
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral Lewis acid catalysis in a visible light-triggered cycloaddition/rearrangement cascade†
Simone
Stegbauer
,
Christian
Jandl
and
Thorsten
Bach
 *
*
Technische Universität München, School of Natural Sciences, Department of Chemistry and Catalysis Research Center, Lichtenbergstrasse 4, Garching 85747, Germany. E-mail: thorsten.bach@ch.tum.de; Web: https://www.ch.nat.tum.de/en/oc1/home/ Fax: +49 (0)89 289 13315
First published on 23rd September 2022
Abstract
Cascade (domino) reactions facilitate the formation of complex molecules from simple starting materials in a single operation. It was found that 1-naphthaldehyde derivatives can be converted to enantioenriched (82–96% ee) polycyclic benzoisochromenes via a cascade of ortho photocycloaddition and ensuing acid-catalysed rearrangement reactions. The cascade was initiated by irradiation with visible light (λ = 457 nm) and catalysed by a chiral AlBr3-activated 1,3,2-oxazaborolidine (14 examples, 65–93% yield). The absolute configuration of the products was elucidated by single crystal X-ray crystallography. Mechanistic experiments suggest that the ortho photocycloaddition occurs on the triplet hypersurface and that the chiral catalyst induces in this step the observed enantioselectivity.
Introduction
A process involving two or more consecutive reactions in which the subsequent reaction is initiated by formation of a new functional group in the previous step is known as cascade or domino reaction. Such a transformation combines high atom economy with a large degree of sustainability and it paves the way to molecules with elevated complexity from simple starting materials in a single operation.1 Arguably, one of the most celebrated cascade reactions is the total synthesis of tropinone by Robinson in 1917.2 Since then, the number of cascade-driven processes has grown tremendously in synthetic chemistry.1 In recent years, several groups have illustrated the power of photochemically initiated cascade reactions (Scheme 1, top).3–6 Hoffmann and co-workers provided the first example for an intramolecular cascade of a naphthalene with a tethered resorcinyl moiety. The transformation of substrate 1 to rac-2 can be understood as an intramolecular ortho photocycloaddition followed by an acid-catalysed ring opening/rearrangement.3 The colours indicate the positions of selected carbon atoms in the substrate and in the product. A two-photon process reported by the Glorius group enabled the formation of polycyclic benzocyclobutanes (e.g.3 → rac-4) in a sequence of an iridium-sensitized intramolecular ortho photocycloaddition and a subsequent vinyl-cyclobutane rearrangement.4 Our group developed a three-photon cascade with the 7-substituted 1-indanone 5 as starting material. The polycyclic scaffold of rac-6 is assembled by an ortho photocycloaddition, followed by a thermal [6π] ring opening, a [4π] photocyclization, and a consecutive di-π-methane rearrangement.5 The three examples provide a glimpse on the structural diversity that cascade reactions can offer when combined with photochemistry.6 A remarkable feature of these transformations is the skeletal rearrangement that follows the initial ortho photocycloaddition. Although mechanistically complex, the predictable creation of three-dimensional structures from arene precursors in these cascade reactions is a concise and attractive pathway to achieve structural product diversity.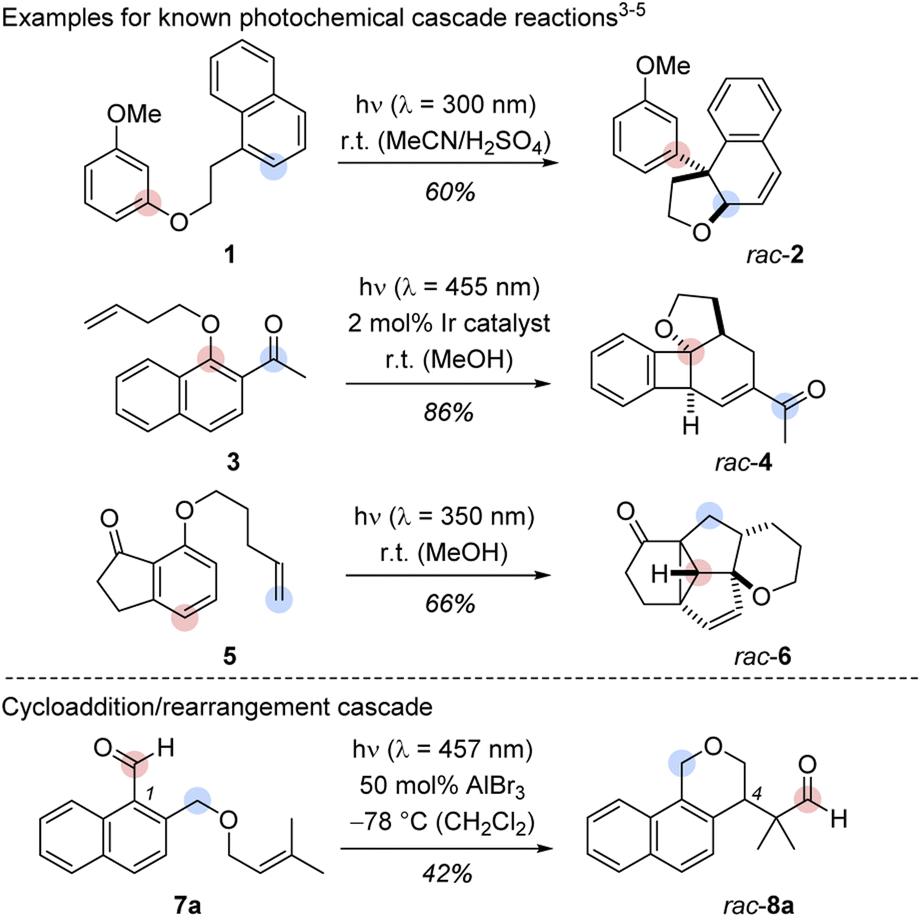 | ||
| Scheme 1 Top: Cascades including a photochemical step as developed by the groups of Hoffmann (1 → rac-2),3 Glorius (3 → rac-4)4 and Bach (5 → rac-6).5 Bottom: Racemic Lewis acid-catalysed cascade starting from 1-naphthaldehyde derivative 7a and yielding benzoisochromene rac-8a. The marked positions indicate the respective carbon positions before and after the cascades. | ||
Despite their obvious utility, enantioselective versions of cascades that include a photochemical step have remained rare.7 In this contribution, we describe a new photochemical cascade reaction which we successfully developed into an enantioselective transformation employing a chiral Lewis acid for chromophore activation.8 The study was triggered by a serendipitous discovery which was made when examining intramolecular ortho photocycloaddition reactions to naphthalene and benzene. Since the reacting aldehydes displayed a binding site for chiral 1,3,2-oxazaborolidine Lewis acids, the latter catalyst class was chosen for the enantioselective variant.
Results and discussion
In initial experiments, we examined the reactivity of 1-naphthaldehyde 7a in the presence of Lewis acidic catalysts under visible light irradiation (λ = 457 nm) at −78 °C in CH2Cl2 as the solvent of choice.8d,e While there was no reaction in the absence of acid, a new product was formed when AlBr3 was added, and benzoisochromene rac-8a with a rearranged carbon skeleton was isolated in a yield of 42% (Scheme 1, bottom). The outcome was unexpected and surprising. The formyl group had moved to the terminal position of the olefin whereas the alkoxymethyl group had migrated to the former C-1 carbon atom of the naphthalene. To understand the photophysical fundamentals of the reaction, the UV/vis spectrum of 1-naphthaldehyde 7a (Fig. 1) was recorded in the absence and presence of a Lewis acid.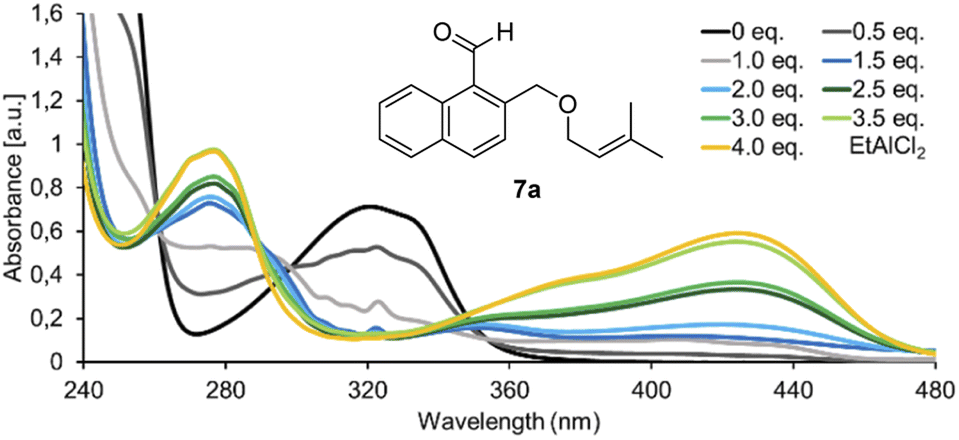 | ||
| Fig. 1 UV/vis spectrum of 1-naphthaldehyde 7a (c = 1.0 mM in CH2Cl2, measured in a 1.0 mm quartz cuvette) in the presence of variable equivalents (eq.) of EtAlCl2. | ||
The spectrum revealed a strong absorption at λ = 248 nm (ε = 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 L mol−1 cm−1), which is assigned to a ππ* transition. A weaker but broad absorption band was observed at longer wavelength with a maximum at λ = 321 nm (ε = 7100 L mol−1 cm−1). The band tails into the longer wavelength region of the spectrum (up to λ = 390 nm) and is assigned to additional arene ππ* transitions (La and Lb band) overlapping with the nπ* transition of the carbonyl group.9,10 Upon successive addition of EtAlCl2, two absorption bands became visible with a maximum at λ = 277 nm and λ = 424 nm.11 Upon addition of 4.0 eq. of EtAlCl2, the absorption reached saturation indicating complete complexation of aldehyde 7a. The extinction coefficients were determined as ε = 9900 L mol−1 cm−1 at λ = 277 nm and ε = 5800 L mol−1 cm−1 at λ = 424 nm. The latter absorption band seems responsible for the photochemical reactivity of 7a observed in the presence of a Lewis acid because it overlaps with the emission spectrum of the light source.
100 L mol−1 cm−1), which is assigned to a ππ* transition. A weaker but broad absorption band was observed at longer wavelength with a maximum at λ = 321 nm (ε = 7100 L mol−1 cm−1). The band tails into the longer wavelength region of the spectrum (up to λ = 390 nm) and is assigned to additional arene ππ* transitions (La and Lb band) overlapping with the nπ* transition of the carbonyl group.9,10 Upon successive addition of EtAlCl2, two absorption bands became visible with a maximum at λ = 277 nm and λ = 424 nm.11 Upon addition of 4.0 eq. of EtAlCl2, the absorption reached saturation indicating complete complexation of aldehyde 7a. The extinction coefficients were determined as ε = 9900 L mol−1 cm−1 at λ = 277 nm and ε = 5800 L mol−1 cm−1 at λ = 424 nm. The latter absorption band seems responsible for the photochemical reactivity of 7a observed in the presence of a Lewis acid because it overlaps with the emission spectrum of the light source.
Based on its UV/vis data, the reaction of aldehyde 7a was examined at shorter wavelength without Lewis acid catalyst. Using an irradiation source with an emission maximum at λ = 366 nm, full conversion of aldehyde 7a was observed after five hours of irradiation at ambient temperature. Under these conditions, an exclusive ortho photocycloaddition12,13 to the naphthalene C1/C2 bond occurred and cyclobutane rac-9a was formed in 72% yield (Scheme 2). A 1H NMR spectrum of the crude product gave no indication for the formation of benzoisochromene rac-8a. However, the formation of product rac-8a from cyclobutane rac-9a was induced by the addition of either AlBr3 (10 mol%) or EtAlCl2 (10 mol%) yielding the rearranged product rac-8a in 86% or 81% yield. The structure of rac-8a was corroborated by single crystal X-ray crystallography.14 Since benzoisochromene rac-8a was formed from cyclobutane rac-9a in a Lewis acid-catalysed rearrangement reaction, the sequence is likely initiated by Lewis acid coordination to the carbonyl oxygen atom of rac-9a. A 1,2-alkyl shift forms the benzylic cation rac-10 which undergoes a second migration to cation rac-11.15 Elimination of the Lewis acid and ring scission restores the aldehyde group and leads to re-aromatisation in the final product of the cascade. All stereogenic centers are formed in the ortho photocycloaddition step and the stereogenic center at the C-4 carbon atom is preserved. It was therefore hypothesized that an enantioselective photocycloaddition step would lead to enantiomerically enriched rearrangement product and we envisioned 1-naphthaldehyde 7a as a suitable substrate for chiral Lewis acid activation.16
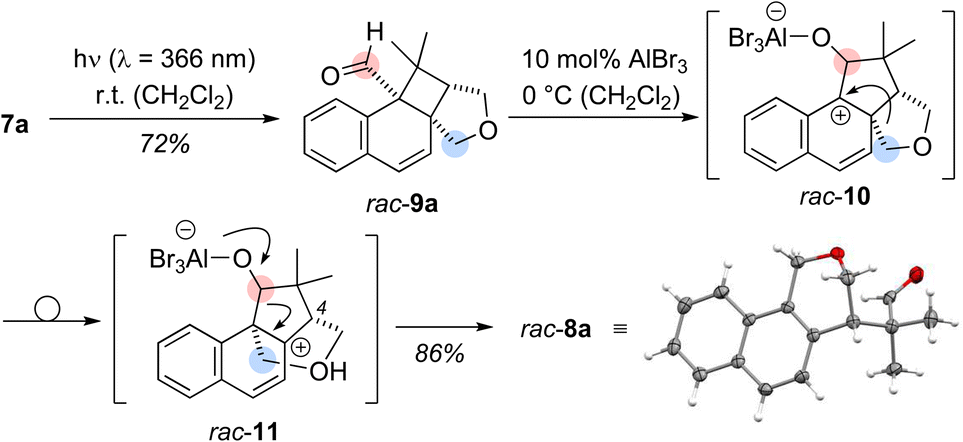 | ||
| Scheme 2 Ortho photocycloaddition of 1-naphthaldehyde 7a to cyclobutane rac-9, followed by the Lewis acid-catalysed rearrangement reaction to benzoisochromene rac-8a. The constitution of rac-8a was established by single crystal X-ray crystallography. | ||
Experiments with chiral AlBr3-activated 1,3,2-oxazaborolidines8c,16,17 commenced with the previously reported catalysts 12a and 12b in CH2Cl2 under the conditions of the racemic reaction (Table 1, entries 1, 2). No conversion was observed. Catalyst 12c bearing a 3-biphenyl group at the boron atom behaved similarly (entry 3). By switching the boron substituent to a 2,6-dimethylphenyl group (12d), full conversion was recorded after 4.5 h and the combined yield of products 8a and 9a was determined as 88% (entry 4). The enantiomeric excess (ee) of both products was high (89% and 90% ee). The influence of substituents Ar′ at the prolinol was probed with catalysts 12e–12g (entries 5–7). When catalyst 12e with a bulky aryl group (Ar′ = 3′,5′-dimethyl-2-biphenyl) was applied, the starting material 7a was fully converted and the yield of the products 8a and 9a was high (90%). However, the enantioface differentiation by the catalyst was poor (37% and 39% ee, entry 5). Likewise, catalysts 12f (Ar′ = 3,5-difluorophenyl) and 12g (Ar′ = 2,3-dimethylphenyl) gave a low enantioselectivity of 54% ee (entry 6) and 65% ee (67%, entry 7) but the yields were in both cases high. It appears as if the aromatic group Ar on the boron atom has a large influence on the reactivity while proper selection of Ar′ is crucial for the enantioselectivity. Catalyst 12d evolved from the screening experiments (for further information, see the ESI†) as the superior Lewis acid that promotes the reaction of 7a to 8a/9a with both high yield and enantioselectivity. Benzoisochromene 8a could be obtained as a single product (88% yield, 89% ee), if the reaction mixture was warmed to 0 °C after irradiation and was subsequently stirred for 30 minutes at this temperature without irradiation (entry 8). Lower catalyst loadings led to a decrease in the enantioselectivity. With 25 mol% 12d, the ee of product 8a was only 83% ee and dropped to 73% ee at a catalyst loading of 10 mol%. It is likely that the high catalyst loading is required because the Lewis acid remains (partially) bound to the final product 8a which also displays a Lewis-basic aldehyde functionality and a tetrahydropyran ring. Dissociation of AlBr3 may occur and initiate an unselective reaction.
|
|
||||
|---|---|---|---|---|
| Entrya | Lewis acid 12b | t [h] at 0 °C | Yield 8a + 9a [%] (ratio)c | ee 8a, (9a) [%]d |
| a The reactions were performed in CH2Cl2 as the solvent with a substrate concentration of c = 20 mM. b 12a–12d: Ar′ = 3,5-dimethylphenyl; 12a: Ar = 2,4,6-trifluorophenyl; 12b: Ar = 2-(trifluoromethyl)phenyl; 12c: Ar = 3-biphenyl; 12d: Ar = 2,6-dimethylphenyl. 12e–12g: Ar = 2,6-dimethylphenyl; 12e: Ar′ = 3′,5′-dimethyl-2-biphenyl; 12f: Ar′ = 3,5-difluorophenyl; 12g: Ar = 2,3-dimethylphenyl. c Yield of isolated mixture 8a and 9a; the ratio was determined by 1H NMR analysis of crude product. d The enantiomeric excess (ee) of 8a and 9a was determined by chiral-phase HPLC analysis. n.d. = not determined. e The reaction was quenched at −78 °C. | ||||
| 1 | 12a | —e | <10% | n.d. |
| 2 | 12b | —e | <10% | n.d. |
| 3 | 12c | —e | <10% | n.d. |
| 4 | 12d | —e | 88 (77:23) |
89 (90) |
| 5 | 12e | —e | 90 (76:24) |
37 (39) |
| 6 | 12f | —e | 87 (100:0) |
54 |
| 7 | 12g | —e | 86 (76:24) |
65 (67) |
| 8 | 12d | 0.5 | 88 (100:0) |
89 |
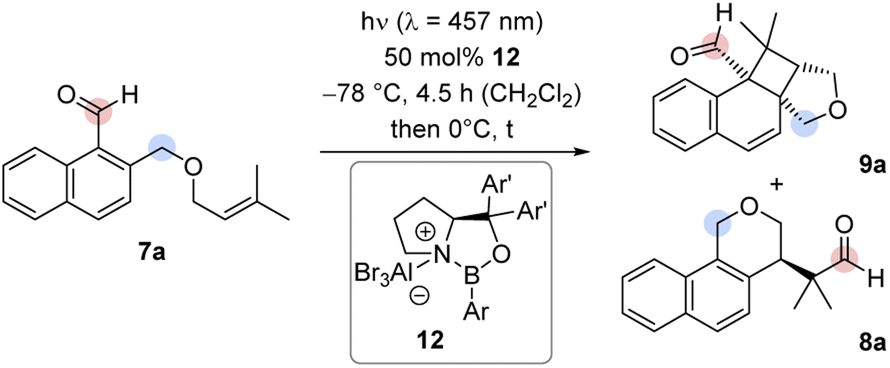
It was possible to apply the method to a variety of substituted 1-naphthaldehydes 7 (Scheme 3). In a first set of experiments, 1-naphthaldehydes with different substituents on the terminal double bond were synthesized by a Williamson ether synthesis18 between 1-bromo-2-(bromomethyl)naphthalene19 and allylic alcohols followed by a formylation reaction20 (see the ESI† for details). In addition to substrate 7a with methyl groups at the terminal position, other alkyl groups in this position were tolerated and the cascade reaction proceeded smoothly with high enantioselectivity (products 8b–8e).
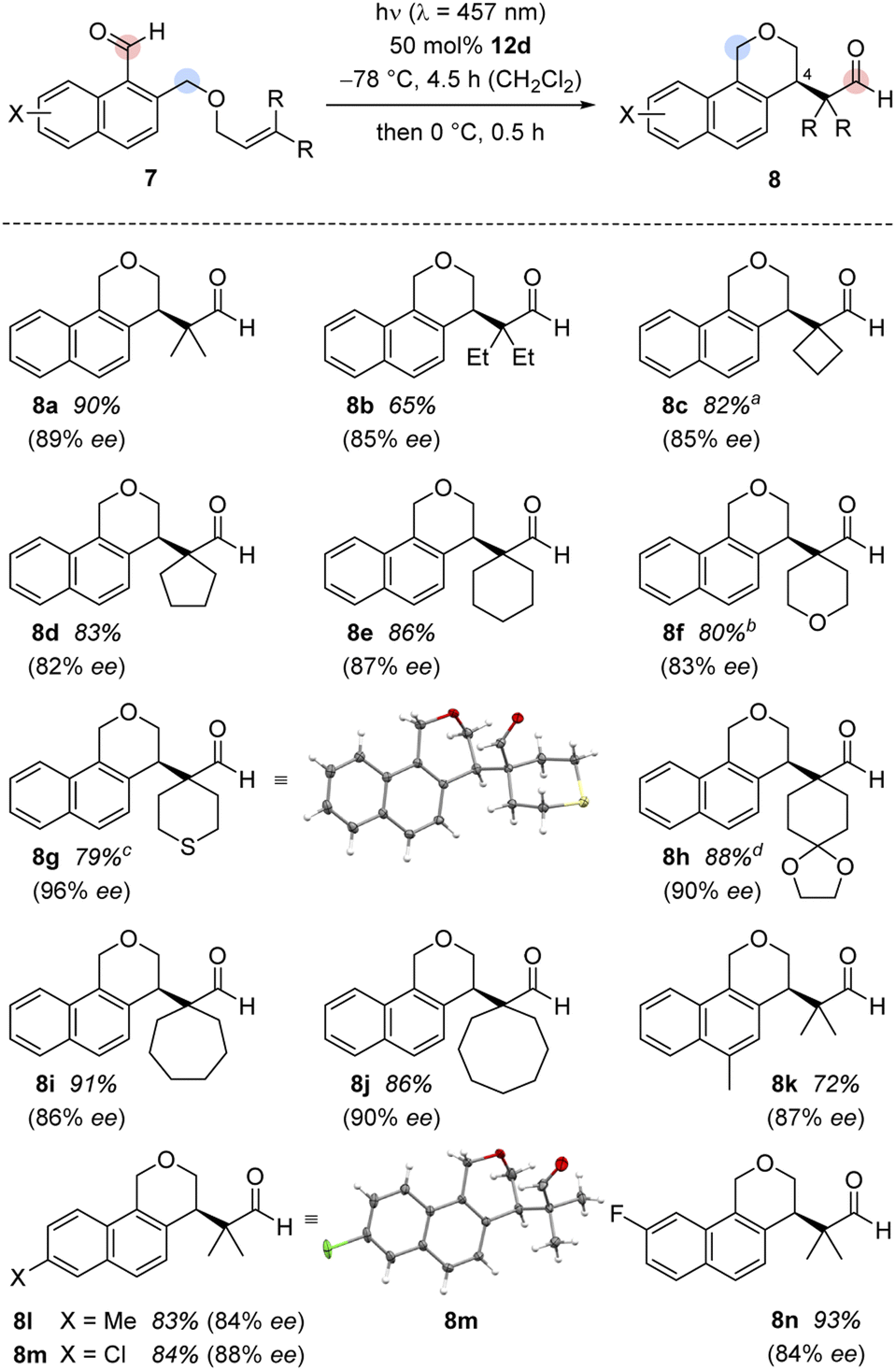 | ||
| Scheme 3 Enantioselective Lewis acid-catalysed cycloaddition/rearrangement cascade of 1-naphthaldehydes 7 to benzoisochromenes 8. a The thermal rearrangement was performed at −78 °C after irradiation. b The irradiation was performed for 16 hours at −78 °C. c The irradiation was performed for 16 hours at −78 °C. The reaction was stopped and the complete rearrangement was done by addition of EtAlCl2 (50 mol%) at 0 °C for 5 hours. d The irradiation was performed for twelve hours at −78 °C. | ||
Different heteroatoms within the cyclic substituents did not alter the consistently high enantioselectivity. Benzoisochromene with tetrahydro-2H-pyran, tetrahydro-2H-thiopyran, and 1,4-dioxaspiro[4.5]decane substituents (8f–8h) were isolated in high yields (79–88%) and excellent enantioselectivities (83–96% ee). The absolute configuration of benzoisochromene 8g was determined by anomalous X-ray diffraction.21 Substrates with larger alkyl rings (7- and 8-membered 7i and 7j) were converted to the respective products, again with high yield (91% and 86%) and in excellent optical purity (86% ee and 90% ee).
In a second set of experiments, the method was applied to 1-naphthaldehydes (7k–7n) with different substituents within the naphthalene core. The synthesis of these substrates started from 1-tetralones followed by a Vilsmeier-type α-formylation reaction,22 oxidative dehydrogenation,23 reduction,24 Williamson ether synthesis,18 and aldehyde formation20 (see the ESI† for details). While the substrates were not as straightforward to access, the enantioselectivities (84–88% ee) as well as the yields (72–93%) for their products 8k–8n remained high. The chloro-substituted product 8m gave suitable crystals for anomalous X-ray diffraction and the configuration at the newly formed stereogenic center could be assigned.25 The absolute configuration was identical to the configuration previously established for 8g and the configuration of the other major product enantiomers was based on analogy. Although the substrate scope is wide regarding substituents R (at the double bond) and X (at the arene), there are significant limitations. Fig. 2 summarizes substrates 13 which did not undergo the cascade reaction. The tether needs to be attached to the 2-position of the naphthalene via a CH2O group. An O-alkyl tether (13a) was insufficient and the respective substrate did not undergo the reaction. Likewise, the intramolecular olefin component requires a specific substitution pattern. Only trisubstituted olefins underwent the reaction while mono- (13b), disubstituted (13d) or tetrasubstituted (13c) olefins did not react. Replacing the formyl group at C1 by an acetyl group led to a complex product mixture (13e).
 | ||
| Fig. 2 A selection of substrates (13) which did not show the desired reactivity under standard reaction conditions (see Scheme 3 and ESI†). | ||
In order to shed light on the reaction course, it was initially tested whether the chiral Lewis acid had any influence on the outcome of the rearrangement 9 → 8. Although it was conceivable that a kinetic resolution26 was partially responsible for the observed enantioselectivity, the rearrangement rac-9a → rac-8a performed in the presence of catalyst ruled out this option (Scheme 4). There was no enrichment of 8a in the process. The reaction was stopped after several time periods and the substrate 9a was always racemic (max. 3% ee). If at all, the Lewis acid led to a preferred formation of ent-8a with minimal ee (see the ESI† for details). After a conversion of 47% for example, there was an ee of 7%. Under the conditions of the reaction, i.e. after warming to 0 °C, the rearrangement went to completion and no ee was notable. It was thus clear that it is the ortho photocycloaddition which governs the enantioselectivity.
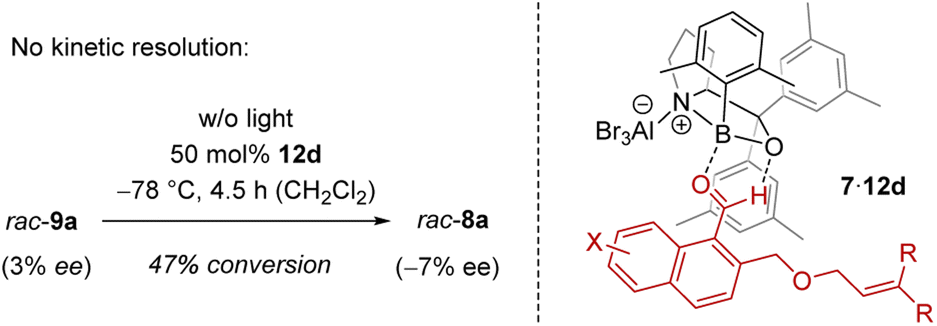 | ||
| Scheme 4 The ortho photocycloaddition step is responsible for the observed enantioselectivity in the reaction 7 → 8 as catalysed by Lewis acid 12d: (Left) no kinetic resolution occurred in the Lewis acid-catalysed rearrangement of the ortho photocycloaddition product rac-9a. (Right) Model for association of Lewis acid 12d to 1-naphthaldehydes 7 and for the enantioface differentiation in the ortho photocycloaddition step. | ||
The explanation for the enantioface differentiation rests on the assumption that cationic oxazaborolidine-based Lewis acids bind to an aldehyde not only via the oxygen atom but that there is a second, non-classical hydrogen bond between the hydrogen atom of the aldehyde group and the oxygen atom of the 1,3,2-oxazaborolidine.17d,27 For 1-naphthaldehydes 7,28 the respective complex 7·12d is depicted in Scheme 4, whereby aldehyde 7 is in a locked conformation via a two-point interaction. One of the two enantiotopic faces of the arene is shielded by the 3,5-dimethylphenyl ring (in gray) of the proline-based backbone, which leads to a selective approach of the tethered olefin from the front face. The following rearrangement forms the benzoisochromenes 8 with the depicted configuration at the future C-4 carbon atom (Scheme 2 and Table 1).
Further mechanistic studies to elucidate the pathway of the ortho photocycloaddition were conducted with 1-naphthaldehyde 7a (Scheme 5). The enantioselective reaction was performed under optimized conditions without warming the reaction mixture. The reaction had previously led to complete conversion and products 8a/9a were isolated in a yield of 88% (Table 1, entry 4). Upon addition of increasing amounts of the known triplet quencher piperylene,29 the reaction slowed down notably. With 2.5 eq. of piperylene, the yield decreased to 34%, with 5.0 eq. of piperylene a further decrease to 23% was observed. Unreacted starting material 7a was recovered in 46% and 55%, indicating that no side reactions occurred and that the reaction rate was reduced. In previous work, we had seen that piperylene does not affect photochemical reactions which are catalysed by AlBr3-activated 1,3,2-oxazaborolidines and which occur on the singlet hypersurface.16f Against this background, the correlation between a higher quencher concentration with a lower reaction rate suggest that the ortho photocycloaddition occurs on the triplet hypersurface.
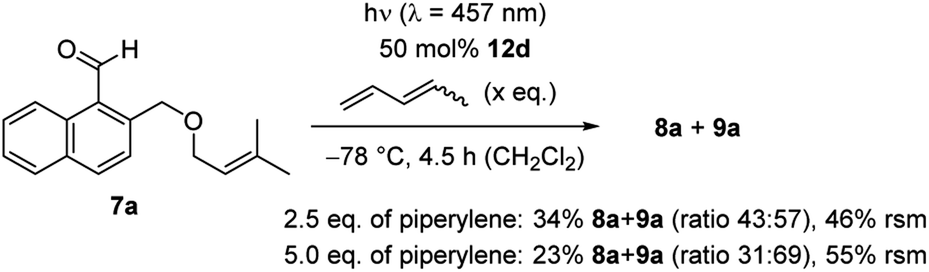 | ||
| Scheme 5 Qualitative triplet quenching experiments of the reaction 7a → 8a/9a by addition of increasing amounts of piperylene. A qualitative decrease of yield is observed which suggests the intermediacy of triplet states (rsm = recovered starting material). | ||
A triplet pathway is also in line with previous work performed on Lewis acid complexes of 1- and 2-naphthaldehyde.11c It had been found that olefins do not quench the fluorescent state of their 1:1 complexes with EtAlCl2 excluding a singlet reaction pathway from this state. Moreover, the AlBr3-catalysed ortho photocycloaddition of 3-hexene to 2-naphthaldehyde had been shown to occur stereoconvergently, i.e. the same major cyclobutane diastereoisomer was formed irrespective of whether the (E)- or (Z)-isomer of 3-hexene was employed. The result rules out a concerted pathway on the singlet hypersurface which should be stereospecific and suggests a triplet reaction.
Conclusions
In summary, we have discovered an enantioselective photocycloaddition/rearrangement cascade triggered by visible light and catalysed by chiral Lewis acid 12d. The transformation enables the formation of structurally unique, polycyclic products and includes a photochemical de-aromatisation followed by a thermal acid-catalysed rearrangement. Catalyst 12d operates by a two-point interaction, whereupon the substrate is precisely oriented and fixed so that the attack can only occur from one of the two enantiotopic faces. The nature of this intermediate and its photophysical properties are currently under investigation. Our results regarding an enantioselective, photochemically triggered cascade suggest the usefulness of this and related processes for synthetic applications and warrant further studies.Data availability
Original NMR datasets (FIDs) are available at Open Science Framework at https://osf.io/mcvrn/.Author contributions
S. S. and T. B. developed the project. Funding was acquired by T. B., S. S. designed and performed the synthetic experiments. C. J. performed the X-ray crystallographic analysis of compounds 8a, 8g, and 8m. S. S. generated and validated the data. T. B. administered the project and supervised the research. S. S. and T. B. wrote, reviewed, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – TRR 325 (project B1) – 444632635 is gratefully acknowledged. We thank O. Ackermann (TU München) for his help with HPLC analyses.Notes and references
- Reviews and books on cascade (domino) reactions: (a) P.-F. Xu and W. Wang, Catalytic Cascade Reaction, Wiley-VCH, Weinheim, 2013 CrossRef; (b) K. C. Nicolaou and J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993 RSC; (c) L. F. Tietze, G. Brasche and K. M. Gericke, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006 CrossRef; (d) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS PubMed; (e) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS PubMed.
- R. Robinson, J. Chem. Soc., 1917, 111, 762 RSC.
- (a) N. Hoffmann, J.-P. Pete, Y. Inoue and T. Mori, J. Org. Chem., 2002, 67, 2315 CrossRef CAS PubMed; (b) N. Hoffmann, Tetrahedron, 2002, 58, 7933 CrossRef CAS.
- M. J. James, J. L. Schwarz, F. Strieth-Kalthoff, B. Wibbeling and F. Glorius, J. Am. Chem. Soc., 2018, 140, 8624 CrossRef CAS PubMed.
- L. Naesborg, C. Jandl, A. Zech and T. Bach, Angew. Chem., Int. Ed., 2020, 59, 5656 CrossRef CAS PubMed.
- For selected additional examples: (a) C. Heinemann and M. Demuth, J. Am. Chem. Soc., 1997, 119, 1129 CrossRef CAS; (b) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 1040 CrossRef CAS PubMed; (c) M. Zhu, K. Zhou, X. Zhang and S.-L. You, Org. Lett., 2018, 20, 4379 CrossRef CAS PubMed; (d) A. Zech, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2019, 58, 14629 CrossRef CAS PubMed; (e) R. C. McAtee, E. A. Noten and C. R. J. Stephenson, Nat. Commun., 2020, 11, 2528 CrossRef CAS PubMed; (f) J. Ma, S. Chen, P. Bellotti, T. Wagener, C. Daniliuc, K. N. Houk and F. Glorius, Nat. Catal., 2022, 5, 405 CrossRef CAS; (g) J. Proessdorf, C. Jandl, T. Pickl and T. Bach, Angew. Chem., Int. Ed., 2022, 61, e202208329 CrossRef CAS PubMed.
- Examples: (a) H.-H. Liao, C.-C. Hsiao, E. Sugiono and M. Rueping, Chem. Commun., 2013, 49, 7953 RSC; (b) Ł. Woźniak, G. Magagnano and P. Melchiorre, Angew. Chem., Int. Ed., 2018, 57, 1068 CrossRef; (c) P. Bonilla, Y. P. Rey, C. M. Holden and P. Melchiorre, Angew. Chem., Int. Ed., 2018, 57, 12819 CrossRef CAS; (d) L. A. Perego, P. Bonilla and P. Melchiorre, Adv. Synth. Catal., 2020, 362, 302 CrossRef CAS; (e) N. Hammer, M. L. Christensen, Y. Chen, D. Naharro, F. Liu, K. A. Jørgensen and K. N. Houk, J. Am. Chem. Soc., 2020, 142, 6030 CrossRef CAS.
- For recent reviews covering enantioselective photochemical reaction mediated by chiral Lewis acids, see: (a) M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2022, 122, 1654 CrossRef CAS PubMed; (b) T. Rigotti and J. Alemán, Chem. Commun., 2020, 56, 11169 RSC; (c) D. P. Schwinger and T. Bach, Acc. Chem. Res., 2020, 53, 1933 CrossRef CAS PubMed; (d) C. Prentice, J. Morrisson, A. D. Smith and E. Zysman-Colman, Beilstein J. Org. Chem., 2020, 16, 2363 CrossRef PubMed; (e) C. Brenninger, J. D. Jolliffe and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14338 CrossRef CAS PubMed.
- E. Pretsch, P. Bühlmann and M. Badertscher, in Structure Determination of Organic Compounds, Springer, Berlin, 2020, pp. 450–463 Search PubMed.
- UV/vis spectrum of 1-naphthaldehyde derivatives: (a) A. I. Privalova, J. P. Morozova, E. R. Kashapova and V. J. Artyukhov, J. Appl. Spectrosc., 2011, 78, 309 CrossRef CAS; (b) S. Ghidinelli, G. Longhi, S. Abbate, C. Hättig and S. Coriani, J. Phys. Chem., 2021, 125, 243 CrossRef CAS.
- (a) S. Fukuzumi, T. Okamoto and J. Otera, J. Am. Chem. Soc., 1994, 116, 5503 CrossRef CAS; (b) S. Fukuzumi, N. Satoh, T. Okamoto, K. Yasui, T. Suenobu, Y. Seko, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 7756 CrossRef CAS PubMed; (c) S. Stegbauer, N. Jeremias, C. Jandl and T. Bach, Chem. Sci., 2019, 10, 8566 RSC.
- For selected intramolecular ortho photocycloaddition reactions of naphthalenes, see: (a) J. J. McCullough, W. K. MacInnis, C. J. L. Lock and R. Faggiani, J. Am. Chem. Soc., 1980, 102, 7780 CrossRef CAS; (b) A. Kashoulis, A. Gilbert and G. Ellis-Davies, Tetrahedron Lett., 1984, 25, 2905 CrossRef CAS; (c) A. Gilbert, P. Heath, A. Kashoulis-Koupparis, G. C. R. Ellis-Davies and S. M. Firth, J. Chem. Soc., 1988, 31 CAS; (d) P. J. Wagner and M. Sakamoto, J. Am. Chem. Soc., 1989, 111, 9254 CrossRef CAS; (e) J. P. Dittami, X. Y. Nie, H. Nie, H. Ramanathan, C. Buntel, S. Rigatti, J. Bordner, D. L. Decosta and P. Williard, J. Org. Chem., 1992, 57, 1151 CrossRef CAS; (f) K. Mizuno, S.-i. Konishi, T. Takata and H. Inoue, Tetrahedron Lett., 1996, 37, 7775 CrossRef CAS; (g) A. B. Rolka and B. König, Org. Lett., 2020, 22, 5035 CrossRef CAS PubMed; (h) M. Zhu, H. Xu, X. Zhang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2021, 60, 7036 CrossRef CAS PubMed.
- For general reviews on the ortho photocycloaddition, see: (a) R. Remy and C. G. Bochet, Chem. Rev., 2016, 116, 9816 CrossRef CAS; (b) N. Hoffmann, Photochem. Photobiol. Sci., 2012, 11, 1613 CrossRef CAS PubMed; (c) U. Streit and C. G. Bochet, Beilstein J. Org. Chem., 2011, 7, 525 CrossRef CAS PubMed; (d) N. Hoffmann, Synthesis, 2004, 481 CrossRef CAS; (e) J. Cornelisse and R. de Haan in Molecular and Supramolecular Photochemistry, eds. V. Ramamurthy and K. Schanze, Dekker, New York, 2001, vol. 8, pp. 1–126 Search PubMed.
- Supplementary crystallographic data for rac-8a have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2158145).
- For reviews on rearrangement of carbocations by a 1,2-shift see: (a) X.-M. Zhang, Y.-Q. Tu, F.-M. Zhang, Z.-H. Chen and S.-H. Wang, Chem. Soc. Rev., 2017, 46, 2272 RSC; (b) R. R. Naredla and D. A. Klumpp, Chem. Rev., 2013, 113, 6905 CrossRef CAS PubMed; (c) V. G. Shubin, Top. Curr. Chem., 1984, 116/117, 267 Search PubMed.
- For the previous use of related Lewis acids in enantioselective photochemical reactions, see: (a) H. Guo, E. Herdtweck and T. Bach, Angew. Chem., Int. Ed., 2010, 49, 7782 CrossRef CAS PubMed; (b) R. Brimioulle and T. Bach, Science, 2013, 342, 840 CrossRef CAS PubMed; (c) S. Poplata and T. Bach, J. Am. Chem. Soc., 2018, 140, 3228 CrossRef CAS PubMed; (d) S. Stegbauer, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14593 CrossRef CAS; (e) M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543 CrossRef CAS PubMed; (f) M. Leverenz, C. Merten, A. Dreuw and T. Bach, J. Am. Chem. Soc., 2019, 141, 20053 CrossRef CAS PubMed.
- For general reviews on the use of chiral oxazaborolidines in asymmetric catalysis, see: (a) S. Y. Shim and D. H. Ryu, Acc. Chem. Res., 2019, 52, 2349 CrossRef CAS PubMed; (b) D. L. Hughes, Org. Process Res. Dev., 2018, 22, 574 CrossRef CAS; (c) Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918 CrossRef CAS; (d) E. J. Corey, Angew. Chem., Int. Ed., 2009, 48, 2100 CrossRef CAS PubMed.
- Y. Yoshimi, S.-i. Konishi, H. Maeda and K. Mizuno, Synthesis, 2001, 1197 CAS.
- K. Sudheendran, C. C. Malakar, J. Conrad and U. Beifuss, J. Org. Chem., 2012, 77, 10194 CrossRef CAS PubMed.
- G. B. Jones and J. E. Mathews, Tetrahedron, 1997, 53, 14599 CrossRef CAS.
- Supplementary crystallographic data for 8g have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2158147).
- (a) Z. Arnold and A. Holý, Collect. Czech. Chem. Commun., 1961, 26, 3059 CrossRef CAS; (b) R. M. Coates, P. D. Senter and W. R. Baker, J. Org. Chem., 1982, 47, 3597 CrossRef CAS.
- S. Paul, S. Samanta and J. K. Ray, Tetrahedron Lett., 2010, 51, 5604 CrossRef CAS.
- B. Satpathi, S. V. Wagulde and S. S. V. Ramasastry, Chem. Commun., 2017, 53, 8042 RSC.
- Supplementary crystallographic data for 8m have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2158146).
- For kinetic resolutions by chiral Lewis acids, see: (a) G. Bringmann and J. Hinrichs, Tetrahedron, 1997, 8, 4121 CrossRef CAS; (b) G. Bringmann, T. Pabst, P. Henschel, J. Kraus, K. Peters, E.-M. Peters, D. S. Rycroft and J. D. Connolly, J. Am. Chem. Soc., 2000, 122, 9127 CrossRef CAS; (c) C. Fehr, J. Galindo and O. Etter, Eur. J. Org. Chem., 2004, 1953 CrossRef CAS; (d) M. D. Barker, R. A. Dixon, S. Jones and B. J. Marsh, Chem. Commun., 2008, 2218 RSC.
- (a) D. H. Ryu, T. W. Lee and E. J. Corey, J. Am. Chem. Soc., 2002, 124, 9992 CrossRef CAS ; computational studies; (b) M. N. Paddon-Row, C. D. Anderson and K. N. Houk, J. Org. Chem., 2009, 74, 861 CrossRef CAS PubMed; (c) R. C. Johnston and P. H.-Y. Cheong, Org. Biomol. Chem., 2013, 11, 5057 RSC; (d) R. S. Paton, Org. Biomol. Chem., 2014, 12, 1717 RSC.
- Mechanistic studies for the preferred all-cis configuration of 1-naphthaldehyde and derivatives: (a) W. B. Smith, D. L. Deavenport and A. M. Ihrig, J. Am. Chem. Soc., 1972, 94, 1959 CrossRef CAS; (b) S. R. Salman, Spectrochim. Acta, 1984, 40, 229 CrossRef; (c) M. Goubet, M.-A. Martin-Drumel, F. Réal, V. Vallet and O. Pirali, J. Phys. Chem., 2020, 124, 4484 CrossRef CAS PubMed.
- (a) G. S. Hammond, J. Saltiel, A. A. Lamola, N. J. Turro, J. S. Bradshaw, D. O. Cowan, R. C. Counsell, V. Vogt and C. Dalton, J. Am. Chem. Soc., 1964, 86, 3197 CrossRef CAS; (b) J. P. Guillory and C. F. Cook, J. Am. Chem. Soc., 1973, 95, 4885 CrossRef CAS; (c) T. Rosenfeld, A. Alchalel and M. Ottolenghi, J. Phys. Chem., 1974, 78, 336 CrossRef CAS; (d) S. Pusch, D. Schollmeyer and T. Opatz, Org. Lett., 2016, 18, 3043 CrossRef CAS; (e) S. G. Modha, A. Pöthig, A. Dreuw and T. Bach, J. Org. Chem., 2019, 84, 1139 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and full characterization for all starting materials and products (7, 8, rac-9a, 12), spectroscopic data, mechanistic studies, crystallographic data. CCDC 215814521581472158146. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03159k |
| This journal is © The Royal Society of Chemistry 2022 |