
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral oxazolidines acting as transient hydroxyalkyl-functionalized N-heterocyclic carbenes: an efficient route to air stable copper and gold complexes for asymmetric catalysis†
Delphine
Pichon-Barré
 a,
Ziyun
Zhang
d,
Aël
Cador
a,
Thomas
Vives
a,
Thierry
Roisnel
a,
Olivier
Baslé
b,
Lucie
Jarrige
a,
Luigi
Cavallo
*d,
Laura
Falivene
*c and
Marc
Mauduit
*a
a,
Ziyun
Zhang
d,
Aël
Cador
a,
Thomas
Vives
a,
Thierry
Roisnel
a,
Olivier
Baslé
b,
Lucie
Jarrige
a,
Luigi
Cavallo
*d,
Laura
Falivene
*c and
Marc
Mauduit
*a
aUniv Rennes, Ecole Nationale Supérieure de Chimie de Rennes, CNRS, ISCR UMR 6226, F-35000 Rennes, France. E-mail: marc.mauduit@ensc-rennes.fr
bLCC-CNRS, Université de Toulouse, CNRS, Toulouse, France
cDipartimento di Chimica, Università di Salerno, Via Ponte Don Melillo, I-84084 Fisciano, Italy. E-mail: lafalivene@unisa.it
dKing Abdullah University of Science and Technology (KAUST), Chemical and Life Sciences and Engineering, Kaust Catalysis Center, Thuwal 23955-6900, Saudi Arabia. E-mail: luigi.cavallo@kaust.edu.sa
First published on 11th July 2022
Abstract
Optically pure oxazolidines were synthesized in nearly quantitative yields from chiral hydroxyalkyl-functionalized imidazolinium salts. Acting as transient chiral diamino N-heterocyclic carbenes (NHCs), these oxazolidines allowed the efficient formation of well-defined copper(I) and gold(I) hydroxyalkyl-NHC complexes, which could be isolated, for the first time, as air stable complexes after silica gel chromatography. Interestingly, X-ray analysis of gold complexes revealed that the hydroxyl-function is not chelated to the metal. Computational studies suggested that both cyclisation to produce oxazolidine and O–H bond elimination to form the transient carbene (prior to coordination) occur through a concerted mechanism. The novel chiral copper-catalysts, as well as oxazolidines alone (copper free), demonstrated excellent performances in asymmetric conjugate addition and allylic alkylation with high regio- and enantio-selectivities (up to 99% ee).
Introduction
Since their discovery in the early 1960s, N-heterocyclic carbenes (NHCs) have become privileged ligands for numerous transition metal (TM) catalysed transformations, in both academic and industrial research environments.1 Due to their remarkable ability to generate a strong sigma-bond with the metal, these ancillary ligands improved drastically the catalyst activities, even surpassing the popular phosphorus-based ligands. With a versatile and highly modular steric environment, exploration of chiral NHCs as stereo-directing ligands has naturally emerged in the early 1990s. With resounding successes, their use in enantioselective catalysis has greatly accelerated in the last decade. Thanks to their easy and straightforward synthetic access, a plethora of chiral NHC ligands featuring various elements of symmetry were developed and studied in asymmetric catalysis.2 Among them, the class of diaminocarbene precursors LH·X bearing a chiral hydroxyalkyl-chelating side chain (Fig. 1a), readily accessible in 1 to 5 steps,3 has proved to be quite efficient in copper-catalysed enantioselective transformations.4 Notably, fruitful applications were achieved in copper-catalysed Asymmetric Conjugated Addition (ACA) and Asymmetric Allylic Alkylation (AAA), two major organometallic reactions for the formation of C–C bonds.5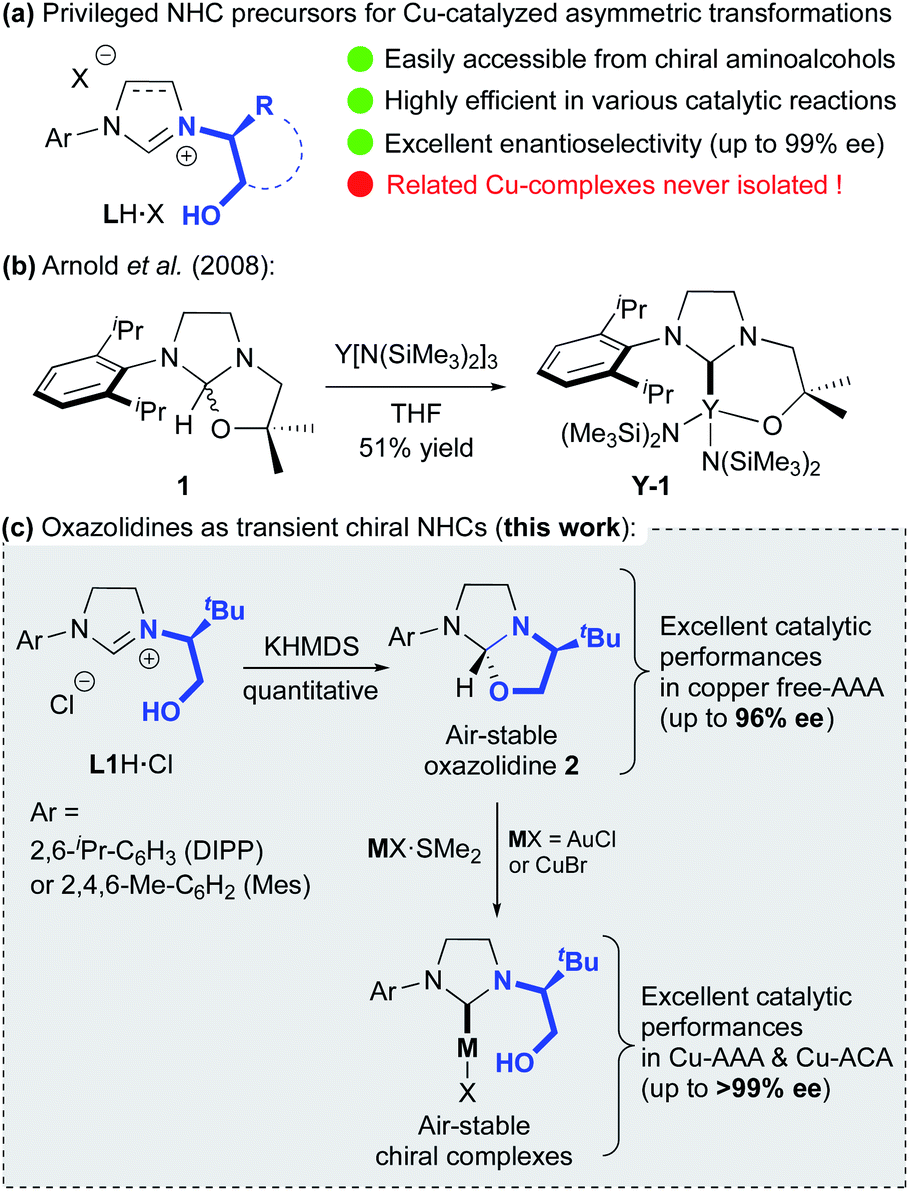 | ||
| Fig. 1 (a) Privileged chelating hydroxyalkyl–NHC ligand precursors for Cu-catalysed asymmetric transformations. (b) Previous synthesis of hydroxyl-chelating NHC–yttrium complex from achiral oxazolidine. (c) Proposed synthetic route to chiral hydroxyalkyl-functionalized NHC copper and gold complexes (this work). | ||
A wide range of Zn-, Mg-, B- and Al-based organometallic nucleophiles and Michael acceptors or allylic substrates were investigated leading to numerous building-blocks, including challenging chiral all-carbon quaternary centers, in good to excellent yields with remarkable enantioselectivities (up to 99% ee).6 Moreover, these highly selective catalytic methodologies were successfully applied in the total synthesis of complex natural products.7 Noteworthy, all catalytic transformations were exclusively done through the in situ formation of related Cu-catalytic species by just mixing LH·X (X = Cl or PF6), a base8 and Cu(I)- or Cu(II)-salts. Nevertheless, several DFT studies were accomplished enabling to propose some mechanistic insights to explain the high levels of regio- and enantioselectivity reached, notably in the case of polyconjugated substrates.9 Unfortunately, all attempts to isolate and characterize a well-defined chiral chelating copper–hydroxyalkyl–NHC complex have failed. In 2008, Arnold and co-workers reported the successful synthesis of chelating alkoxy-NHC yttrium complex Y-1 (Fig. 1b) from achiral oxazolidine 1, suggesting the formation of a transient carbene.10 We report herein that chiral hydroxyalkyl–NHC copper and gold complexes may be readily formed from oxazolidine intermediate 2 derived from optically pure imidazolinium salts L1H·Cl (Fig. 1c). The solid-state structure of gold complexes show that hydroxyl-function is not coordinated to the metal. The mechanisms for both the oxazolidine and the transient carbene formation as well as the metal insertion are supported by DFT calculations. Additionally, the resulting well-defined chiral copper-catalysts, as well as the oxazolidines, demonstrate excellent catalytic performances in Cu-AAA and Cu-ACA.
Results and discussion
Our study began with the preparation of oxazolidines 2 from the well-known hydroxyalkyl-imidazolinium chloride salts L1aH·Cl and L1bH·Cl featuring either a N-2,6-diisopropylphenyl (DIPP) or a N-2,4,6-trimethylphenyl (Mes) substituent in association with a N-(S)tert-leucinol moiety (Scheme 1).3a After a rapid screening of several bases (see ESI† for details), potassium hexamethyldisilazide (KHMDS) appeared the most effective one producing exclusively oxazolidines 2a and 2b within 5 minutes at ambient temperature. These latter were then isolated in quantitative yields after a simple filtration through a pad of Celite. Interestingly, similar high yields were also obtained at 500 mg scale (see ESI† for details). In accordance with Marazano works,11 the cyclisation occurred in opposite way to the tert-butyl group leading to a trans relationship between the two stereogenic carbons. This structural feature was confirmed by X-ray diffraction analysis of oxazolidine 2a, allowing to attribute the absolute configuration of the new stereogenic center (S, Fig. 2). As two possible pathways could be envisaged involving either the deprotonation of the hydroxyl function followed by the nucleophile addition of the resulting alcoholate to the iminium (path A, Scheme 2)12 or the deprotonation of imidazolinium proton and the subsequent carbene insertion to the OH bond (path B), mechanistic insights were investigated by DFT calculations (Fig. 3).13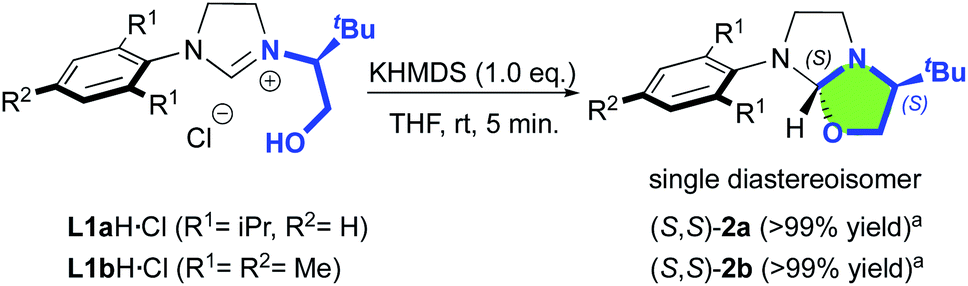 | ||
| Scheme 1 KHMDS-mediated diastereoselective formation of oxazolidines 2a,b. aIsolated yield after silica gel filtration. | ||
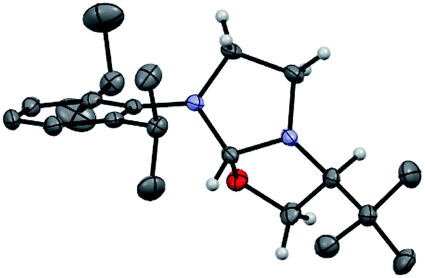 | ||
| Fig. 2 Solid-state structure of oxazolidine (S,S)-2a from single crystal X-ray diffraction. Displacement ellipsoids are drawn at 50% probability. Most hydrogen atoms have been omitted for clarity (N in purple, C in grey, O in red, H in white). | ||
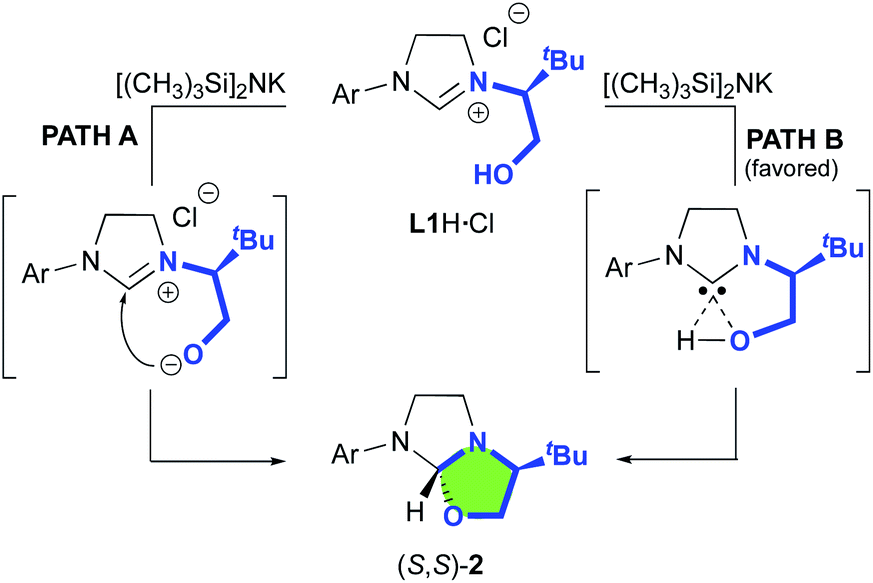 | ||
| Scheme 2 Plausible pathways for cyclisation leading to oxazolidine (S,S)-2. | ||
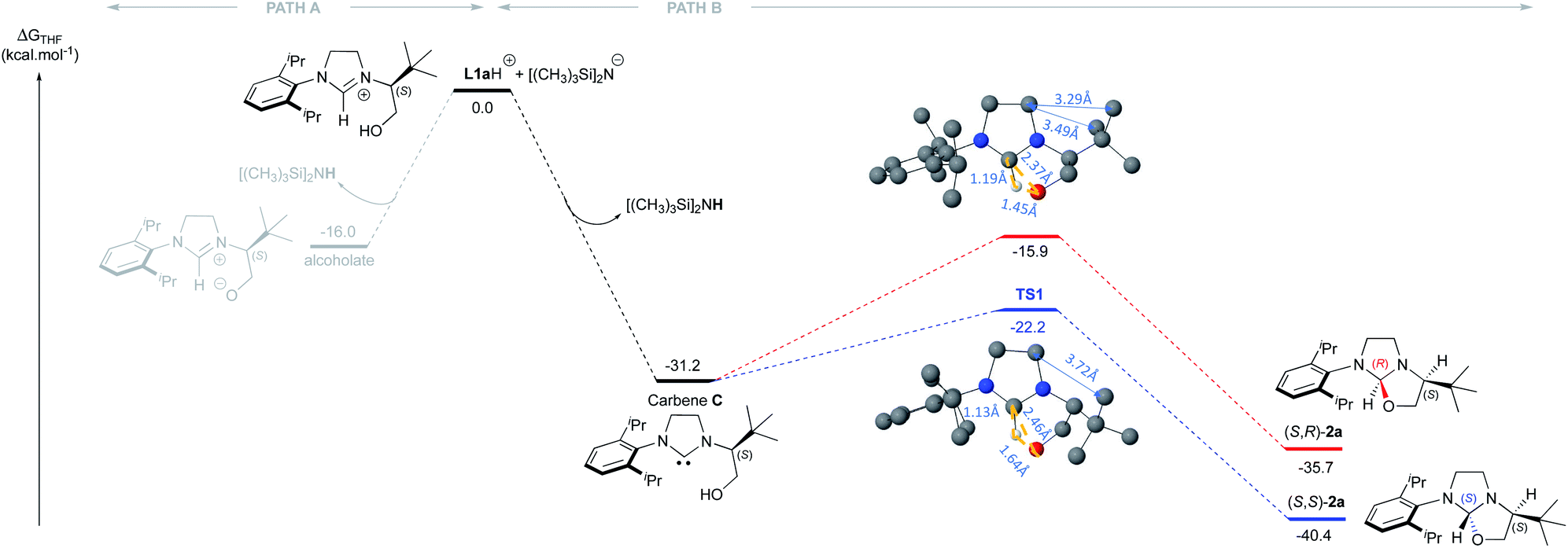 | ||
| Fig. 3 DFT calculations regarding the mechanism of the oxazolidine (S,S)-2a formation from L1aH. (The Cl− and K+ were not considered in the calculations). | ||
Starting from L1aH, set as zero-point energy, both deprotonated species are energetically favored, with the carbene intermediate C being 31.2 kcal mol−1 more stable than L1aH and the alternative alcoholate lying at −16 kcal mol−1, i.e. 15.2 kcal mol−1 higher in energy with respect to carbene C (Fig. 3). Starting from the thermodynamically favored carbene species C, the formation of the C–O bond and the transfer of the proton from oxygen to carbon are synchronous and lead directly to the formation of oxazolidine 2a in a concerted event. Any attempts to localize a stepwise mechanism failed. The reductive elimination transition state TS1 requires an energy barrier relative to C of 9.1 and 15.3 kcal mol−1 for the formation of S,S and S,R product, respectively. The resulting oxazolidine (S,S)-2a at −40.4 kcal mol−1 is thermodynamically more favored by almost 5 kcal mol−1 respect to (S,R)-2a lying at −35.7 kcal mol−1. The calculated ΔΔG≠ of 6.3 kcal mol−1 between the two enantiomeric transitions states clearly indicates the exclusive formation of the (S,S)-2a product, in agreement with experiments. The analysis of transition state geometries highlights that the unfavored (S,R)-TS1 is destabilized by steric repulsions between the NHC backbone and the tert-butyl group (see short distances in Fig. 3).
Having these optically pure oxazolidine (S,S)-2a,b in hand, the introduction of transition metals to yield corresponding chelating hydroxyalkyl–NHC–metal complexes was next investigated (Scheme 3). While 2a,b remained totally inert toward CuCl salt, we were delighted to observe that CuBr·SMe2 enabled the formation of novel air-stable copper complexes CuBr-1a and CuBr-1b in respectively 54% and 55% yields after silica gel purification.
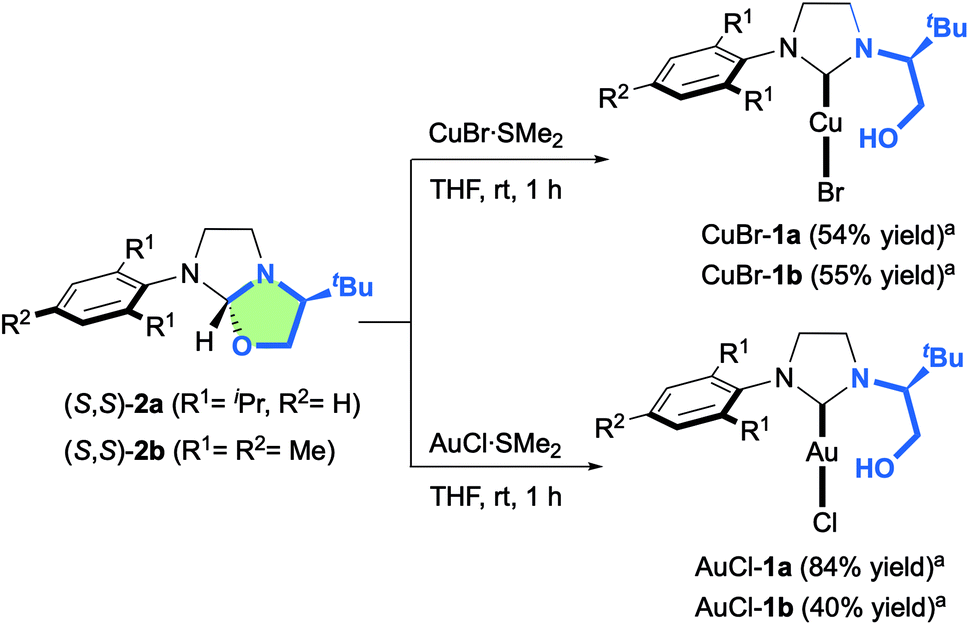 | ||
| Scheme 3 Synthesis of chiral hydroxyalkyl–NHC CuBr-1a,b and AuCl-1a,b complexes from oxazolidines 2a,b. aIsolated yields after silica gel filtration. | ||
Similarly, the reaction with AuCl·SMe2 produced the NHC-gold complexes AuCl-1a and AuCl-1b in 84% and 40% isolated yields after purification. Interestingly, the 1H-NMR analysis showed that the hydroxyl-functionality was not chelated to the metal. This structural feature was unambiguously confirmed by solid-state structure analysis of AuCl-1a,b complexes by X-ray diffraction (Fig. 4). Intrigued by the presence of the free hydroxyl function, DFT calculations about the transient carbene formation and the gold-metal insertion were next investigated (Fig. 5). Starting from oxazolidine (S,S)-2a, the backward reductive elimination transition state TS1 requires an energy barrier of 18.3 kcal mol−1 with the transient carbene C being almost 9 kcal mol−1 higher than the reactant. However, the following gold insertion step occurs easily with a relatively low energy barrier of only 5.4 kcal mol−1 with respect to carbene C, pushing the reaction towards the product. Overall, the thermochemistry of the reaction is estimated to be highly exergonic, with AuCl-1a complex plus SMe2 being at −21.4 kcal mol−1 respect to (S,S)-2a plus AuCl·SMe2. Any attempts to localize transition states for coordination of gold to the carbene carbon before oxygen leaves were ruled out due to the higher energy barriers involved.
 | ||
| Fig. 4 Solid-state structure of AuCl-1a (left) and AuCl-1b (right) complexes from single crystal X-ray diffraction. Displacement ellipsoids are drawn at 50% probability. Most hydrogen atoms have been omitted for clarity (N in purple, C in grey, O in red, H in white, Cl in green and Au in yellow). For AuCl-1b, only one molecule of the asymmetric unit is shown (see ESI† for details). | ||
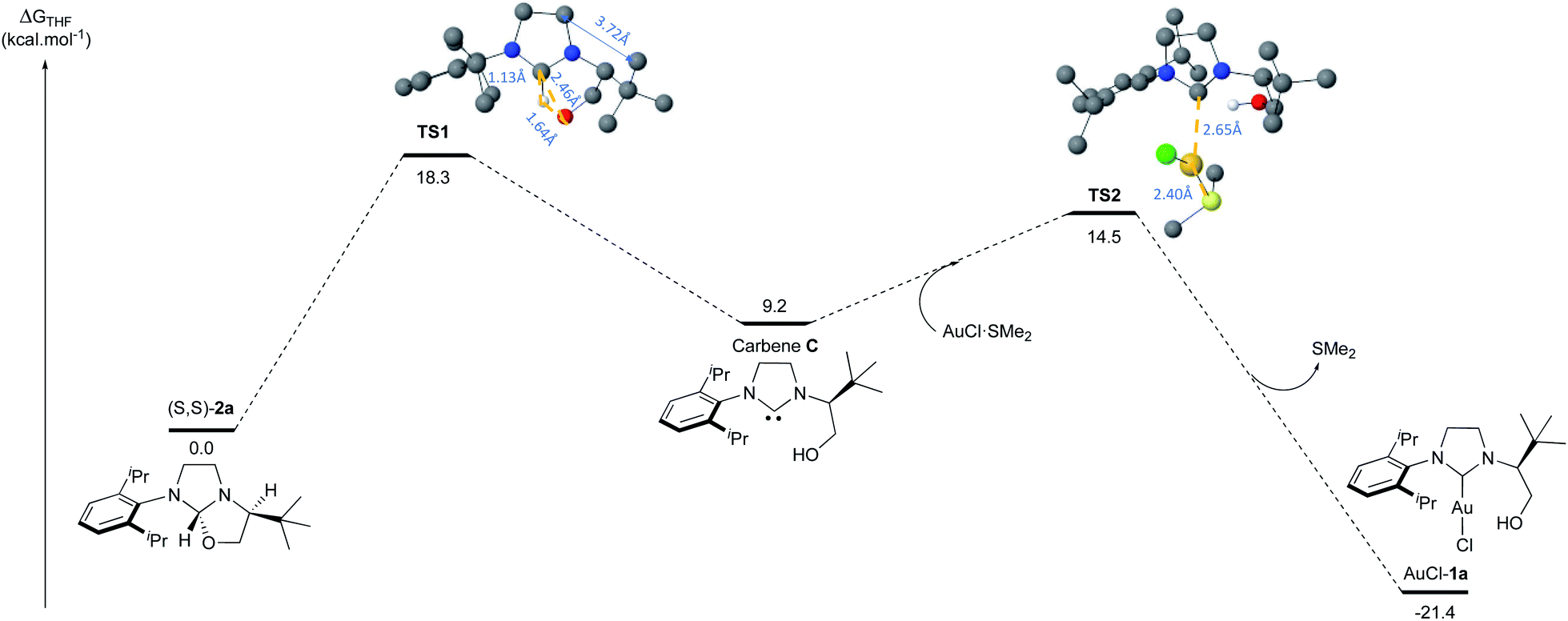 | ||
| Fig. 5 DFT calculations regarding the mechanism of the transient carbene formation from oxazolidine (S,S)-2a followed by the gold insertion to produce AuCl-1a complex. | ||
The catalytic performances of these new transient carbene precursors (S,S)-2a,b and their related copper catalysts CuBr-1a,b were further evaluated in asymmetric catalysis. The allylic alkylation of cinnamyl phosphate S1 with diethylzinc was first studied (Table 1). In comparison with the previously reported catalytic system L1aH·PF6/(CuOTf)2·toluene which required the prior deprotonation of the imidazolinium salt by n-BuLi to form the corresponding Cu–NHC catalytic species,6a,14 we were delighted to observe a similar catalytic performance with oxazolidine 2a in presence of (CuOTf)2·toluene complex (entries 1 and 2). A full-conversion occurred within 30 minutes at ambient temperature with the exclusive formation of the expected product γ-P1 in 92% isolated yield and 89% ee. With CuBr·SMe2, oxazolidine (S,S)-2a led also to γ-P1 in a slightly higher 97% yield and 88% ee (entry 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalytic system (mol%) | Time (h) | Conv.a (yield)b (%) | γ/α-P1 ratioc | eed (%) |
| a Conversions were monitored by TLC. b Isolated yields of γ-P1 after silica gel chromatography. c Molar ratio of γ/α adduct were monitored by 1H NMR spectroscopy analysis of the crude mixture. d Enantiomeric excesses were determined by chiral-phase GC analysis. e Reaction conditions: L1aH·PF6 (1.2 mol%), n-BuLi (2.5 mol%), (CuOTf)2·tol (0.5 mol%), THF, 0 °C, Et2Zn (3 equiv.), then S1, rt. f Conversion and yield were determined by 1H NMR spectroscopy (see ESI for details). Nr = no reaction. Nd = not determined. | |||||
| 1e | L1aH·PF6/(CuOTf)2·tol (1.2/1) | 0.5 | >99 (62) | >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
90 |
| 2 | 2a/(CuOTf)2·tol (1.2/1) | 0.5 | >99 (92) | >99:1 |
89 |
| 3 | 2a/CuBr·SMe2 (1.2/1) | 0.5 | >99 (97) | >99:1 |
88 |
| 4 | CuBr-1a (1) | 0.5 | >99 (99) |
>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
89 |
| 5 | 2a (5) | 12 | >99 (98) |
>99:1
|
90 |
| 6 | 2a (1) | 23 | 87 (76)f | 97:3 |
91 |
| 7 | CuBr-1b (1) | 0.5 | >99 (88) | >99:1 |
87 |
| 8 | 2b (5) | 12 | Nr | Nd | Nd |
| 9 | CuBr-1a (1) with EtMgBr | 0.5 | >99 (92) |
>99:1
|
90 |
| 10 | 2a (5) with EtMgBr | 0.5 | >99 (Nd) | 21:79 |
39 |
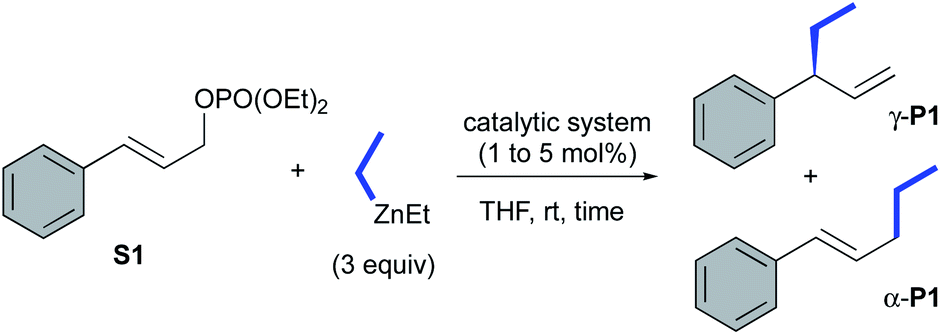
As expected, the well-defined and air stable copper catalyst CuBr-1a (1 mol%) gave similar high efficiency affording γ-P1 in quantitative yield and 89% ee (entry 4). Interestingly, without copper,152a was also able to catalyse efficiently the addition of Et2Zn, producing the desired γ-P1 in excellent 98% yield with the same performance of chiral induction (90% ee, entry 5).16,17 Remarkably, 2a remained active as low as 1 mol% affording the ramified product γ-P1 in respectable 76% yield and up to 91% ee (entry 6); nevertheless, a prolonged reaction time (23 h) were required. Changing from Dipp to Mes N-substituent did not influence dramatically the reaction as CuBr-1b complex gave similar results in terms of chiral induction as well as reactivity (entries 4 and 7). However, the chiral oxazolidine (S,S)-2b surprisingly remained inert, as no conversion was observed (entry 8 vs. entry 5). It is worth mentioning that Grignard reagent (EtMgBr) also provided similar excellent results when CuBr-1a was employed as catalytic system affording expected product γ-P1 with 90% ee and 92% isolated yield (entry 9). In contrast, a dramatic drop of selectivity was observed in copper-free conditions with 2a, leading to a mixture of non-separable regioisomers γ-:α-P1 in a 21:79 ratio (entry 10). Of note, AuCl-1a,b complexes were inactive in AAA.18
Next, the scope of the AAA was explored with both CuBr-1a and oxazolidine 2a as catalytic systems across a broad range of allylic phosphates and dialkylzinc reagents (Scheme 4). As previously observed, the same excellent levels of efficiency, regio- and stereoselectivity were obtained with or without copper for 2-naphthyl and 1-naphthyl-substituted substrates S2 and S3 giving corresponding γ-adducts P2 and P3a in 89 and 94% ees, respectively. Interestingly, the methodology was successfully extended to dimethylzinc reagent that led to expected methylated compound γ-P3b in good to excellent yields and very high enantioselectivities (96% ee vs. 94% ee with Et2Zn).
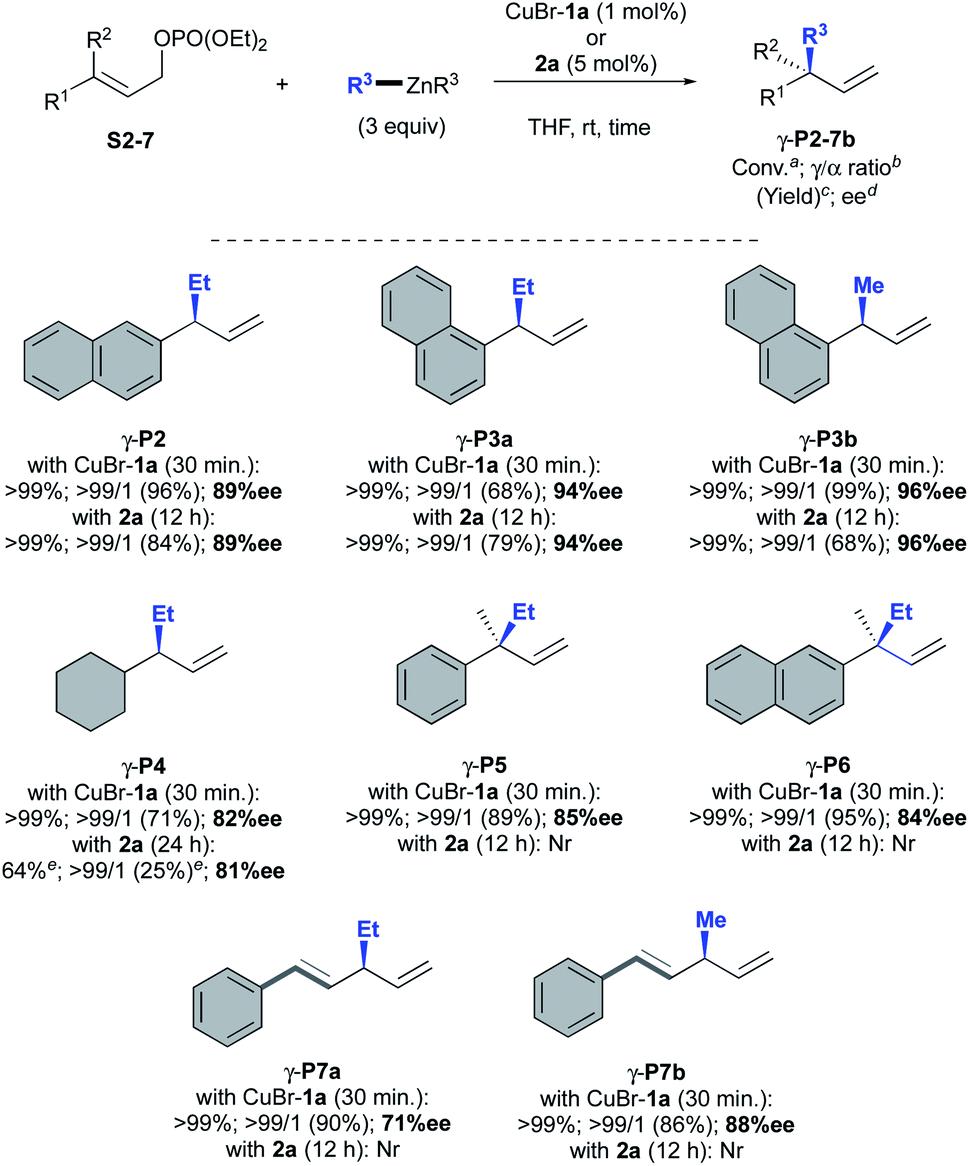 | ||
| Scheme 4 Scope of AAA catalysed by oxazolidine 2a and CuBr-1a. aConversions were monitored by TLC. bMolar ratio of γ/α adduct were monitored by 1H NMR spectroscopy analysis on the crude mixture. cIsolated yields after silica gel chromatography. dee were determined by chiral-phase GC or HPLC analysis. eConversion and yield were determined by 1H NMR spectroscopy (see ESI† for details). Nr = no reaction. | ||
In the case of the cyclohexyl-derived substrate S4, however, the copper-free catalytic system showed less efficiency than CuBr-1a as a significant drop of conversion was observed for γ-P4 (64 vs. 99%) but similar good enantiomeric excesses were obtained (81 and 82% ee, respectively). Unfortunately, oxazolidine 2a alone was unable to promote the formation of chiral all-carbon quaternary centers from trisubstituted allylic phosphate S5–6. On the other hand, well-defined CuBr-1a complex furnished the desired products γ-P5 and γ-P6 with good to excellent isolated yields (89 and 95%, respectively) and satisfactory enantioselectivities (85 and 84% ee). To our delight, a remarkable full γ-selectivity was also observed when diethyl- or dimethylzinc were reacted to the more challenging dienic conjugate substrate S7 in the presence of CuBr-1a. Highly desirable 1,3-skipped dienes γ-P7a and γ-P7b were thus isolated in 90–86% yield with good to excellent enantioselectivity (71 and 88% ee, respectively). Nevertheless, oxazolidine 2a remained unreactive toward S7 without cooper salts.
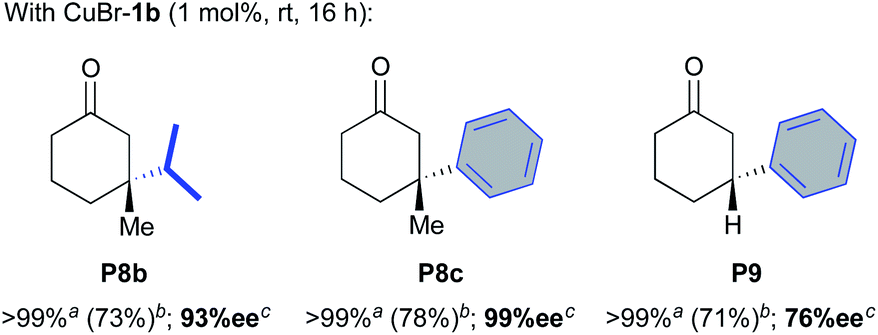
The good catalytic performances demonstrated in AAA prompted us to further evaluate oxazolidines 2a,b and copper-catalysts CuBr-1a,b in ACA of diethylzinc to 3-methylcyclohex-2-enone S8 (Table 2). Surprisingly, although an excellent 97% ee was obtained for 1,4-product P8a, the combination of oxazolidine 2b with (CuOTf)2·tol appeared significantly less productive (35% conv., 23% yield, entry 2) than the in situ catalytic system involving the NHC precursor L1bH·PF6 and (CuOTf)2·tol (>99% conv., 80% yield, entry 1). On the other hand, when 2b was used in presence of CuBr·SMe2, an excellent 97% isolated yield was obtained in combination with a full enantioselectivity (>99% ee, entry 3). To our delight, air stable CuBr-1b gave also a good catalytic performance while unfortunately, oxazolidine 2b appeared totally inactive without copper salts (entries 4 and 5). Similar behaviours were observed with oxazolidine 2a and CuBr-1a featuring a DIPP N-moiety (entries 6 and 7). The latter afforded P8a in good 84% isolated yield despite a slight drop of selectivity (85% ee, entry 6). Ethylmagnesium bromide was also evaluated in the ACA reaction. Once again, the desired compound P8a was exclusively formed when CuBr-1b complex was used as catalyst, with a good 86% yield despite a lower enantioselectivity (77% ee). Additionally, although a complete conversion of the substrate was observed during the reaction catalysed by the oxazolidine 2b, no trace of the expected compound P8a were detected in the messy crude mixture. Finally, as aforementioned above, AuCl-1a,b were also inefficient to catalyse the ACA reaction.
|
|
|||
|---|---|---|---|
| Entry | Catalytic system (mol%) | Conv.a (yield)b (%) | eec (%) |
| a Conversions were monitored by TLC. b Isolated yields after silica gel chromatography. c Enantiomeric excesses were determined by chiral-phase GC analysis. d Reaction conditions: L1bH·PF6 (1.2 mol%), n-BuLi (2.5 mol%), (CuOTf)2·tol (0.5 mol%), THF, 0 °C, Et2Zn, then S8, rt. e Conversion and yield were determined by 1H NMR spectroscopy (see ESI for details). Nr = no reaction. Nd = not determined. Mr = Messy reaction. | |||
| 1d | L1bH·PF6/(CuOTf)2·tol (1.2/1) | >99 (80) | 99 |
| 2 | 2b/(CuOTf)2·tol (1.2/1) | 35 (23)e | 97 |
| 3 | 2b/CuBr·SMe2 (1.2/1) | >99 (97) | >99 |
| 4 | CuBr-1b (1) | >99 (67) | 99 |
| 5 | 2b (5) | Nr | Nd |
| 6 | CuBr-1a (1) | >99 (84) | 85 |
| 7 | 2a (5) | Nr | Nd |
| 8 | CuBr-1b (1) with EtMgBr | >99 (86) | 77 |
| 9 | 2b (5) with EtMgBr | >99 (Mr) | Nd |
|
|||
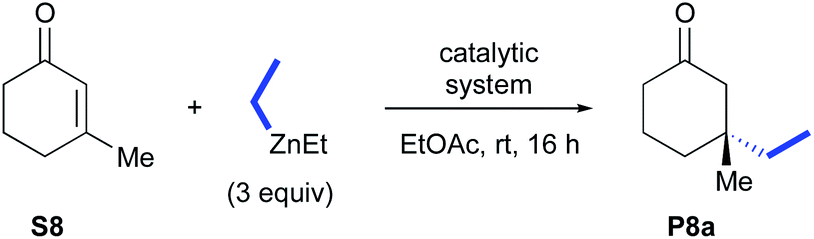
We next investigated the catalytic performances of the CuBr-1b complex regarding the ACA of diisopropyl- and diphenyl-zinc to 3-methylcyclohex-2-enone S8 and cyclohex-2-enone S9. Both of conjugate additions implying substrate S8 delivered the desired products P8b and P8c with good isolated yields (73 and 78%, respectively) and excellent 93–99% enantioselectivities. It is worth noting that in the case of the less hindered cyclohex-2-enone S9, a notable drop of the stereoselectivity occurred (76% ee) although the desired compound P9 was isolated with a satisfying 71% yield.
The more challenging 1,6-selective Cu-catalysed ACA of organozincs to cyclic dienone S10 was then investigated (Table 3). Curiously, the in situ catalytic system involving the NHC precursor L1b·PF6 and Cu(OTf)2 led only to a messy reaction with no detection of the expected 1,6-product P10a (entry 1). With oxazolidine (S,S)-2b either in combination with Cu(OTf)2 or CuBr·SMe2, 1,6-P10a was detected but embedded in a large amount of side products which rendered its isolation unsuccessful (entries 2 and 3). Nevertheless, moderate enantiomeric excesses of respectively 53 and 74% were observed. Advantageously, the use of the well-defined CuBr-1b complex allowed us to isolate 1,6-P10a in a satisfying 68% yield and a higher enantioselectivity (85% ee, entry 4). Moreover, albeit CuBr-1a bearing a bulkier N-DIPP substituent demonstrated a better catalytic performance as the expected 1,6-product was isolated in excellent 90% yield, a severe drop of the chiral induction was observed (65% ee, entry 5). Of note, by analogy with the aforementioned results in Table 2, oxazolidines 2a,b were also inefficient under copper-free conditions as no reaction occurred in either cases (entries 6 and 7). Different organozinc reagents were next engaged in this 1,6-ACA reaction with n-butyl-chain substituted cyclic dienone S11. First, addition of the diphenyl zinc furnished the expected product P11a in a regiospecific way with a good 70% isolated yield and an excellent enantioselectivity (90% ee). However, the chiral induction was affected by the use of diisopropylzinc since the corresponding desired compound P11b was isolated in 74% yield as a single regioisomer with only 66% ee.
|
|
||||
|---|---|---|---|---|
| Entry | Catalytic system (mol%) | Conv.a (yield)b (%) | Selectivityc 1,6:1,4 |
eed (%) |
|
a Conversions were monitored by TLC.
b Isolated yields after silica gel chromatography.
c Ratios between 1,6:1,4-adducts were determined by 1H NMR spectroscopy.
d Enantiomeric excesses were determined by chiral-phase GC or HPLC analysis.
e Reaction conditions: (1) L1bH·PF6 (3 mol%), n-BuLi (8 mol%), Cu(OTf)2 (2 mol%), THF, 0 °C, Et2Zn, then S10, rt. (2) DBU (1 equiv.), CH2Cl2, rt, 5 h.
f Large amount of side products were formed. Mr = Messy reaction. Nd = not determined. Nr = no reaction.
|
||||
| 1e | L1bH·PF6/Cu(OTf)2 (3/2) | Mr | Nd | Nd |
| 2 | 2b/Cu(OTf)2 (3/2) | >99 (Nd)f | >99:1 |
53 |
| 3 | 2b/CuBr·SMe2 (3/2) | >99 (Nd)f | >99:1 |
74 |
| 4 | CuBr-1b (2) | >99 (68) |
>99:1
|
85 |
| 5 | CuBr-1a (2) | > 99 (90) | >99:1 |
65 |
| 6 | 2b (5) | Nr | Nd | Nd |
| 7 | 2a (5) | Nr | Nd | Nd |

|
||||

Finally, we directed our attention to Grignard reagents, which led to a complete inversion of the regioselectivity of the conjugate addition6d,f in the presence of the CuBr-1b complex as catalyst (Scheme 5). Indeed, the exclusive formation of the 1,4 regioisomer was observed and the resulting product 1,4-P10a was isolated in a good 84% yield and an excellent 95% ee. Noteworthy, in comparison to the in situ catalytic system that required a high catalyst loading (up to 6 mol% of Cu(OTf)2 and 9 mol% of NHC precursor L1bH·Cl),6d,f a drastic improvement of reactivity was reached here, thanks to use of a well-defined copper complex, as only 3 mol% of CuBr-1b was necessary to complete the addition. At last, the 1,4-regiospecificity of the reaction was also confirmed with but-3-en-1-ylmagnesium bromide as nucleophile leading to desired compound 1,4-P10b in a similar high enantioselectivity (97%) despite a lower 24% isolated yield.
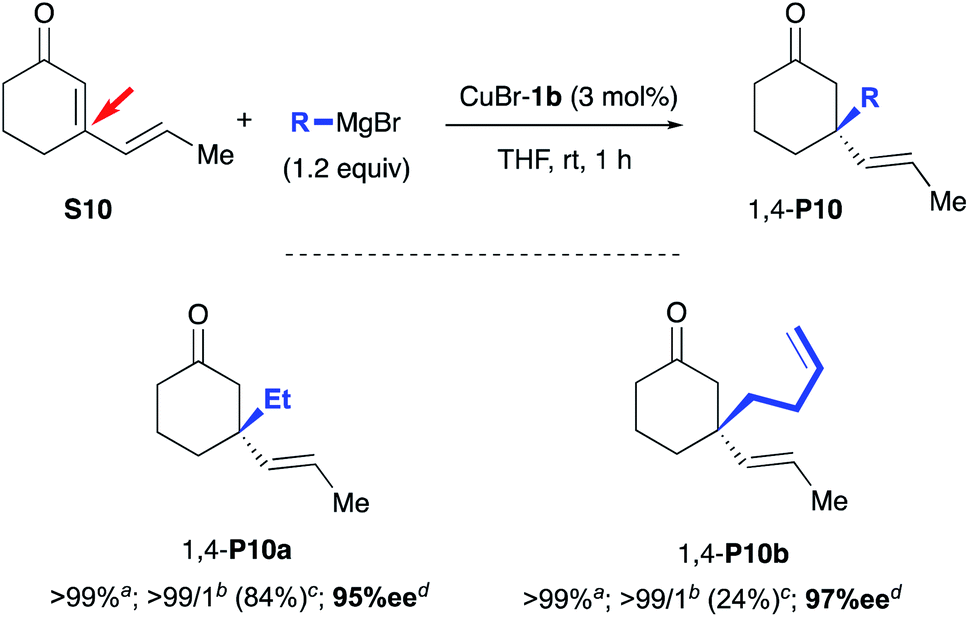 | ||
| Scheme 5 1,4-ACA of Grignard reagents to cyclic dienone S10 catalysed by CuBr-1b complex. aConversions were monitored by TLC. bRatios between 1,4:1,6-adducts were determined by 1H NMR spectroscopy analysis on the crude mixture. cIsolated yields after silica gel chromatography. dee were determined by chiral-phase GC analysis. | ||
Conclusions
In conclusion, well-defined copper(I) and gold(I) complexes containing chiral hydroxyalkyl-functionalized diamino N-heterocyclic carbenes were synthesized for the first time. The strategy involved the formation of optically pure oxazolidines (S,S)-2a,b, isolated in quantitative yield from chiral hydroxyalkyl-imidazolinium salts L1a,bH·Cl with a complete diastereoselectivity. Advantageously, in the presence of Cu(I) and Au(I) salts, these oxazolidines were converted to corresponding air-stable CuBr- and AuCl–NHC complexes in good isolated yields. Interestingly, the solid-state structures of the gold-complexes showed that hydroxyl-function is not coordinated to the metal. Computational studies suggested that formation of the oxazolidine operates by concerted O–H bond insertion and that the transient carbene is formed prior to coordination trough concerted O–H bond elimination. Furthermore, resulting copper-catalysts CuBr-1a,b afforded excellent catalytic performances in both asymmetric allylic alkylation and 1,6- or 1,4-conjugate addition involving organozinc or Grignard reagents. Excellent regioselectivities were obtained in association with high chiral inductions (up to 99% ee). Noteworthy, air-stable oxazolidines (S,S)-2a,b have also demonstrated similar catalytic performances in copper-free AAA transformations. These results will pave the way for the development of more sophisticated hydroxyalkyl-NHC catalytic systems (with or without transition metals19) and could offer new opportunities for the discovery of new catalytic reactions.Data availability
All experimental and computational data associated with this work are available in the ESI.†Author contributions
M. M. conceived, designed and directed the project. D. P.-B. and L. J. conducted all the experiments. A. C., Z. Z., L. F. and L. C. performed the DFT calculations. T. V. developed GC analysis methods while T. R. accomplished of X-ray diffraction analysis. The manuscript was written and reviewed by L. J., O. B., L. C., L. F. and M. M. The ESI† was written by D. P.-B. and L. J.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the CNRS, the Ecole Nationale Supérieure de Chimie de Rennes. Umicore AG & Co is acknowledged for a generous gift of gold complexes. This work was supported by the Agence Nationale de la Recherche (ANR-16-CE07-0019 “Hel-NHC” grant to D. P.-B.). L. J. thanks Rennes Metropole for supporting this work through the AIS grant for young researchers. L. C., L. F. and Z. Z. thank the King Abdullah University of Science and Technology (KAUST) for supporting this work. For computer time, this research used the resources of the KAUST Super-computing Laboratory (KSL) at KAUST. We are grateful to Philippe Jéhan and the CRMPO core facility for HMRS of Au- and Cu-complexes.Notes and references
- (a) For a recent book on NHCs, see: S. Díez-González, N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, Royal Society of Chemistry, Cambridge, 2011 Search PubMed; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed . For a review dealing with NHC/TM in catalysis, see:; (c) S. D. Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (d) E. Peris, Chem. Rev., 2018, 118, 9988 CrossRef CAS PubMed.
- For recent reviews on chiral NHCs in catalysis, see: (a) F. Wang, L.-J. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804 CrossRef CAS; (b) D. Janssen-Müller, C. Schlepphorst and F. Glorius, Chem. Soc. Rev., 2017, 46, 4845 RSC; (c) J. Thongpaen, R. Manguin and O. Baslé, Angew. Chem., Int. Ed., 2020, 59, 10242 CrossRef CAS PubMed.
- (a) H. Clavier, L. Coutable, J.-C. Guillemin and M. Mauduit, Tetrahedron: Asymmetry, 2005, 16, 921 CrossRef CAS; (b) H. Clavier, L. Coutable, J.-C. Guillemin and M. Mauduit, J. Organomet. Chem., 2005, 690, 5237 CrossRef CAS; (c) D. Rix, S. Labat, L. Toupet, C. Crévisy and M. Mauduit, Eur. J. Inorg. Chem., 2009, 1989 CrossRef CAS . For the one-step synthetic route, see:; (d) C. Jahier-Diallo, M. S. T. Morin, P. Queval, M. Rouen, I. Artur, P. Querard, L. Toupet, C. Crévisy, O. Baslé and M. Mauduit, Chem.–Eur. J., 2015, 21, 993 CrossRef CAS PubMed; (e) J. Wang, X. Cheng, Y. Liu and J. Zhang, J. Org. Chem., 2021, 86, 6278 CrossRef CAS PubMed.
- For a review dealing with chiral hydroxyalkyl-NHCs in Copper catalysed-ACA, see: J. Wencel, H. Hénon, S. Kehrli, A. Alexakis and M. Mauduit, Aldrichimica Acta, 2009, 42, 43 CAS.
- For a book chapter or review dealing with metal catalysed-ACA, see: (a) M. Mauduit, O. Baslé, H. Clavier, C. Crévisy and A. Denicourt-Nowicki, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, second edn, 2014, vol. 4, pp. 186–341 Search PubMed; (b) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC; (c) D. Vargova, I. Nemethova and R. Sebesta, Org. Biomol. Chem., 2020, 18, 3780 RSC . For a book chapter or reviews dealing with Cu-AAA, see:; (d) O. Baslé, A. Denicourt-Nowicki, C. Crévisy and M. Mauduit, in Copper-Catalysed Asymmetric Synthesis, ed. A. Alexakis, N. Krause and S. Woodward, Wiley-VCH, Weinheim, 2014, chapter 4, pp. 85–126 Search PubMed; (e) A. Alexakis, J. E. Backvall, N. Krause, O. Pamies and M. Dieguez, Chem. Rev., 2008, 108, 2796 CrossRef CAS PubMed; (f) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnard and B. L. Feringa, Chem. Rev., 2008, 108, 2824 CrossRef CAS PubMed.
- For selected examples with diorganozinc reagents, see: (a) T. Jennequin, J. Wencel-Delord, D. Rix, J. Daubignard, C. Crévisy and M. Mauduit, Synlett, 2010, 11, 1661 Search PubMed; (b) S. Drissi-Amraoui, M. Morin, C. Crévisy, O. Baslé, R. Marcia de Figueiredo, M. Mauduit and J.-M. Campagne, Angew. Chem., Int. Ed., 2015, 54, 11830 CrossRef CAS PubMed . For selected examples with Grignard reagents, see:; (c) D. Martin, S. Kehli, M. d'Augustin, H. Clavier, M. Mauduit and A. Alexakis, J. Am. Chem. Soc., 2006, 128, 8416 CrossRef CAS PubMed; (d) H. Henon, M. Mauduit and A. Alexakis, Angew. Chem., Int. Ed., 2008, 47, 9122 CrossRef CAS PubMed; (e) N. Germain, M. Magrez, S. Kehrli, M. Mauduit and A. Alexakis, Eur. J. Org. Chem., 2012, 5301 CrossRef CAS; (f) M. Tissot, D. Poggiali, H. Hénon, D. Müller, L. Guénée, M. Mauduit and A. Alexakis, Chem.–Eur. J., 2012, 18, 8731 CrossRef CAS PubMed; (g) M. Magrez, Y. Le Guen, O. Baslé, C. Crévisy and M. Mauduit, Chem.–Eur. J., 2013, 19, 1199 CrossRef CAS PubMed . For selected examples with boron nucleophiles, see:; (h) R. Shintani, K. Takatsu, M. Takeda and T. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 8656 CrossRef CAS PubMed; (i) F. Meng, X. Li, S. Torker, Y. Shi, X. Shen and A. H. Hoveyda, Nature, 2016, 537, 387 CrossRef CAS PubMed; (j) C.-Y. Shi, Z.-Z. Pan, P. Tian and L. Yin, Nat. Commun., 2020, 11, 5480 CrossRef CAS PubMed . For selected examples with aluminium reagents, see:; (k) D. Gillingham and A. H. Hoveyda, Angew. Chem., Int. Ed., 2007, 46, 3860 CrossRef CAS PubMed; (l) Y. Lee, K. Akiyama, D. G. Gillingham, M. K. Brown and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 446 CrossRef CAS PubMed.
- (a) J. Y. W. Mak and C. M. Williams, Chem. Commun., 2012, 48, 287 RSC; (b) F. Meng, K. P. McGrath and A. H. Hoveyda, Nature, 2014, 513, 367 CrossRef CAS PubMed; (c) Y. Kanda, H. Nakamura, S. Umemiya, R. K. Puthukanoori, V. R. M. Appala, G. K. Gaddamanugu, B. R. Paraselli and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 10526 CrossRef CAS PubMed.
- When Grignard reagents are involved, the use of base is not required, see ref. 6c and 6d.
- S. Drissy-Amraoui, T. E. Schmid, J. Lauberteaux, C. Crévisy, O. Baslé, R. Maria de Figueiredo, S. Halbert, H. Gérard, M. Mauduit and J.-M. Campagne, Adv. Synth. Catal., 2016, 358, 2519 CrossRef.
- P. L. Arnold, I. J. Casely, Z. R. Turner and C. D. Carmichael, Chem.–Eur. J., 2008, 14, 10415 CrossRef CAS PubMed.
- Y.-S. Wong, C. Marazano, D. Gnecco, Y. Génisson, A. Chiaroni and B. C. Das, J. Org. Chem., 1997, 62, 729 CrossRef CAS PubMed.
- For the intermolecular insertion of alcohols, see for instance: S. Csihony, D. A. Culkin, A. C. Sentman, A. P. Dove, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 9079 CrossRef CAS PubMed.
- An oxidative/reductive elimination process was recently reported with cyclic(alkyl)(amino)carbenes, see: D. R. Tolentino, S. E. Neale, C. J. Isaac, S. A. Macgregor, M. K. Whittlesey, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2019, 141, 9823 CrossRef CAS PubMed.
- R. Tarrieu, A. Dumas, J. Thongpaen, T. Vives, T. Roisnel, V. Dorcet, C. Crévisy, O. Baslé and M. Mauduit, J. Org. Chem., 2017, 82, 1880 CrossRef CAS PubMed.
- For selected examples of copper-free AAA, see: (a) Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2006, 128, 15604 CrossRef CAS PubMed; (b) Y. Lee, B. Li and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 11625 CrossRef CAS PubMed; (c) O. Jackowski and A. Alexakis, Angew. Chem., Int. Ed., 2010, 49, 3346 CrossRef CAS PubMed; (d) D. Grassi and A. Alexakis, Adv. Synth. Catal., 2015, 357, 3171 CrossRef CAS.
- Any attempt to identify/characterize the active catalytic species involved in the copper-free AAA failed. Nevertheless, based on ref. 15b, we speculated that a Zn(II)hydroxylakyl-chelating NHC species is formed.
- For seminal mechanistic insights highlighting the difference between NHC-Cu and the NHC Cu-free catalytic system in AAA, see ref. 15b.
- Similar Au-complexes were recently found to catalyse efficiently alkoxycyclization of 1,6-enynes: H. F. Jónsson, A. Orthaber and A. Fiksdahl, Dalton Trans., 2021, 50, 5128 RSC.
- Racemic NHC-alcohol adducts demonstrated efficiency in organocatalytic ring-opening polymerisation of lactide, see ref. 12.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures NMR spectra, GC analyses. CCDC 2090324–2090326. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02908a |
| This journal is © The Royal Society of Chemistry 2022 |