
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical CO2 reduction catalyzed by atomically precise alkynyl-protected Au7Ag8, Ag9Cu6, and Au2Ag8Cu5 nanoclusters: probing the effect of multi-metal core on selectivity†
Xiaoshuang
Ma‡
a,
Fang
Sun‡
b,
Lubing
Qin
a,
Yonggang
Liu
a,
Xiongwu
Kang
 a,
Likai
Wang
c,
De-en
Jiang
d,
Qing
Tang
*b and
Zhenghua
Tang
*ae
a,
Likai
Wang
c,
De-en
Jiang
d,
Qing
Tang
*b and
Zhenghua
Tang
*ae
aNew Energy Research Institute, Guangdong Provincial Key Laboratory of Atmospheric Environment and Pollution Control, School of Environment and Energy, South China University of Technology, Guangzhou Higher Education Mega Centre, Guangdong 510006, China. E-mail: zhht@scut.edu.cn
bSchool of Chemistry and Chemical Engineering, Chongqing Key Laboratory of Theoretical and Computational Chemistry, Chongqing University, Chongqing, 401331, China. E-mail: qingtang@cqu.edu.cn
cSchool of Chemistry and Chemical Engineering, Shandong University of Technology, Shandong 255049, China
dDepartment of Chemistry, University of California, Riverside, CA 92521, USA
eGuangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications, Jinan University, Guangdong 510632, China
First published on 15th August 2022
Abstract
Doping metal nanoclusters (NCs) with another metal usually leads to superior catalytic performance toward CO2 reduction reaction (CO2RR), yet elucidating the metal core effect is still challenging. Herein, we report the systematic study of atomically precise alkynyl-protected Au7Ag8, Ag9Cu6, and Au2Ag8Cu5 NCs toward CO2RR. Au2Ag8Cu5 prepared by a site-specific metal exchange approach from Ag9Cu6 is the first case of trimetallic superatom with full-alkynyl protection. The three M15 clusters exhibited drastically different CO2RR performance. Specifically, Au7Ag8 demonstrated high selectivity for CO formation in a wide voltage range (98.1% faradaic efficiency, FE, at −0.49 V and 89.0% FE at −1.20 V vs. RHE), while formation of formate becomes significant for Ag9Cu6 and Au2Ag8Cu5 at more negative potentials. DFT calculations demonstrated that the exposed, undercoordinated metal atoms are the active sites and the hydride transfer as well as HCOO* stabilization on the Cu–Ag site plays a critical role in the formate formation. Our work shows that, tuning the metal centers of the ultrasmall metal NCs via metal exchange is very useful to probe the structure–selectivity relationships for CO2RR.
Introduction
The electrochemical CO2 reduction reaction (CO2RR) has been attracting increasing research efforts continuously, as it can convert CO2 into valuable fuels and balance the carbon cycle.1–4 So far, various metals including Au, Ag, Cu, etc. as catalytic materials have been investigated for CO2RR.5,6 Bimetallic or trimetallic catalysts usually exhibit superior catalytic performance than their homometallic counterparts due to the catalytic synergistic effects.7,8 To improve the catalytic efficiency and advance the fundamental mechanistic understanding, one of the major challenges is the polydispersity of the catalyst. Specifically, despite the size, morphology, composition, even the coordination environment seems to be uniform in bulk or at a large-scale dimension, it can't offer a homogeneous chemical environment at the atomic level, making it extremely challenging to profoundly elucidate the mechanism and establish the structure–function relationship.The emergence of atomically precise coinage metal nanoclusters (NCs) offers great opportunities to resolve the above problem due to their definitive size, morphology, composition, and more importantly, the crystallographically resolved structure can provide well-defined chemical environment to correlate the structure–performance relationship.9–16 Pioneering work has been extensively conducted on thiolate-protected bimetallic NCs. For instance, in an early study, Jin group discovered that, compared to homogold Au25 NC, monopalladium-doped Pd1Au24 NC can drastically inhibit the H2 evolution, and had much higher CO product selectivity (faradaic efficiency for CO, FECO = ∼100%) at high potentials.17 Zhuang et al. found that, compared with the parent Au44 NC, Au47Cd2(TBBT)31 (TBBT: 4-tert-butylbenzenelthiol) NC exhibited not only higher selectivity for CO (FECO up to 96% at −0.57 V), but also a higher CO partial current density (jCO = −3.67 mA cm−2) with a stronger suppression of the hydrogen evolution reaction (HER) (FEH2 = ∼3.8%).18 In another study, by only substituting four surface Au atoms in Au23(SR)16 with two Cd atoms, Au19Cd2(SR)16 was prepared by Li et al., and such modification greatly enhanced the selectivity of CO in CO2RR (FECO = ∼90 to 95% at −0.5 to −0.9 V), which is doubled compared to the undoped Au23 NC.19 Recently, Sun et al. devised a strategy to control the cleavage of Au–S or S–C bonds by introducing Cd atoms, and identified the reaction sites of Au25(SR)18, Au24Cd1(SR)18, Au19Cd4(SR)18, and Au38Cd4(SR)30 for CO2RR.20 In the above cases, DFT calculations disclosed that, the Cd doping altered the surface geometry and electronic structure of the NCs, which further changed the intermediate binding energy.
Noteworthily, for Au-based NCs, CO is the main product in CO2RR test. Copper-based catalysts have demonstrated to be effective to convert CO2 into highly valuable products including formate,21 methanol,22 methane,23 and so on. Tang et al. synthesized a Cu32H20L12 (L: a dithiophosphate ligand) NC, which can offer a unique selectivity of formate (FEformate = 90%) for CO2RR at low overpotentials.24 DFT calculations revealed that, the presence of the negatively charged hydrides in the NC played a critical role in determining the selectivity of the product, while the formate formation proceeded via the lattice-hydride mechanism.24 Thanks to the versatile metal–ligand bonding moieties,25–27 alkynyl ligands have been attracting more and more attentions to prepare coinage metal NCs in the past decade,25,28,29 and homoleptic alkynyl-protected coinage metal NCs possess unique physicochemical properties and have found broad applications in semiconductor,30 hypergolic fuels,31 and biomedical regime.32 Until so far, significant progress has been made on structure determination and formation mechanism study,25,28 yet the cases on alkynyl-protected metal NCs for CO2RR are still quite rare. Recently, our group reported the quite small all-alkynyl-protected [Ag15(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-tBu)12]+ NC, which was able to convert CO2 into CO with a FECO of ∼95% at −0.6 V.33 Also, the first case on homoleptic alkynyl-protected AgCu superatom of [Ag9Cu6(CC-tBu)12]+ was prepared to compare the physicochemical properties with [Au7Ag8(CC-tBu)12]+, and the two M15 clusters exhibited distinctly different optical properties due to the metal core difference.34 The following questions arise immediately: will these two clusters have different CO2RR performance as well? Furthermore, as both clusters are belonging to the M15 series, if the metal core is atomically tailored, how does the CO2RR performance change? In another word, can we atomically tailor the core to probe the metal core effect of the M15 series toward CO2RR? The above questions form the primary aim and goal of our current study.
C-tBu)12]+ NC, which was able to convert CO2 into CO with a FECO of ∼95% at −0.6 V.33 Also, the first case on homoleptic alkynyl-protected AgCu superatom of [Ag9Cu6(CC-tBu)12]+ was prepared to compare the physicochemical properties with [Au7Ag8(CC-tBu)12]+, and the two M15 clusters exhibited distinctly different optical properties due to the metal core difference.34 The following questions arise immediately: will these two clusters have different CO2RR performance as well? Furthermore, as both clusters are belonging to the M15 series, if the metal core is atomically tailored, how does the CO2RR performance change? In another word, can we atomically tailor the core to probe the metal core effect of the M15 series toward CO2RR? The above questions form the primary aim and goal of our current study.
Herein, we report the CO2RR performance and comprehensive mechanistic study of atomically precise alkynyl-protected [Au7Ag8(CC-tBu)12]SbF6 (Au7Ag8 in short hereafter), [Ag9Cu6(CC-tBu)12]SbF6 (Ag9Cu6 in short hereafter), and [Au2Ag8Cu5(CC-tBu)12]SbF6 (Au2Ag8Cu5 in short hereafter) NCs. As a note, in a recent study, Kang et al. reported a shortening of the A3-coupling reaction time from hours to minutes at higher temperatures (175 °C) catalyzed by a thermally robust, trimetallic Au1Ag16Cu12(SSR)12(PPh3)4 NC (SSR: benzene-1,3-dithiolate), demonstrating the unique potential of trimetallic alloying in catalytic enhancement.35 By using a chiral reducing agent, Hakkinen and Zheng groups reported a novel phosphine and thiolate ligand co-protected trimetallic [Au7Ag6Cu2(R- or S-BINAP)3(SCH2Ph)6]SbF6 (BINAP: 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) NC with tertiary chiral nanostructure.36 In this study, Au2Ag8Cu5 is the first case of all-alkynyl-protected trimetallic superatom documented so far. It can be synthesized by a metal exchange approach from Ag9Cu6, and X-ray crystallography reveals a body-centered-cubic (BCC) structure with an Au@AuAg4Cu3@Ag4Cu2 core configuration. Interestingly, the three M15+ NCs exhibited significantly different CO2RR properties. Au7Ag8 can convert CO2 into CO exclusively with FECO reaching 98.1% at −0.49 V, while CO and formate are the main products for Ag9Cu6 and Au2Ag8Cu5 at more negative potentials, in which the highest FEformate value is 47.0% at −1.19 V and 28.3% at −0.99 V, respectively. In addition, Ag9Cu6 and Au7Ag8 can inhibit H2 evolution effectively with FEH2 less than 10% in the whole tested potential range. Density functional theory (DFT) calculations disclosed that –CCR removal from the intact NC can expose the undercoordinated metal atom as the catalytic site to significantly promote the activity and selectivity of CO2RR. In particular, the formation of negative hydride is the key for the exclusive formate formation on Ag9Cu6 and Au2Ag8Cu5.
Results and discussion
Preparation and characterization of Au7Ag8, Ag9Cu6, and Au2Ag8Cu5 NCs
Au7Ag8 and Ag9Cu6 NCs were first synthesized by following the method in our previous report, in which the crystal structure and optical properties of the two NCs were compared.34 Note that, the fabrication and total structure of Au7Ag8 NC was first reported by Wang et al. in 2016,37 and our anti-galvanic synthetic approach can improve the yield drastically.34 In this study, Au2Ag8Cu5 NC was synthesized by a site-specific metal exchange method by a reaction between Me2SAu(I)Cl and Ag9Cu6 NC with a controlled stoichoiometric ratio. The detailed synthetic procedure can be found in ESI,† and the relevant elucidation of the process will be discussed next.Subsequently, the chemical composition of the three NCs were verified by electrospray ionization mass spectrometry (ESI-MS). As illustrated in Fig. 1a, the sharp peak at m/z = 3214.8510 and 2325.5677 is assigned to Au7Ag8 and Ag9Cu6, respectively, and the well-matched experimental/simulated isotopic pattern (Fig. S1a and b†) confirmed the molecular composition of Au7Ag8 and Ag9Cu6. In addition, the main peak at m/z = 2548.6640 corresponds well with [Au2Ag8Cu5(C6H9)12]+ (cal.: 2548.6634 Da, deviation: 0.0006 Da), and the isotopic patterns of the NC match perfectly with the simulated results (Fig. S1c†). There is also the one Au atom exchanged product of AuAg8Cu6 NC (indicated by ▲, see enlarged spectra in Fig. S1f†), and the fragments of Au7Ag8 and Ag9Cu6 NCs are identified in Fig. S1d and e,† respectively. To further confirm the metallic ratio in Au2Ag8Cu5, X-ray photoelectron spectroscopy (XPS, Fig. S2†) and energy-dispersive X-ray spectroscopy (EDS, Fig. S3†) were conducted. The atomic ratio of Au/Ag/Cu is 1.9/8.1/5.3 (2.0/8.7/5.7) and 1.5/6.7/4.2 (2.0/8.9/5.5) from XPS (Table S1†) and EDS (Fig. S3†), respectively, both are in agreement with the theoretical value (2/8/5). The XPS survey scan spectra confirmed the presence of the essential elements (Fig. S2a†). The binding energy of the Au 4f7/2 electrons is located at 84.43 eV, between bulk Au (84.0 eV)38 and Au(I) (84.5 to 86.0 eV)38 (Fig. S2b†). Furthermore, the binding energy of the Ag 3d5/2 electrons is located at 368.65 eV (Fig. S2c†), indicating that the valence state of Ag atoms in Au2Ag8Cu5 is +1.39 In addition, the binding energy of Cu 2p3/2 (933.40 eV) agrees well with that of Cu(I) (933.3 eV),40 implying that Cu atoms are present as Cu(I) (Fig. S2d†). Moreover, as illustrated in Fig. 1b, the fingerprint absorbance peaks of Au2Ag8Cu5 NC are located at 335, 461, 484, 521, and 571 nm, quite different from that of Au7Ag8 (313, 339, 422, 477, and 506 nm) and Ag9Cu6 (333, 357, 422, 544, 579, and 620 nm) NCs. Nevertheless, we monitored the absorbance change in the formation process of Au2Ag8Cu5. As shown in Fig. S4a,† the absorbance feature of Ag9Cu6 NC disappeared immediately upon the addition of Me2SAu(I)Cl, while a new absorption band at ∼484 nm arose. There is an obvious colour change at the timing point of Me2SAu(I)Cl addition (Fig. S4b†). In 1 h, the characteristic peak at 484 nm from Au2Ag8Cu5 gradually arose, meanwhile the absorbance peak at 571 nm can be identified. The metal exchange process occurs very fast, and as manifested by the two visualized video records (see Video 1† under room light and Video 2† under 365 nm UV-light as additional ESI†). In addition, we also studied the photoluminescence property of the Au2Ag8Cu5 NC. As shown in Fig. S5,† Au2Ag8Cu5 NC strongly emits in the near-infrared region (λmax = 825 nm), which is much stronger than that of Au7Ag8 NC, while Ag9Cu6 NC is not photoluminescent.34 Given the standard absorbance curve (Fig. S6a†) of Au2Ag8Cu5 NC, according to Lambert–Beer's law, the molecular absorptivity (ε) of Au2Ag8Cu5 NC can be determined (ε = 1.88 × 104 M−1 cm−1), as summarized in Table S2.† Subsequently, the yield of Au2Ag8Cu5 NC was calculated as ∼66.85% (based on Cu). The details of the calculation process can be found in ESI† (Fig. S6b and Table S3†).
 | ||
| Fig. 1 (a) Positive-mode ESI-MS and (b) absorbance spectra of Au7Ag8, Ag9Cu6, and Au2Ag8Cu5 NCs. The asterisk (*) and octothorpe (#) indicate the fragment ion and molecular ion of Au7Ag8, Ag9Cu6, and Au2Ag8Cu5 NCs, respectively, and the triangle (▲) indicates the AuAg8Cu6 NC product in the Au2Ag8Cu5 sample. | ||
Elucidating metal exchange process from Ag9Cu6 to Au2Ag8Cu5
Tailoring the metal core while retaining the other parts has been proven as an effective approach to modify the physicochemical property and enhance the functionality of thiolate-protected metal NCs.41 Such metal core tailoring is actually part of the tailoring chemistry of metal nanoclusters.42 To the best of our knowledge, no case on metal core tailoring has been reported for homoleptic alkynyl-protected coinage metal NCs. Inspired by the findings in thiolate-protected metal NCs, herein, Au2Ag8Cu5 was synthesized by a controlled stoichiometry of Me2SAu(I)Cl-to-Ag9Cu6 (=2) through atomic-level tailoring by metal exchange (Fig. 2a). The total structure comparison between Au2Ag8Cu5, Ag9Cu6, and Au7Ag8 will be discussed in the next session, nevertheless, the detailed five-step transformation from Ag9Cu6 to Au2Ag8Cu5 is presented in Fig. 2b, and the corresponding chemical reaction equations are shown in Fig. 2c.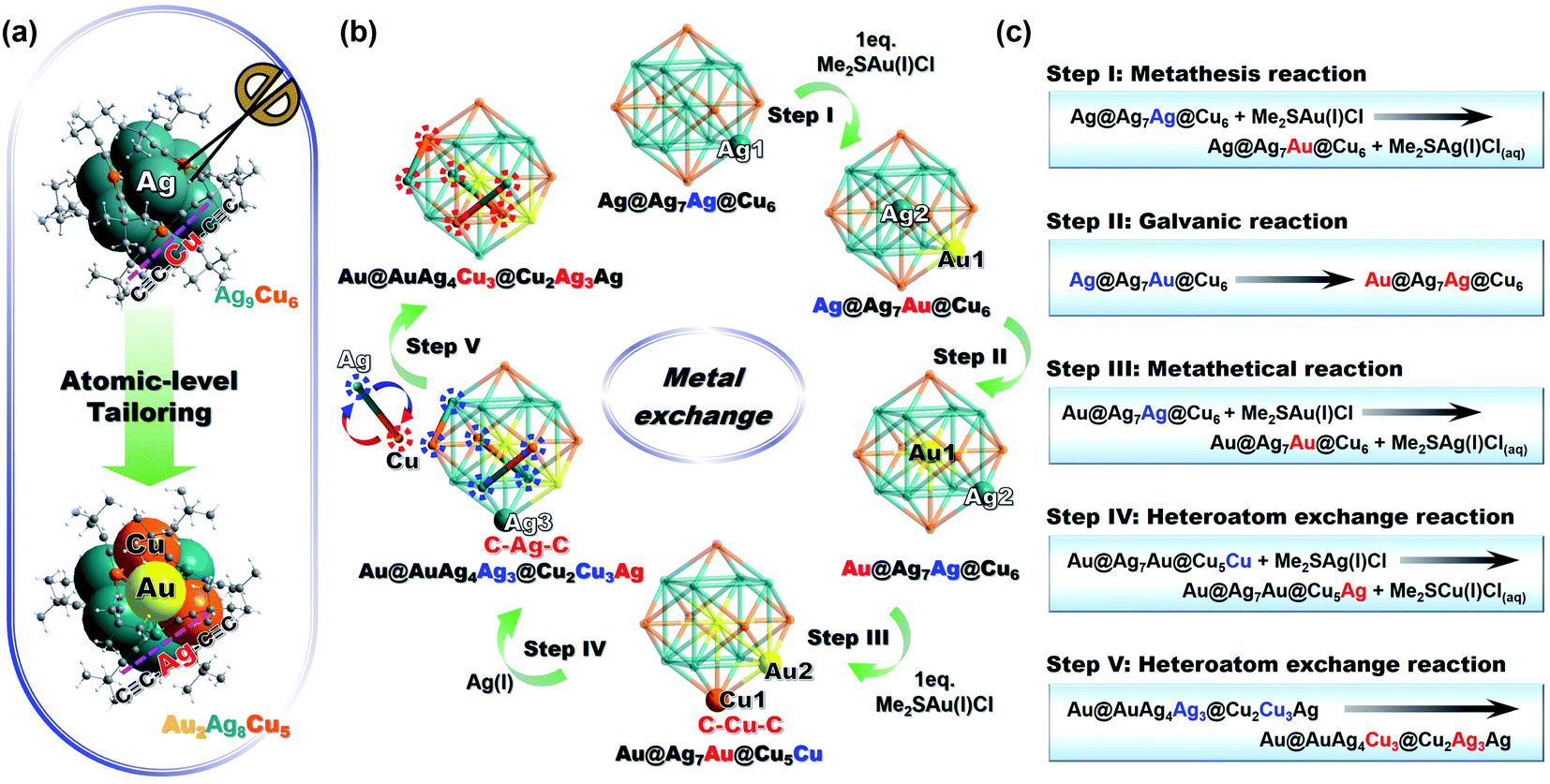 | ||
| Fig. 2 (a) Synthesis of the [Au2Ag8Cu5(CC-tBu)12]+ NC by atomic-level tailoring. (b) Metal exchange process from Ag9Cu6 NC to Au2Ag8Cu5 NC via a five-step process. Color legend: Au, yellow; Ag, cyan; Cu, orange; C, gray, H, white. (c) Equations of the Au2Ag8Cu5 formation by metal exchange from Ag9Cu6 NC. The atoms before (blue) and after (red) the reaction. | ||
Firstly, 1 eq. of Me2SAu(I)Cl was added to react with Ag9Cu6, and one Au(I) atom replaces one Ag(I) atom to form a Ag@Ag7Au@Cu6 kernel (Step I). It is a metathesis reaction, and Me2SAg(I)Cl is also generated in the solution. Note that, the driving force of such heteroatom exchange is probably the interaction between the Cl− ion and the Ag(I) atoms on the Ag8 cube. Subsequently, the as-formed intermediate was transformed into more stable molecule (kernel: Au@Ag8@Cu6) via galvanic reaction, in which the Au(I) atom is reduced by the central Ag(0) atom (determined by DFT structures with Mulliken charges in Table S4†),34 and the two atoms exchanged the position with each other (Step II). As a note, such Au heteroatom diffusion phenomenon has been previously documented in thiolate-protected alloy NCs,43,44 for instance, Xie and coworkers discovered that, the Au heteroatom diffuses into the surface layer of the Ag13 icosahedron kernel and finally is reduced by the central Ag(0) atom, forming thermodynamically stable AuAg24(MHA)18 molecule.45 Then, in the presence of another 1 eq. of Me2SAu(I)Cl, like the first step, Au2Ag7Cu6 (kernel: Au@Ag7Au@Cu6) NC was formed by the metal exchange reaction (Step III). Consequently, the Cu atoms around the Au atom in the M8 cube are activated and can react with Me2SAg(I)Cl generated in the previous steps, and one Ag(I) atom exchanges with one Cu(I) atom in the Cu6 octahedron (Step IV). Finally, three Ag(I) atoms on the M8 cube were exchanged by three Cu(I) atoms to form Au2Ag8Cu5 NC with the optimal thermodynamic stability (Step V).
It is worth pointing out that, the precise stoichiometric ratio of Me2SAu(I)Cl-to-Ag9Cu6 (=2) is critical for yielding the optimal amount of Au2Ag8Cu5 NC. In fact, different ratios of Me2SAu(I)Cl (0.4 eq., 1.0 eq., 1.6 eq., 2.0 eq., and 2.4 eq. per Ag9Cu6) were tested, and the results are shown in Fig. S7.† As depicted in the absorbance change in Fig. S7a,† with the increasing of the Me2SAu(I)Cl amount (from 0.4 to 2.0 eq.), the intensity of the characterstic peak (at 579 nm) from Ag9Cu6 decreased gradually (totally disappeared with 2 eq.), while the characteristic peak (at 484 nm) from Au2Ag8Cu5 gradually became intensified. However, when it increased to 2.4 eq., the intensity of the peak at 484 nm slightly decreased. Such trend can be more clearly observed in Fig. S7b and c,† that is, with lower amounts of Me2SAu(I)Cl (0.4 to 1.6 eq.), Ag9Cu6 NC is not fully converted, and with extra amount of Me2SAu(I)Cl, Ag9Cu6 can be fully converted but polydispersed mixture was obtained. For instance, in the presence of 8 eq. Me2SAuCl, a series of [AuxAg8Cu7−x (CC-tBu)12]+ (x = 1 to 7) molecules including Au2Ag8Cu5 and Au7Ag8 NCs were acquired, as confirmed by the ESI-MS spectra in Fig. S8.† Unfortunately, several attempts were conducted to separate the intermediate but was not successful, mainly due to that, this reaction occurs too fast (the whole process is finished in 1 h). We also noticed that, in the previous report, Wang et al. employed Cu atoms to react with Au7Ag8 clusters, and a series of cluster mixture [CunAg8Au7−n (CC-tBu)12]+ (n = 0 to 6) including Au2Ag8Cu5 NC (n = 5) was identified by mass spectrometry but the separation was also not performed neither.37 Therefore, the exact stoichiometric ratio of 2 is the optimal value and also very critical.
Structural comparison of the three M15 NCs
Subsequently, the atomic packing structure of Au2Ag8Cu5 was examined by single crystal X-ray diffractometer (SC-XRD). As illustrated in Fig. S9,† Au2Ag8Cu5 crystallizes in space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , and each unit cell has a SbF6− counterion, indicating that Au2Ag8Cu5 NC possesses a +1 charge. The detailed structural parameters are summarized in Table S5.† The overall structure of monocationic Au2Ag8Cu5 is shown in Fig. 3a, which contains two Au atoms, eight Ag atoms, five Cu atoms, and twelve tert-butylacetylene ligands, hence the molecule can be formulated as [Au2Ag8Cu5(CC-tBu)12]SbF6. As illustrated in the space-filling structure, one Au site, five Cu sites, and eight Ag sites on the surface of Au2Ag8Cu5 are exposed partially, which might result in the differences in catalytic performance compared with the other M15 NCs (Ag9Cu6 and Au7Ag8). As a note, all the currently reported alkynyl-protected M15 NCs such as Ag9Cu6, Au7Ag8, and Ag15 have only one type of tBu-CC-M-CC-tBu (M = Cu/Au/Ag) linear motif.33,34 However, for Au2Ag8Cu5, there are five types of tBu-CC-M-CC-tBu (M = Cu/Ag) motifs on the surface, in which the coordination mode of tBu-CC– ligands are μ2-η1 (Ag/Cu), η1 (Ag1/Ag2) for motif 1; μ2-η1 (Au/Ag/Cu), η1 (Ag3) for motif 2; μ2-η1 (Au/Ag/Cu), η1 (Ag4) for motif 3; μ2-η1 (Au/Ag/Cu), η1 (Cu1) for motif 4 and μ2-η1 (Au/Ag), η1 (Cu2) for motif 5, respectively (Fig. 3b). As a result, the σ (Cu2–C) and π (Ag/Cu–C) bond lengths of motif 5 (average value: 1.870 Å and 2.389 Å) are shorter than those of motifs 1, 2, 3, and 4 (average value: 1.894 Å and 2.392 Å; 1.893 Å and 2.395 Å; 1.894 Å and 2.398 Å; and 1.873 Å and 2.394 Å, Table S6†). Motif 5 in Au2Ag8Cu5 is nearly identical with the motif in Ag9Cu6. The average σ and π bond lengths of motifs on the surface of Au2Ag8Cu5 (average 1.886 Å and 2.394 Å) are longer than those of Ag9Cu6 (average 1.870 Å and 2.381 Å) and shorter than those of Au7Ag8 (average 1.982 Å and 2.513 Å) (Table S7†). Note that, such different types of motifs can lead to the distortion of the kernel structure of Au2Ag8Cu5. Furthermore, Au2Ag8Cu5 adopts structural feature from both Au7Ag8 and Ag9Cu6, as it has the same central Au atom with Au7Ag8 and a more similar outlayer (Ag4Cu2vs. Cu6) with Ag9Cu6 (Fig. 3c). Specifically, the anatomical structure of Au2Ag8Cu5 is compared with the two bimetallic NCs. As shown in Fig. 3d, Au2Ag8Cu5 adopts a core–shell–shell configuration (Mcore@Mcube@Moctahedron) of Au@AuAg4Cu3@Ag4Cu2, similar to the other two NCs, but there are some difference in the Mcube and Moctahedron layers. For Au2Ag8Cu5, the two layers consist of the heteroatoms (AuAg4Cu3 and Cu2Ag4), while there are the homoatoms in the middle layer (Ag8) and outer layer (Au6 and Cu6) of Au7Ag8 and Ag9Cu6. Such structural difference leads to the difference in average bond lengths spread on different layers of the three NCs (Table S8†). Compared with Au7Ag8 and Ag9Cu6, the doped Au and Cu heteroatoms lead to the Mcube in Au2Ag8Cu5 slightly relaxed, as the average adjacent Mcube–Mcube bond (3.414 Å) length is larger than those in the other two NCs (3.333 Å and 3.283 Å). However, there are four Ag atoms in the Moctahedron of Au2Ag8Cu5, and the bonding lengths of the outer layer in Au2Ag8Cu5 are slightly longer than those of Ag9Cu6 (e.g. Moctahedron–Cligand: 1.886 Å vs. 1.870 Å; Mcube–Cligand: 2.394 Å vs. 2.381 Å), further attesting the similarity of the outer layer between Au2Ag8Cu5 and Ag9Cu6.
, and each unit cell has a SbF6− counterion, indicating that Au2Ag8Cu5 NC possesses a +1 charge. The detailed structural parameters are summarized in Table S5.† The overall structure of monocationic Au2Ag8Cu5 is shown in Fig. 3a, which contains two Au atoms, eight Ag atoms, five Cu atoms, and twelve tert-butylacetylene ligands, hence the molecule can be formulated as [Au2Ag8Cu5(CC-tBu)12]SbF6. As illustrated in the space-filling structure, one Au site, five Cu sites, and eight Ag sites on the surface of Au2Ag8Cu5 are exposed partially, which might result in the differences in catalytic performance compared with the other M15 NCs (Ag9Cu6 and Au7Ag8). As a note, all the currently reported alkynyl-protected M15 NCs such as Ag9Cu6, Au7Ag8, and Ag15 have only one type of tBu-CC-M-CC-tBu (M = Cu/Au/Ag) linear motif.33,34 However, for Au2Ag8Cu5, there are five types of tBu-CC-M-CC-tBu (M = Cu/Ag) motifs on the surface, in which the coordination mode of tBu-CC– ligands are μ2-η1 (Ag/Cu), η1 (Ag1/Ag2) for motif 1; μ2-η1 (Au/Ag/Cu), η1 (Ag3) for motif 2; μ2-η1 (Au/Ag/Cu), η1 (Ag4) for motif 3; μ2-η1 (Au/Ag/Cu), η1 (Cu1) for motif 4 and μ2-η1 (Au/Ag), η1 (Cu2) for motif 5, respectively (Fig. 3b). As a result, the σ (Cu2–C) and π (Ag/Cu–C) bond lengths of motif 5 (average value: 1.870 Å and 2.389 Å) are shorter than those of motifs 1, 2, 3, and 4 (average value: 1.894 Å and 2.392 Å; 1.893 Å and 2.395 Å; 1.894 Å and 2.398 Å; and 1.873 Å and 2.394 Å, Table S6†). Motif 5 in Au2Ag8Cu5 is nearly identical with the motif in Ag9Cu6. The average σ and π bond lengths of motifs on the surface of Au2Ag8Cu5 (average 1.886 Å and 2.394 Å) are longer than those of Ag9Cu6 (average 1.870 Å and 2.381 Å) and shorter than those of Au7Ag8 (average 1.982 Å and 2.513 Å) (Table S7†). Note that, such different types of motifs can lead to the distortion of the kernel structure of Au2Ag8Cu5. Furthermore, Au2Ag8Cu5 adopts structural feature from both Au7Ag8 and Ag9Cu6, as it has the same central Au atom with Au7Ag8 and a more similar outlayer (Ag4Cu2vs. Cu6) with Ag9Cu6 (Fig. 3c). Specifically, the anatomical structure of Au2Ag8Cu5 is compared with the two bimetallic NCs. As shown in Fig. 3d, Au2Ag8Cu5 adopts a core–shell–shell configuration (Mcore@Mcube@Moctahedron) of Au@AuAg4Cu3@Ag4Cu2, similar to the other two NCs, but there are some difference in the Mcube and Moctahedron layers. For Au2Ag8Cu5, the two layers consist of the heteroatoms (AuAg4Cu3 and Cu2Ag4), while there are the homoatoms in the middle layer (Ag8) and outer layer (Au6 and Cu6) of Au7Ag8 and Ag9Cu6. Such structural difference leads to the difference in average bond lengths spread on different layers of the three NCs (Table S8†). Compared with Au7Ag8 and Ag9Cu6, the doped Au and Cu heteroatoms lead to the Mcube in Au2Ag8Cu5 slightly relaxed, as the average adjacent Mcube–Mcube bond (3.414 Å) length is larger than those in the other two NCs (3.333 Å and 3.283 Å). However, there are four Ag atoms in the Moctahedron of Au2Ag8Cu5, and the bonding lengths of the outer layer in Au2Ag8Cu5 are slightly longer than those of Ag9Cu6 (e.g. Moctahedron–Cligand: 1.886 Å vs. 1.870 Å; Mcube–Cligand: 2.394 Å vs. 2.381 Å), further attesting the similarity of the outer layer between Au2Ag8Cu5 and Ag9Cu6.
 | ||
| Fig. 3 Structural analysis in body-centered cubic (BCC) M15 NCs. (a) Overall and space-filling structure of monocationic Au2Ag8Cu5, (b) the five types of linear tBu-CC–CuCC-tBu staple motifs on the metal surface. Coordination modes of tBu-CC ligands: μ2-η1 (Ag/Cu), η1 (Ag); μ2-η1 (Au/Ag/Cu), η1 (Ag); μ2-η1 (Au/Ag/Cu), η1 (Cu) and μ2-η1 (Ag/Cu), η1 (Cu). (c) Structural analysis and (d) anatomy of BCC M15 kernel in Au2Ag8Cu5, Ag9Cu6, and Au7Ag8, respectively. Color legend: Au, yellow; Ag, cyan; Cu, orange; C, gray; H, white. | ||
Electrocatalytic CO2 reduction performance of the three M15 NCs in flow cell
As the three NCs possess a M15 configuration yet different metal core and significant discrepancies are observed on the physiochemical properties (e.g. optical absorbance), we wonder whether they have different catalytic properties. To probe the metal core effect, we next examined the electrocatalytic performance of the three catalysts on gas diffusion electrode (GDL, 2 × 1.5 cm2) toward CO2RR by constant-potential electrolysis (CPE) measurements at various applied potentials in a custom-designed flowcell (Fig. 4a) (the electrochemical measurement details can be found in ESI†). The linear scanning voltammetry (LSV) was first conducted for Au7Ag8/GDL, Ag9Cu6/GDL, and Au2Ag8Cu5/GDL. As depicted in Fig. S10,† for all the samples, a sudden decrease in the reduction current can be observed after the first potential sweep (black line) along with the onset potential shifted positively. In the second and third sweeps (red and blue line), the resulting current and onset potential remained unchanged. It suggests that, all the catalysts were activated, and such ligand stripping phenomenon has been recorded in thiolate-protected Au25 NCs in the CO2 electroreduction process as well.46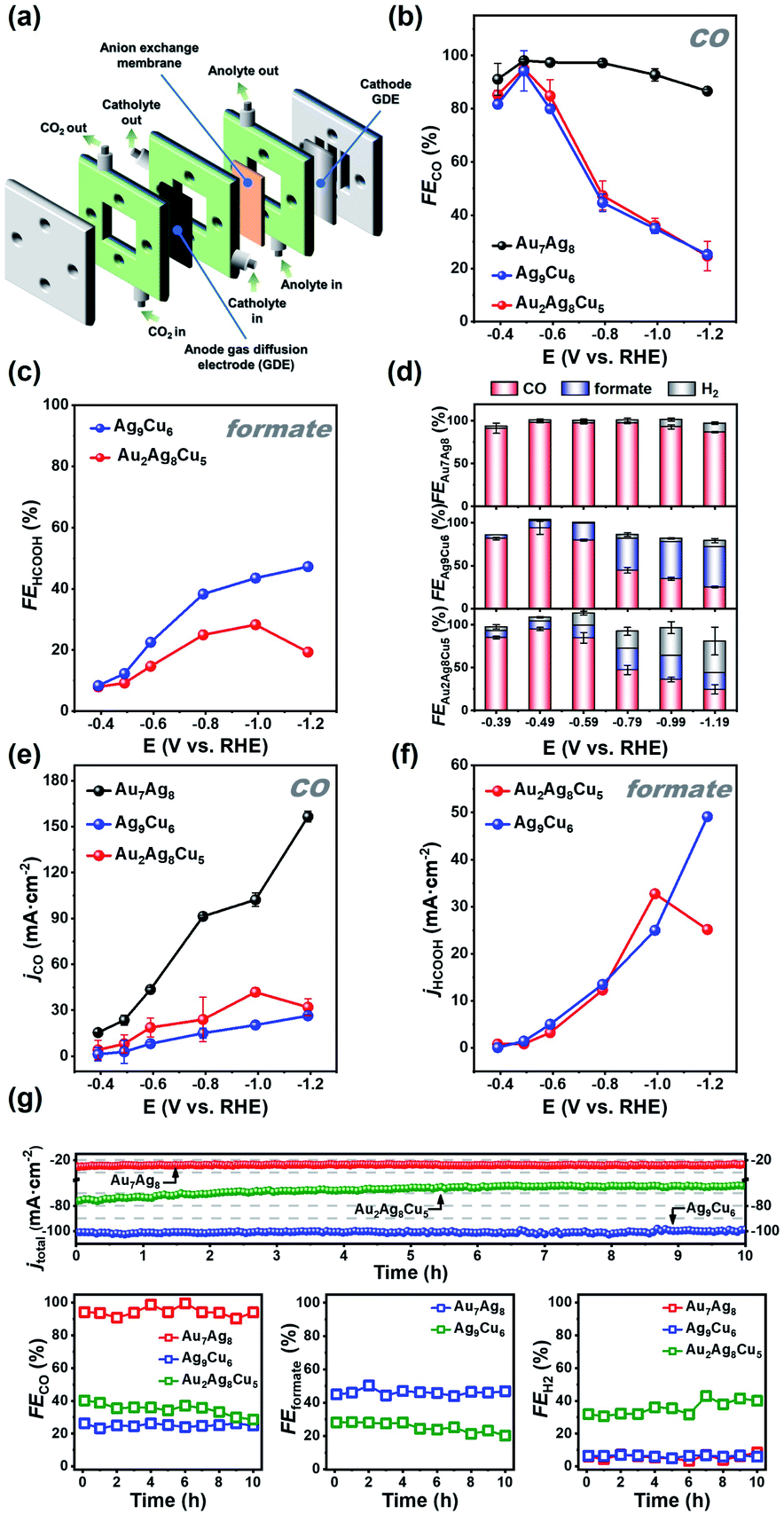 | ||
| Fig. 4 (a) Exploded diagrams of the electrochemical reactors for CO2 electroreduction in flow cell. (b) CO and (c) formate faradaic efficiency for Au7Ag8, Ag9Cu6, and Au2Ag8Cu5 NC/GDLs examined at different applied potentials. (d) FEs for various CO2RR products obtained on the three NC/GDLs. The corresponding (e) CO and (f) formate partial current density. (g) Long-term stability of Au7Ag8/GDL, Ag9Cu6/GDL, and Au2Ag8Cu5/GDL at −0.49 V, −1.19 V, and −0.99 V (vs. RHE), respectively. (top) i–t curve; (bottom) FEs of CO, H2, and formate at different time. | ||
For all the catalysts, CO is the main product at more positive potentials, and formate at more negative potentials was also detected in the liquid phase by 1H-NMR for the two Cu-containing catalysts (Fig. S11 and S12†). As shown in Fig. 4b, Au7Ag8 exhibited high selectivity for CO formation, evidenced by the higher FECO values at all tested potentials, ranging from ∼86.6% at −0.39 V to ∼98.1% at −1.19 V (vs. RHE). In contrast, for both Ag9Cu6 and Au2Ag8Cu5, a similar volcanic shape on the FECO value is observed, in which the highest FECO value of ∼94.2% and ∼95.0% at −0.49 V is obtained for Ag9Cu6 and Au2Ag8Cu5, respectively. However, for these two NCs, CO has higher FEs at more positive potentials, whereas the FE for formate (FEformate) increases rapidly when the potential goes more negatively (Fig. 4c). The largest FEformate value for Ag9Cu6 and Au2Ag8Cu5 is 47.0% at −1.19 V and 28.3% at −0.99 V, respectively. However, the FEformate value for Au2Ag8Cu5 decreased to less than 20% at −1.19 V. Impressively, for both bimetallic NCs of Ag9Cu6 and Au7Ag8, the H2 evolution can be significantly suppressed, as the FEH2 is less than 10% in the whole tested potential range (Fig. S13a†). However, for Au2Ag8Cu5, when the potential goes more negatively, the HER becomes more dominant, and the highest FEH2 can reach 37.0% at −1.19 V. The total FE values of the products for the three catalysts were presented as a function of applied potential from −0.59 V to −1.19 V, showing that CO, H2, and formate are the main products with a total FE value close to 100% in the whole potential range (Fig. 4d). No other product was detected by NMR or GC. By deducting H2, at −0.49 V, the highest FECO for Au7Ag8 is ∼98.1%, while the highest FECO+formate for Ag9Cu6 and Au2Ag8Cu5 is ∼100.0% and ∼97.4%, respectively, indicating the three NCs can efficiently convert CO2 into value-added carbon products (Fig. 4d). Meanwhile, the CO partial current density (jCO) increased with the increasing of applied potential for the Au7Ag8 and Ag9Cu6 catalysts, and jCO reached the maximal value at −0.99 V then diminished at −1.19 V for Au2Ag8Cu5 (Fig. 4e). Au7Ag8 had a much larger jCO value than Ag9Cu6 and Au2Ag8Cu5 at all potentials, further manifesting its unique advantage for converting CO2 into CO exclusively. Furthermore, the partial current density of formate (jformate) for Ag9Cu6 and Au2Ag8Cu5 exhibited the same trend with the FEformate in the tested potential range (Fig. 4f), that is, jformate increased from −0.39 V to −1.19 V, reaching the maximal value of 49.1 mA cm−2 at −1.19 V for Ag9Cu6, however, for Au2Ag8Cu5, it first increased then decreased, and the maximal value is 32.7 mA cm−2 at −0.99 V. This is mainly due to that the HER process became dominant at very high negative potentials (Fig. S13b†).
Stability is another important criterion to evaluate the catalytic property of the electrocatalyst, hence the long-term stability of Au7Ag8, Ag9Cu6, and Au2Ag8Cu5 was tested at −0.49 V, −1.19 V, and −0.99 V, respectively. As illustrated in Fig. 4g, the current density and corresponding FE value of Au7Ag8 and Ag9Cu6 remained almost unchanged after 10 h's continuous operation, indicating robust long-term durability, however, under the same conditions, the current density of Au2Ag8Cu5 decreased about 15% (from 75.4 to 64.1 mA cm−2), meanwhile the FECO+formate decreased and FEH2 increased gradually, presumably due to that the asymmetric metal core of Au2Ag8Cu5 is easier to decompose and/or aggregate during the electrocatalytic process. We conducted the MS measurement of the three NCs before and after CO2RR to examine the change. As shown in Fig. S14,† for the Cu-containing NCs, the molecular ions with strong signal (m/z = 2548.6640 for Au2Ag8Cu5, m/z = 2325.5677 for Ag9Cu6) are still dominant, indicating both clusters are rather robust. However, for both clusters, some peaks in the lower m/z region appeared, suggesting some of the cluster molecules decomposed. For Au2Ag8Cu5, after CO2RR, two peaks with m/z = 1112.4193 (Fragment A) and m/z = 1471.6617 (Fragment B) can be assigned to Au4L4 (L: C6H9, cal. MW: 1112.4183) and Au5L6+ (cal. MW: 1471.6608), respectively. In addition, compared to product A, the peak intensity of Au2Ag8Cu5 decreased, suggesting that some Au2Ag8Cu5 molecules might decompose to Au-alkynyl complexes and/or metal nanoparticles; for Ag9Cu6, after CO2RR, there are three peaks appearing at m/z = 1020.8771 (Fragment A), 1099.9494 (Fragment B), and 1183.0180 (Fragment C), which can be assigned to Ag2Cu5L6+ (cal. MW: 1020.8762), Ag2Cu5L7+ (cal. MW: 1099.9485), and Ag2Cu5L8+ (cal. MW: 1183.0171), respectively. Also, there is one peak with m/z at 2476.3047 (D), which can be assigned to Ag12Cu2L13+ (cal. MW: 2476.3039), and it is probably formed in the chamber during the MS measurement. For Au7Ag8, after CO2RR, there are two minor peaks appearing at m/z = 3053.0904 (Fragment A) and 2937.2619 (Fragment B), which can be assigned to Au7Ag8L10+ (cal. MW: 3053.0910) and Au6Ag8L11+ (cal. MW: 2937.2625), respectively. These results indicate that, the majority of the cluster molecules can be well preserved during the CO2RR process.
Furthermore, we also tested the recover capability of the Ag9Cu6 and Au2Ag8Cu5 catalysts for CO2RR. Using the 579 and 484 nm fingerprint absorbance peak as the metric, the absorbance change can be quantified and employed to estimate the recovery rate (Fig. S15 and S16†). It is worth noting that, besides the intensity of the characteristic peak decreased with different extents at different potentials, the whole absorbance feature of these two NCs remained intact. The calculated results are summarized in Tables S9 and S10.† Specifically, from −0.39 V to −1.19 V, the intensity of the absorbance peak at 579 nm decreased gradually, and the recovery rate of these two NCs decreased as well. Also, the recovery rate of Ag9Cu6 ranges from 30.4% to 96.6%, higher than that of Au2Ag8Cu5 (21.0% to 89.7%) at all applied potentials, in good agreement with the finding in the i–t test.
The observation that all M15 NCs exhibited high catalytic selectivity of CO2 electroreduction to CO at the low potentials and that the clusters containing Cu metals, namely Ag9Cu6 and Au2Ag8Cu5, were found to generate formate products is interesting. We then compared the formation selectivity of formate and CO of the three M15 NCs with recently reported atomically precise metal nanoclusters in CO2RR, as summarized in Table S11 and S12†, respectively. Although the reports were conducted in different cell type such as H-cell, flow-cell and MEA-cell, it can be noted that, the formate selectivity of the Ag9Cu6 and Au2Ag8Cu5 clusters is lower than the Cu32 cluster, but much higher than the Au25 cluster and all the AuCd alloy clusters. For CO formation selectivity, the highest FECO value of the Au7Ag8, Ag9Cu6, and Au2Ag8Cu5 clusters are all over 94%, at least comparable with, if not superior to, the Au, Ag clusters and the AuCd, AuPd, AuAg alloy clusters. Particularly, the FECO value of the Au7Ag8 cluster can reach as high as 98.1%, larger than most of the recent reports, quite close to the Au25(PET)18 and Au24Pd1(PET)18 clusters (∼100% for both).
CO2RR mechanistic study by DFT calculations
To deeply comprehend the electrocatalytic mechanism, we next performed DFT calculations (see ESI† for computational details) to determine the optimal catalytic site and analyze the selectivity difference. To simplify the calculation, all –CC-tBu ligands are replaced with –CC-CH3. The optimized structure based on the crystal structure of Au@AuAg4Cu3@Ag4Cu2 is used as a model for DFT calculation, as shown in Fig. S17.† On the intact [Au7Ag8(CC-CH3)12]+ (Fig. S18a†) and [Ag9Cu6(CC-CH3)12]+ (Fig. S18b†), CO2RR and HER compete on the same staple metal site (Au for [Au7Ag8(CC-CH3)12]+ and Cu for [Ag9Cu6(CC-CH3)12]+). While for [Au2Ag8Cu5(CC-CH3)12]+ NC, the staple Cu acts as the active site for HCOO* binding, while *COOH, *CO, and *H tend to bond with the sub-surface Au atom (Fig. S18c†). The free energy diagrams of CO2RR and HER on these three intact systems are depicted in Fig. 5a, c, and e, and the H2 pathway is thermodynamically more favourable than CO2RR. In addition, we found that the bonding of *H on clusters containing copper is stronger. To uncover this phenomenon, we performed Bader charge analysis of metal active site (Table S13†), which intuitively shows that *H on Ag9Cu6 has the most negative charge (−0.29 |e|), indicating a stronger adsorption. However, *H has the strongest bonding on Au2Ag8Cu5 cluster, which is possibly due to the special coordination environment of active Au atom, so that Au with greater electronegativity can rob electrons from the surrounding Ag or Cu. Therefore, the active Au here is negatively charged (−0.15 |e|) and interacts strongly with *H.
 | ||
| Fig. 5 (a, c and e) Comparison of CO2RR vs. HER on three intact NCs. (b, d and f) Reaction scheme for CO2 electroreduction on single ligand-removed clusters to form CO via the proton mechanism (blue region) and to form formate via the hydride–proton mechanism (green region) at zero applied potential. The reaction step with the highest free-energy change step is framed in red. | ||
Inspired by related studies17,19,46 and our recent finding on Ag15 NC for CO2RR,33 ligand removal to expose undercoordinated metal atom may serve as the electrocatalytic active centers. As depicted in Fig. S19,† the release of one –CC-CH3 results in exposure of four shell–metal atoms to form two (111)-like triangular faces. Due to the highly symmetrical structures of [Ag9Cu6(CC-CH3)12]+ and [Au2Ag8Cu5(CC-CH3)12]+, the removal of either –CC-CH3 ligand is equivalent. However, it is predicted that, the removal of alkynyl ligand bonded to two Ag atoms near the shell Au atom on the [Au2Ag8Cu5(CC-CH3)12]+ cluster is more thermodynamically supported (the red circle marked in Fig. S19c†), thereby exposing 111-like surfaces (Fig. S19d†). In this context, the H* would readily adsorb to the hollow position of the triangle in a bridging manner (Fig. S20†). The calculated Bader charge (Fig. S20†) shows that the adsorbed *H has a negative charge of −0.11 to −0.22 |e|, suggesting that the adsorbed *H functions as a hydride and may provide the hydrogen source for CO2 reduction.24,47 As a consequence, there are four possible reaction channels: (1) proton mechanism of reacting CO2 with the proton from solution; (2) hydride mechanism of reacting CO2 with the capping hydride (H*); (3) the hydride–proton or (4) proton–hydride mechanism, where hydride and proton alternately participate in the catalytic process. The free energy difference (ΔG) of each reduction step can be found in Fig. S21 to S23.† On the ligand-removed NCs the proton-reduction channel is preferred for CO; whereas the hydride–proton channel is more favoured for formate, that is, the first adsorbed H* (marked in green) is easily transferred to the C atom to form HCOO*, the second adsorbed H*(marked in blue) is difficult to transfer, but can occupy the active site to facilitate subsequent protonation. The overall mechanism of CO formation via the proton mechanism and formate formation via the hydride–proton mechanism from CO2 reduction on three NCs are summarized in Fig. S24 to S26.† The corresponding free energy profile for generating CO and formate are shown in Fig. 5b, d, and f. Apparently, for the CO pathway, the formation of *COOH is the potential-determining step (PDS), the same as on intact NCs; for the formate pathway, the PDS corresponds to the electrochemical protonation of *HCOO to formate or the transfer of H* to the C atom to form HCOO*. Their comparable ΔG for PDS (CO is slightly preferred) indicates that the CO and formate formation is competitive, which is consistent with the experimental observation that CO and formate are the main products on Ag9Cu6 and Au2Ag8Cu5. The corresponding optimal configurations of key intermediates are depicted in Fig. 6. On both [Ag9Cu6(CC-CH3)11]+ and [Au2Ag8Cu5(CC-CH3)11]+, the two O atoms of HCOO* bind tightly with one Cu atom and one Ag atom on the metal triangle. The active site for CO formation differs from each other, where the trans-COOH* prefers to bind to Ag atom on [Au7Ag8(CC-CH3)11]+, to Cu atom on [Ag9Cu6(CC-CH3)11]+, and to Au atom on [Au2Ag8Cu5(CC-CH3)11]+. The H* on all three clusters easily occupy the hollow sites of the triangle in a bridging manner. Note that, the attraction between the negatively charged H* and the positively charged C of the CO2 reactant can trigger the favourable H* transfer to form *HCOO, and the participation of metallic Cu as the active center is also important in stabilizing the *HCOO intermediate. Based on the high CO and formate selectivity observed in experiments, the exposure of more active surface metal site upon ligand removal could be the real reason for the feasible CO2RR pathway. It is worth noting that we use a simplified –CC-CH3 group for our simulation, while in experiment, much bulkier butyl groups are employed for protection. To further illustrate the feasibility of this simplification, we investigated the CO2RR and HER performance of [Ag9Cu6(CC-tBu)12]+ synthesized in the actual experiment and compared it with [Ag9Cu6(CC-CH3)12]+. As shown in Fig. S27,† the bulkiness brought by –CC-tBu groups slightly weakens the adsorption strength for intermediate state. However, the predicted response and the PDS are basically the same. Thus, the simplification of the butyl ligand can provide valid prediction on the performance.
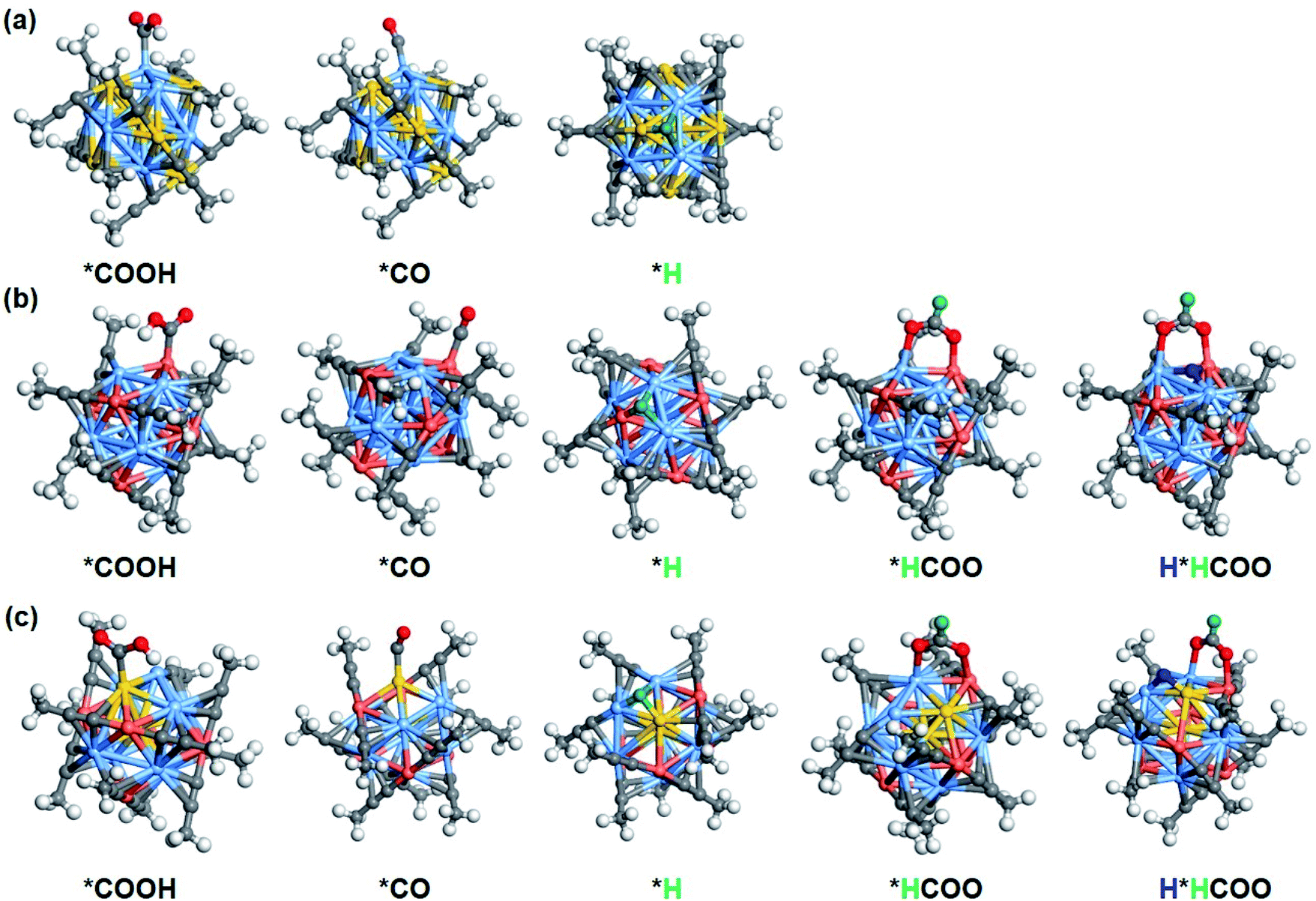 | ||
| Fig. 6 Schematic presentation of key intermediates on (a) [Au7Ag8(CC-CH3)11]+, (b) [Ag9Cu6(CC-CH3)11]+, and (c) [Au2Ag8Cu5(CC-CH3)11]+ NCs, respectively. Color legend: Au, gold; Ag, blue; Cu, brick-red; C, gray; O, red; H, white (mark the first H* in green and the second H* in blue). | ||
Discussion on metal core effect of the M15 NCs toward CO2RR
Finally, with the combined experimental and theoretical results of the three M15 clusters for CO2RR, plus the reported Ag15 one,33 we would like to discuss the metal core effect of the M15 series toward CO2RR. Note that, Ag15 and Au7Ag8 clusters can exclusively convert CO2 into CO with very high FE values, however, for Au2Ag8Cu5 and Ag9Cu6 clusters, formate can be generated. Apparently, the presence of Cu atoms is critical for generating formate, and more importantly, with two-atom difference (Au2Ag8Cu5vs. Ag9Cu6), the catalytic performance is drastically different (the FEformate value for Ag9Cu6 is higher than that of Au2Ag8Cu5, and the latter one has stronger H2 evolution than formate formation at −1.19 V). That means, strong core size effect toward CO2RR is observed in the M15 series, and metal exchange is an effective strategy to fine-tune the electrocatalytic performance of homoleptic alkynyl-protected metal nanoclusters. The theoretical calculations also support the above findings. For both Ag15 and Au7Ag8 clusters, the undercoordinated Ag and/or Au atoms upon one intact ligand stripping are the active sites for CO formation. However, for Ag9Cu6 and Au2Ag8Cu5 NCs, the undercoordinated Cu atoms can serve as the active sites for CO and formate formation. As revealed from Fig. S19,† the different electrocatalytic performance can be ascribed not only from the core atom difference (Ag vs. Au), but more importantly, the exposed (111)-like Ag2Cu2 surface and Au1Cu1Ag2 surface for Ag9Cu6 and Au2Ag8Cu5, respectively.Conclusions
In conclusion, the first all-alkynyl-protected trimetallic superatom of Au2Ag8Cu5 is synthesized through a metal exchange approach, of which the formation process is elucidated. Au2Ag8Cu5 NC has a similar M@M8@M6 metal core configuration with Ag9Cu6 and Au7Ag8 NCs, but quite different absorbance feature. Moreover, the three NCs exhibited drastically different catalytic performance toward CO2RR, in which Au7Ag8 can convert CO2 into CO exclusively, while CO and formate are the main products for Ag9Cu6 and Au2Ag8Cu5 at more negative potentials with the highest FEformate of 47.0% and 28.3%, respectively. DFT calculations revealed that ligand stripping can expose more active surface metal atoms to boost CO2RR activity and selectivity. The formation of surface hydride plays a critical role in triggering the formation and stabilization of HCOO* on the Ag–Cu active center, leading to the exclusive formation of formate in the Cu-containing NCs. Strong core effect toward CO2RR is observed. This study not only provides an ingenious strategy to tailor the metal core of alkynyl-protected metal NCs at atomic level, but also highlights the unique advantages of employing metal NCs as model catalysts to advance the fundamental mechanistic understanding toward CO2RR and beyond.Data availability
All the data in this study are provided in the main text and ESI.†Author contributions
Z. T. conceived the idea, X. M. conducted most of the experiments, L. Q. and Y. L. helped the characterization, F. S. and Q. T. conducted the DFT calculations, X. K. and L. W. provided the technique support for CO2RR test, D. J. offered guidance for theoretical calculations, X. M. and Z. T. wrote up the draft, Q. T. and Z. T. provided the funding support, and all the authors contributed to the final proof of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported by the Open Fund of Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications (No. 2021A07). Z. T. acknowledges the financial support from Guangdong Natural Science Funds (No. 2022A1515011840). L. W. acknowledges the funding from National Natural Science Foundation of China (No. 21805170). Q. T. thanks the grants from the National Natural Science Foundation of China (No. 21903008) and the Chongqing Science and Technology Commission (cstc2020jcyj-msxmX0382).Notes and references
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439–456 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- Y. Quan, J. Zhu and G. Zheng, Small Sci., 2021, 1, 2100043 CrossRef.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- K. D. Gilroy, A. Ruditskiy, H. C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS.
- L. Qin, G. Ma, L. Wang and Z. Tang, J. Energy Chem., 2021, 57, 359–370 CrossRef.
- S. Li, A. V. Nagarajan, Y. Li, D. R. Kauffman, G. Mpourmpakis and R. Jin, Nanoscale, 2021, 13, 2333–2337 RSC.
- D. R. Kauffman, D. Alfonso, C. Matranga, H. Qian and R. Jin, J. Am. Chem. Soc., 2012, 134, 10237–10243 CrossRef CAS PubMed.
- S. Zhao, N. Austin, M. Li, Y. B. Song, S. D. House, S. Bernhard, J. C. Yang, G. Mpourmpakis and R. C. Jin, ACS Catal., 2018, 8, 4996–5001 CrossRef CAS.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC.
- X. Lin, W. Ma, K. Sun, B. Sun, X. Fu, X. Ren, C. Liu and J. Huang, J. Phys. Chem. Lett., 2020, 12, 552–557 CrossRef PubMed.
- N. Austin, S. Zhao, J. R. McKone, R. Jin and G. Mpourmpakis, Catal. Sci. Technol., 2018, 8, 3795–3805 RSC.
- D. R. Alfonso, D. Kauffman and C. Matranga, J. Chem. Phys., 2016, 144, 184705 CrossRef PubMed.
- S. Li, D. Alfonso, A. V. Nagarajan, S. D. House, J. C. Yang, D. R. Kauffman, G. Mpourmpakis and R. Jin, ACS Catal., 2020, 10, 12011–12016 CrossRef CAS.
- S. Zhuang, D. Chen, L. Liao, Y. Zhao, N. Xia, W. Zhang, C. Wang, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 3073–3077 CrossRef CAS.
- S. Li, A. V. Nagarajan, D. R. Alfonso, M. Sun, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2021, 60, 6351–6356 CrossRef CAS PubMed.
- Y. N. Sun, X. Liu, K. Xiao, Y. Zhu and M. Y. Chen, ACS Catal., 2021, 11, 11551–11560 CrossRef CAS.
- Z. X. Tao, Z. S. Wu, Y. S. Wu and H. L. Wang, ACS Catal., 2020, 10, 9271–9275 CrossRef CAS.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS.
- Y. F. Wang, Z. Chen, P. Han, Y. H. Du, Z. X. Gu, X. Xu and G. F. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- Q. Tang, Y. Lee, D. Y. Li, W. Choi, C. W. Liu, D. Lee and D. E. Jiang, J. Am. Chem. Soc., 2017, 139, 9728–9736 CrossRef CAS PubMed.
- Z. Lei, X. K. Wan, S. F. Yuan, Z. J. Guan and Q. M. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS PubMed.
- Z. Lei and Q.-M. Wang, Coord. Chem. Rev., 2019, 378, 382–394 CrossRef CAS.
- P. Maity, S. Takano, S. Yamazoe, T. Wakabayashi and T. Tsukuda, J. Am. Chem. Soc., 2013, 135, 9450–9457 CrossRef CAS PubMed.
- X. Ma, Y. Tang, G. Ma, L. Qin and Z. Tang, Nanoscale, 2021, 13, 602–614 RSC.
- Z. Lei, X. K. Wan, S. F. Yuan, J. Q. Wang and Q. M. Wang, Dalton Trans., 2017, 46, 3427–3434 RSC.
- P. Yuan, R. Zhang, E. Selenius, P. Ruan, Y. Yao, Y. Zhou, S. Malola, H. Hakkinen, B. K. Teo, Y. Cao and N. Zheng, Nat. Commun., 2020, 11, 2229 CrossRef CAS PubMed.
- Z. Y. Wang, M. Q. Wang, Y. L. Li, P. Luo, T. T. Jia, R. W. Huang, S. Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2018, 140, 1069–1076 CrossRef CAS PubMed.
- T. T. Jia, G. Yang, S. J. Mo, Z. Y. Wang, B. J. Li, W. Ma, Y. X. Guo, X. Chen, X. Zhao, J. Q. Liu and S. Q. Zang, ACS Nano, 2019, 13, 8320–8328 CrossRef CAS PubMed.
- L. Qin, F. Sun, X. Ma, G. Ma, Y. Tang, L. Wang, Q. Tang, R. Jin and Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 26136–26141 CrossRef CAS PubMed.
- X. Ma, L. Xiong, L. Qin, Y. Tang, G. Ma, Y. Pei and Z. Tang, Chem. Sci., 2021, 12, 12819–12826 RSC.
- X. Kang, X. Wei, X. Liu, S. Wang, T. Yao, S. Wang and M. Zhu, Nat. Commun., 2021, 12, 6186 CrossRef.
- H. Shen, Z. Xu, L. Wang, Y. Z. Han, X. Liu, S. Malola, B. K. Teo, H. Hakkinen and N. Zheng, Angew. Chem., Int. Ed., 2021, 60, 22411–22416 CrossRef CAS.
- Y. Wang, H. Su, L. Ren, S. Malola, S. Lin, B. K. Teo, H. Hakkinen and N. Zheng, Angew. Chem., Int. Ed., 2016, 55, 15152–15156 CrossRef CAS PubMed.
- Z. Tang, D. A. Robinson, N. Bokossa, B. Xu, S. Wang and G. Wang, J. Am. Chem. Soc., 2011, 133, 16037–16044 CrossRef CAS PubMed.
- W. Du, S. Jin, L. Xiong, M. Chen, J. Zhang, X. Zou, Y. Pei, S. Wang and M. Zhu, J. Am. Chem. Soc., 2017, 139, 1618–1624 CrossRef CAS PubMed.
- X. Zou, Y. Li, S. Jin, X. Kang, X. Wei, S. Wang, X. Meng and M. Zhu, J. Phys. Chem. Lett., 2020, 11, 2272–2276 CrossRef CAS.
- T. Higaki, Q. Li, M. Zhou, S. Zhao, Y. Li, S. Li and R. Jin, Acc. Chem. Res., 2018, 51, 2764–2773 CrossRef CAS PubMed.
- Q. Li, T. Y. Luo, M. G. Taylor, S. Wang, X. Zhu, Y. Song, G. Mpourmpakis, N. L. Rosi and R. Jin, Sci. Adv., 2017, 3, e1603193 CrossRef.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- X. Kang, X. Wei, S. Jin, Q. Yuan, X. Luan, Y. Pei, S. Wang, M. Zhu and R. Jin, Proc. Natl. Acad. Sci., 2019, 116, 18834–18840 CrossRef CAS PubMed.
- K. Zheng, V. Fung, X. Yuan, D. E. Jiang and J. Xie, J. Am. Chem. Soc., 2019, 141, 18977–18983 CrossRef CAS PubMed.
- H. Seong, V. Efremov, G. Park, H. Kim, J. S. Yoo and D. Lee, Angew. Chem., Int. Ed., 2021, 60, 14563–14570 CrossRef CAS PubMed.
- F. Li and Q. Tang, J. Catal., 2020, 387, 95–101 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: synthesis, characterization, supporting figures and tables. Details and crystal data of [Au2Ag8Cu5(CC-tBu)12]SbF6 (CIF). CCDC 2072663. The videos for the metal exchange process to synthesize Au2Ag8Cu5 from Ag9Cu6. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02886g |
| ‡ X. Ma and F. Sun contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |