
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect observation of reversible bond homolysis by 2D EXSY NMR†
Satoshi
Takebayashi
 *a,
Robert R.
Fayzullin
b and
Richa
Bansal
a
*a,
Robert R.
Fayzullin
b and
Richa
Bansal
a
aScience and Technology Group Okinawa Institute of Science and Technology Graduate University 1919-1 Tancha, Onna-son, Okinawa 904-0495, Japan. E-mail: satoshi.takebayashi@oist.jp
bArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center, Russian Academy of Sciences, 8 Arbuzov Street, Kazan 420088, Russian Federation
First published on 1st August 2022
Abstract
Bond homolysis is one of the most fundamental bond cleavage mechanisms. Thus, understanding of bond homolysis influences the development of a wide range of chemistry. Photolytic bond homolysis and its reverse process have been observed directly using time-resolved spectroscopy. However, direct observation of reversible bond homolysis remains elusive. Here, we report the direct observation of reversible Co–Co bond homolysis using two-dimensional nuclear magnetic resonance exchange spectroscopy (2D EXSY NMR). The characterization of species involved in this homolysis is firmly supported by diffusion ordered NMR spectroscopy (DOSY NMR). The unambiguous characterization of the Co–Co bond homolysis process enabled us to study ligand steric and electronic factors that influence the strength of the Co–Co bond. Understanding of these factors will contribute to rational design of multimetallic complexes with desired physical properties or catalytic activity.
Introduction
Bond homolysis and heterolysis are the most common mechanisms for the cleavage of covalent bonds. Therefore, a fundamental understanding of these mechanisms is important for the development of a wide range of chemistry. Many studies have been reported to understand the fundamental aspects of bond homolysis, and studies are ongoing to utilize this process in a variety of chemistry, biology, and interplay of them,1 including catalysis,2,3 natural product synthesis,4 polymer5 and material chemistry.6,7 The majority of bond homolysis reactions are irreversible reactions that favour the formation of covalent bonds. However, it is known that steric crowding around the radical center and delocalization of spin density shift this equilibrium towards the generation of persistent radical species,8 and by virtue of this stabilization, many radical species have been isolated.7,9,10 Conventionally (Fig. 1a), reversible bond homolysis has been studied most frequently using a temperature-dependent reversible change of UV-Vis,11–22 IR,15,23,24 EPR,17,18,20–22,25–29 or NMR16–18,21–24,30–37 spectra. In a few cases, reversible bond homolysis has been observed in the solid state by powder X-ray diffractometry.38–40 Direct observation of photolytic bond homolysis and irreversible recombination and/or decomposition of transient radical species has been reported in detail.41–45 However, direct spectroscopic detection of reversible bond homolysis remains elusive.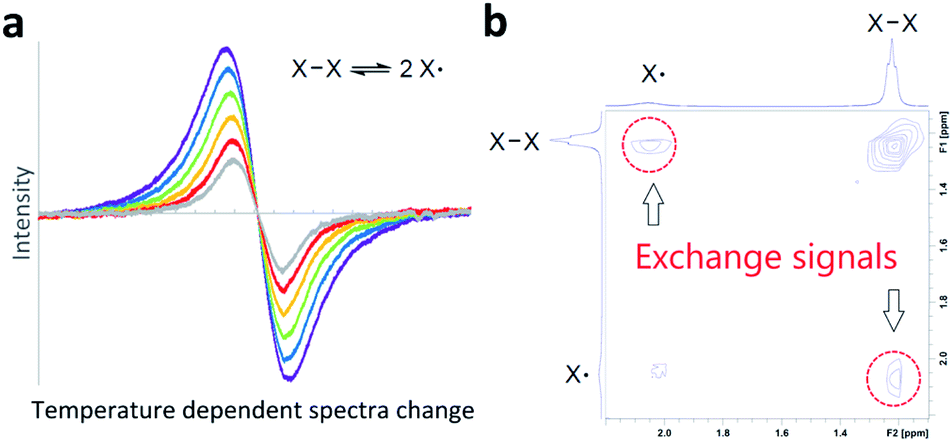 | ||
| Fig. 1 Methods to observe and prove reversible bond homolysis. (a) A conventional method: temperature-dependent reversible change of spectra. (b) This work: direct observation of exchange signals by 2D EXSY NMR. | ||
Two-dimensional NMR exchange spectroscopy46–48 (2D EXSY NMR) and 2D IR49–52 spectroscopy are unique methods to detect real-time chemical exchange as exchange signals. A large variety of chemical exchanges with a wide range of exchange time scales have been studied using these methods;48,51 however, their application in direct observation of reversible bond homolysis is unknown (Fig. 1b). A possible reason for this lack of observation is the incompatibility of the exchange rates of reversible bond homolysis. Thus, reversible bond homolysis is too slow to be observed using a 2D IR method.53,54 In contrast, it is usually too fast to obtain resolved NMR signals from both the radical species and dimer of the radical species at NMR compatible temperatures (−150 to 150 °C).18,32,33
Few reports24,37,55 are known where the exchange rates are slow enough to resolve monomeric and dimeric species in an NMR spectrum. However, real-time 2D EXSY NMR observation of exchange between these monomers and dimers has not been reported, possibly due to the broadening of NMR signals caused by paramagnetic radical species. Therefore, in theory, if the rate of reversible bond homolysis is slow and sufficiently sharp NMR signals from both paramagnetic radical species and diamagnetic radical dimers are detectable, direct observation of reversible bond homolysis is possible using 2D EXSY NMR.
Recently, we have isolated formal 17-electron Co(0) metalloradical complex 1 utilizing a bulky ring-expanded N-heterocyclic carbene (reNHC) ligand (Fig. 2).56 This class of Co(0) carbonyl complexes usually exists as dimers to fulfill the formal 18-electron configuration; however, 1 was isolated as a monomer as the bulky reNHC ligand prevents dimerization. Furthermore, some of the 1H NMR signals from 1 were relatively sharp (linewidth at half maximum < 100 Hz). From this finding, we envisioned that direct 2D EXSY NMR observation of reversible bond homolysis is possible if less bulky NHC ligands, which promote reversible homolysis of Co–Co bonds, were employed. Here, we report the direct observation of reversible Co–Co bond homolysis using 2D EXSY NMR.
 | ||
| Fig. 2 Isolable Co-metalloradical 1. | ||
Results and discussion
Preparation and characterization of a cobalt dimer
At the onset of this study, 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene (SIMes) was chosen as a less bulky NHC ligand with respect to the ring-expanded analog in 1 since the steric bulk of NHC ligands is known to increase by increasing the ring size of the NHC ligands.57–61 The reaction between SIMes and Co2(CO)8 formed dimeric cobalt complex 2-Mes in a 95% yield (Fig. 3a). The diamagnetic complex 2-Mes was characterized using single-crystal X-ray diffractometry (SC-XRD), 1H, 13C{1H}, and 2D NMR, FTIR, and UV-Vis spectroscopy, and elemental analysis. The SC-XRD structures of 2-Mes revealed a dimeric structure with a long Co–Co bond of 2.71989(18) Å, which is one of the longest Co(0)–Co(0) bonds known62,63 due to the steric effect of the SIMes ligand (Fig. 3b). There is another crystallographically independent, centrosymmetric molecule of 2-Mes in the same unit cell; however, the Co–Co bond length of this molecule is even longer (2.7520(2) Å). The presence of a Co–Co bond is also supported by UV-Vis absorption at 369 nm. This absorption was assigned as a σ–σ* transition of the Co–Co bonds based on a previous report.64 At first glance, sharp, intense 1H and 13C{1H} NMR signals from 2-Mes at 24.0 °C support a diamagnetic, dimeric structure in a solution as observed in the solid state (Fig. 3c). However, a close inspection of the 1H NMR spectra revealed the presence of a small broad signal at 4.62 ppm in all batches of 2-Mes (Fig. 3c). Initially, we assumed this species (2-Mes*) to be a trace impurity in 2-Mes since the relative integration of this species was roughly 30 times smaller than that of 2-Mes. However, we found that the intensity of this signal increased reversibly with increasing temperature. Thus, in C6D6, the initial 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 ratio at 24.0 °C changed to a 1:3 ratio at 69.9 °C, along with a significant broadening of signals from 2-Mes (Fig. 3c). In comparison, no broadening of the residual C6D6 signal was observed. Four very broad 1H NMR signals were detectable at 60.6 °C; however, an accurate integration ratio could not be obtained due to signal overlapping and broadening. Monitoring of magnetic susceptibility of C6D6 solution of 2-Mes using Evans' method also revealed a reversible increase of magnetic susceptibility with increasing temperature (Table S1†). Thus, paramagnetic species is formed on heating.
:30 ratio at 24.0 °C changed to a 1:3 ratio at 69.9 °C, along with a significant broadening of signals from 2-Mes (Fig. 3c). In comparison, no broadening of the residual C6D6 signal was observed. Four very broad 1H NMR signals were detectable at 60.6 °C; however, an accurate integration ratio could not be obtained due to signal overlapping and broadening. Monitoring of magnetic susceptibility of C6D6 solution of 2-Mes using Evans' method also revealed a reversible increase of magnetic susceptibility with increasing temperature (Table S1†). Thus, paramagnetic species is formed on heating.
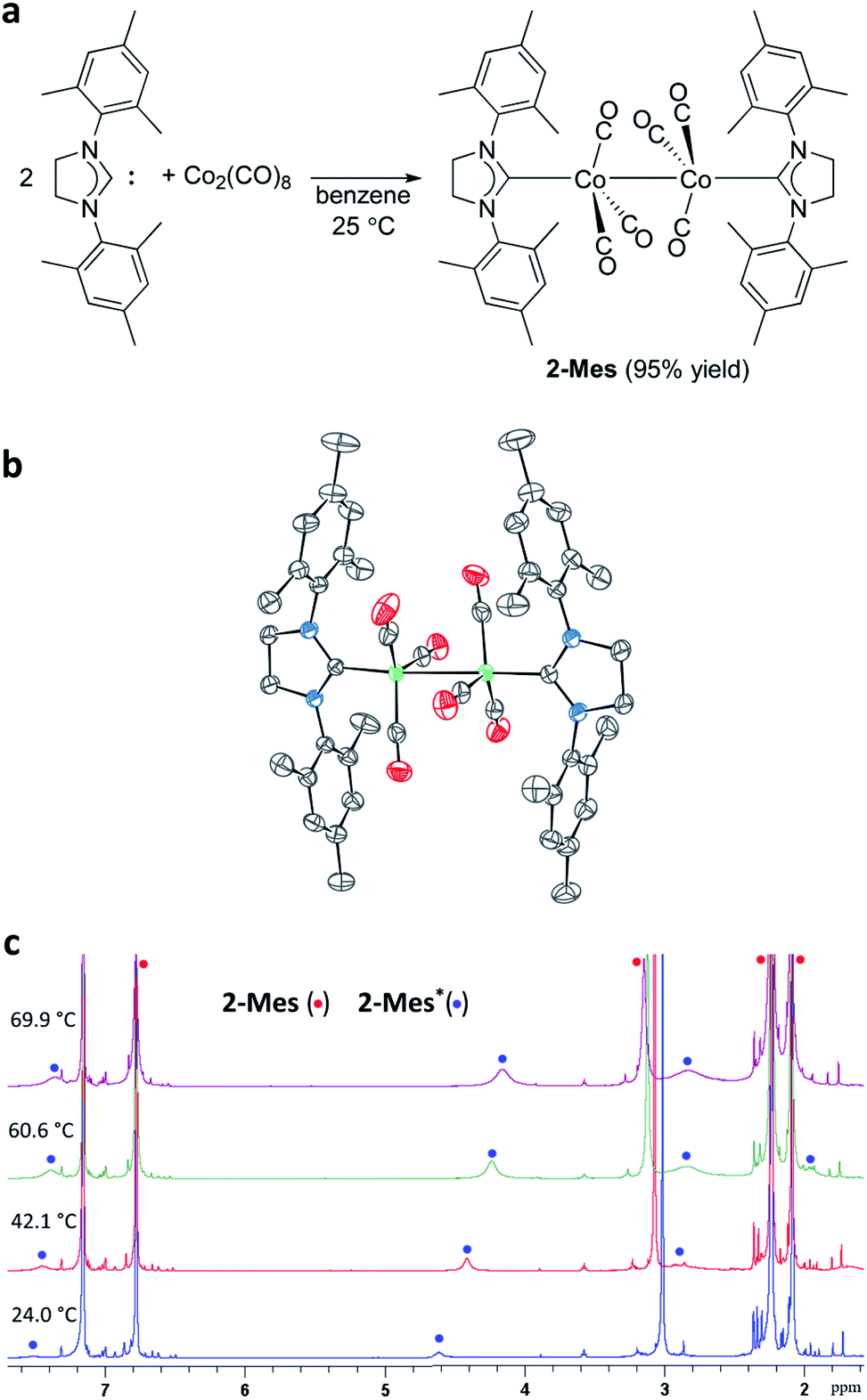 | ||
| Fig. 3 Preparation and characterization of 2-Mes. (a) Preparation of 2-Mes. (b) SC-XRD structure of 2-Mes with 80% thermal ellipsoids. Hydrogen atoms, solvent molecules, and the second crystallographically independent 2-Mes molecule are omitted for clarity. (c) Variable temperature 1H NMR spectra (500 MHz, C6D6) of 2-Mes showing the increasing concentration of radical species 2-Mes* on increasing the temperature. | ||
Characterization of the paramagnetic species 2-Mes*
The identity of the paramagnetic species 2-Mes was confirmed by several control experiments and spectroscopic observations. First, trapping of the paramagnetic species by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) resulted in the formation of TEMPO adducts 3 (Fig. 4a). This result suggests homolysis of the Co–Co bond in 2-Mes based on analogous reactivity between the isolated cobalt radical 1 and TEMPO.56 Second, the 1H NMR spectra showed that the formation of 2-Mes* is not accompanied by the formation of a free SIMes ligand, and hence, dissociation of the SIMes ligand is not involved in this process. Third, the formation of 2-Mes* is not affected by the presence of CO pressure (3 bar) (Fig. S44†). Thus, dissociation of the CO ligand is also not involved in the formation of 2-Mes*. Fourth, the 2-Mes*/2-Mes ratio increases with increasing dilution of the solution (Table S2†). This observation is consistent with the favored formation of monomeric species in a dilute solution. Fifth, the EPR measurement of the C6D6 solution of 2-Mes at 20 to 62 °C showed the presence of a cobalt-centered radical species with an isotropic g-value of 2.08. The observed isotropic g-value is the same as the one observed for isolated Co radical 1.56 As in the case of the NMR observations, EPR signal intensity increased reversibly with increasing temperature (Fig. 4b and S46†). Variable temperature UV-Vis spectroscopy revealed a reversible decrease or increase of the Co–Co σ–σ* transition absorption band at 369 nm on increasing or decreasing the solution temperature, respectively12 (Fig. 4c and S47†). Thus, the Co–Co bond cleaves reversibly according to temperature change. Collectively, these observations support the temperature-dependent formation of a cobalt-centered radical via the cleavage of the Co–Co bond in 2-Mes. Finally, the characterization of 2-Mes* was strongly supported by 2D diffusion ordered NMR spectroscopy65 (2D DOSY NMR) measurement of a mixture of 2-Mes and 2-Mes* in the presence of [Ni(SIMes)(CO)3](4)66 as an internal standard (Fig. 4d). The molecular weight (449.17 g mol−1), tetrahedral geometry, and charge neutrality of 4 make this molecule the closest structural analog to the tetrahedral56 and neutral [Co(SIMes)(CO)3] radical (449.41 g mol−1) with practically identical molecular weights. 2D DOSY NMR spectra recorded at 24 °C showed complete sets of signals from 2-Mes and 4 (Fig. 4d), and one signal from 2-Mes* at 4.62 ppm due to a lower concentration of 2-Mes* and broadening of signals from paramagnetic 2-Mes*.65 A 2D DOSY NMR study revealed that 2-Mes* (1.25(5) × 10−9 m2 s−1) and 4 (1.27 × 10−9 m2 s−1) have nearly identical diffusion coefficients. Consistent with this observation, the diffusion coefficient of the dimeric complex 2-Mes (0.895 × 10−9 m2 s−1) was significantly smaller than that of 4 and 2-Mes*. To the best of our knowledge, DOSY NMR characterization of monomeric and dimeric species involved in reversible bond homolysis is unprecedented. Together, these observations strongly support that the paramagnetic species 2-Mes* is a monomeric [Co(SIMes)(CO)3] radical that is in equilibrium with dimeric 2-Mes.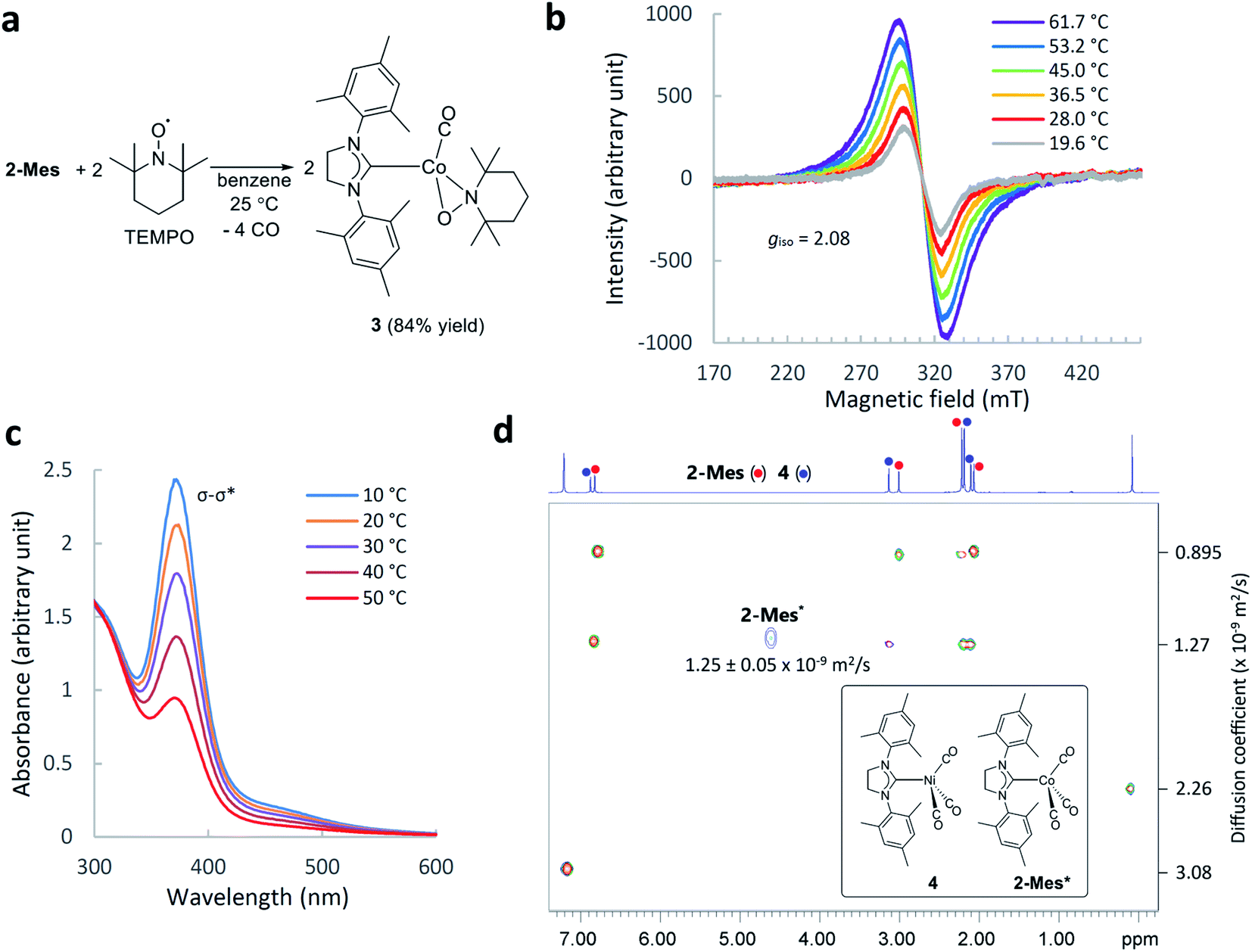 | ||
| Fig. 4 Characterization of 2-Mes*. (a) Trapping of 2-Mes* by TEMPO. (b) Variable temperature EPR spectra (X-band, in C6D6) of 2-Mes*. (c) Change of UV-Vis spectra (in C6H6) of a mixture of 2-Mes and 2-Mes* on increasing the temperature from 10 to 50 °C. (d) 2D DOSY NMR spectrum (500 MHz, C6D6, 24.0 °C) of an equilibrium mixture of 2-Mes and 2-Mes* in the presence of 4 and hexamethyldisiloxane (a signal at around 0 ppm) as internal standards. | ||
Observation of reversible Co–Co bond homolysis by 2D EXSY NMR
With the reliable characterization of both 2-Mes and 2-Mes* in hand, 2D EXSY NMR observation of reversible Co–Co bond homolysis was examined. To our delight, the 2D EXSY NMR spectra (46.7 °C, in C6D6) of the mixture of 2-Mes and 2-Mes* unequivocally showed exchange signals for the homolysis of the Co–Co bond in 2-Mes (Fig. 5). More specifically, 2D EXSY NMR showed four exchange signals between four 1H signals from 2-Mes and four 1H signals from 2-Mes*. The phase difference between these exchange signals (black) and the nuclear Overhauser effect (NOE) signals (red) clearly showed that these signals are due to an exchange between 2-Mes and 2-Mes*. Consistent with the 2D EXSY NMR observation, the 1H NMR signals from 2-Mes and 2-Mes* broadened upon increasing the temperature and merged into four broad signals at 137 °C (recorded in toluene-d8 under 5 bar N2) (Fig. S48†).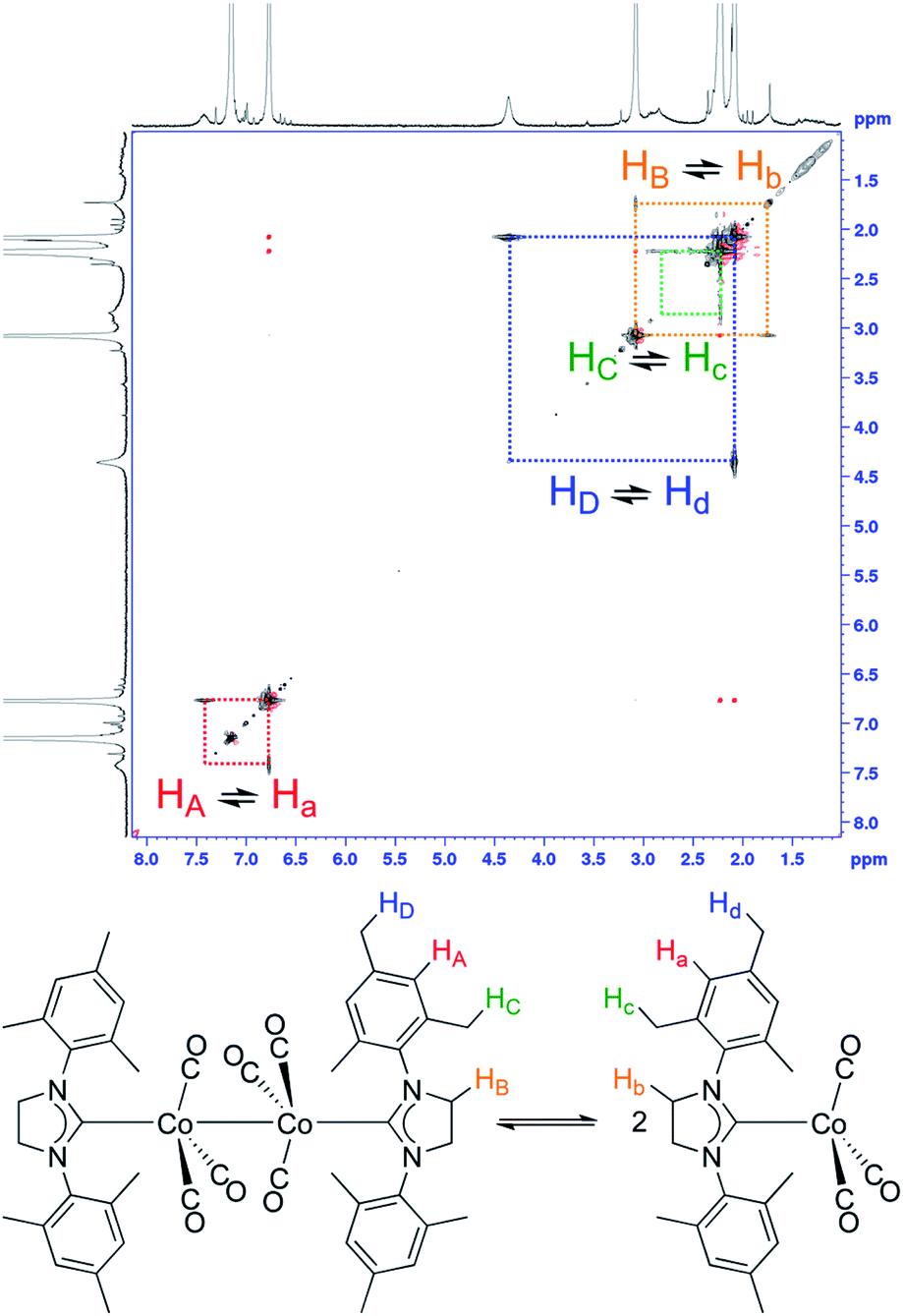 | ||
| Fig. 5 Direct 2D EXSY NMR observation of Co–Co bond homolysis. 2D EXSY NMR spectra (500 MHz, C6D6, 46.7 °C) of an equilibrium mixture of 2-Mes and 2-Mes*, and assignment of exchanging signals. Red off-diagonal signals are due to NOE. | ||
2D EXSY NMR enabled us to assign all 1H NMR signals from paramagnetic 2-Mes*, and hence concentration ratios between 2-Mes and 2-Mes* were determined at variable temperatures using the integration ratio of 1H NMR signals. The van't Hoff plot (Fig. 6) based on the obtained ratio of 2-Mes and 2-Mes* revealed an experimental Co–Co bond dissociation enthalpy (BDE) of 20.4(2) kcal mol−1, entropy (ΔS°) of 50.1(7) cal mol−1 K−1 (averages of three independent measurements and standard deviations in brackets), and bond dissociation free energy (BDEF) of 5.5 ± 0.02 kcal mol−1. The observed BDE is within the range of the reported Co–Co BDE of Co2(CO)8 (19(2) or 20(7) kcal mol−1).32,67 The observed large positive ΔS° is in line with other bond homolysis processes.12,18,21,23,25,29,32–34,37,55 It is proposed that ΔS° increases with increasing steric factors due to more restricted metal–metal bond rotation.23,25
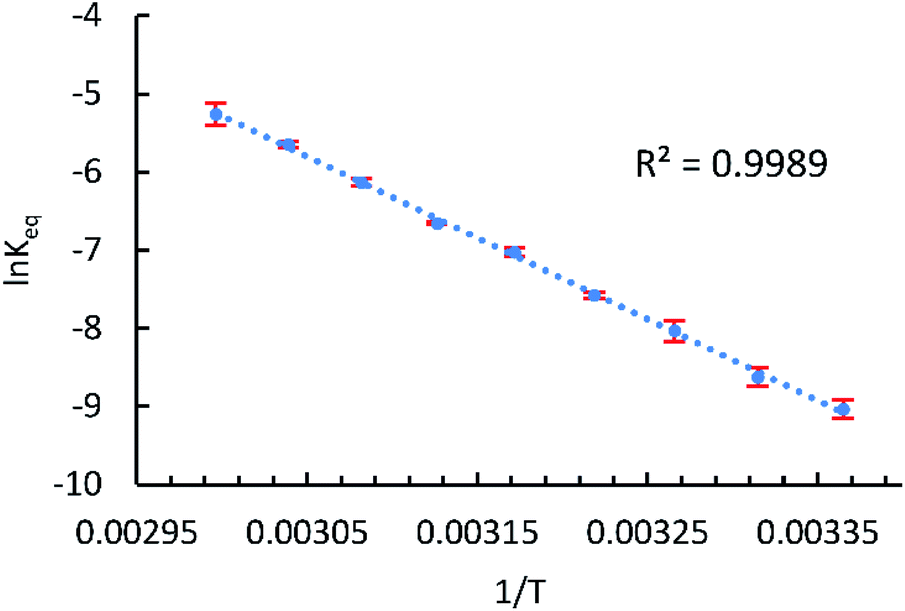 | ||
| Fig. 6 van't Hoff plot for the Co–Co bond homolysis of 2-Mes plotted using NMR data. T: temperature (K), Keq: [2-Mes*]2/[2-Mes] (mol L−1), and error bar: standard deviation of three independent measurements. | ||
BDE and ΔS° for the homolysis of the Co–Co bond in 2-Mes were also determined by double integration of variable temperature EPR spectra using a solution of 1 as an external standard (see the ESI for more details). The van't Hoff plot based on the EPR measurement in C6D6 gave a BDE of 20.3(5) kcal mol−1 and ΔS° of 48.2(17) cal mol−1 K−1 (averages and standard deviations of two independent measurements). These values are consistent with BDE and ΔS° determined by the NMR method. A Co–Co BDFE of 5.5 ± 0.02 kcal mol−1 is significantly lower than that of Co2(CO)8 (10(3) kcal mol−1) despite the relatively large experimental uncertainty of the latter value. This large decrease of Co–Co BDFE can be explained by a combination of the bulky NHC ligand and electron donation from the NHC ligand to a Co–Co antibonding orbital.68
Elucidation of ligand steric and electronic effects on Co–Co bond strength
The study of metal–metal bonds and their application are currently among the most active areas of study in catalysis,69,70 and inorganic and organometallic chemistry.71 The ligand steric effect is a reliable way to control metal–metal bond strength.13,56,72–74 In contrast, the ligand electronic effect on metal–metal bond strength is not fully understood23,68,75 partly due to the lack of an appropriate system to study ligand steric and electronic effects separately.23 Thus, we addressed this question by investigating BDE, ΔS°, and BDFE of Co–Co bond homolysis in several [Co(NHC)(CO)3]2 complexes (Fig. 7) using our NMR method and systematic change of steric and electronic properties of the NHC ligands.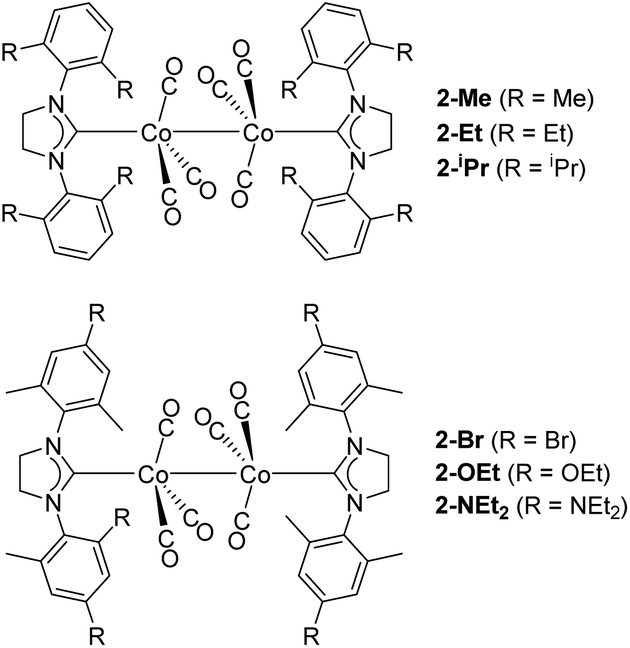 | ||
| Fig. 7 Structures of [Co(NHC)(CO)3]2 complexes. | ||
As in the case of 2-Mes, in all 1H NMR spectra of [Co(NHC)(CO)3]2 complexes, one set of sharp diamagnetic signals from [Co(NHC)(CO)3]2 and another set of broader paramagnetic signals from monomeric [Co(NHC)(CO)3] species were observed (Fig. S54–59†). The 2D EXSY NMR spectra confirmed exchange between [Co(NHC)(CO)3]2 and [Co(NHC)(CO)3]. Experimental Co–Co BDE, ΔS°, and BDEF were determined using the van't Hoff plot based on the NMR integration ratio of dimeric and monomeric species. Comparison of thermodynamic parameters between 2-Me, 2-Et, and 2-iPr revealed a small decrease of BDFE (<1.0 kcal mol−1) upon increasing the steric bulk due to a combination of a slight (<0.8 kcal mol−1) decrease of BDE and a slight increase of ΔS° (<1.1 cal mol−1 K−1) (Table 1). This observation is consistent with the study by Watkins et al.,23 where they found that bulkier [CrCp*(CO)3]2 (Cp*: pentamethylcyclopentadienyl) has a smaller BDE and larger positive ΔS° than [CrCp(CO)3]2 (Cp: cyclopentadienyl) for the cleavage of Cr–Cr bonds, even though their study could not separate the contribution from steric and electronic effects.
| Complex | Co–Co (Å)a | BDE (kcal mol−1)b | ΔS° (cal mol−1 K−1)b | BDFE (kcal mol−1)b,c | σ–σ* (nm)d | ΔG‡298 (kcal mol−1)b,e |
|---|---|---|---|---|---|---|
| a Data from SC-XRD recorded at 100(2) K. b Average of three independent measurements and their standard deviations in brackets or after ± sign. c Calculated using BDE and ΔS° obtained from the van't Hoff plot at a temperature of 298.15 K. d Data from UV-Vis spectra measured in THF. e Calculated using the Eyring plot at a temperature of 298.15 K. f Values for each crystallographically independent molecule in the unit cell. | ||||||
| 2-Me | 2.7183(6) | 21.1(2) | 51.3(7) | 5.8 ± 0.01 | 372 | 18.1(7) |
| 2-Et | 2.7461(4), 2.7492(4)f | 20.5(2) | 50.9(6) | 5.3 ± 0.04 | 372 | 18.5(1) |
| 2-iPr | 2.7512(3) | 20.3(3) | 52.0(10) | 4.8 ± 0.03 | 381 | 18.4(6) |
| 2-Br | 2.7153(3) | 23.1(4) | 53.9(11) | 7.0 ± 0.1 | 371 | 19.2(3) |
| 2-Mes | 2.71989(18), 2.7520(2)f | 20.4(2) | 50.1(7) | 5.5 ± 0.02 | 369 | 18.5(7) |
| 2-OEt | 2.7431(3) | 19.3(1) | 47.0(3) | 5.3 ± 0.06 | 370 | 18.9(3) |
| 2-NEt2 | 2.72357(18) | 17.9(5) | 45.3(13) | 4.4 ± 0.06 | 364 | 18.9(1) |
When the electronic effect of the NHC ligands was examined using 2-Me, 2-Mes, 2-Br, 2-OEt, and 2-NEt2 (Table 1), a larger decrease of BDFE (<2.6 kcal mol−1) was observed upon increasing electron-donating ability of NHC ligands. In this case, a combination of a larger decrease of BDE (<5.2 kcal mol−1) and a large decrease of ΔS° (<8.6 cal mol−1 K−1) resulted in the overall larger decrease of BDFE. From this experiment, we conclude that Co–Co bonds in [Co(ligand)(CO)3]2 complexes are weakened not only by steric bulk but also by electron donation from the ligand. This conclusion is consistent with the previous argument made by comparing Co–Co bond lengths.76 It is worth pointing out that Co(III)–alkyl bonds in cobaloxime complexes are strengthened by electron donation from the trans-axial ligands.77 This observation was explained by the fact that homolysis of the Co(III)–alkyl bond involves the reduction of the Co(III) complex to a Co(II) complex that is destabilized in the presence of an electron-donating ligand.77 Such an oxidation state change is not involved in Co–Co bond homolysis. However, the observed weakening of the Co–Co bond by an electron-donating ligand can be explained by an analogous argument. Thus, stabilization of the electron-deficient, formal 17-electron species by an electron-donating ligand will result in the preferred formation of 17-electron species and consequently weaker Co–Co bonds. Considering that all homolysis of bonds between transition metals forms species with a less formal electron count, we expect that the observed electronic effect is common in bonds between transition metals and can be applicable to rational design of transition metal complexes with various metal–metal bond strengths. Interestingly, in line with our observation, C–C bonds in substituted dicyanomethyl radical dimers are weaker in the presence of electron-donating substituents.29 We propose that the reported23,33 weakening of the Cr–Cr bond in [CrCp*(CO)3]2 is due to the combination of steric and electronic effects. For comparison, we also examined the 1H NMR spectra of [Co(PBu3)(CO)3]2, [Co(PCy3)(CO)3]2, and [Co(P(OAr)3)(CO)3]2 (Bu: n-butyl, Cy: cyclohexyl, Ar: 2,4-di-tert-butylphenyl). However, these phosphine analogs did not show signals from monomeric species even at 75 °C. This result is consistent with the crucial role of both steric hindrance60,78 and electron-donating properties79,80 of NHC ligands in weakening Co–Co bonds. Indeed, the BDE of a Co–Co bond in [Co(PBu3)(CO)3]2 is estimated to be > 23 kcal mol−1.81
Examination of the correlation between solid-state Co–Co bond length and BDE revealed a general trend of increasing Co–Co bond length with decreasing BDE. However, an anomalously shorter Co–Co bond was observed in 2-NEt2, probably due to crystal packing effects. Crystallographically determined metal–metal bond length does not always reflect metal–metal bond strength.70 We also estimated Gibbs free energy of activation (ΔG‡298) for the formation of a Co–Co bond (reverse homolysis) using a method based on NMR line broadening.33,82 In this case, however, all [Co(NHC)(CO)3]2 complexes studied showed a ΔG‡298 between 18.1 and 19.2 kcal mol−1 with a larger experimental error and no significant steric nor electronic effect on ΔG‡298 was observed. This observation is consistent with a relatively narrow range of UV-Vis absorption maxima (369–381 nm) that corresponds to the σ–σ* transition of Co–Co bonds in [Co(NHC)(CO)3]2 complexes. Thus, rate of Co–Co bond homolysis can be controlled by the relative thermodynamic stability of radical species.
Conclusions
We reported direct observation of Co–Co bond homolysis using 2D EXSY NMR. The unambiguous observation of this process was supported by variable temperature NMR, EPR, and UV-Vis measurements and a series of control experiments, including unprecedented characterization of the dimeric and monomeric species by 2D DOSY NMR. 2D EXSY NMR characterization of Co–Co bond homolysis of [Co(NHC)(CO)3]2 complexes enabled us to study ligand steric and electronic effects that influence Co–Co bond strength. This study revealed that Co–Co bonds in [Co(NHC)(CO)3]2 complexes are weakened by steric bulk as well as electron donation. The observed electronic effect was explained based on the stabilization of the electron-deficient formal 17-electron species by an electron-donating ligand. We expect that the observed steric and electronic effects on Co–Co bond strength are applicable to designing other multimetallic complexes with various metal–metal bond strengths for applications including catalysis and materials chemistry.Data availability
All experimental data associated with this work are available in the ESI.†Author contributions
S. T. conceived the research program, synthesized, and characterized NHC ligands and complexes, discovered the 2D EXSY NMR observation, collected SC-XRD data, and conducted experiments to elucidate the thermodynamic and kinetic parameters. R. R. F analyzed the SC-XRD data and wrote the SC-XRD part of the ESI.† R. B. synthesized the precursors of NHC ligands. S. T. wrote the manuscript with revisions provided by the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Japan Society for the Promotion of Science (18K14230 and 22K05134, S. T.). This work was supported by Okinawa Institute of Science and Technology Graduate University instrumental analysis and engineering sections. S. T. thanks Dr Hajime Sato (Bruker Japan) for assistance with the 2D DOSY NMR measurement. R. R. F. performed crystal structure determination within the government statements for the Kazan Scientific Center of RAS.Notes and references
- Encyclopedia of radicals in chemistry, biology, and materials, ed. C. Chatgilialoglu and A. Studer, Wiley, Hoboken, 2014 Search PubMed.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- J. Stubbe and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 13463–13472 CrossRef CAS PubMed.
- K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC.
- Y. Gao, D. Zhou, J. Lyu, S. A, Q. Xu, B. Newland, K. Matyjaszewski, H. Tai and W. Wang, Nat. Rev. Chem., 2020, 4, 194–212 CrossRef CAS.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- D. Sakamaki, S. Ghosh and S. Seki, Mater. Chem. Front., 2019, 3, 2270–2282 RSC.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13–19 CrossRef CAS.
- C. D. Hoff, Coord. Chem. Rev., 2000, 206–207, 451–467 CrossRef CAS.
- R. G. Hicks, Org. Biomole. Chem., 2007, 5, 1321–1338 RSC.
- J. Halpern and M. Pribanic, Inorg. Chem., 1970, 9, 2616–2618 CrossRef CAS.
- S. J. McLain, J. Am. Chem. Soc., 1988, 110, 643–644 CrossRef CAS.
- I. Kuksis and M. C. Baird, Organometallics, 1996, 15, 4755–4762 CrossRef CAS.
- T. R. Dugan, E. Bill, K. C. MacLeod, G. J. Christian, R. E. Cowley, W. W. Brennessel, S. Ye, F. Neese and P. L. Holland, J. Am. Chem. Soc., 2012, 134, 20352–20364 CrossRef CAS PubMed.
- J. M. Wittman, R. Hayoun, W. Kaminsky, M. K. Coggins and J. M. Mayer, J. Am. Chem. Soc., 2013, 135, 12956–12959 CrossRef CAS PubMed.
- D. C. Lacy, G. M. Roberts and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 4860–4864 CrossRef CAS PubMed.
- K. Uchida, Z. Mou, M. Kertesz and T. Kubo, J. Am. Chem. Soc., 2016, 138, 4665–4672 CrossRef CAS PubMed.
- B. Liu, T. Yoshida, X. Li, M. Stępień, H. Shinokubo and P. J. Chmielewski, Angew. Chem., Int. Ed., 2016, 55, 13142–13146 CrossRef CAS PubMed.
- T. Kobashi, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2016, 55, 8634–8638 CrossRef CAS PubMed.
- N. M. Bonanno, P. K. Poddutoori, K. Sato, K. Sugisaki, T. Takui, A. J. Lough and M. T. Lemaire, Chem.– Eur. J., 2018, 24, 14906–14910 CrossRef CAS PubMed.
- B. Adinarayana, D. Shimizu, K. Furukawa and A. Osuka, Chem. Sci., 2019, 10, 6007–6012 RSC.
- B. Adinarayana, K. Kato, D. Shimizu, T. Tanaka, K. Furukawa and A. Osuka, Angew. Chem., Int. Ed., 2020, 59, 4320–4323 CrossRef CAS PubMed.
- W. C. Watkins, T. Jaeger, C. E. Kidd, S. Fortier, M. C. Baird, G. Kiss, G. C. Roper and C. D. Hoff, J. Am. Chem. Soc., 1992, 114, 907–914 CrossRef CAS.
- Y. Sunada, S. Ishida, F. Hirakawa, Y. Shiota, K. Yoshizawa, S. Kanegawa, O. Sato, H. Nagashima and T. Iwamoto, Chem. Sci., 2016, 7, 191–198 RSC.
- E. L. Muetterties, B. A. Sosinsky and K. I. Zamaraev, J. Am. Chem. Soc., 1975, 97, 5299–5300 CrossRef CAS.
- W. P. Neumann, A. Penenory, U. Stewen and M. Lehnig, J. Am. Chem. Soc., 1989, 111, 5845–5851 CrossRef CAS.
- B. B. Wayland, A. E. Sherry, G. Poszmik and A. G. Bunn, J. Am. Chem. Soc., 1992, 114, 1673–1681 CrossRef CAS.
- V. Zaitsev, S. V. Rosokha, M. Head-Gordon and J. K. Kochi, J. Org. Chem., 2006, 71, 520–526 CrossRef CAS PubMed.
- J. P. Peterson, M. R. Geraskina, R. Zhang and A. H. Winter, J. Org. Chem., 2017, 82, 6497–6501 CrossRef CAS PubMed.
- B. B. Wayland, V. L. Coffin and M. D. Farnos, Inorg. Chem., 1988, 27, 2745–2747 CrossRef CAS.
- L. Y. Goh, S. K. Khoo and Y. Y. Lim, J. Organomet. Chem., 1990, 399, 115–123 CrossRef CAS.
- R. J. Klingler and J. W. Rathke, J. Am. Chem. Soc., 1994, 116, 4772–4785 CrossRef CAS.
- D. C. Woska, Y. Ni and B. B. Wayland, Inorg. Chem., 1999, 38, 4135–4138 CrossRef CAS.
- A. G. Bunn, Y. Ni, M. Wei and B. B. Wayland, Inorg. Chem., 2000, 39, 5576–5578 CrossRef CAS PubMed.
- E. F. van der Eide, T. Liu, D. M. Camaioni, E. D. Walter and R. M. Bullock, Organometallics, 2012, 31, 1775–1789 CrossRef CAS.
- G. H. Imler, M. J. Zdilla and B. B. Wayland, Inorg. Chem., 2013, 52, 11509–11513 CrossRef CAS PubMed.
- G. Nocton, W. W. Lukens, C. H. Booth, S. S. Rozenel, S. A. Medling, L. Maron and R. A. Andersen, J. Am. Chem. Soc., 2014, 136, 8626–8641 CrossRef CAS PubMed.
- K. Lekin, S. M. Winter, L. E. Downie, X. Bao, J. S. Tse, S. Desgreniers, R. A. Secco, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2010, 132, 16212–16224 CrossRef CAS PubMed.
- K. Lekin, H. Phan, S. M. Winter, J. W. L. Wong, A. A. Leitch, D. Laniel, W. Yong, R. A. Secco, J. S. Tse, S. Desgreniers, P. A. Dube, M. Shatruk and R. T. Oakley, J. Am. Chem. Soc., 2014, 136, 8050–8062 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, A. S. Filatov, R. T. Oakley, X. Li, K. Lekin, A. Huq, C. Pak, S. M. Greer, J. McKay, M. Jo, J. Lengyel, I. Hung, E. Maradzike, A. E. DePrince, S. A. Stoian, S. Hill, Y.-Y. Hu and M. Shatruk, J. Am. Chem. Soc., 2019, 141, 17989–17994 CrossRef CAS PubMed.
- J. Zhang, D. C. Grills, K.-W. Huang, E. Fujita and R. M. Bullock, J. Am. Chem. Soc., 2005, 127, 15684–15685 CrossRef CAS PubMed.
- W. Li, X. Zhou, R. Lock, S. Patchkovskii, A. Stolow, C. Kapteyn Henry and M. Murnane Margaret, Science, 2008, 322, 1207–1211 CrossRef CAS PubMed.
- H. J. Wörner, J. B. Bertrand, D. V. Kartashov, P. B. Corkum and D. M. Villeneuve, Nature, 2010, 466, 604–607 CrossRef PubMed.
- J. P. Lomont and C. B. Harris, Inorg. Chim. Acta, 2015, 424, 38–50 CrossRef CAS.
- S. Yoshidomi, M. Mishima, S. Seyama, M. Abe, Y. Fujiwara and T.-a. Ishibashi, Angew. Chem., Int. Ed., 2017, 56, 2984–2988 CrossRef CAS PubMed.
- J. Jeener, B. H. Meier, P. Bachmann and R. R. Ernst, J. Chem. Phys., 1979, 71, 4546–4553 CrossRef CAS.
- C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90, 935–967 CrossRef CAS.
- K. Nikitin and R. O'Gara, Chem.– Eur. J., 2019, 25, 4551–4589 CrossRef CAS PubMed.
- P. Hamm, M. Lim and R. M. Hochstrasser, J. Phys. Chem. B, 1998, 102, 6123–6138 CrossRef CAS.
- J. Zheng, K. Kwak and M. D. Fayer, Acc. Chem. Res., 2007, 40, 75–83 CrossRef CAS PubMed.
- J. P. Kraack, Top. Curr. Chem., 2017, 375, 86 CrossRef PubMed.
- L. M. Kiefer and K. J. Kubarych, Coord. Chem. Rev., 2018, 372, 153–178 CrossRef CAS.
- C. R. Baiz, R. McCanne and K. J. Kubarych, J. Am. Chem. Soc., 2009, 131, 13590–13591 CrossRef CAS PubMed.
- R. Kania, A. I. Stewart, I. P. Clark, G. M. Greetham, A. W. Parker, M. Towrie and N. T. Hunt, PCCP, 2010, 12, 1051–1063 RSC.
- S. Rösel, C. Balestrieri and P. R. Schreiner, Chem. Sci., 2017, 8, 405–410 RSC.
- S. Takebayashi and R. R. Fayzullin, Organometallics, 2021, 40(4), 500–507 CrossRef CAS.
- M. Mayr, K. Wurst, K.-H. Ongania and M. R. Buchmeiser, Chem.– Eur. J., 2004, 10, 1256–1266 CrossRef CAS PubMed.
- M. Iglesias, D. J. Beetstra, A. Stasch, P. N. Horton, M. B. Hursthouse, S. J. Coles, K. J. Cavell, A. Dervisi and I. A. Fallis, Organometallics, 2007, 26, 4800–4809 CrossRef CAS.
- J. J. Dunsford, K. J. Cavell and B. M. Kariuki, Organometallics, 2012, 31, 4118–4121 CrossRef CAS.
- A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
- A. Kumar, D. Yuan and H. V. Huynh, Inorg. Chem., 2019, 58, 7545–7553 CrossRef CAS PubMed.
- P. N. Bungu and S. Otto, Dalton Trans., 2011, 40, 9238–9249 RSC.
- J. Du, W. Chen, Q. Chen, X. Leng, Y.-S. Meng, S. Gao and L. Deng, Organometallics, 2020, 39, 729–739 CrossRef CAS.
- H. B. Abrahamson, C. C. Frazier, D. S. Ginley, H. B. Gray, J. Lilienthal, D. R. Tyler and M. S. Wrighton, Inorg. Chem., 1977, 16, 1554–1556 CrossRef CAS.
- M. P. Crockett, H. Zhang, C. M. Thomas and J. A. Byers, Chem. Commun., 2019, 55, 14426–14429 RSC.
- R. Dorta, E. D. Stevens, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2003, 125, 10490–10491 CrossRef CAS PubMed.
- S. Goebel, C. L. Haynes, F. A. Khan and P. B. Armentrout, J. Am. Chem. Soc., 1995, 117, 6994–7002 CrossRef CAS.
- G. G. Christoph and Y. B. Koh, J. Am. Chem. Soc., 1979, 101, 1422–1434 CrossRef CAS.
- I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS.
- J. Campos, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS.
- J. F. Berry and C. C. Lu, Inorg. Chem., 2017, 56, 7577–7581 CrossRef CAS PubMed.
- R. J. Hoobler, M. A. Hutton, M. M. Dillard, M. P. Castellani, A. L. Rheingold, A. L. Rieger, P. H. Rieger, T. C. Richards and W. E. Geiger, Organometallics, 1993, 12, 116–123 CrossRef CAS.
- G. W. Margulieux, N. Weidemann, D. C. Lacy, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2010, 132, 5033–5035 CrossRef CAS PubMed.
- D. W. Agnew, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Angew. Chem., Int. Ed., 2015, 54, 12673–12677 CrossRef CAS PubMed.
- R. H. Duncan Lyngdoh, H. F. Schaefer and R. B. King, Chem. Rev., 2018, 118, 11626–11706 CrossRef CAS PubMed.
- L. D. Brown, K. N. Raymond and S. Z. Goldberg, J. Am. Chem. Soc., 1972, 94, 7664–7674 CrossRef CAS.
- F. T. T. Ng, G. L. Rempel, C. Mancuso and J. Halpern, Organometallics, 1990, 9, 2762–2772 CrossRef CAS.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- R. J. Klingler, M. J. Chen, J. W. Rathke and K. W. Kramarz, Organometallics, 2007, 26, 352–357 CrossRef CAS.
- F. A. L. Anet, I. Yavari, I. J. Ferguson, A. R. Katritzky, M. Moreno-Mañas and M. J. T. Robinson, J. Chem. Soc., Chem. Commun., 1976, 399–400, 10.1039/C39760000399.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, spectroscopic and X-ray diffraction data (pdf). Raw data used for the van't Hoff plot (excel). Raw data used for the Eyring plot (excel). CCDC 2165654–2165661. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03028d |
| This journal is © The Royal Society of Chemistry 2022 |