
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric higher-order [10 + n] cycloadditions of palladium-containing 10π-cycloaddends†
Ao
Li‡
a,
Yang
Gao‡
a,
Jian-Bin
Lu
a,
Zhi-Chao
Chen
a,
Wei
Du
 *a and
Ying-Chun
Chen
*ab
*a and
Ying-Chun
Chen
*ab
aKey Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry and Sichuan Province, Sichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu 610041, China. E-mail: duweiyb@scu.edu.cn; ycchen@scu.edu.cn; Fax: +86 28 85502609
bCollege of Pharmacy, Third Military Medical University, Shapingba, Chongqing 400038, China
First published on 15th July 2022
Abstract
We uncovered an asymmetric higher-order [10 + 2] cycloaddition reaction between diverse activated alkenes and a new type of π-allylpalladium complex-containing dipole-type 10π-cycloaddend, which was generated in situ from 2-methylene-1-indanols via a dehydrative insertion and deprotonation strategy under double activation of Pd(0) and phosphoric acid. A similar strategy was applied to an asymmetric higher-order [10 + 8] cycloaddition reaction or [10 + 4] cycloaddition reaction by using a heptafulvene derivative or a cyclic enone, respectively, as the acceptor. A variety of polycyclic frameworks imbedding an indene core were generally furnished in moderate to excellent yields with high levels of enantioselectivity by employing a newly designed chiral phosphoramidite ligand.
Introduction
Higher-order cycloaddition involving conjugated systems with more than 6π-electrons, which enables rapid construction of complicated cyclic frameworks,1 has received considerable attention since its discovery in the 1960s.2 In spite of its charm in organic synthesis, higher-order cycloaddition generally suffers from low reactivity, and poor periselectivity and stereoselectivity; thus a variety of pre-prepared cyclic polyenes (>4π) have been commonly utilised, including fulvenes,3 Cr(0)-cycloheptatriene complexes,4 tropone and its analogues,5 and 3H-pyrrolizines,6 as well as amino-stabilised isobenzofulvenes,7 in combination with diverse 2π- or 4π-systems under different conditions (Scheme 1a). Recently, significant progress in the field of higher-order cycloaddition reactions has been made with some well-designed carbonyl substrates, which could be activated by a suitable organocatalyst to generate several types of 6π, 8π, 10π or even 12π-cycloaddends in situ, typically featuring HOMO-raised polyenamine8 or polyenolate species9 (Scheme 1b). However, the development of relevant cycloaddends embedding a reactive metal-complexed motif catalytically, which can be successfully applied in higher-order cycloaddition reactions, has not been disclosed yet. | ||
| Scheme 1 Summary of typical cycloaddends (>4π) used in higher-order cycloadditions and our design. (a) Diverse pre-prepared cycloaddends. (b) Diverse in situ formed cycloaddends via organocatalysis. (c) This work: in situ formed π-allylpalladium-containing 10π-cycloaddends. | ||
Over the past few decades, the zwitterionic reagents bearing a π-allylmetal moiety, usually generated in situ from allylic alcohol derivatives under palladium or iridium catalysis, have been extensively utilised as valuable 1,n-dipoles for various asymmetric formal cycloaddition reactions.10 However, such a reaction strategy, through designing suitable conjugated π-systems, has not been envisioned for potentially developing higher-order cycloadditions.11 As a result, it would be particularly intriguing to uncover a new type of metal-embedding dipole, which could participate in asymmetric higher-order cycloadditions as cycloaddends with more than 6π-electrons. With these considerations, we envisaged that allylic alcohol or its derivative 1, readily available from 1-indanone, would undergo oxidative addition under Pd(0) catalysis. The resultant π-allylpalladium complex I, having an indene-type structure,12 would feasibly increase the acidity of the benzylic C–H.13 Thus, zwitterionic intermediate II would be generated after deprotonation, as outlined in Scheme 1c, which might perform as a unique metal-containing 10π-cycloaddend to undertake higher-order [10 + n] cycloaddition reactions with suitable electrophilic counterparts.
Results and discussion
Condition optimisation
The initial attempt with allylic carbonate 1a and activated alkene α-cyano chalcone 2a proved to be unsuccessful in toluene at 50 °C under the catalysis of Pd(PPh3)4 (Table 1, entry 1). It was speculated that enhancing the acidity of the benzylic C–H, by introducing an electron-withdrawing group at the indane ring, would be helpful for the formation of the desired dipole intermediate. Pleasingly, 6-nitro-substituted substrate 1b showed good reactivity under the identical catalytic conditions. The expected formal [10 + 2] cycloadduct 3a′ was detected, whereas product 3a, after isomerisation of the double bond, was found to be the more thermally stable one. Moreover, by simple treatment with catalytic amounts of Et3N in one pot, pure 3a was efficiently isolated in a moderate yield with excellent diastereoselectivity (entry 2). We next explored the asymmetric version by utilising Pd2(dba)3 and chiral ligands. Unfortunately, only moderate enantioselectivity was attained with phosphoramidite L1 after extensively screening diverse ligands (entry 3).14 We turned to explore the reaction by using a new substrate and a new catalytic system. Compared to allylic carbonates or esters, free allylic alcohols represent more atom-economic and environmentally benign precursors for generating the corresponding π-allyl species under transition metal catalysis, as water is the sole side product.15 Nevertheless, alcohol 1c suffered from low reactivity under the catalysis of Pd(0) due to the poor leaving ability of the hydroxyl group (entry 4). Subsequently, we tried to add a phosphoric acid as a co-catalyst, as it has been demonstrated to be beneficial for the oxidative addition of Pd(0) towards allylic alcohols and also might be beneficial for enantiocontrol.16 To our gratification, the cycloaddition of 1c and 2a proceeded smoothly by using BINOL-derived (R)-A1 as an additive and PPh3 as the ligand, and 3a was afforded in a moderate yield albeit with low enantioselectivity (entry 5). It should be noted that such a dehydration strategy has not been previously utilised in Pd-based dipole chemistry.10 Moreover, it also suggested that acid (R)-A1 would play dual roles in sequential activation of both substrates 1c and 2a. Consequently, some chiral ligands were investigated in combination with (R)-A1. Chiral phosphoramidites L1 and L2 showed moderate reactivity but still with low enantiocontrol (entries 6 and 7). While the newly designed phosphoramidite L3 gave fair enantioselectivity (entry 8), (R)-BINOL derived L4, a diastereomer of L3, exhibited much higher enantioselectivity (entry 9). Moreover, an improved yield with a better ee value was obtained by using the combination of acid (S)-A1 and L4 (entry 10). Nevertheless, ligand L5 with a smaller TES group delivered reduced enantioselectivity (entry 11). A slightly higher yield was attained at 30 °C (entry 12). It was further found that the chiral acid was not necessary, and even higher catalytic activity and exclusive regioselectivity with retained enantioselectivity was achieved with simple phosphoric acid A2 (entry 13).|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | L | A | Yieldb (%) | eec (%) |
a Unless noted otherwise, reactions were performed with substrate 1 (0.1 mmol), α-cyano chalcone 2a (0.12 mmol), Pd2(dba)3 (5 mol%), L (20 mol%), additive (20 mol%) and 4 Å MS (100 mg) in toluene (1 mL) at 50 °C under Ar for 24 h. After completion, Et3N (20 mol%) was added, and the mixture was stirred at rt for 2 h.
b Yield of isolated product 3a.
c Determined by HPLC analysis on a chiral stationary phase, and dr > 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by 1H NMR analysis.
d Pd(PPh3)4 (10 mol%) was used.
e At 30 °C. :1 by 1H NMR analysis.
d Pd(PPh3)4 (10 mol%) was used.
e At 30 °C.
|
|||||
| 1d | 1a | — | — | — | — |
| 2d | 1b | — | — | 65 | — |
| 3 | 1b | L1 | — | 45 | −65 |
| 4 | 1c | PPh3 | — | <5% | — |
| 5 | 1c | PPh3 | (R)-A1 | 65 | −12 |
| 6 | 1c | L1 | (R)-A1 | 40 | −5 |
| 7 | 1c | L2 | (R)-A1 | 65 | −25 |
| 8 | 1c | L3 | (R)-A1 | 65 | −30 |
| 9 | 1c | L4 | (R)-A1 | 60 | 86 |
| 10 | 1c | L4 | (S)-A1 | 68 | 91 |
| 11 | 1c | L5 | (S)-A1 | 75 | 76 |
| 12e | 1c | L4 | (S)-A1 | 72 | 91 |
| 13e | 1c | L4 | A2 | 99 | 91 |
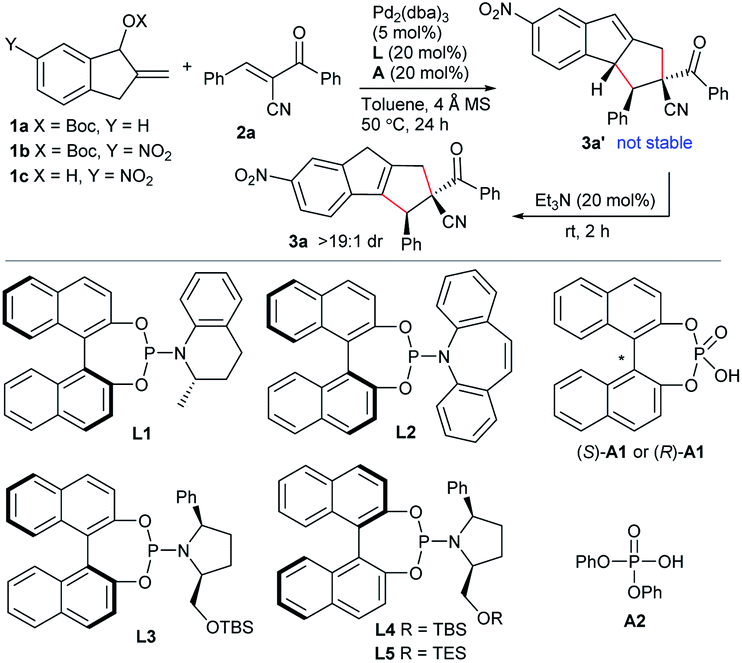
Substrate investigation of asymmetric [10 + 2] cycloadditions
Subsequently, we explored the substrate scope and limitations of the asymmetric formal [10 + 2] cycloaddition reaction under the cooperative catalysis of Pd/L4 and phosphoric acid A2. As summarised in Table 2, an array of α-cyano chalcones 2 were first tested in the reactions with benzocyclopentenol 1c. Acceptors 2 with different aromatic substituents at the β-position, including electron-deficient and -rich ones, smoothly gave corresponding products 3a–3j in moderate to good yields with high stereoselectivity (Table 2, entries 1–10). The one with a p-nitrophenyl group showed low reactivity under the standard conditions, but desired product 3e was obtained in a good yield with excellent enantioselectivity by using (S)-A1 as the additive (entry 5). Notably, the halogen-substituted ones were compatible with the reactions (entries 2–4). Similarly good results were generally produced for substrates 2 with diverse α′-aroyl groups (entries 11–18), whereas (S)-A1 was found to be helpful in some cases. Unfortunately, the activated alkenes with aliphatic substituents failed to afford the cycloadducts, probably because of the undesired acidic vinylogous C–H of these substrates.14 On the other hand, benzocyclopentenols 1 with a nitro group at different positions on the phenyl ring were applied in the reactions with 2a. When the one with a 4-nitro substituent was utilised, cycloadduct 3s was afforded in good yield with good enantioselectivity (entry 19).14 Nevertheless, the alcohol with a 5-nitro group also delivered product 3a, same as that from 1c (6-nitro), indicating an isomerisation process of the 10π-intermediate would be involved (entry 20).14 In addition, a 6-cyano-substituted alcohol showed comparable reactivity, and the expected cycloadduct 3t was attained quantitatively with high enantioselectivity (entry 21). We also conducted the reaction on a larger scale, and similar good results were afforded (entry 22).|
|
||||
|---|---|---|---|---|
| Entry | EWG | R1, R2 | Yieldb (%) | eec (%) |
|
a Unless noted otherwise, reactions were performed with allylic alcohol 1 (0.1 mmol), activated alkene 2 (0.12 mmol), Pd2(dba)3 (5 mol%), L4 (20 mol%), acid A2 (20 mol%) and 4 Å MS (100 mg) in toluene (1 mL) at 30 °C under Ar for 2–24 h. After completion, Et3N (20 mol%) was added, and the mixture was stirred at rt for 0.5–2 h.
b Yield of the isolated product.
c Determined by HPLC analysis on a chiral stationary phase; dr > 19:1 by 1H NMR analysis.
d The absolute configuration of enantiopure 3a was determined by X-ray analysis. The other products were assigned by analogy.
e Data in parentheses were obtained with acid (S)-A1 (20 mol%).
f On a 1.0 mmol scale.
|
||||
| 1 | 6-NO2 | Ph, Ph | 3a, 99 | 91d |
| 2 | 6-NO2 | Ph, 2-ClC6H4 | 3b, 96 | 93 |
| 3e | 6-NO2 | Ph, 3-ClC6H4 | 3c, 89 (80) | 82 (92) |
| 4 | 6-NO2 | Ph, 4-BrC6H4 | 3d, 67 | 89 |
| 5e | 6-NO2 | Ph, 4-NO2C6H4 | 3e, — (70) | — (90) |
| 6 | 6-NO2 | Ph, 2-MeOC6H4 | 3f, 99 | 95 |
| 7e | 6-NO2 | Ph, 3-MeOC6H4 | 3g, 99 (75) | 87 (93) |
| 8 | 6-NO2 | Ph, 4-MeOC6H4 | 3h, 99 | 88 |
| 9 | 6-NO2 | Ph, 2-naphthyl | 3i, 99 | 91 |
| 10 | 6-NO2 | Ph, 2-thienyl | 3j, 70 | 88 |
| 11e | 6-NO2 | 2-BrC6H4, Ph | 3k, — (73) | — (91) |
| 12e | 6-NO2 | 3-BrC6H4, Ph | 3l, — (75) | — (82) |
| 13 | 6-NO2 | 4-ClC6H4, Ph | 3m, 99 | 93 |
| 14 | 6-NO2 | 4-BrC6H4, Ph | 3n, 76 | 94 |
| 15e | 6-NO2 | 2-MeC6H4, Ph | 3o, 86 (90) | 83 (89) |
| 16e | 6-NO2 | 3-MeC6H4, Ph | 3p, 88 (72) | 82 (91) |
| 17 | 6-NO2 | 4-MeC6H4, Ph | 3q, 99 | 92 |
| 18 | 6-NO2 | 2-Naphthyl, Ph | 3r, 91 | 90 |
| 19 | 4-NO2 | Ph, Ph | 3s, 99 | 89 |
| 20 | 5-NO2 | Ph, Ph | 3a, 61 | 88 |
| 21 | 6-CN | Ph, Ph | 3t, 99 | 89 |
| 22f | 6-NO2 | Ph, Ph | 3a, 91 | 88 |
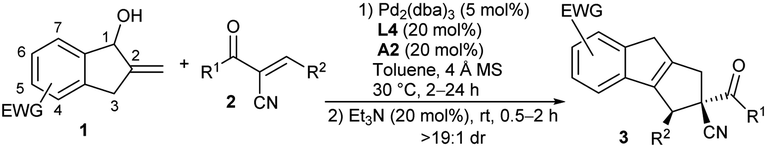
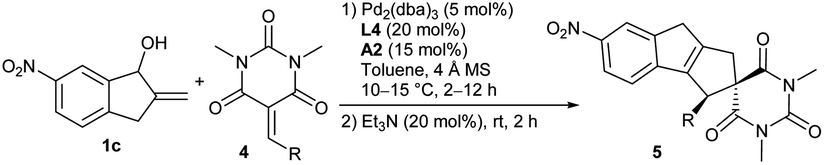
More substrate exploration
Apart from α-cyano chalcones, we successfully extended the asymmetric [10 + 2] cycloaddition reactions to other types of activated alkenes for constructing polycyclic frameworks with more structural diversity. It was found that barbiturate-derived alkenes 4 could be well assembled with alcohol 1c under the standard catalytic conditions. As summarised in Table 3, alkenes 4 with a different aryl or heteroaryl substituent underwent the cycloaddition reaction smoothly, furnishing corresponding spirocyclic architectures 5a–5e in moderate to good yields with high stereoselectivity (entries 1–7). Besides, the one with a 2-styryl group also worked well, and product 5h was obtained in moderate yield and enantioselectivity (entry 8). (S)-A1 was further tested when the reactions did not work well (entries 2 and 7).|
|
|||
|---|---|---|---|
| Entry | R | Yieldb (%) | eec (%) |
| a Unless noted otherwise, reactions were performed with allylic alcohol 1c (0.1 mmol), alkene 4 (0.12 mmol), Pd2(dba)3 (0.005 mmol, 5 mol%), L4 (20 mol%), A2 (15 mol%) and 4 Å MS (100 mg) in toluene (1 mL) at 10–15 °C for 2–12 h under Ar. After completion, Et3N (20 mol%) was added, and the mixture was stirred at rt for 2 h. b Yield of the isolated product. c Determined by HPLC analysis on a chiral stationary phase. d Data in parentheses were obtained with acid (S)-A1 (20 mol%). | |||
| 1 | Ph | 5a, 99 | 93 |
| 2d | 4-BrC6H4 | 5b, — (60) | — (94) |
| 3 | 4-MeC6H4 | 5c, 99 | 93 |
| 4 | 4-MeOC6H4 | 5d, 99 | 80 |
| 5 | 2-Naphthyl | 5e, 75 | 93 |
| 6 | 2-Furyl | 5f, 89 | 96 |
| 7d | 2-Thienyl | 5g, 76 (64) | 80 (87) |
| 8 | 2-Styryl | 5h, 91 | 80 |
Moreover, the formal [10 + 2] cycloaddition reaction could be extended to benzylidene Meldrum's acid 6 by using the combination of Pd/L4 and (S)-A1, delivering product 7 in a moderate yield with high enantioselectivity, whereas a higher yield with lower enantiocontrol was observed with acid A2 (Scheme 2). Interestingly, when barbiturate–heptafulvene 8 was employed,17 an asymmetric [10 + 8] higher-order cycloaddition reaction was applicable, and polycyclic 9 was constructed in a moderate yield with excellent enantiocontrol. Moreover, the assembly of carbonate 1g and 2-benzylidenebenzo[b]thiophen-3(2H)-one 10 was successful under the cooperative catalysis of Pd/L4 and benzoic acid, and [10 + 4] cycloadduct 11 embedding an oxepine motif was isolated in a moderate yield with high enantiocontrol, along with the observation of minor [10 + 2] product 12. Interestingly, using o-fluorobenzoic acid as the additive, cycloadduct 12 with similar enantiocontrol was delivered as the major one after the treatment with Et3N, albeit in a slightly lower yield.14
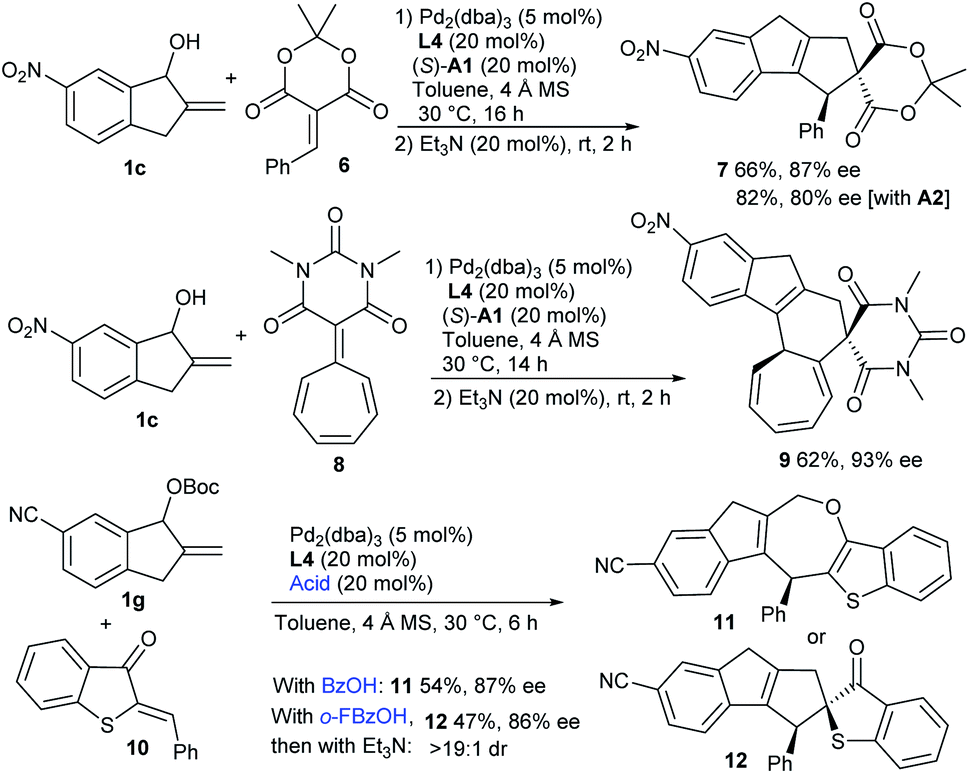 | ||
| Scheme 2 More exploration of higher-order cycloadditions. | ||
Mechanistic proposal
To gain more insight into the mechanism, a control experiment with the analogous acyclic alcohol 13 was conducted. As shown in Scheme 3a, no reaction occurred in combination with acceptor 2a in the presence of Pd/L4 and acid A2, which suggested that expected zwitterionic intermediate III might not be generated via a similar oxidative addition/deprotonation process under the standard conditions.18 These results indicated that the indene-based structure was crucial for the vinylogous activation of the benzylic C–H group to generate the active dipole species. Therefore, as outlined in Scheme 3b, it was proposed that allylic alcohol 1c would be partially protonated in the presence of phosphoric acid A2 and undergo oxidative addition with complex Pd(0)/L4 along with the release of H2O. The π-allylpalladium complex moiety of the resultant intermediate I would further enhance the acidity of the benzylic C–H group, and a deprotonation process would occur to give dipole II, which would more reasonably exist as a polyconjugated 10π-type cycloaddend. Subsequently, acid A2 would act as a Brønsted acid to activate α-cyano chalcone 2a, rendering the assembly with dipole II to deliver adduct IV. An intramolecular allylic alkylation would be followed to afford 3a′ together with the regeneration of Pd(0). Finally, an isomerisation process took place with the assistance of Et3N to furnish the thermally more stable product 3a.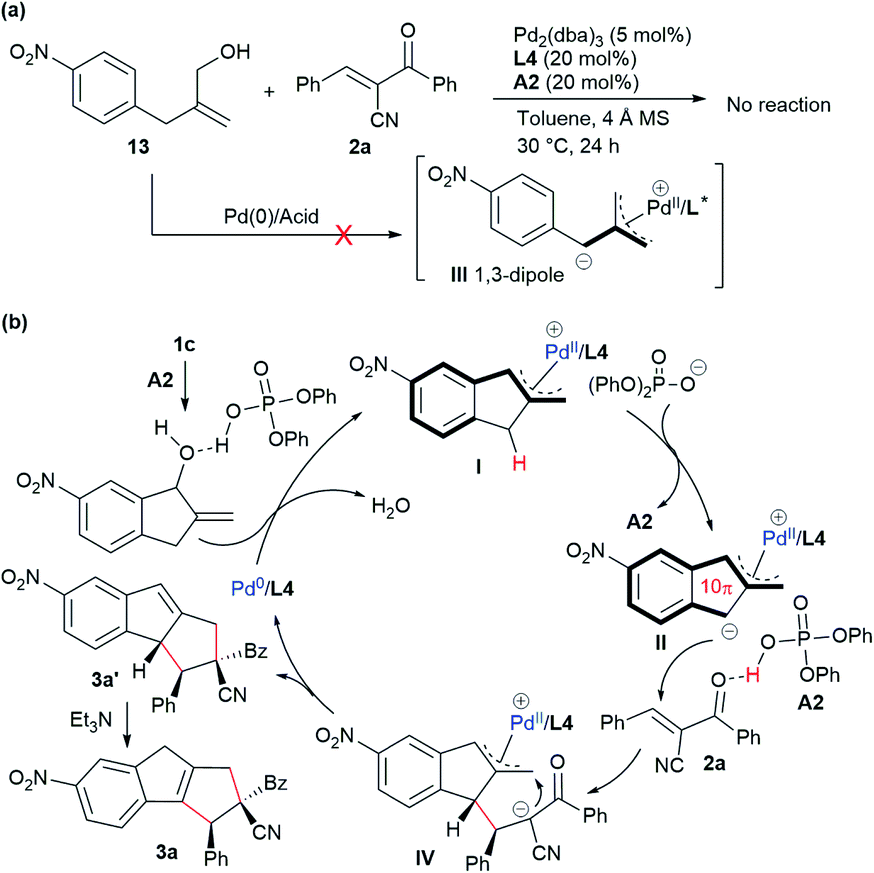 | ||
| Scheme 3 Mechanistic proposal. (a) Control experiment. (b) Proposed catalytic mechanism. | ||
Conclusions
A new type of metal-containing 10π-cycloaddend could be generated from benzocyclopentenols with an electron-withdrawing group via a deprotonation strategy under the cooperative catalysis of Pd(0) and phosphoric acid. The subsequent asymmetric higher-order [10 + 2] cycloaddition reactions with diverse activated alkenes were efficiently accomplished by employing a newly designed phosphoramidite ligand, producing polycyclic frameworks embedding an indene core in moderate to excellent yields with good to excellent enantioselectivity. In addition, [10 + 8] and [10 + 4] higher-order cycloaddition reactions were similarly realised with a heptafulvene derivative or a cyclic enone substrate, respectively. The newly designed metal-containing 10π-synthons would have more potential in asymmetric reactions, and the results will be reported in due course.Data availability
The data that support the findings of this study are available in the ESI† or on request from the corresponding author.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from NSFC (21971166, 21961132004, 21931006 and 21921002) and 111 Project (B18035).Notes and references
- (a) D. McLeod, M. K. Thøgersen, N. I. Jessen, K. A. Jørgensen, C. S. Jamieson, X.-S. Xue, K. N. Houk, F. Liu and R. Hoffmann, Acc. Chem. Res., 2019, 52, 3488 CrossRef CAS PubMed; (b) S. Frankowski, M. Romaniszyn, A. Skrzyńska and Ł. Albrecht, Chem.–Eur. J., 2020, 26, 2120 CrossRef CAS; (c) N. I. Jessen, D. McLeod and K. A. Jørgensen, Chem, 2022, 8, 20 CrossRef CAS; (d) M.-M. Zhang, B.-L. Qu, B. Shi, W.-J. Xiao and L.-Q. Lu, Chem. Soc. Rev., 2022, 51, 4146 RSC.
- (a) K. N. Houk and R. B. Woodward, J. Am. Chem. Soc., 1970, 92, 4143 CrossRef CAS; (b) K. N. Houk and R. B. Woodward, J. Am. Chem. Soc., 1970, 92, 4145 CrossRef CAS For related initial contributions, see: (c) R. C. Cookson, B. V. Drake, J. Hudec and A. Morrison, Chem. Commun., 1966, 15 RSC; (d) S. Ito, Y. Fujise, T. Okuda and Y. Inoue, Bull. Chem. Soc. Jpn., 1966, 39, 1351 CrossRef CAS; (e) L. A. Pa-quette and J. H. Barrett, J. Am. Chem. Soc., 1966, 88, 2590 CrossRef CAS.
- (a) K. N. Houk, L. J. Luskus and N. S. Bhacca, J. Am. Chem. Soc., 1970, 92, 6392 CrossRef CAS; (b) T. C. Wu and K. N. Houk, J. Am. Chem. Soc., 1985, 107, 5308 CrossRef CAS; (c) W. Friedrichsen, W. Seidel and T. Debaerdemaeker, J. Heterocycl. Chem., 1983, 20, 1621 CrossRef CAS; (d) D. McLeod, A. CherubiniCelli, N. Sivasothirajah, C. H. McCulley, M. L. Christensen and K. A. Jørgensen, Chem.–Eur. J., 2020, 26, 11417 CrossRef CAS PubMed; (e) Y. Hayashi, H. Gotoh, M. Honma, K. Sankar, I. Kumar, H. Ishikawa, K. Konno, H. Yui, S. Tsuzuki and T. Uchimaru, J. Am. Chem. Soc., 2011, 133, 20175 CrossRef CAS PubMed.
- For a review, see: (a) J. H. Rigby, Acc. Chem. Res., 1993, 26, 579 CrossRef CAS For selected examples, see: (b) J. H. Rigby and H. S. Ateeq, J. Am. Chem. Soc., 1990, 112, 6442 CrossRef CAS; (c) K. Chaffee, P. Huo, J. B. Sheridan, A. Barbieri, A. Aistars, R. A. Lalancette, R. L. Ostrander and A. L. Rheingold, J. Am. Chem. Soc., 1995, 117, 1900 CrossRef CAS; (d) J. H. Rigby, K. M. Short, H. S. Ateeq and J. A. Henshilwood, J. Org. Chem., 1992, 57, 5290 CrossRef CAS.
- For selected examples, see: (a) G. Bertuzzi, D. McLeod, L.-M. Mohr and K. A. Jørgensen, Chem.–Eur. J., 2020, 26, 15491 CrossRef CAS PubMed; (b) M. Xie, X. Liu, X. Wu, Y. Cai, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2013, 52, 5604 CrossRef CAS PubMed; (c) B. M. Trost, P. J. McDougall, O. Hartmann and P. T. Wathen, J. Am. Chem. Soc., 2008, 130, 14960 CrossRef CAS PubMed; (d) P. Yu, C. Q. He, A. Simon, W. Li, R. Mose, M. K. Thøgersen, K. A. Jørgensen and K. N. Houk, J. Am. Chem. Soc., 2018, 140, 13726 CrossRef CAS PubMed; (e) M. Romaniszyn, K. Gronowska and Ł. Albrecht, J. Org. Chem., 2019, 84, 9929 CrossRef CAS PubMed; (f) S. Frankowski, A. Skrzyńska and Ł. Albrecht, Chem. Commun., 2019, 55, 11675 RSC; (g) R. Mose, G. Preegel, J. Larsen, S. Jakobsen, E. H. Iversen and K. A. Jørgensen, Nat. Chem., 2017, 9, 487 CrossRef CAS PubMed; (h) H. Liu, Y. Wu, Y. Zhao, Z. Li, L. Zhang, W. Yang, H. Jiang, C. Jing, H. Yu, B. Wang, Y. Xiao and H. Guo, J. Am. Chem. Soc., 2014, 136, 2625 CrossRef CAS PubMed.
- (a) D. Johnson and G. Jones, J. Chem. Soc., Perkin Trans. 1, 1972, 840 RSC; (b) N. I. Jessen, M. Bura, G. Bertuzzi and K. A. Jørgensen, Angew. Chem., Int. Ed., 2021, 60, 18527 CrossRef CAS PubMed.
- K. Hafner and W. Bauer, Angew. Chem., Int. Ed. Engl., 1968, 7, 297 CrossRef CAS.
- (a) Z. Zhou, Z.-X. Wang, Y.-C. Zhou, W. Xiao, Q. Ouyang, W. Du and Y.-C. Chen, Nat. Chem., 2017, 9, 590 CrossRef CAS PubMed; (b) G. Bertuzzi, M. K. Thøgersen, M. Giardinetti, A. Vidal-Albalat, A. Simon, K. N. Houk and K. A. Jørgensen, J. Am. Chem. Soc., 2019, 141, 3288 CrossRef CAS PubMed; (c) B. S. Donslund, A. Monleón, T. A. Palazzo, M. L. Christensen, A. Dahlgaard, J. D. Erickson and K. A. Jørgensen, Angew. Chem., Int. Ed., 2018, 57, 1246 CrossRef CAS PubMed; (d) B. S. Donslund, N. I. Jessen, G. Bertuzzi, M. Giardinetti, T. A. Palazzo, M. L. Christensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2018, 57, 13182 CrossRef CAS PubMed; (e) D. McLeod, J. A. Izzo, D. K. B. Jørgensen, R. F. Lauridsen and K. A. Jørgensen, ACS Catal., 2020, 10, 10784 CrossRef CAS; (f) J. Zhao, X. Zheng, Y.-S. Gao, J. Mao, S.-X. Wu, W.-L. Yang, X. Luo and W.-P. Deng, Chin. J. Chem., 2021, 39, 3219 CrossRef CAS; (g) N. I. Jessen, G. Bertuzzi, M. Bura, M. L. Skipper and K. A. Jørgensen, J. Am. Chem. Soc., 2021, 143, 6140 CrossRef CAS PubMed; (h) G. Bertuzzi, V. Corti, J. A. Izzo, S. Ričko, N. I. Jessen and K. A. Jørgensen, J. Am. Chem. Soc., 2022, 144, 1056 CrossRef CAS PubMed.
- (a) Q. Peng, S.-J. Li, B. Zhang, D. Guo, Y. Lan and J. Wang, Commun. Chem., 2020, 3, 177 CrossRef CAS; (b) Y. Yang, Y. Jiang, W. Du and Y.-C. Chen, Chem.–Eur. J., 2020, 26, 1754 CrossRef CAS PubMed.
- (a) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366 CrossRef CAS PubMed; (b) J.-J. Feng and J. Zhang, ACS Catal., 2016, 6, 6651 CrossRef CAS; (c) B. D. W. Allen, C. P. Lakeland and J. P. A. Harrity, Chem.–Eur. J., 2017, 23, 13830 CrossRef CAS PubMed; (d) N. De and E. J. Yoo, ACS Catal., 2018, 8, 48 CrossRef CAS; (e) T.-R. Li, Y.-N. Wang, W.-J. Xiao and L.-Q. Lu, Tetrahedron Lett., 2018, 59, 1521 CrossRef CAS.
- For selected examples with non-conjugated dipole species, see: (a) L.-C. Yang, Y.-N. Wang, R. Liu, Y. Luo, X. Q. Ng, B. Yang, Z.-Q. Rong, Y. Lan, Z. Shao and Y. Zhao, Nat. Chem., 2020, 12, 860 CrossRef CAS PubMed; (b) Y.-N. Wang, L.-C. Yang, Z.-Q. Rong, T.-L. Liu, R. Liu and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 1596 CrossRef CAS; (c) Q.-L. Zhang, Q. Xiong, M.-M. Li, W. Xiong, B. Shi, Y. Lan, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 14096 CrossRef CAS PubMed.
- (a) T. Hayashi, T. Suzuka, A. Okada and M. Kawatsura, Tetrahedron: Asymmetry, 2004, 15, 545 CrossRef CAS; (b) H.-L. Cui, X.-H. Sun, L. Jiang, L. Dong and Y.-C. Chen, Eur. J. Org. Chem., 2011, 7366 CrossRef CAS; (c) J. Zhang, H.-H. Wu and J. Zhang, Org. Lett., 2017, 19, 6080 CrossRef CAS PubMed.
- Z. Wang, Z. He, L. Zhang and Y. Huang, J. Am. Chem. Soc., 2018, 140, 735 CrossRef CAS PubMed.
- Please see the ESI† for more details.
- (a) B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467 RSC; (b) A. Baeza and C. Nájera, Synthesis, 2014, 46, 25 CrossRef; (c) N. A. Butt and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929 RSC.
- (a) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed; (b) G. Jiang and B. List, Angew. Chem., Int. Ed., 2011, 50, 9471 CrossRef CAS PubMed; (c) Z.-L. Tao, W.-Q. Zhang, D.-F. Chen, A. Adele and L.-Z. Gong, J. Am. Chem. Soc., 2013, 135, 9255 CrossRef CAS PubMed; (d) C.-C. Tsai, C. Sandford, T. Wu, B. Chen, M. S. Sigman and F. D. Toste, Angew. Chem., Int. Ed., 2020, 59, 14647 CrossRef CAS PubMed.
- X. Chen, M. K. Thøgersen, L. Yang, R. F. Lauridsen, X.-S. Xue, K. A. Jørgensen and K. N. Houk, J. Am. Chem. Soc., 2021, 143, 934 CrossRef CAS PubMed.
- The corresponding 1,3-dipole could be formed from the allylic carbonate under the catalysis of Pd(0) and special ligands, see: (a) B. M. Trost and Z. Zuo, Angew. Chem., Int. Ed., 2020, 59, 1243 CrossRef CAS PubMed; (b) B. M. Trost, Z. Jiao and C.-I. Hung, Angew. Chem., Int. Ed., 2019, 58, 15154 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data for new compounds, NMR and HRMS spectra and HPLC chromatograms, and the CIF file of enantiopure product 3a. CCDC 2174555. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02985e |
| ‡ A. Li and Y. Gao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |