
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlexible organic crystals. Understanding the tractable co-existence of elastic and plastic bending†
Indira S.
Divya
ab,
Saravanan
Kandasamy
c,
Shodai
Hasebe
d,
Toshiyuki
Sasaki
 e,
Hideko
Koshima
*f,
Krzysztof
Woźniak
*c and
Sunil
Varughese
*ab
e,
Hideko
Koshima
*f,
Krzysztof
Woźniak
*c and
Sunil
Varughese
*ab
aChemical Science and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology, Thiruvananthapuram, 695019, India
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India. E-mail: s.varughese@niist.res.in
cCrystallochemistry Laboratory, University of Warsaw, Warsaw 02-093, Poland. E-mail: kwozniak@chem.uw.edu.pl
dDepartment of Advanced Science and Engineering, Waseda University, Tokyo 162-8480, Japan
eGraduate School of Nanobioscience, Yokohama City University, Kanagawa 236-0027, Japan
fResearch Organization for Nano and Life Innovation, Waseda University, Tokyo 162-0041, Japan. E-mail: h.koshima@kurenai.waseda.jp
First published on 5th July 2022
Abstract
As an emerging class of flexible materials, mechanically bendable molecular crystals are broadly classified as elastic or plastic. Nevertheless, flexible organic crystals with mutually exclusive elastic and plastic traits, with contrasting structural requirements, co-existing under different stress settings are exceptional; hence, it is imperative to establish the concurring factors that beget this rare occurrence. We report a series of halogen-substituted benzil crystals showing elastic bending (within ∼2.45% strain), followed by elastoplastic deformation under ambient conditions. Under higher stress settings, they display exceptional plastic flexibility that one could bend, twist, or even coil around a capillary tube. X-ray diffraction, microscopy, and computational data reveal the microscopic and macroscopic basis for the exciting co-existence of elastic, elastoplastic, and plastic properties in the crystals. The layered molecular arrangement and the weak dispersive interactions sustaining the interlayer region provide considerable tolerance towards breaking and making upon engaging or releasing the external stress; it enables restoring the original state within the elastic strain. Comparative studies with oxalate compounds, wherein the twisted diketo moiety in benzil was replaced with a rigid and coplanar central oxalate moiety, enabled us to understand the effect of the anisotropy factor on the crystal packing induced by the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯C tetral interactions. The enhanced anisotropy depreciated the elastic domain, making the oxalate crystals more prone to plastic deformation. Three-point bending experiments and the determined Young's moduli further corroborate the co-existence of the elastic and plastic realm and highlight the critical role of the underlying structural elements that determine the elastic to plastic transformation. The work highlights the possible co-existence of orthogonal mechanical characteristics in molecular crystals and further construed the concurrent role of microscopic and macroscopic elements in attaining this exceptional mechanical trait.
O⋯C tetral interactions. The enhanced anisotropy depreciated the elastic domain, making the oxalate crystals more prone to plastic deformation. Three-point bending experiments and the determined Young's moduli further corroborate the co-existence of the elastic and plastic realm and highlight the critical role of the underlying structural elements that determine the elastic to plastic transformation. The work highlights the possible co-existence of orthogonal mechanical characteristics in molecular crystals and further construed the concurrent role of microscopic and macroscopic elements in attaining this exceptional mechanical trait.
Introduction
Organic crystalline materials attract immense academic and industrial interest because of their exciting physicochemical properties and unique utilities in an ample repertoire of applications such as optoelectronics,1,2 mechanical actuators,3,4 pharmaceuticals,5,6 and explosives.7,8 From the practical application perspective, however, the materials need to attain excellent workability; in this regard, mechanical deformability (plastic or elastic) is indispensable.9,10 The mechanically inflexible and brittle attributes of molecular crystalline materials at the macroscopic level au contraire limit their utility; it makes them far from the stage of materials development compared to flexible polymers exhibiting viscoelasticity.11,12 Not surprisingly, immense interest has been devoted to comprehending the mechanical properties of molecular materials in recent times.13–17 Momentous progress attained in this field owes much to the systematic and earnest efforts made to gain significant insights into precise mechanisms that govern the structural and mechanical properties of molecular materials.18 Such understanding is imperative to attain predictable mechanical properties in crystalline materials.Simple and easy-to-characterize molecular crystals of a related set of compounds with limited variables provide a good starting point for a regressive analysis to evaluate various contributing elements to the observed mechanical traits.19 To date, elasticity and plasticity in crystals are perceived in terms of molecular packing and the relative distribution of interaction types having varying strengths within the crystal. The isotropic distribution of robust electrostatic interactions and close-packed molecular arrangements add brittleness to the crystals, whereas weak dispersive interactions favor elasticity.20,21 Layered molecular arrangement with an orthogonal distribution of strong and weak interactions ensues plasticity in the system.22,23 Further to the aforesaid critical structural determinants, other factors such as the solvent of crystallization,24,25 structural rugosity,26 globularity of the molecules,16,27 and aspect ratio of the crystals3 also could contribute to the mechanical properties of molecular crystals. Also, one could employ crystal engineering strategies to tune the mechanical properties of molecular crystals.28–30
Interestingly the elasticity and plasticity are orthogonal mechanical properties, having contrasting structural requirements and contributing factors. Their co-existence hence appears contradictory and is exceptional.31–36 The genesis of the present study is rooted in an earlier report on the co-existence of elasticity and plasticity in the crystals of bromobenzil,31 wherein crystals notably undergo reversible to irreversible bending under the varying extent of applied stress. Thus, the deformation becomes irreversible beyond the elastic domain, rather than breaking. In the present work, we explore this unique property and the scope of the generality of the observation. We studied a series of benzil compounds (Scheme 1) and a related set of oxalate compounds having halogen substituents in the p-carbon atom; most compounds have comparable global packing mode—layered structures. In the course of the study, we identified a novel crystal form of the F-derivative (BZF), and the dimorphs exhibit distinct mechanical responses under applied stress. The benzil crystals show intense emission at the crystal ends, in both straight and bent states, compared to the central areas, hinting their optical wave guiding properties. Such molecular crystals with exceptional flexibility and emitting characteristics are rare. This work aims at expounding the microscopic and macroscopic factors that contribute to the rare and exceptional co-existence of mutually orthogonal mechanical traits.
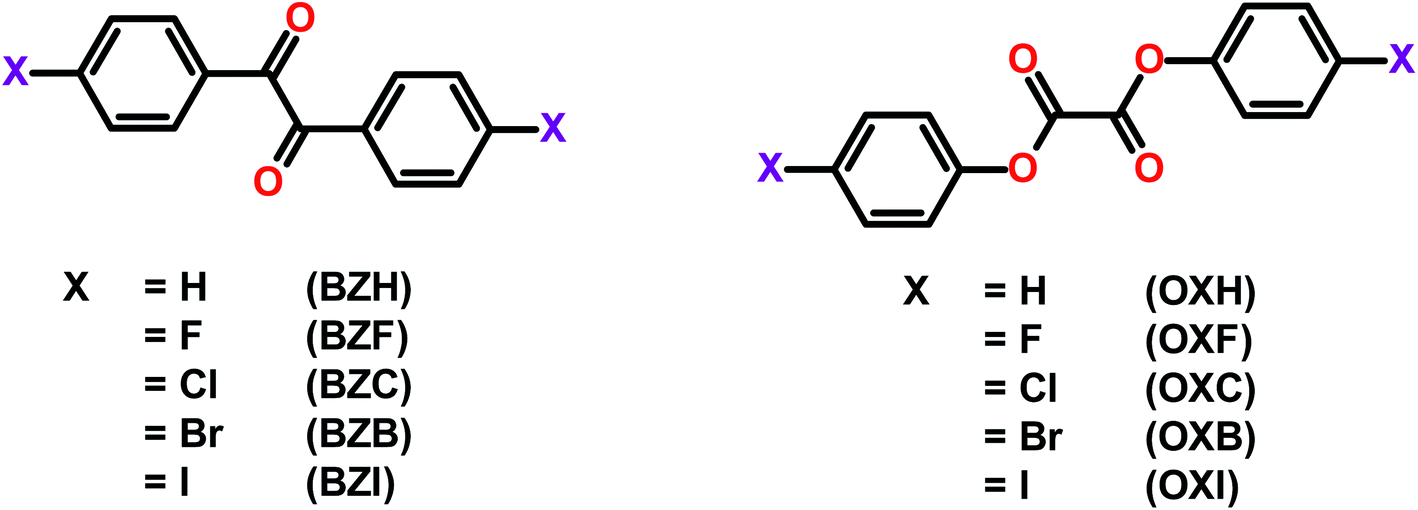 | ||
| Scheme 1 The molecular structures of the benzil and oxalate compounds studied in the present contribution. | ||
Results and discussion
To verify the structural relationships and the mechanical properties, we investigated the crystal structure of the benzil compounds with a halogen substituted in the p-position of the phenyl rings, analogous to the Br-derivative reported in a previous publication. Slow evaporation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dichloromethane:methanol/ethanol solutions yielded the long tape-like crystals of BZC and BZB. The methanol solution of the fluoro-derivative yielded pale yellow block-like crystals of BZF_O, while a 1:1 dichloromethane:methanol mixture yielded long tape-like crystals of BZF (Table S1†).
:1 dichloromethane:methanol/ethanol solutions yielded the long tape-like crystals of BZC and BZB. The methanol solution of the fluoro-derivative yielded pale yellow block-like crystals of BZF_O, while a 1:1 dichloromethane:methanol mixture yielded long tape-like crystals of BZF (Table S1†).
Structure and mechanical properties of benzil compounds
Under applied mechanical stress, thin tape-like crystals of the benzil compound BZB exhibit good elasticity. The crystals of BZB could bend elastically into a loop by three-point bending methods.Small deformation stress, leading to a bending strain (ε) of ∼2.45% on a crystal with a thickness of ∼65 μm, resulted in elastic deformation (Table S4†). The bending in the BZB crystal is associated with the {020} face, while it cracked when other faces were deformed. The bending behavior is repeatable without fatigue. At excessive ε beyond ∼2.45%, the bending of the crystal does not lead to its breakage but macroscopic plastic deformation (Fig. S1†). Akin to the recent observation of plastic bending with some extent of elastic recovery,37 the crystals of BZB show partial elastic recovery even after the crystals are plastically deformed (ε = 1.26%). The elastic to plastic bending characteristics and the elastoplastic trait of BZB can be intuitively observed in the movie S1 of the ESI.† Thus, the crystal is an archetype system wherein the two distinct mechanical traits with an orthogonal structural origin co-exist. The BZC crystals exhibit similar behavior, wherein both the elastic and plastic properties co-exist. The elasticity in BZC is associated with the {002} crystal face, and akin to BZB, excess mechanical stress (>ε = 2.39%) leads to plastic deformation (Fig. S2†). The benzil crystals can be looped, and the bending–relaxation process is repeatable, highlighting the excellent elasticity. Beyond the elastic strain, however, plastic deformation ensues, and the crystals deform irreversibly into contorted shapes, twisted, or even coiled around a capillary tube in the plastic regime (Fig. 1c; movie S2†).
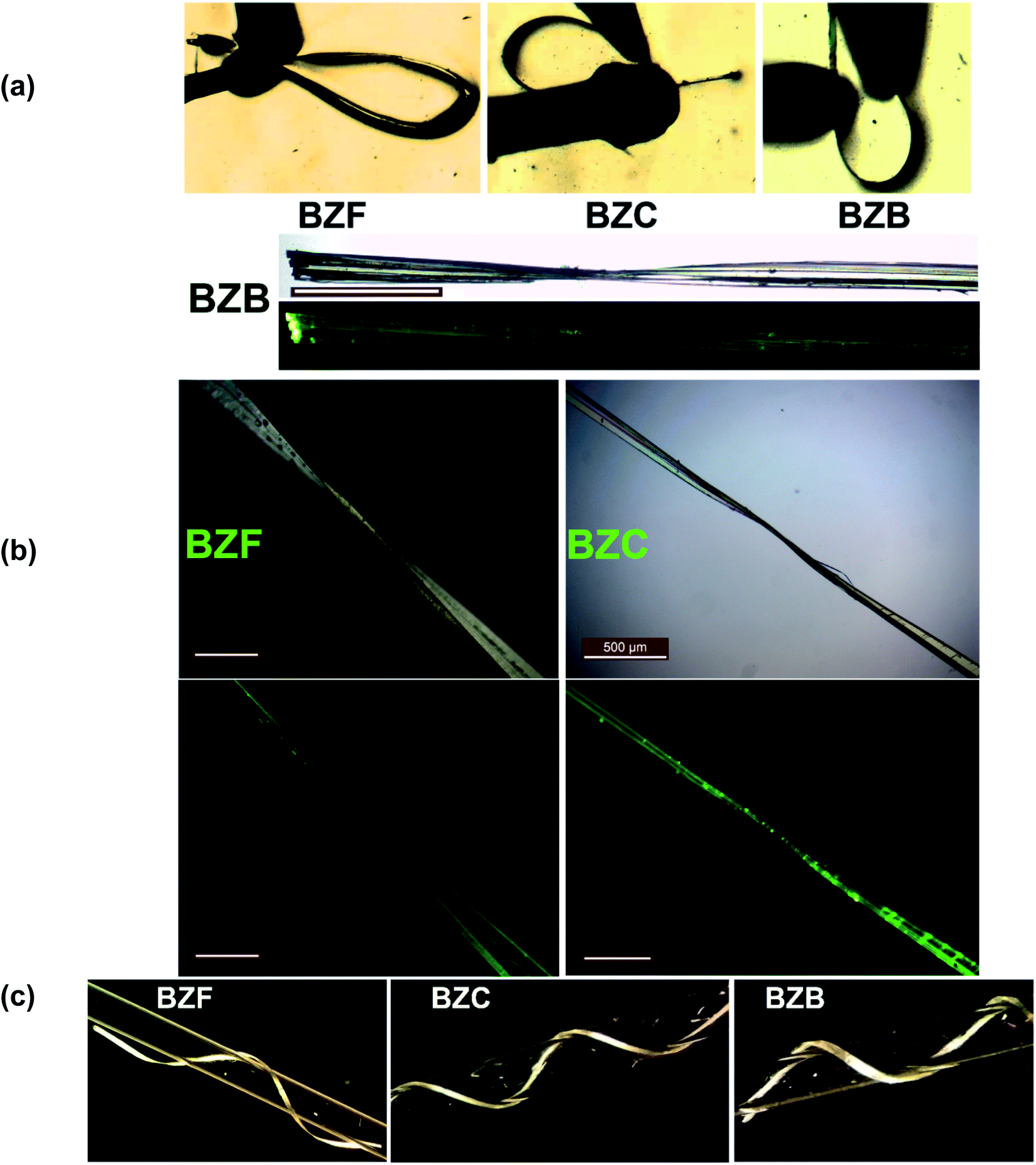 | ||
| Fig. 1 The crystals of BZF, BZC, and BZB under different deformation stages. (a) Elastic, (b) twisted and (c) coiled around a capillary tube. The scale bar corresponds to 500 μm. | ||
The polymorphs of the F-derivative (BZF_O and BZF; Fig S4†) exhibit distinct mechanical properties. While block-like crystals of BZF_O are brittle, the long tape-like crystals of BZF could be mechanically deformed based on the extent of the applied stress. Besides getting deformed elastically to a loop (ε = 2.36%) (Fig. S3†), the crystals of BZF can also be plastically twisted (twist angle of ∼180°; Fig. 1b), wrapped (out of plane) (Fig. S6b†), or coiled around a capillary (Fig. 1c). The crystal face associated with the bending in BZF is {020}. BZF_O, in contrast, has a close-packed molecular arrangement and an isotropic distribution of the interactions, making it devoid of any energetically viable slip plane, and hence brittle (Fig. 2b).
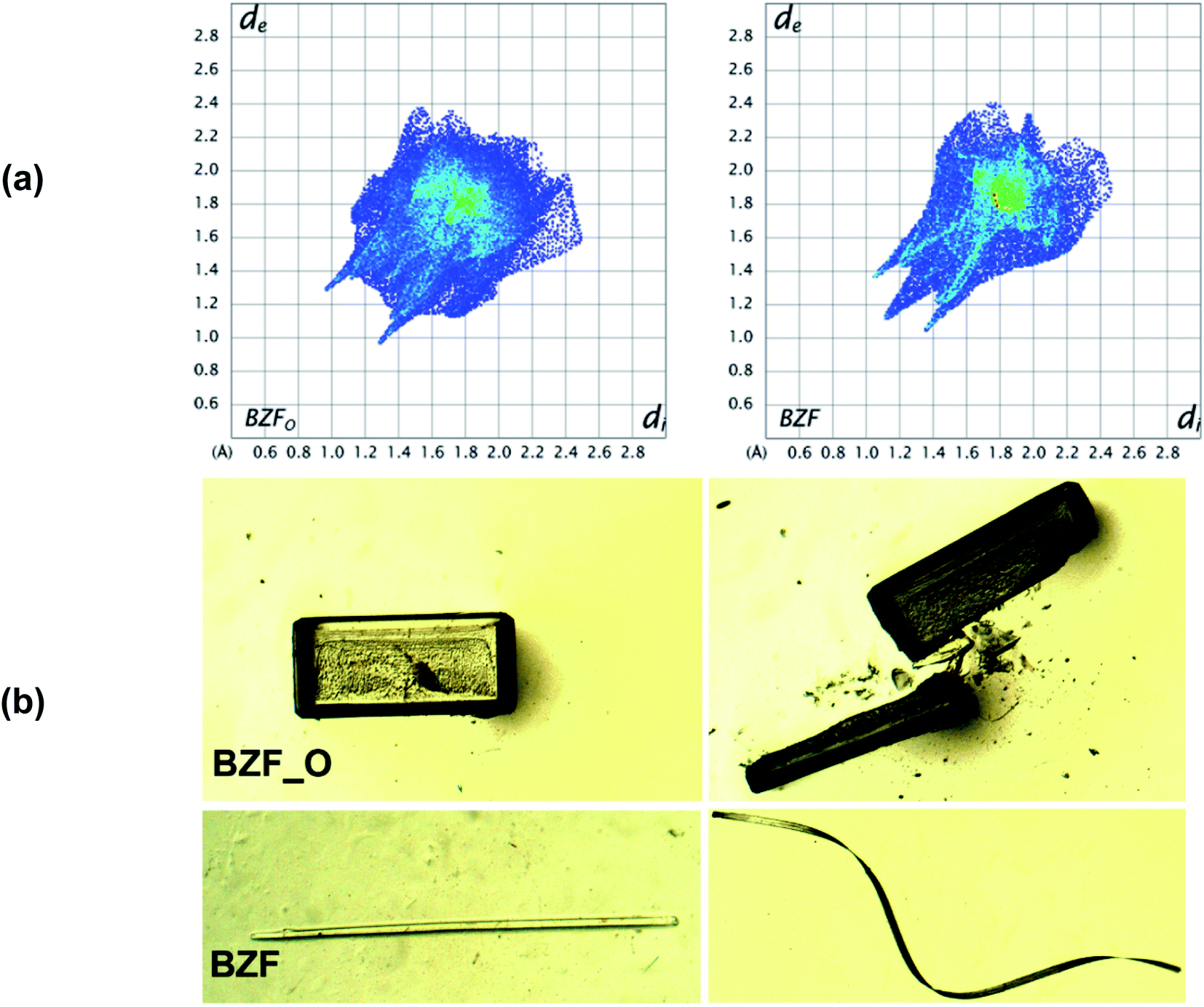 | ||
| Fig. 2 The dimorphs of the benzil F-derivative (BZF_O and BZF): (a) HS fingerprint plot highlights the difference in the interaction characteristics; (b) distinct mechanical characteristics of the dimorphs. | ||
To gain insights into the molecular-level mechanism of the intricate co-existence of elasticity and plasticity in the crystals, we analyzed the crystal structure of the benzil compounds in terms of crystal packing and interactions. In the Br-derivative (BZB), the measured torsion about the diketo (OC–CO) moiety is 116°, whereas the planes containing the aromatic rings describe an angle of 57° (Table S5†). The molecules adopt a stacked-layered arrangement. Within a layer, the molecules form a grid-like arrangement in the ac-plane, and along [010], adjacent grids interweave through a network of C–H⋯O (2.49 Å) and π⋯π (3.54 Å) interactions (Fig. 3b). The interlayer region with type-II Br⋯Br (θ1 = 162 and θ2 = 95°) and C–H⋯Br (3.19 Å) interactions form potential slip planes parallel to {010}. The I-derivative (BZI) is isostructural to BZB and has similar interaction types and packing arrangements. In the chloro-compound (BZC), the overall packing adopted by the molecule is comparable to BZB and can be described as a stacked-layered type. In a unique molecule of BZC, the diketo moiety forms bifurcated C–H⋯O (2.47 and 2.70 Å) hydrogen bonds with three molecules (two from the same grid and one from the adjacent one). Further to C–H⋯O hydrogen bonds, π⋯π (average 3.45 Å) interactions also significantly contribute to layer formation. The (001) defines the interlayer region dominated by Type I Cl⋯Cl (θ1 = θ2 = 164°) and C–H⋯Cl (3.06 Å) interactions, which form the slip plane.
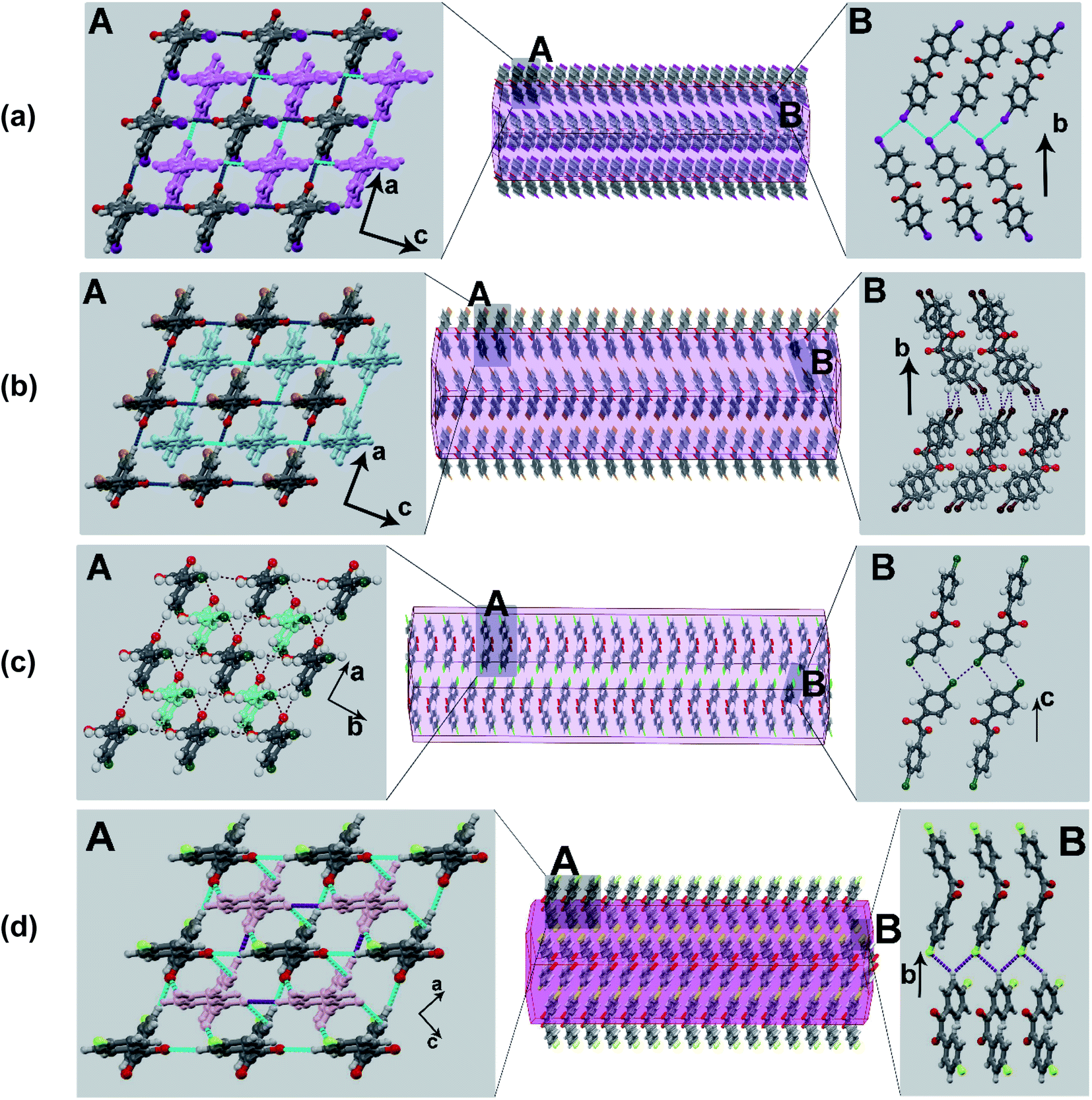 | ||
| Fig. 3 The computed crystal morphology and molecular packing. (a) BZI; (b) BZB; (c) BZC; (d) BZF. Note the similar stacking arrangement though the interlayer region shows distinct interaction types. | ||
The known crystal form of the F-derivative (BZF_O) has a close-packed structure, distinct from the rest in the series. As the diffraction studies establish, the acicular crystals of the novel polymorph BZF are isostructural to the rest of the halo-derivatives. The compound crystallizes in an orthorhombic P21212 space group with an overall packing described as a stacked-layered type. Within a layer, molecules form square grid-like structures along the (101) plane, and the adjacent grid structures interweave through C–H⋯O hydrogen bonds along [001]. Each diketo moiety in the compound form bifurcated C–H⋯O (2.54 and 2.78 Å) hydrogen bonds. Further to C–H⋯O hydrogen bonds, π⋯π (average 3.60 Å) interactions also significantly contribute to layer formation. In [010], the layers are connected through C–H⋯F (2.73 and 2.75 Å) hydrogen bonds, wherein each F atom forms bifurcated interactions. The twistability of BZF crystals is rooted in the two-directional molecular orientation and multifaceted bending, which enable the crystals to bend in two different directions.
Thus, in the halo-derivatives of benzils, the overall structural topology remains comparable, though there is a notable variation in the interaction types when analyzed in greater detail.38 The CO of the diketo moiety forms either bifurcated C–H⋯O hydrogen bonds (as in BZC and BZF) or single C–H⋯O interaction (as in BZB and BZI). Besides the C–H⋯O hydrogen bonds, all the compounds have π⋯π interactions as a major stabilizing force, though their average distance increases as one moves from BZB to BZF in the series. The interlayer region that constitutes the slip planes has distinct interactions. BZB has type-II Br⋯Br together with C–H⋯Br interactions, while type I Cl⋯Cl and C–H⋯Cl interactions occupy the interlayer region in BZC. In BZF, C–H⋯F interactions organize the interlayer region. Thus, the benzil compounds show a systematic transition in the interaction type and energetics in the interlayer region. In contrast, the stacked-layered arrangement and the interlayer region that sustained through weaker interactions provide the impetus to deform irreversibly beyond the elastic realm. The weak X⋯X and C–H⋯X interactions afford initial resistance to a long-range slippage of layers along the slip planes, facilitating initial elasticity followed by plasticity at higher strains.
To further explore the nuance and the intricate relationship of the structure and mechanical properties, we correlated them with a series of oxalate compounds with a halogen substituted at the p-position. In the series, we wanted to explore the possible anisotropy brought about by the π-hole interactions by the central oxalate moiety. The oxalate functionality remains planar, though it is near orthogonal to the coplanar aromatic rings.39 The coplanar aromatic rings are optimal for forming π⋯π interactions. Thus with weaker interactions in the interlayer region, the system develops enough anisotropy to alter the subtle balance between elasticity and plasticity. The torsional shifts in the compounds are provided in Table S6.†
Structure and mechanical properties of oxalate compounds
Oxalate crystals also exhibit the property of the co-existence of elastic and plastic deformation, though with a considerably depreciated elastic domain compared with the benzil analogs. The block-like crystals of OXH and needles of OXF are brittle and crack under applied mechanical stress. However, the crystals of OXC and OXB can be elastically bent into semicircles; but they fail to curl into a loop (movies 3 and 4†). The crystals do not crack beyond the elastic strain limit but deform plastically (Fig. S9 and S10†). The plastic deformation is dominant in the oxalate series with notably less elastoplastic characteristics than the benzil compounds. Furthermore, an increase in the thickness of crystals diminishes the elastic region making the crystals undergo plastic deformation directly. Though the crystals of oxalates deform plastically under applied stress, they are not viable for twisting. The OXI crystals do not bend; instead, they can be sheared along the {002} plane, which is defined by I⋯I interactions (Fig. 4). The observed mechanical properties in the oxalate can be correlated with microscopic structural factors such as crystal packing and interaction types. | ||
| Fig. 4 The mechanical properties in the oxalate compounds. The OXF crystals are brittle, while OXC and OXB show plastic bending. The block-like crystals of OXI undergo shearing under applied stress. | ||
OXB and OXC have similar structures with the formation of a stacked structure and the morphology as long thin tape-like crystals with {100} as the prominent face (Fig. 5). Along the [010], OXB molecules form linear chains through π⋯π (3.46 Å) and CO⋯C (3.20 Å) interactions. The structure is defined by the distribution of relatively more robust interactions along the bc-plane; type-I Br⋯Br (θ1 = θ2 = 153°) and C–H⋯Br (3.34 Å and 3.48 Å) define the interlayer region, the (100) plane, which acts as the slip plane. OXC has a layered structure wherein adjacent chains are connected along [001] through bifurcated C–H⋯O (2.65 and 2.71 Å) and C–H⋯Cl (2.93 Å) interactions. The (100), which corresponds to the slip plane, contains the type-I Cl⋯Cl (θ1 = θ2 = 162°) contacts and weak dispersive C–H⋯Cl (3.11 Å) and C–H⋯π (3.10 Å) interactions. In OXF, molecules form linear chains through centric R22(12) C–H⋯O (2.66 Å) hydrogen bonds along [100] and further stacked through π⋯π (3.54 Å) interactions along [001]. A network of weak dispersive interactions (C–H⋯O (2.66 Å), F⋯C (3.05 Å), and C–H⋯π (3.03 Å)) along [011] leads to an intercalated structure, wherein the stacking of linear chains leads to the formation of a close-packed structure.
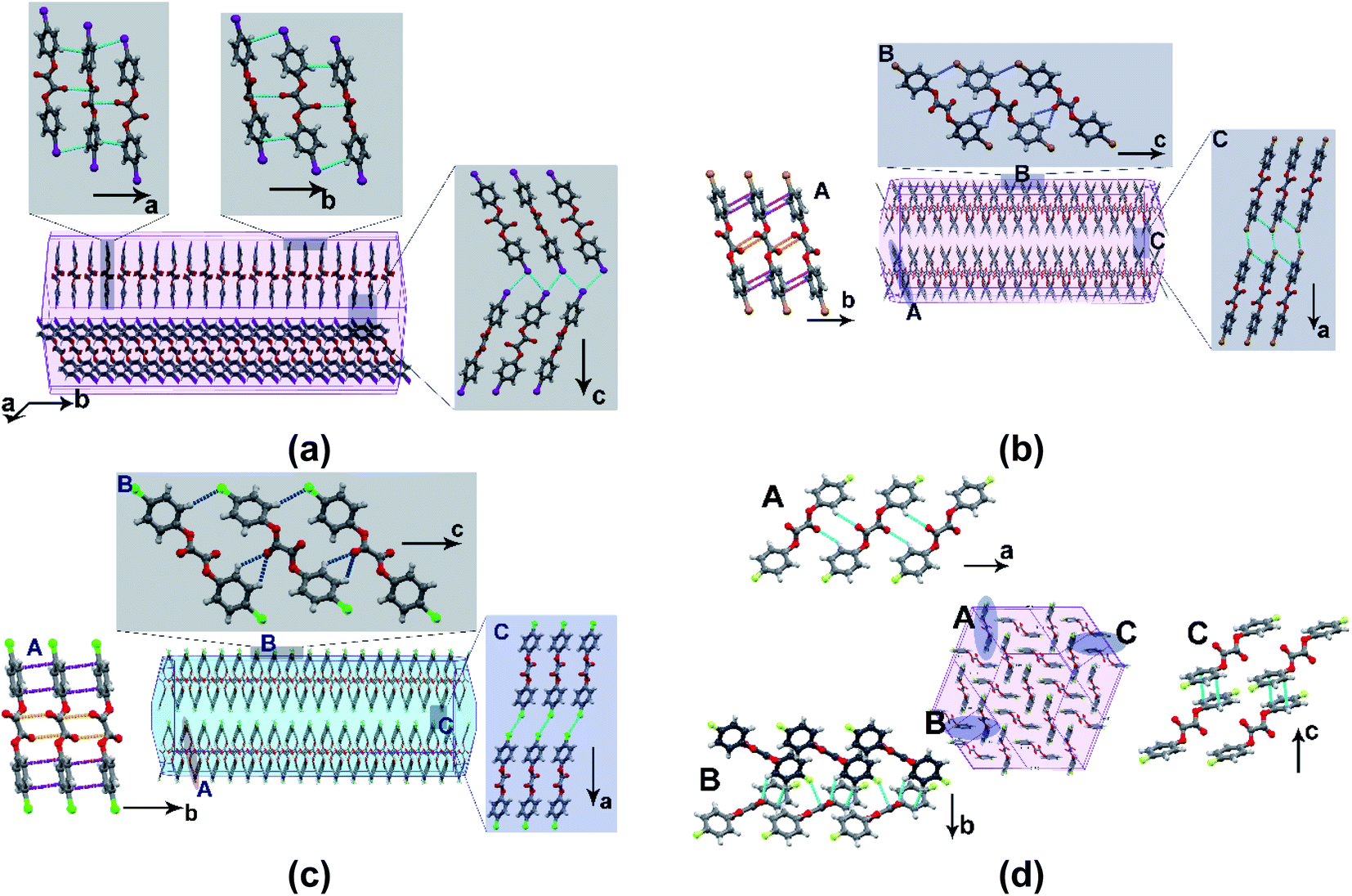 | ||
| Fig. 5 The crystal morphology and molecular packing: (a) OXI; (b) OXB; (c) OXC; (d) OXF. | ||
In the stacked-layered structure of OXI, a layer consists of zig-zag chains (along [010]) of the molecules connected through π⋯π (3.37 and 3.25 Å), CO⋯C (2.95 and 2.97 Å), and C–H⋯O (2.79 and 2.86 Å) interactions. These zig-zag chains further connect through π⋯π (3.47 and 3.41 Å), CO⋯C (3.07 and 3.17 Å), and C–H⋯O (2.80 Å) interactions along [100] to form a two-dimensional layer structure. The interlayer region is stabilized through a network of type-II I⋯I (C–I⋯I angle of 165 and 114°) interactions. OXH, unlike the rest of the series, has the phenyl substitution in syn-conformation (Table S5†), and the aromatic rings are twisted with respect to each other (22.56°). The brick-wall crystal packing and the criss-cross arrangement of interactions make OXH brittle.
Quantifying the mechanically induced flexibility: three-point bending method
To complement the qualitative analysis of the crystals' flexible nature and quantify the stress required to enable elastic to plastic transformation, we did three-point bending experiments using a universal material testing machine (the experimental details are provided in the ESI†) under ambient conditions. The three-point bending studies utilized acicular crystals with a dimension of ∼3.5 mm as the longest axis and a thickness of ∼80 μm. The calculated stress–strain curve (Fig. 6) shows a typical linear strain–stress profile corresponding to the elastic regime. The elastic domain ends with a plastic realm rather than a fracture; it validates the elastic to plastic transformation of the crystals under higher stress conditions.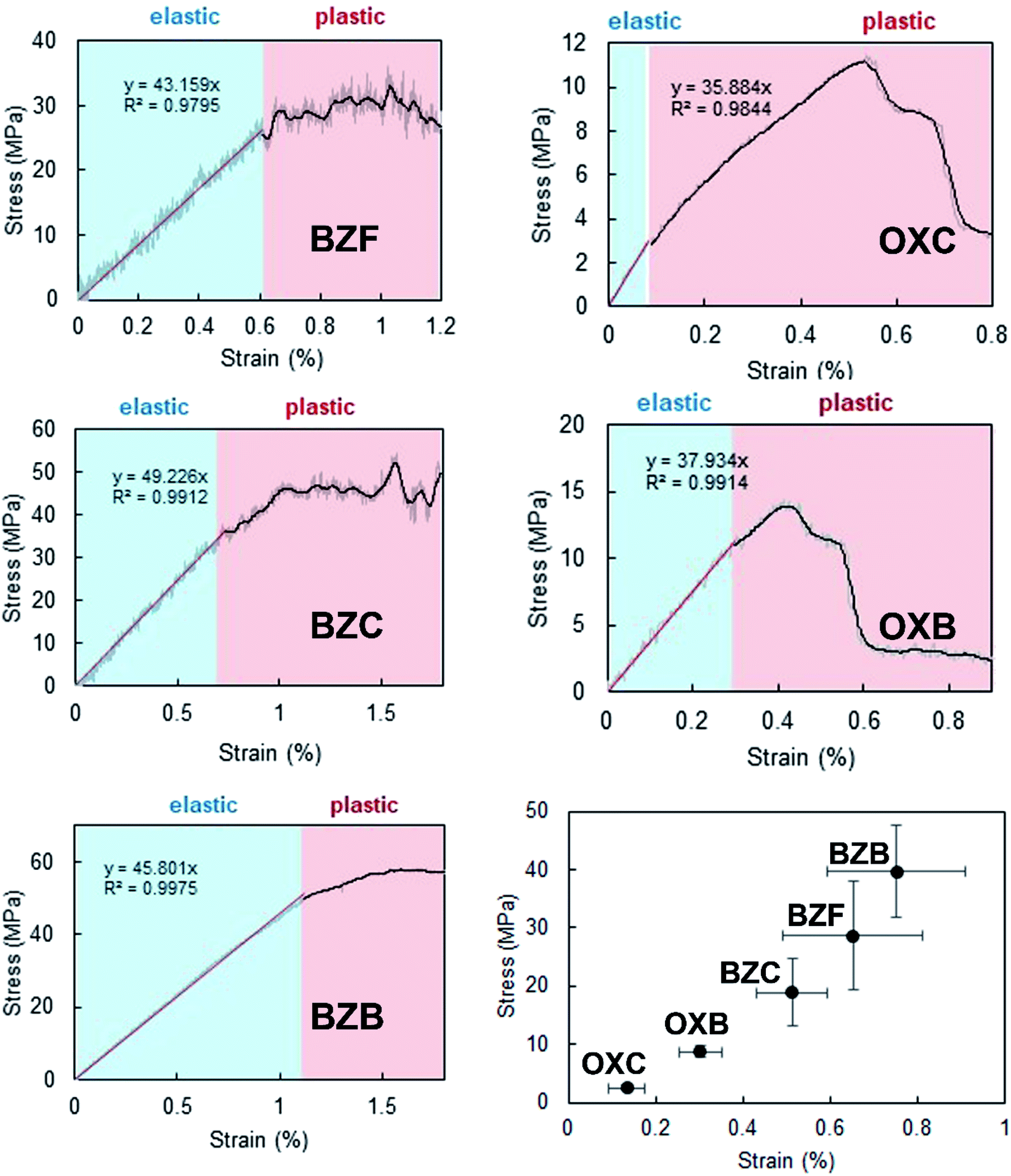 | ||
| Fig. 6 Representative stress–strain curves of different crystals. The elastic and plastic regimes are highlighted in blue and red, respectively. The high magnitude of stress against strain for the benzil crystals highlights their significant elastic recovery before the plastic transformation compared to the oxalate systems (OXC and OXB). | ||
The computed mean Young's moduli of benzil crystals are larger than those of the oxalate crystals (Table 1). Hence, more significant strain and stress are required to deform the benzil crystals plastically than the oxalate counterparts. Also, the results highlight a more extensive elastic regime for the benzil crystals vis-à-vis the oxalate crystals, enabling their elastic looping before deforming plastically (Fig. 6). The smaller elastic domain associated with the crystals of oxalate compounds validates their greater tendency to deform plastically. While considering the benzil crystals, Young's moduli are in the order BZB > BZF > BZC and lie in the range of 3.5 to 5.5 GPa, indicating that the crystals are soft and conducive to elastic deformation. The trend correlates with a similar topology of the energy framework in BZB and BZF. The interwoven network of dispersive C–H⋯F interactions sustaining the interlayer region (Fig. 7) provides BZF with an attractive force to maintain reversibility in the transformation within the elastic realm (vide post). Furthermore, the layer separation about the slip regions is in the order BZB > BZC > BZF; the shorter layer separation in BZF leads to a greater extent of molecular friction, resulting in a constrained relative layer movement under lower stress conditions.
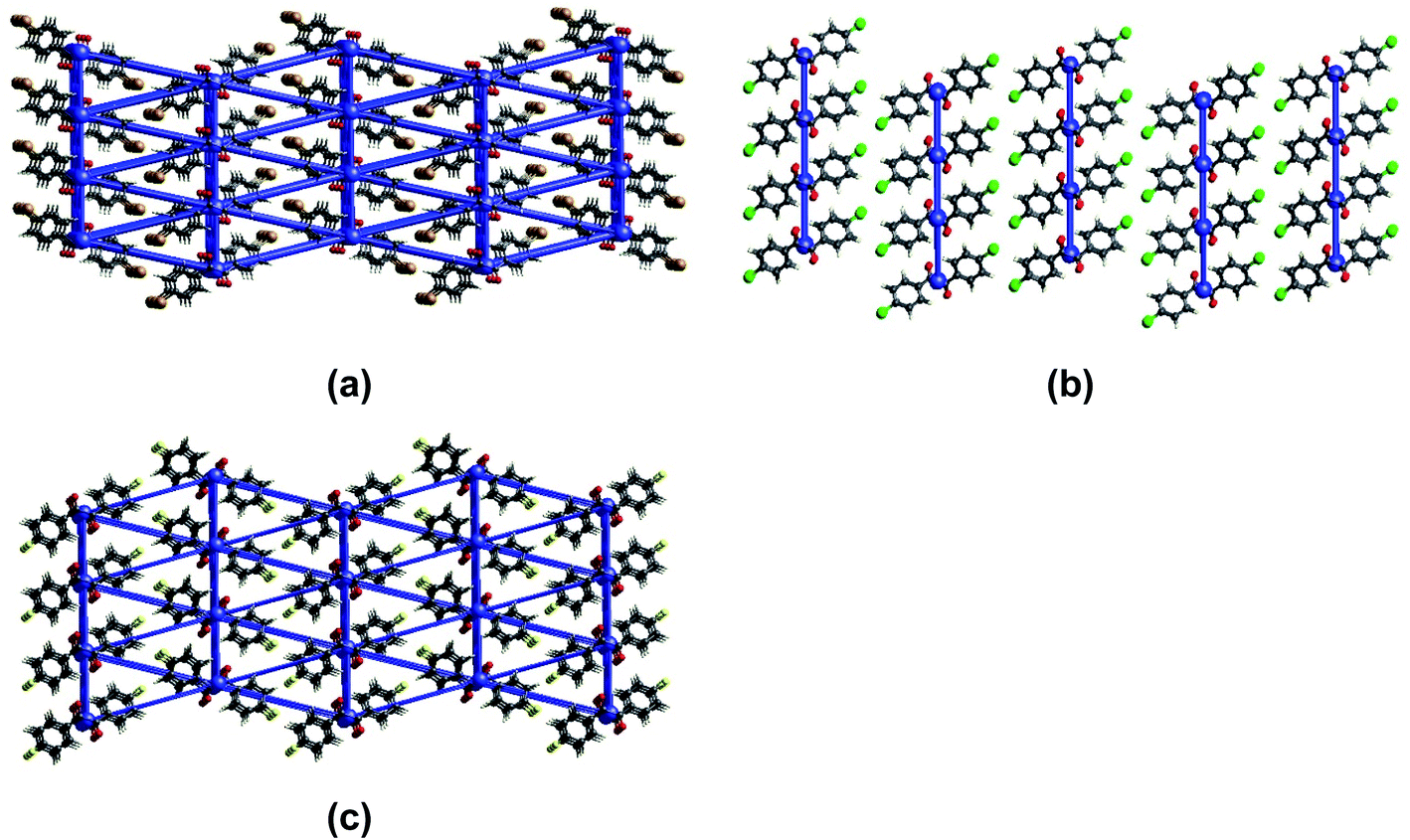 | ||
| Fig. 7 The energy frameworks calculated for (a) BZB, (b) BZC, and (c) BZF. Note the interlayer regions being interwoven by weak dispersive interactions in BZB and BZF, as against a 2D topology observed in BZC. | ||
The structural elements that contribute to the mechanical properties
As manifested by twistable crystals, multiple bendable faces involve more crystal isotropy—a requirement for elasticity; simultaneously, it opposes the prerequisite of plastic bending—the interaction anisotropy. Thus, two contrasting mechanical characteristics co-exist in the crystals. We explored various microscopic structural and macroscopic morphological factors to gain insights into this unique co-existence. The structures are devoid of classical electrostatic interactions; hence, it is critical to tune the relative distribution and the subtle balance of interactions and further comprehend the anisotropy factor in determining the macroscopic mechanical properties. Towards this, we explored the role of the linker moiety (Z) in the X–Ar–Z–Ar–X compound types in attaining a non-planar conformation, modifying the interaction types, and inducing their influence on the structural anisotropy.O⋯C interactions compared to the twisted diketo moiety in benzil compounds. Thus, the O⋯C contribution is >50% higher in a unique oxalate compound in comparison to its analogous benzil derivative (Fig. S12†).
The interaction types established by the halogen atoms secure the interlayer region. The fluorine in BZF forms a network of C–H⋯F interactions in the interlayer region with a prominent contribution in the structure formation (26.9%) as against 16.2% in OXF—a notable 66% increase. Considering the interactions involving the Cl atom, the Cl⋯Cl interaction stands out in OXC as against C–H⋯Cl in BZC. The contribution of Br⋯Br in both OXB and BZB is comparable; the benzil compound, in addition, has a significant contribution of C–H⋯Br interaction (20%). The enhanced contribution of the X⋯H interactions in the benzil compounds can be attributed to their twisted conformation, and these interactions in the interlayer region help to ease the structural anisotropy.
The HS, when mapped along with unique interactions, provides information on the nature and strength of interactions, which is critical in determining the mechanical properties (Fig. 8). The HS of OXH has more contact points with the neighboring molecules than BZH. The electron-rich F-atom in OXF forms π-hole C–F⋯CO (3.05 Å) interactions with the sp2 C-atom of the carbonyl group (Fig. 8). In the rest of the halo-derivatives of oxalates, the carbonyl group forms carbonyl–carbonyl (CO⋯CO) tetral bonds. The OXC and OXB form CO⋯CO contacts, and depending on the relative orientation of the interacting carbonyl groups, they are classified as motif-II: C⋯OC distances (dCO) are 3.24 and 3.10 Å and have an O⋯CO angle of 84 and 83°, respectively.41 The measured angle of O⋯CO is lower than the average value of 92.6° and that of the Burgi–Dunitz (BD) angle (107°).42,43 In OXI, the symmetry-independent molecules form CO⋯CO interaction of the motif-I type but with distinct parameters; the dCO is 2.95 and 3.20 Å, with angles of 110 and 103°.
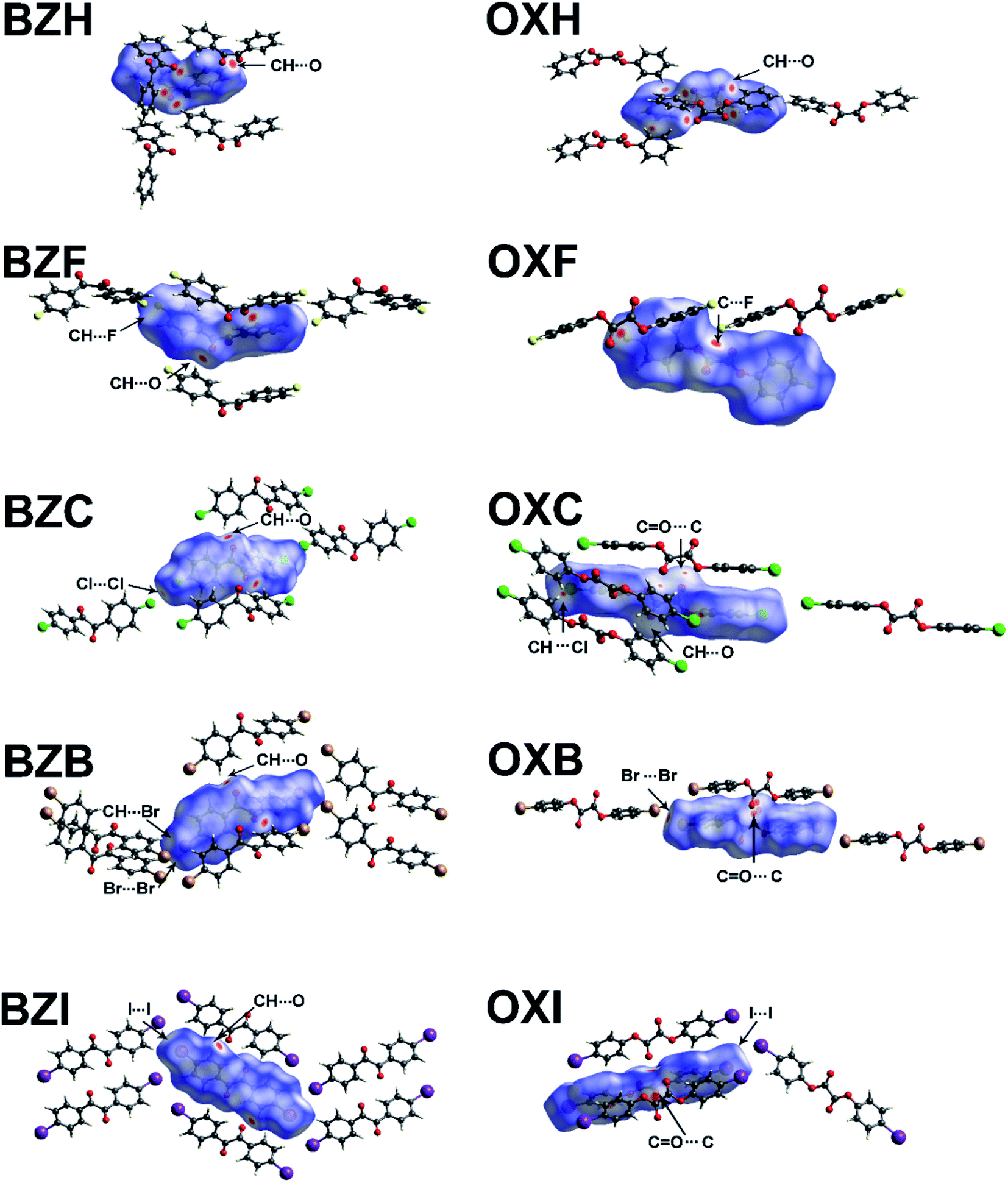 | ||
| Fig. 8 Hirshfeld surface with dnorm mapped over it using the universal red, white and blue color code that indicates strong, medium, and weak interactions, respectively. The prominent interaction types are highlighted. | ||
The characteristic close-packed molecular arrangement and isotropic distribution of interactions of comparable strength account for the brittleness in the crystals of OXH and OXF. Because of the relatively weak halogen···halogen interactions describing the weakest plane, OXC and OXB have two-dimensional EFs (Fig. S19 and S20†). The type-I (θ1 = θ2 = 161°) Cl⋯Cl interaction in OXC, which is longer (by ∼0.1 Å) than the sum of the van der Waals radii, could be described as a crystal packing effect rather than with a chemical connotation. In OXB, the computed EF shows no apparent evidence for Br⋯Br interactions; instead, the neighboring layers connect through C–H⋯Br interactions with a total energy component of ∼−9.2 kJ mol−1. The observed difference in the interactions in the interlayer region led to the higher magnitude of Young's modulus for OXB than OXC. In OXI, the I⋯I interaction is quasi type-II (θ1 = 176° and θ2 = 124°) and is predominantly dispersive. In OXC, OXB, and OXI, the two-dimensional EF results from the robust interactions established by the oxalate and aromatic C–H moieties within a layer and the weak dispersive halogen interactions in the interlayer region. Furthermore, the calculated normalized contact (Nc) values of the halogen interactions in the interlayer region are close to or higher than unity, which signifies their weak characteristics (Table S7†).
As demonstrated in Fig. 9, the most prominent negative MESPs are confined to the sites around the central oxalate or diketo moieties. At the same time, the aromatic H atoms account for the positive MESPs. In the halogen series, the σ-hole trait and dimension increase, though they are less prominent than classic XB-forming compounds such as tetrafluoro-1,4-diiodobenzene or 1,4-dibromo tetrafluorobenzene.48,49 Of note, the positive region on the MESPs corresponding to the halogen σ-holes is less significant than the aromatic C–H regions and it corroborates the weak attributes of the halogen⋯halogen interactions observed in the structures. The central oxalate moiety with positive MESPs validates the CO⋯C π-hole interactions in the crystal structures. Nevertheless, the lower magnitude of the positive MESPs in the benzil diketo moiety and its twisted conformations deter the formation of CO⋯C bonds. In a related set of compounds, the magnitude of the MESP maxima in the halogen polar region is higher for the benzil compounds than its corresponding oxalate analog. In OXF, the C–F⋯C π-hole interaction can also be understood based on the complementarity of MESP charges.
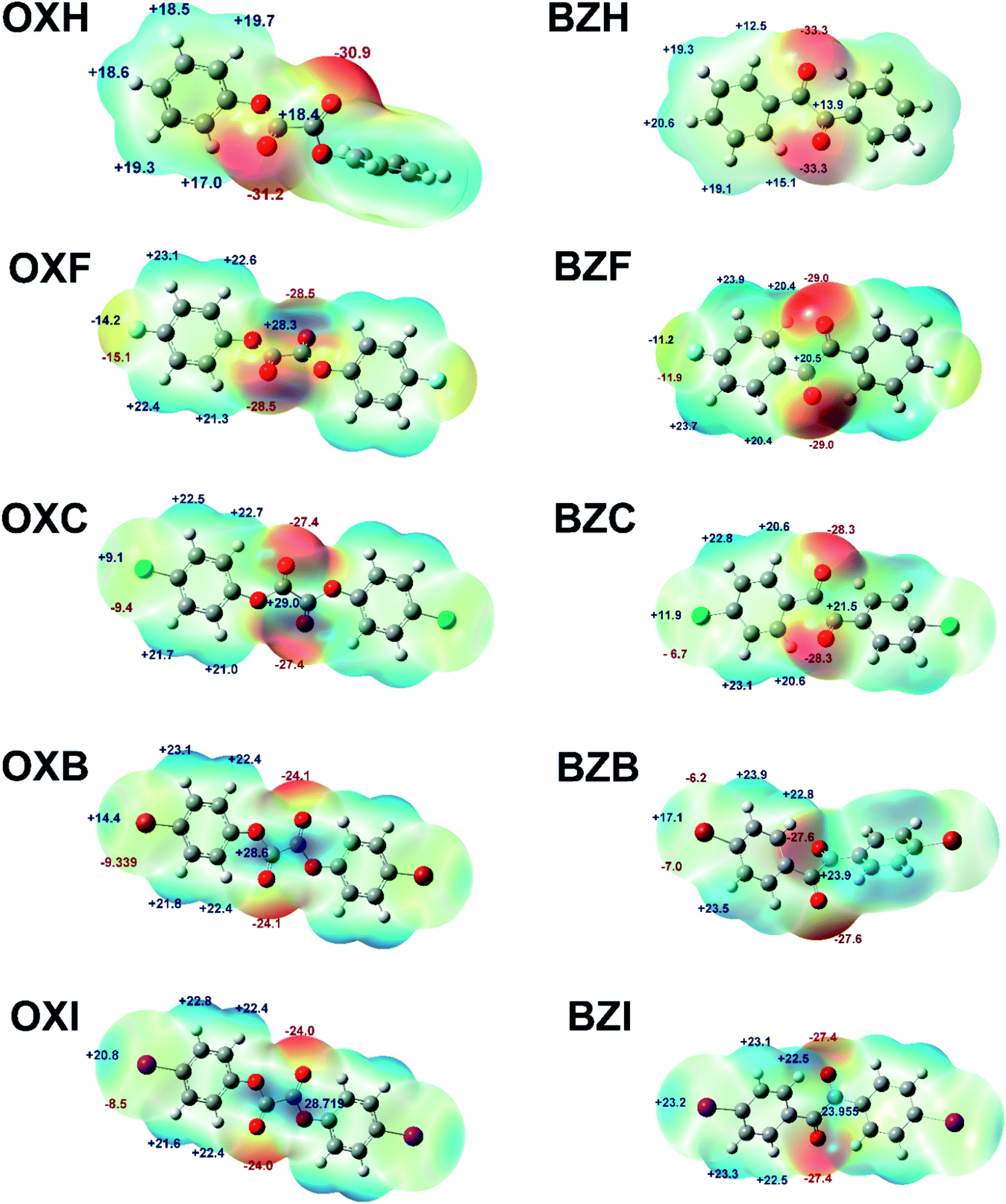 | ||
| Fig. 9 MESP surfaces of the oxalate and benzil derivatives at the B3LYP/6-311 + G(d,p)/DGDZVP level of theory. Isosurface 0.001 a.u. The MESP maximum and minimum energies are indicated in kcal mol−1. | ||
The NCI plot index analyses show isosurfaces located between the halogen atoms in the structures and validate the weak nature of the X⋯X interactions in the interlayer region (Fig. 10). In both OXC and BZC, the type-I C–Cl⋯Cl–C intermolecular topology is described by an irregular RDG isosurface, highlighting them as chemically less significant. These isosurfaces correspond to the broad green in the sign(λ2) < 0 regions of the ρ × sign(λ2) vs. RDG plot in 2D, where λ2 is the second eigenvalue of the Hessian of the charge density matrix. In both cases, the compounds show weak van der Waals interactions (>−0.01 a.u.); the energy difference between the two systems is negligible. Nevertheless, in BZC and BZB, the C–H⋯X interactions play a complementary role in strengthening the interlayer region and implied by the corresponding isosurfaces located between the CH and halogen atoms. The broad red spike in the sign(λ2) > 0 regions, indicative of steric repulsion, corresponds to an RDG domain appearing at the center of the phenyl rings. The other oxalate and benzil compounds exhibit similar RDG scatter plots, indicating similar interaction characteristics. The isosurface becomes increasingly prominent in a unique series while moving down the halogen group. Thus in the I-derivatives, both benzil and oxalate compounds show comparatively prominent isosurface corresponding to the I⋯I interactions, though the RDG scatterplot identifies them as weak. The MESP and the RDG scatterplot highlight the critical role of the interlayer region sustained through weak dispersive X⋯X bonds or their combination with C–H⋯X interactions in determining the macromechanical properties.
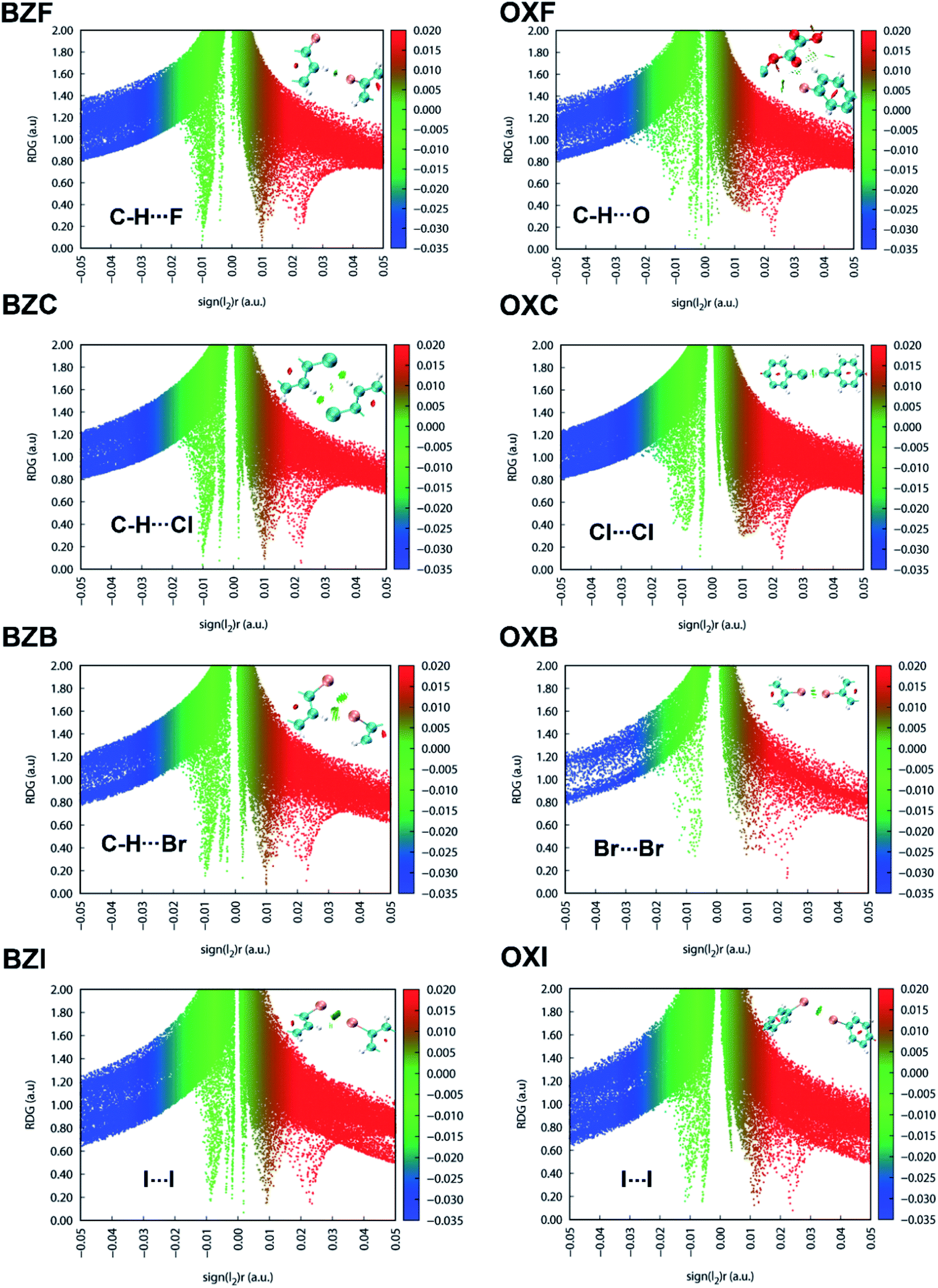 | ||
| Fig. 10 Scatter plot of the reduced density gradient (RDG) against the sign of second Hessian eigenvalue times the electron density. The corresponding RDG electron density surface plots for the benzil and oxalate compounds are also provided. | ||
Structure–mechanical property correlations
The twisted conformation of the central diketo moiety enables the molecules to form interwoven grid-like networks sustained through weak dispersive C–H⋯O and C–H⋯π interactions. Thus formed layers act as a ‘structural buffer’ with considerable tolerance towards breaking and making upon engaging and releasing the external stress. Hence, weak dispersive interactions are critical in attaining the elastic bending behavior. It provides a spring effect, permitting regular structural changes at the bent site and accommodating the excess strain generated within the crystal. The interactions in the outer arc elongate to accommodate the applied strain, whereas those in the inner arc get compressed. The moderately weak and dispersive X⋯X and C–H⋯X interactions sustaining the interlayer region maintain the system in an attractive regime and provide the stabilizing attractive force to retain the system in a thermodynamic equilibrium—elastic deformation. Thus, dispersive interactions in the interlayer region provide enough flexibility and impetus to the system to adopt partial molecular movements enabling it to restore the original state with the withdrawal of the applied stress. The inclined orientation of the molecules with respect to the surface also contributes to the minimal molecular movement. The near isotropic distribution of weak interactions allows them to have a larger elastic regime (Fig. 6) but beyond which the layers slip, thereby ensuing a plastic character.The oxalate crystals also exhibit the property of the co-existence of elastic and plastic deformation, though the elastic domain is considerably less vis-à-vis the benzil compounds. The robust π-hole carbonyl–carbonyl interactions within the layer induce an added degree of anisotropy in the system, even though the structure lacks any classical electrostatic interactions. However, the X⋯X interactions in the interlayer region are either type-I or longer than the sum of van der Waals radii, making them relatively weak. Furthermore, the layer separation in the slip regions in the oxalates is slightly bigger (OXB: 2.34 Å and OXC: 2.11 Å) than in the benzil series (BZB: 2.25 Å, BZC: 2.03 Å, and BZF: 1.79 Å). Consequently, significant anisotropy in the oxalate systems makes the layer slipping energetically viable and thus favoring the plastic deformation.
In general, plastic deformation in molecular crystals is defined by either slip or twinning. All the studied systems in the present work have stacked-layered arrangements with weak interactions in the interlayer region. However, in the absence of classic electrostatic interactions, the observed anisotropy in benzils is not large enough to induce extensive plastic characteristics in the crystals. Thinner benzil crystals exhibit better elastic flexibility compared to the thicker ones. The observation corroborates some of the earlier reports that highlighted the elastic properties of thinner crystals compared to thicker ones.31 Such size-dependent mechanical response could be correlated with possible defects also. With a relatively smaller number of defects than the thicker ones, the mechanical properties of the thinner crystals could be the manifestation of the intrinsic structural factors. However, defect concentration and mobility in thicker crystals contribute significantly to the mechanical response.52,53 The hypothesis conforms with an earlier observation that the crystals in the nanometer regime have significantly higher stiffness (∼85%) compared to the millimeter size crystals.54
Upon cutting or smudging with a blunt substrate, the ends of the long tape-like crystals of BZF, BZC, and BZB exfoliate into bundles of fibrils (Fig. 11 and S29†). The fluorescence images of the crystals evident the characteristic observation. The resultant fibrils exhibit high flexibility. Such crystal delamination could be a critical parameter in ensuing plasticity in the benzil crystals. We further analyzed the macroscopic crystal delamination properties in the benzil crystals using confocal microscopy (Fig. S28†). The crystals of BZC show excellent plastic deformation and delaminate into thin fibrils in the bent and the end regions (Fig. S28a†). In crystals with inherent anisotropy, the plane with the least attachment energy provides an energetically viable route to delaminate under applied bending strain. For example, in BZC, (002) containing the interplanar region sustained through weak Cl⋯Cl interactions could give in under bending strain. Thus, apart from the microscopic structural contribution, crystal delamination could be considered a possible macroscopic factor contributing to attaining the plastic deformation at higher applied stress. Within the elastic limit, the microscopic structural elements that favor the reversible deformation dominate; at higher stress, along with the long-range molecular movements (microscopic factors), defect formation and propagation, and macroscopic factors, such as crystal delamination, contribute to irreversible deformation. In oxalate systems also, crystal delamination is observed. Hence the observed plastic deformation in the oxalate crystals could be understood in terms of structural anisotropy and macroscopic contribution (Fig. S29†).
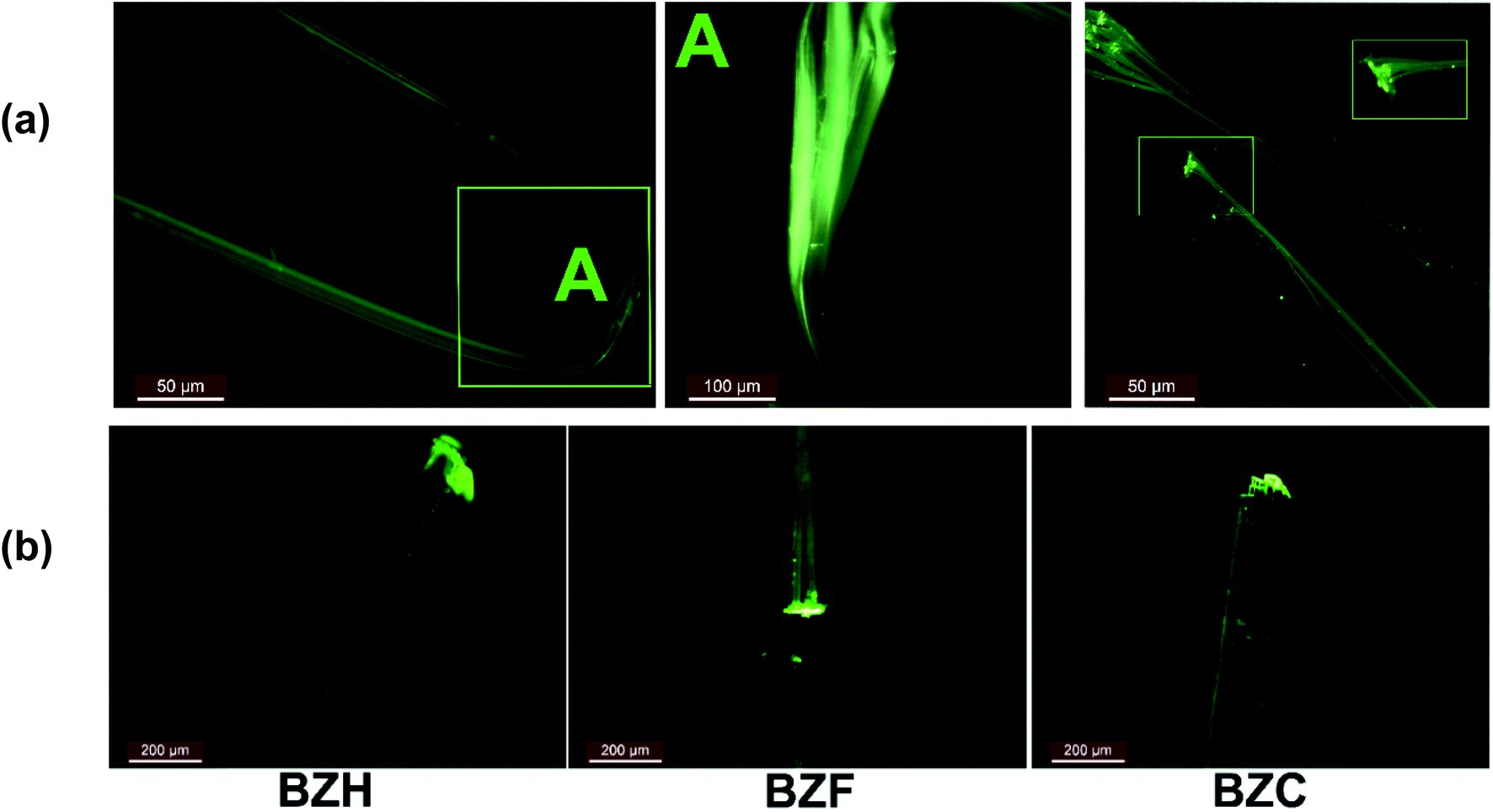 | ||
| Fig. 11 The fluorescence microscopy images of the benzil crystals. (a) The delamination behavior in the BZC crystals. (b) The higher emission intensities at the ends of the crystals than the central parts, suggesting a conventional photowaveguide effect. | ||
To validate the possible defect generation, structural transformation, or partial amorphization resulting from the mechanical deformation of the crystals, we carried out diffraction studies of the bend crystals. The diffused and deformed diffraction patterns indicate a possible disruption of long-range structural order due to the crystal deformation and further highlight the strain developed within (Fig. S30–S32†). Nevertheless, none of the systems exhibited a complete loss of the diffraction despite their extensive deformation in the plastic regime, indicating the possible retention of the crystalline ordering even in their bent state.
The benzil compounds have interesting emission properties: besides their intense green emission characteristics, they exhibit crystallization-induced phosphorescence (CIP).55 Accordingly, BZH, BZF, and BZB emit at 521, 500, and 526 nm, respectively, upon excited at 365 nm. Of note, the dimorphs of the F-derivative (BZF_O and BZF) exhibit distinct emission spectra (Fig. S33b†). The benzil crystals (BZF, BZC, and BZB) are comparable because of the isostructural characteristics and the recorded absorption and emission spectra (Fig. S33†). A recent report highlighted the waveguiding properties of BZB.31 We noted that in all the members of the benzil series (BZH, BZF, BZC, and BZB), both ends of the crystals show higher emission intensities than the central parts, suggesting a conventional photo-waveguide effect (Fig. 11). The benzil compounds, including BZB, exhibit room-temperature phosphorescence (RTP); BZC and BZF are also likely to show the effect.31,55,56 However, we lack access to facilities that could quantify the RTP and the waveguiding properties. Mechanically exfoliated thin fibrils also fluoresce intensely at their ends (Fig. 11). Long crystal edges look similar to a bundle of optical fibers under UV irradiation. Deformed crystals in the bent state also show appreciable intense emission at the crystal ends. The delaminated crystals, by excessive bending, retained the property. Thus, combining the mechanical flexibility in tandem with photonic traits in organic crystals could give enormous opportunity for fabricating miniature organic optoelectronic materials.57,58
Conclusions
The preceding sections demonstrated the co-existence of two mutually exclusive mechanical deformation properties—elastic and plastic—in a set of halogen-substituted benzil and oxalate compounds. Though the studies related to elastic and plastic crystals are known, the intermediate type examples that display elasticity and plasticity under different stress settings are exceptional. The Hirshfeld surface, energy frameworks, MESP, RDG, and NCI plot analyses corroborate the dispersive characteristics of the interactions and their relative distribution to correlate with the observed macroscopic mechanical deformation. The isotropic deployment of the interactions of dominant dispersive characteristics in the interlayer and intralayer regions in unison with the inclined orientation of the molecules provides enough flexibility and impetus to the system to adopt partial molecular movements and restore the original state with the withdrawal of the applied stress. Thus, the unique structural features observed in the benzil compounds are crucial to enabling the co-existence of ‘continuity’ and ‘deformation’ synergically under external stimuli. At intermediate stress, the crystals of benzil compounds show elastoplastic characteristics, which is rare in molecular crystals. At higher stress, further to the long-range molecular movements (microscopic factors), defect formation and propagation, as well as macroscopic factors such as crystal delamination, contribute to irreversible deformation in crystals. Replacing the twisted diketo moiety in benzil with a rigid and coplanar central oxalate moiety enabled an enhanced contribution of CO⋯C interactions, furthering the anisotropic element in the crystal packing. In contrast to a robust combination of intralayer interactions having both the electrostatic and dispersive components, weak X⋯X interactions that sustain the interlayer region allow long-range slippage of layers. The added anisotropy reduced the elastic domain of the oxalate crystals to a large extent, making them susceptible to plastic deformation even at lower stress.
The work highlights the structural basis for the co-existence of elastic and plastic bending with contrasting structural requirements in molecular crystals and the possibility of fine-tuning the extent of anisotropy to modulate the macroscopic mechanical response. In the absence of a universal supramolecular strategy to improve upon a specific mechanical property of a unique crystalline material, we need to analyze an extensive library of materials with comparable contributing factors to derive better correlations between the structures and properties.
Data availability
All experimental and characterisation data in this article are available in the ESI.†Author contributions
S. V., K. W. and H. K. conceived and designed the project. I. S. D. did the synthesis and crystallization experiments and also analyzed the data; S. K. performed the diffraction studies and data analysis, guided by K. W.; S. H. did the three-point bending experiments, guided by H. K.; T. S. discussed, compiled and commented on the mechanical properties; S. V. did the computational studies; S. V., K. W., T. S. and H. K. co-wrote the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. V. thanks SERB, the Government of India, for a Core Research Grant (CRG/2020/000460). I. S. D. is grateful to UGC, the Government of India, for a research fellowship. We acknowledge the Council of Scientific and Industrial Research (CSIR), Government of India, for the infrastructure and research facilities. We thank M. S. R. N. Kiran (SRM University, Chennai) for the fruitful discussion. We thank Jagadeesh Babu Nanubolu (CSIR-IICT, Hyderabad) for the X-ray data.References
- H. Jiang and W. Hu, Angew. Chem., Int. Ed., 2020, 59, 1408 CrossRef CAS PubMed.
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379 CrossRef CAS PubMed.
- P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440 CrossRef CAS PubMed.
- K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377 CrossRef CAS PubMed.
- S. Varughese, M. S. R. N. Kiran, K. A. Solanko, A. D. Bond, U. Ramamurty and G. R. Desiraju, Chem. Sci., 2011, 2, 2236 RSC.
- B. P. A. Gabriele, C. J. Williams, M. E. Lauer, B. Derby and A. J. Cruz-Cabeza, CrystEngComm, 2021, 23, 5826 RSC.
- M. Reichel, D. Dosch, T. Klapötke and K. Karaghiosoff, J. Am. Chem. Soc., 2019, 141, 19911 CrossRef CAS PubMed.
- C. Zhang, X. Xue, Y. Cao, Y. Zhou, H. Li, J. Zhou and T. Gao, CrystEngComm, 2013, 15, 6837 RSC.
- A. J. Thompson, A. Worthy, A. Grosjean, J. R. Price, J. C. McMurtrie and J. K. Clegg, CrystEngComm, 2021, 23, 5731 RSC.
- X. Chu, Z. Lu, B. Tang, B. Liu, K. Ye and H. Zhang, J. Phys. Chem. Lett., 2020, 11, 5433 CrossRef CAS PubMed.
- T. Aida and E. W. Meijer, Isr. J. Chem., 2020, 60, 33 CrossRef CAS.
- R. Geitner, F.-B. Legesse, N. Kuhl, T. W. Bocklitz, S. Zechel, J. Vitz, M. Hager, U. S. Schubert, B. Dietzek, M. Schmitt and J. Popp, Chem.–Eur. J., 2018, 24, 2493 CrossRef CAS PubMed.
- M. Pisacic, I. Biljan, I. Kodrin, N. Popov, Z. Soldin and M. Dakovic, Chem. Mater., 2021, 33, 3660 CrossRef CAS.
- S. Varughese, M. S. R. N. Kiran, U. Ramamurty and G. R. Desiraju, Angew. Chem., Int. Ed., 2013, 52, 2701 CrossRef CAS PubMed.
- P. Naumov, D. P. Karothu, E. Ahmed, L. Catalano, P. Commins, J. Mahmoud Halabi, M. B. Al-Handawi and L. Li, J. Am. Chem. Soc., 2020, 142, 13256 CrossRef CAS PubMed.
- A. Hasija and D. Chopra, CrystEngComm, 2021, 23, 5711 RSC.
- S. Varughese, M. S. R. N. Kiran, U. Ramamurty and G. R. Desiraju, Chem.–Asian J., 2012, 7, 2118 CrossRef CAS PubMed.
- E. Ahmed, D. P. Karothu and P. Naumov, Angew. Chem., Int. Ed., 2018, 57, 8837 CrossRef CAS PubMed.
- M. K. Mishra, S. Varughese, U. Ramamurty and G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 8121 CrossRef CAS PubMed.
- A. J. Thompson, A. I. Chamorro Orué, A. J. Nair, J. R. Price, J. McMurtrie and J. K. Clegg, Chem. Soc. Rev., 2021, 50, 11725 RSC.
- B. Tang, B. Liu, H. Liu and H. Zhang, Adv. Funct. Mater., 2020, 30, 2004116 CrossRef CAS.
- C. M. Reddy, R. C. Gundakaram, S. Basavoju, M. T. Kirchner, K. A. Padmanabhan and G. R. Desiraju, Chem. Commun., 2005, 3945 RSC.
- K. Zhang, C. C. Sun, Y. Liu, C. Wang, P. Shi, J. Xu, S. Wu and J. Gong, Chem. Mater., 2021, 33, 1053 CrossRef CAS.
- F. Liu, D. E. Hooks, N. Li, N. A. Mara and J. A. Swift, Chem. Mater., 2018, 30, 3798 CrossRef CAS.
- T. Sasaki and S. Takamizawa, Cryst. Growth Des., 2020, 20, 6990 CrossRef CAS.
- M. J. Bryant, A. G. P. Maloney and R. A. Sykes, CrystEngComm, 2018, 20, 2698 RSC.
- S. P. Thomas, M. W. Shi, G. A. Koutsantonis, D. Jayatilaka, A. J. Edwards and M. A. Spackman, Angew. Chem., Int. Ed., 2017, 56, 8468 CrossRef CAS PubMed.
- S. Saha and G. R. Desiraju, Chem. Commun., 2016, 52, 7676 RSC.
- S. Li and D. Yan, ACS Appl. Mater. Interfaces, 2018, 10, 22703 CrossRef CAS PubMed.
- S. SeethaLekshmi, M. S. R. N. Kiran, U. Ramamurty and S. Varughese, Chem.–Eur. J., 2019, 25, 526 CrossRef CAS PubMed.
- H. Liu, Z. Bian, Q. Cheng, L. Lan, Y. Wang and H. Zhang, Chem. Sci., 2019, 10, 227 RSC.
- C. M. Reddy, G. R. Krishna and S. Ghosh, CrystEngComm, 2010, 12, 2296 Search PubMed.
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957 CrossRef CAS PubMed.
- P. Gupta, S. Allu, D. P. Karothu, T. Panda and N. K. Nath, Cryst. Growth Des., 2021, 21, 1931 CrossRef CAS.
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, J. Am. Chem. Soc., 2016, 138, 13561 CrossRef CAS PubMed.
- K. Naim, M. Singh, S. Sharma, R. V. Nair, P. Venugopalan, S. C. Sahoo and P. P. Neelakandan, Chem.–Eur. J., 2020, 26, 11979 CrossRef CAS PubMed.
- S. Das, S. Saha, M. Sahu, A. Mondal and C. M. Reddy, Angew. Chem., Int. Ed., 2021, e202115359 Search PubMed.
- M. Felsmann, F. Eissmann, A. Schwarzer and E. Weber, Cryst. Growth Des., 2011, 11, 982 CrossRef CAS.
- D. E. Wu, Y. H. Luo, M. N. Wang, Q. L. Liu, G. J. Wen, L. J. Zhu, C. P. Fan and B. W. Sun, Struct. Chem., 2017, 28, 1731 CrossRef CAS.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814 RSC.
- F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320 CrossRef.
- B. Sahariah and B. K. Sarma, Chem. Sci., 2019, 10, 909 RSC.
- A. Rahim, P. Saha, K. K. Jha, N. Sukumar and B. K. Sarma, Nat. Commun., 2017, 8, 78 CrossRef PubMed.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735 RSC.
- S. Saha and G. R. Desiraju, Chem. Commun., 2017, 53, 6371 RSC.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178 RSC.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541 CrossRef CAS PubMed.
- Y. Zhao, Y. Cotelle, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 4270 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
- G. Saleh, C. Gatti and L. Lo Presti, Comput. Theor. Chem., 2012, 998, 148 CrossRef CAS.
- S. Bhandary, A. J. Thompson, J. C. McMurtrie, J. K. Clegg, P. Ghosh, S. R. N. K. Mangalampalli, S. Takamizawa and D. Chopra, Chem. Commun., 2020, 56, 12841 RSC.
- M. Li, C. Zhang, M. Li, F. Liu, L. Zhou, Z. Gao, J. Sun, D. Han and J. Gong, Chem. Eng. J., 2022, 429(1), 132450 CrossRef CAS.
- C. Karunatilaka, D. K. Bucar, L. R. Ditzler, T. Friscic, D. C. Swenson, L. R. MacGillivray and A. V. Tivanski, Angew. Chem., Int. Ed., 2011, 50, 8642 CrossRef CAS PubMed.
- Y. Y. Gong, Y. Tan, H. Li, Y. Zhang, W. Z. Yuan, Y. M. Zhang, J. Z. Sun and B. Z. Tang, Sci. China: Chem., 2013, 56, 1183 CrossRef CAS.
- S. Yamada, T. Higashida, Y. Wang, M. Morita, T. Hosokai, K. Maduwantha, K. R. Koswattage and T. Konno, Beilstein J. Org. Chem., 2020, 16, 1154 CrossRef CAS PubMed.
- A. L. Briseno, R. J. Tseng, M. M. Ling, E. H. L. Falcao, Y. Yang, F. Wudl and Z. Bao, Adv. Mater., 2006, 18, 2320 CrossRef CAS.
- E. Ahmed, A. L. Briseno, Y. Xia and S. A. Jenekhe, J. Am. Chem. Soc., 2008, 130, 1118 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2142844. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02969c |
| This journal is © The Royal Society of Chemistry 2022 |