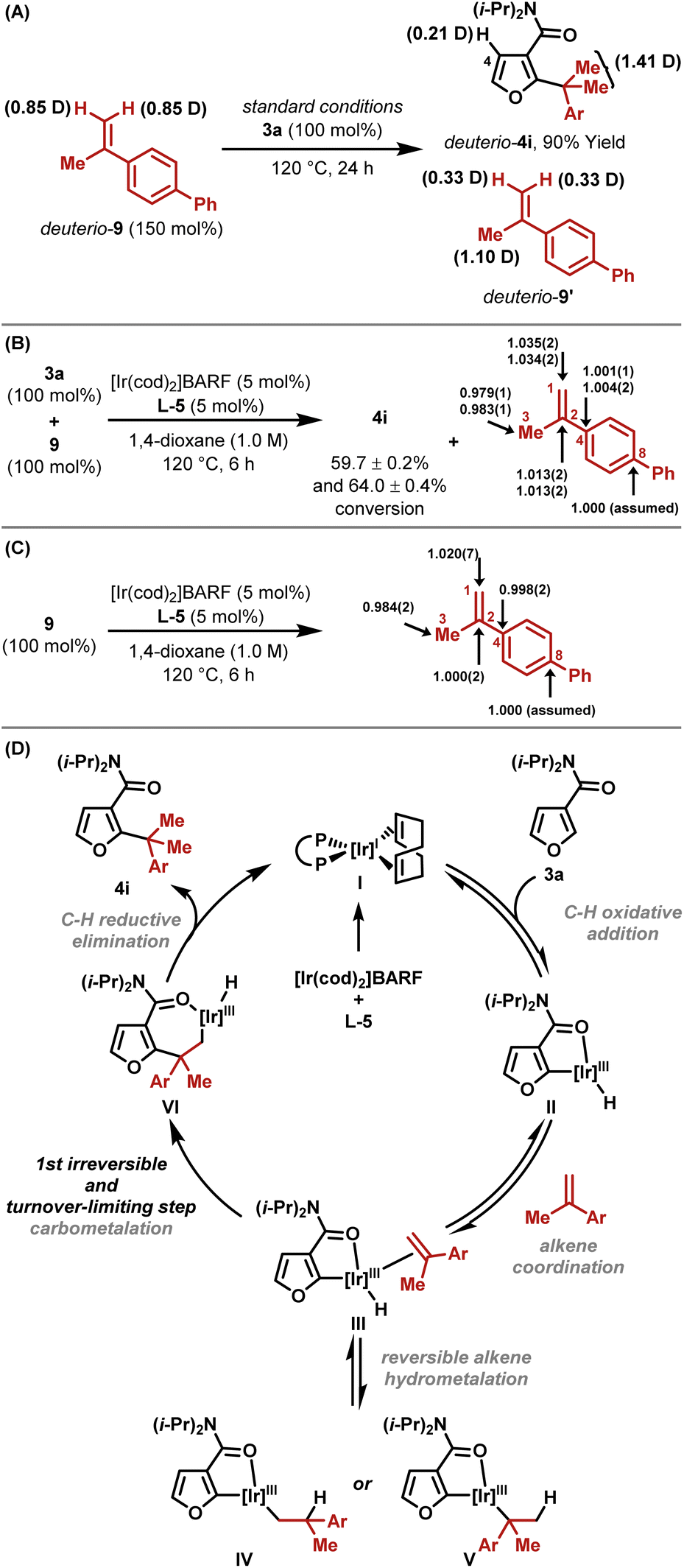 | ||
| Scheme 2 Mechanistic studies. | ||
Ligand effects and computational studies
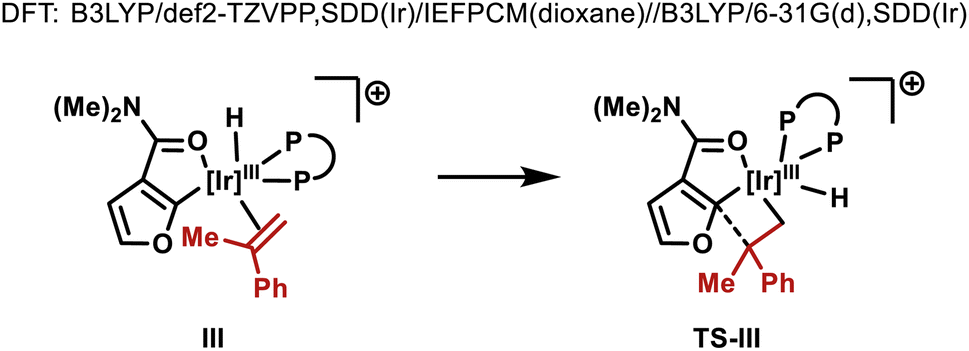
As mentioned earlier, the structural features of the central biaryl unit of L-5 are critical for efficient reactivity. To probe this, structurally varied ligands L-6–8 were synthesized and compared for the hydroheteroarylation of 9 with 3a (Table 3). L-5 (R1 = Me, R2 = t-Bu) affords 4i in 86% yield (entry 1). Reducing the size of the R2 substituent (L-6, R1 = R2 = Me) decreased the yield slightly (76%), whereas removal of this unit (L-7) significantly lowered the yield (19%) (entry 2–3). Removal of the R1 substituent (L-8, R1 = H, R2 = H) had a less significant effect (entry 4). NMR profiling indicated that the yields are reflected in the reaction rates (see the ESI†).|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Yield of 4i | Rel. barrier to TS-III (ΔΔG; kcal mol−1) | δ θ III to TS-III (°) |
| 1 | L-5 | 86% | 0 | −9.2 |
| 2 | L-6 | 76% | 2.5 | −7.8 |
| 3 | L-7 | 19% | 5.7 | 13.9 |
| 4 | L-8 | 12% | 6.5 | 30.5 |
|
||||
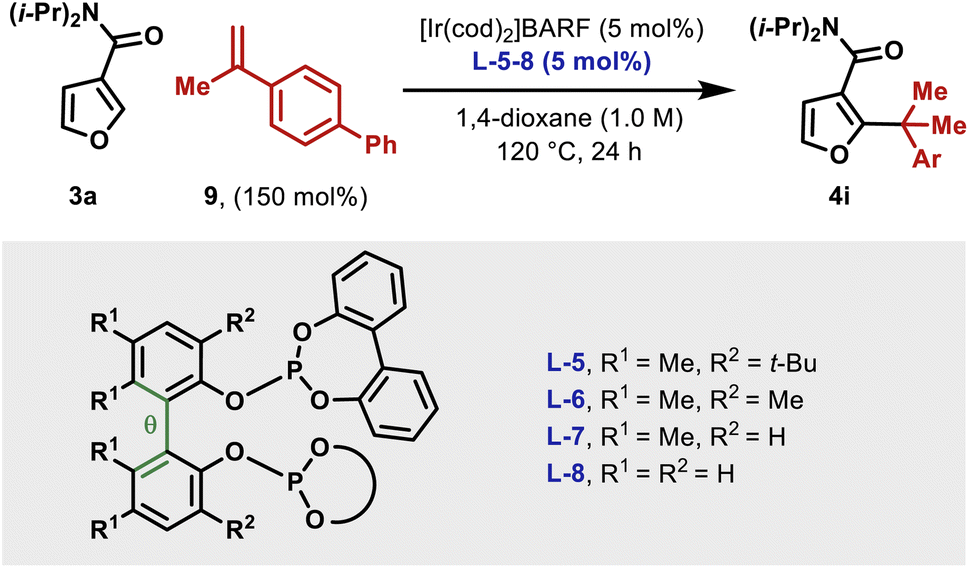
Given the observed significance of the R2 substituent, the turnover-limiting carbometallation event (III to TS-III) was modelled with DFT using Gaussian 16 (full details are given in the ESI†).20 A thorough conformational analysis was performed to identify the lowest energy conformations of III-L-5–8 and TS-III-L-5–8; as expected, their energies were found to be significantly lower with the amide oxygen coordinated to the Ir(III)-center. The lowest energy conformations of each species and the relative barriers to transition state TS-III from intermediate III are provided in Scheme 3A. The relative barriers show an increase upon progression from L-5 to L-8, which correlates with the trend in yield for 4i. As shown by superimposing the transition states of type TS-III for ligands L-5-8, the Ir(III) reaction center is nearly identical for each ligand (Scheme 3B). However, there are differences in the dihedral angle (labelled θ in Scheme 3A and Table 3) between the conformation of ligands L-5–8 in each of the transition states. The difference in the dihedral angle between intermediates of type III and transition states TS-III has an approximately linear relationship with the relative reaction barrier for carbometallation, and hence the yield of furan 4i. When ligands with more sterically demanding substituents are used, this difference is smaller. As a result, the energy required to distort the intermediate into its transition state geometry, a principal contributor to the reaction barrier, decreases.21 Thus, the barrier to carbometallation is determined primarily by the extent to which the conformation of the ligand is predistorted in intermediate III.
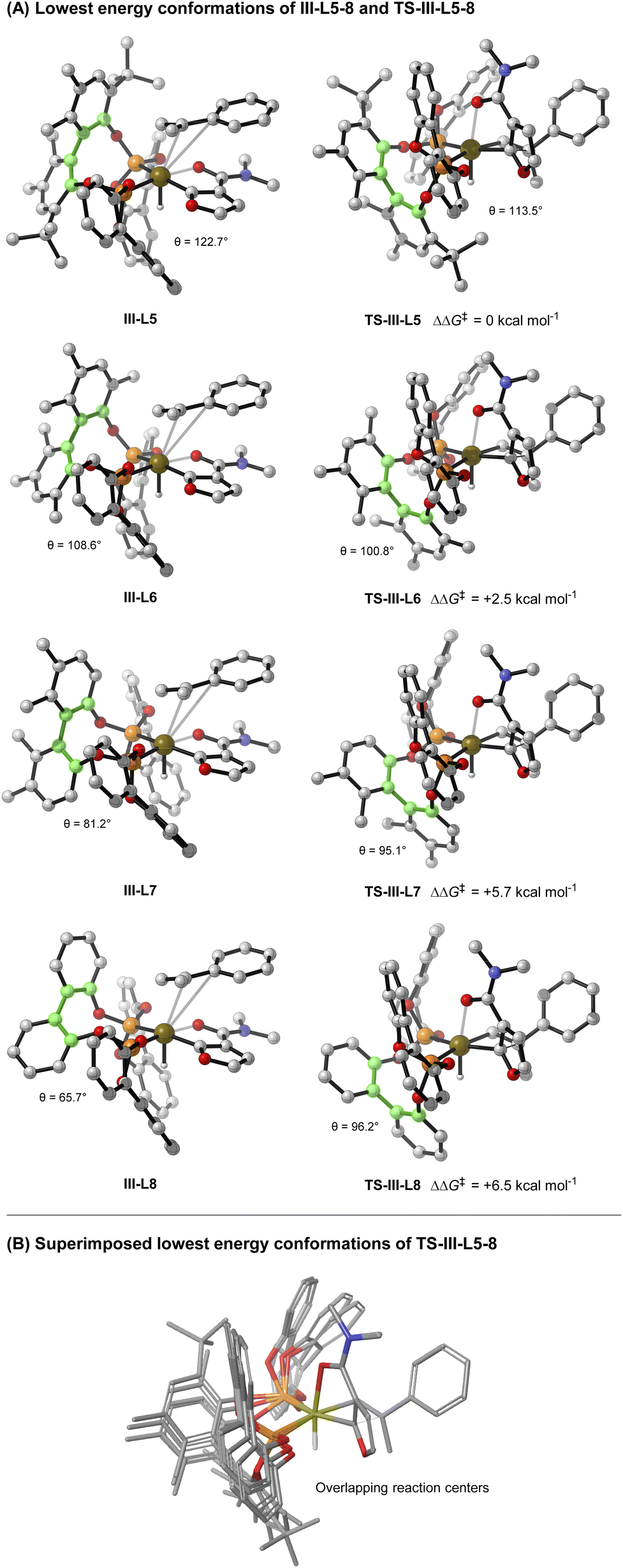 | ||
| Scheme 3 Computational studies (B3LYP/def2-TZVPP,SDD(Ir)/IEFPCM(dioxane)//B3LYP/6-31G(d),SDD(Ir)). Green highlighted atoms indicate measured dihedral angle θ. | ||
Conclusions
In summary, we report unique examples of branch selective Murai-type hydro(hetero)arylations of non-polarized 1,1-disubstituted alkenes. The chemistry is enabled by key structural features of L-5, a ligand that is accessed in just one step. Experimental trends in yield over a variety of ligands were examined by DFT calculations, which revealed how more sterically demanding ligands reduce the reaction barrier via predistortion of the structure of the reacting intermediate. The protocol provides a broad range of challenging quaternary benzylic centers in an atom and step economical manner, and addresses key problems associated with Friedel–Crafts alkylation (i.e. tolerance of electron poor arenes, mild conditions, mono-alkylation only, regioselective, high yields). Consequently, the method can be considered “enabling”, and so we anticipate that it will be of broad interest. Ongoing efforts towards (a) an enantioselective variant and (b) catalysts that can accommodate internal alkenes will be guided by the intriguing ligand design insights outlined here.Data availability
Compound characterisation data and Gaussian 16 output files for all computed structures are openly available in the ESI.†Author contributions
P. C., A. G. D, T. P. A., S. G. and E. L. performed the experimental work, which was directed by L. J. F., P. D. K. and J. F. B. The computational studies were conducted by E. H. E. F. and M. N. G. The manuscript was written by J. F. B. and M. N. G. with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Professor Barry Carpenter for insightful discussions. The Bristol Chemical Synthesis Centre for Doctoral Training, funded by the EPSRC (EP/L015366/1), EPSRC (EP/M507994/1) and AstraZeneca (studentships to P. C. and T. P. A.), the ERC (Grants 639594 and 863799), the Leverhulme Trust, the EPSRC DTP (EP/R513155/1, studentship to E. H. E. F.), the EPSRC (EP/W003724/1), and the University of Bath provided funding. The Balena High Performance Computing (HPC) Service at the University of Bath was used.Notes and references
- (a) J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564 CrossRef CAS; (b) T. Ling and F. Rivas, Tetrahedron, 2016, 72, 6729 CrossRef CAS; (c) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS PubMed; (d) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593 RSC.
- T. P. Stockdale and C. M. Williams, Chem. Soc. Rev., 2015, 44, 7737 RSC.
- (a) R. R. Naredla and D. A. Klumpp, Chem. Rev., 2013, 113, 6905 CrossRef CAS PubMed; (b) L. L. Anderson, J. Arnold and R. G. Bergman, J. Am. Chem. Soc., 2005, 127, 14542 CrossRef CAS; (c) G. Evano and C. Theunissen, Angew. Chem., Int. Ed., 2019, 58, 7202 CrossRef CAS.
- S. A. Green, S. Vásquez-Céspedes and R. A. Shenvi, J. Am. Chem. Soc., 2018, 140, 11317 CrossRef CAS PubMed.
- G. Helmchen, U. Kazmaier and S. Förster, Enantioselective Allylic Substitutions with Carbon Nucleophiles, in Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley & Sons, 3rd edn, Hoboken, NJ, 2010, pp. 497–641 Search PubMed.
- Review: (a) Selected examples: C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC; (b) R. Shintani, Y. Tsutsumi, M. Nagaosa, T. Nishimura and T. Hayashi, J. Am. Chem. Soc., 2009, 131, 13588 CrossRef CAS; (c) C. Hawner, D. Müller, L. Gremaud, A. Felouat, S. Woodward and A. Alexakis, Angew. Chem., Int. Ed., 2010, 49, 7769 CrossRef CAS PubMed; (d) J. A. Dabrowski, M. T. Villaume and A. H. Hoveyda, Angew. Chem., Int. Ed., 2013, 52, 8156 CrossRef CAS PubMed.
- (a) A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584 CrossRef CAS PubMed; (b) J. Llaveria, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10958 CrossRef CAS PubMed; (c) M. Odachowski, A. Bonet, S. Essafi, P. Conti-Ramsden, J. N. Harvey, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2016, 138, 9521 CrossRef CAS PubMed; (d) V. Ganesh, A. Noble and V. K. Aggarwal, Org. Lett., 2018, 20, 6144 CrossRef CAS PubMed; (e) C. M. Wilson, V. Ganesh, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 16318 CrossRef CAS.
- (a) Q. Zhou, K. M. Cobb, T. Tan and M. P. Watson, J. Am. Chem. Soc., 2016, 138, 12057 CrossRef CAS PubMed; (b) J. Xu, O. P. Bercher and M. P. Watson, J. Am. Chem. Soc., 2021, 143, 8608 CrossRef CAS PubMed.
- Enantioselective hydrovinylations fulfill these ideals in a complementary fashion: (a) W.-J. Shi, Q. Zhang, J.-H. Xie, S.-F. Zhu, G.-H. Hou and Q.-L. Zhou, J. Am. Chem. Soc., 2006, 128, 2780 CrossRef CAS PubMed; (b) A. Zhang and T. V. RajanBabu, J. Am. Chem. Soc., 2006, 128, 5620 CrossRef CAS; (c) C. R. Smith and T. V. Rajanbabu, Org. Lett., 2008, 10, 1657 CrossRef CAS PubMed; (d) C. R. Smith, H. J. Lim, A. Zhang and T. V. RajanBabu, Synthesis, 2009, 2089 CAS.
- Enantioselective redox relay Heck reactions of 1,1-disubstituted alkenes can access quaternary benzylic stereocenters, while retaining the alkene derived unsaturation: T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed.
- (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS; (b) G. E. M. Crisenza and J. F. Bower, Chem. Lett., 2016, 45, 2 CrossRef CAS; (c) T. P. Aldhous, R. W. M. Chung, A. G. Dalling and J. F. Bower, Synthesis, 2021, 53, 2961 CrossRef CAS . As outlined in ref. 11c, intramolecularization is an alternate strategy for enforcing branch selectivity and this has been used for the formation of quaternary centers. Selected examples: ; (d) B. Ye, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 507 CrossRef CAS; (e) T. Shibata, H. Kurita, S. Onoda and K. S. Kanyiva, Asian J. Org. Chem., 2018, 7, 1411 CrossRef CAS; (f) G. Li, Q. Liu, L. Vasamsetty, W. Guo and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 3475 CrossRef CAS PubMed; (g) A. Arribas, M. Calvelo, D. F. Fernández, C. A. B. Rodrigues, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2021, 60, 19297 CrossRef CAS PubMed.
- (a) G. Tran, K. D. Hesp, V. Mascitti and J. A. Ellman, Angew. Chem., Int. Ed., 2017, 56, 5899 CrossRef CAS PubMed; (b) For linear selective and enantioselective hydroheteroarylations of α-substituted acrylates, see: G. Tran, D. Confair, K. D. Hesp, V. Mascitti and J. A. Ellman, J. Org. Chem., 2017, 82, 9243 CrossRef CAS PubMed; (c) C. M. Fillouz and T. Rovis, J. Am. Chem. Soc., 2015, 137, 508 CrossRef.
- S. Grélaud, P. Cooper, L. J. Feron and J. F. Bower, J. Am. Chem. Soc., 2018, 140, 9351 CrossRef PubMed.
- For a review on iridium(I) catalyzed hydrocarbonations of unsaturated systems, see: D. F. Fernández, J. L. Mascareñas and F. López, Chem. Soc. Rev., 2020, 49, 7378 RSC.
- Under optimized conditions replacement of 1,4-dioxane with 1,2-DCB or PhMe generated 2a in 52% and 63% yield, respectively. Use of 7.5 mol% Ir/L-5 gave 2a in 45% yield.
- G. E. M. Crisenza, N. G. McCreanor and J. F. Bower, J. Am. Chem. Soc., 2014, 136, 10258 CrossRef CAS PubMed.
- (a) D. A. Singleton and A. A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357 CrossRef CAS; (b) L. K. Vo and D. A. Singleton, Org. Lett., 2004, 6, 2469 CrossRef CAS PubMed.
- Different methods for the synthesis of alkene 9 gave different carbon-13 ratios (see the ESI†).
- Under “isomerization-free” conditions, the KIE associated with C3 of 9 is expected to be negligible for both C–C bond forming mechanistic options. The information in Scheme 2C is indicative but the absolute values are not directly comparable to Scheme 2B because different batches of alkene 9 were used.
- M. J. Frisch, Gaussian 16, Revision A.03, 2016.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all compounds are provided. CCDC 2084176. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02790a |
| This journal is © The Royal Society of Chemistry 2022 |