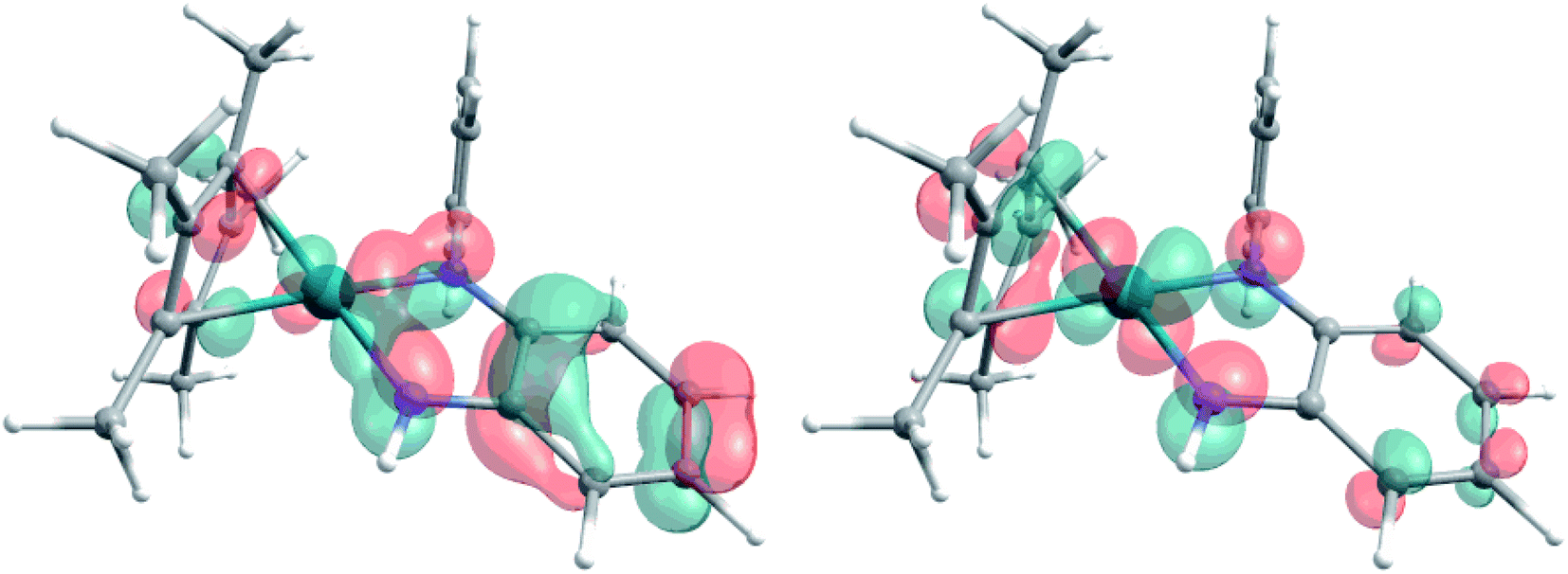DOI:
10.1039/D2SC03227A
(Edge Article)
Chem. Sci., 2022,
13, 10532-10545
Rhodium diamidobenzene complexes: a tale of different substituents on the diamidobenzene ligand†
Received
8th June 2022
, Accepted 15th July 2022
First published on 21st July 2022
Abstract
Diamidobenzene ligands are a prominent class of redox-active ligands owing to their electron reservoir behaviour, as well as the possibility of tuning the steric and the electronic properties of such ligands through the substituents on the N-atoms of the ligands. In this contribution, we present Rh(III) complexes with four differently substituted diamidobenzene ligands. By using a combination of crystallography, NMR spectroscopy, electrochemistry, UV-vis-NIR/EPR spectroelectrochemistry, and quantum chemical calculations we show that the substituents on the ligands have a profound influence on the bonding, donor, electrochemical and spectroscopic properties of the Rh complexes. We present, for the first time, design strategies for the isolation of mononuclear Rh(II) metallates whose redox potentials span across more than 850 mV. These Rh(II) metallates undergo typical metalloradical reactivity such as activation of O2 and C–Cl bond activations. Additionally, we also show that the substituents on the ligands dictate the one versus two electron nature of the oxidation steps of the Rh complexes. Furthermore, the oxidative reactivity of the metal complexes with a [CH3]+ source leads to the isolation of a unprecedented, homobimetallic, heterovalent complex featuring a novel π-bonded rhodio-o-diiminoquionone. Our results thus reveal several new potentials of the diamidobenzene ligand class in organometallic reactivity and small molecule activation with potential relevance for catalysis.
Introduction
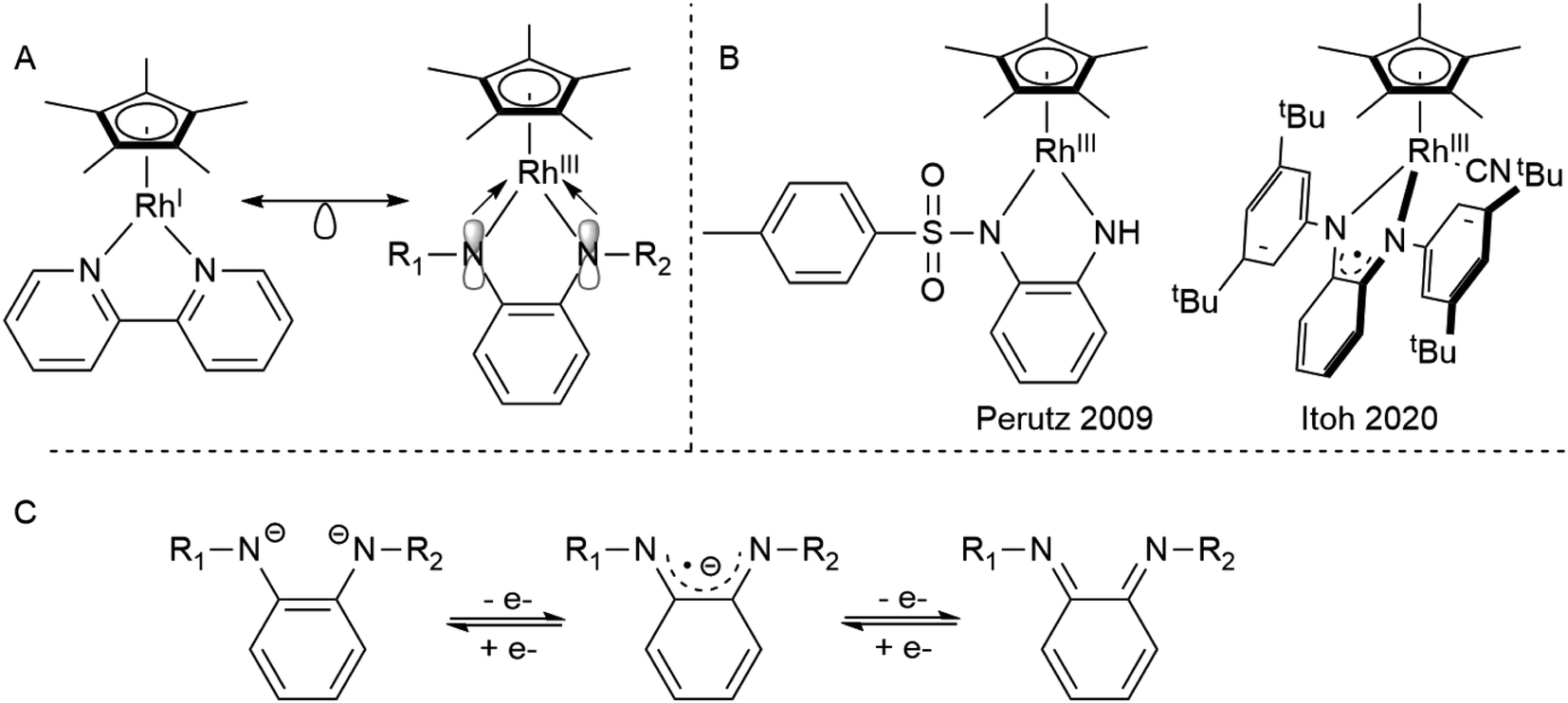
Rhodium half-sandwich complexes are a staple of organometallic chemistry.1 The 18 valence electron species [Cp*RhIL2] act as prototypical metallobases capable of oxidative addition of electrophiles.2 This reactivity has been used maybe most famously in the electro- and photochemical reduction of protons by [Cp*Rh] complexes bearing chelating diimine ligands.3 Therefore, the electrochemistry of these systems has been studied extensively and minute details regarding the influence of structure and substitution pattern of the diimines on the redox properties have been reported.4–7 In most cases, the electrochemistry is dominated by two-electron events in which both the chelating ligand and the metal center participate due to the covalent nature of the metal–ligand bond.
Diamidobenzene complexes of [Cp*RhIII] can be considered isoelectronic analogues to their diimine [Cp*RhI] counterparts, with electron density shifted from the metal center to the chelating π-donor ligand. However, since their first description by Maitlis in 1978,8 only very few studies (Scheme 1) have been concerned with the reactivity and electrochemistry of the diamidobenzene systems – their use in aminations reported by Itoh9 and as model compounds for transfer hydrogenation as described by Perutz10,11 are notable exceptions. This scarcity is surprising, considering that diamidobenzenes as redox-active ligands12–17 may impart novel reactivity on rhodium half-sandwich complexes. Diamidobenzene ligands can support three different oxidation states18–24 and therefore offer access to an open-shell reactivity which may complement the more commonplace two-electron redox reactions of Rh half-sandwich complexes.25,26 Particularly in the diimine state, they form strong, highly covalent bonds to transition metals.27–29 Additionally, the [NR] handle allows steric and electronic tuning in the direct vicinity of the reaction center. This has also enabled the development of redox-active tri30–34- and tetradentate35–38 ligand systems based on diamidobenzene. In this context, it is worthwhile mentioning that a majority of work that deals with such ligands has focussed on R = aryl substituents on the [NR] group.39–43 This fact is surprising considering the extreme variations that fundamentally different kinds of R substituents would allow in terms of tuning the steric, and in particular the electronic properties of the compounds. Various studies have proven the utility of o-diiminoquinonoid ligands in diverse areas such as photochemistry,44,45 molecular magnetism,46–49 electrocatalysis50,51 and activation of small molecules.52–55 A pertinent example is the aforementioned work by the group of Itoh, who used a RhIII half-sandwich complex with a singly-oxidized, open-shell ligand in the amination of trisylazide.9 We have recently shown the ligand-centered redox activity and the ensuing reactivity of diamidobenzene complexes of [Cp*IrIII].56,57 Finally, apart from the expected ligand-centered reactivity, the [RhIII] moiety in principle offers access to reductive chemistry (and hence formally mononuclear RhII complexes) and thus to a novel class of metal-ate complexes without strong acceptors.58 Both reversible and irreversible reductions to anionic species with putative metalloradical character have been described (spectro-)electrochemically for a variety of [Cp*Rh] diimines,59,60 but they have generally eluded isolation. Diamidobenzenes offer rational access to such reactive species, as their donor properties are easily tunable via the N-bound substituents. In this work, we provide a comprehensive investigation into the structure, bonding, electrochemistry and spectroelectrochemistry of diamidobenzene complexes of [Cp*Rh] and prove that they offer access to rare and uncommon molecular species in organometallic rhodium chemistry. By systematically investigating four differently substituted complexes, we show that
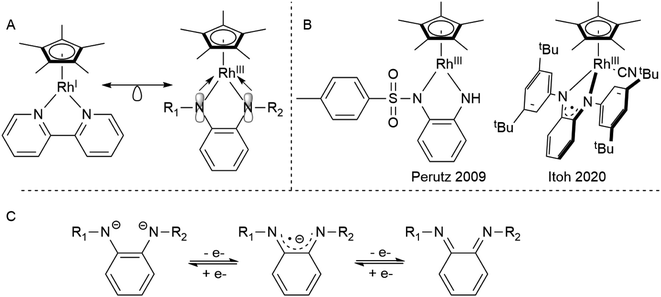 |
| | Scheme 1 (A) The isoelectronic analogy between RhI diimine and RhIII diamido complexes. (B) Catalytically active Rh diamidobenzene complexes. (C) The different oxidation states attainable by diamidobenzenes. | |
(a) The nature of substituents allows switching between separated one-electron and concerted two-electron oxidations centered on the diamidobenzene ligands.
(b) The complexes offer access to reactive, yet isolable mononuclear RhII metalates with tunable potential; such large tuning in isolable mononuclear RhII metalates is to the best of our knowledge unprecedented.
(c) An unprecedented reactivity towards electrophilic [CH3]+ groups allows the direct formation of homobimetallic, heterovalent complexes featuring a novel π-bonded rhodio-o-diiminoquionone. To the best of our knowledge, such a reactivity as well as the π-bonded rhodio-o-diiminoquionone have never been observed previously.
Results and discussion
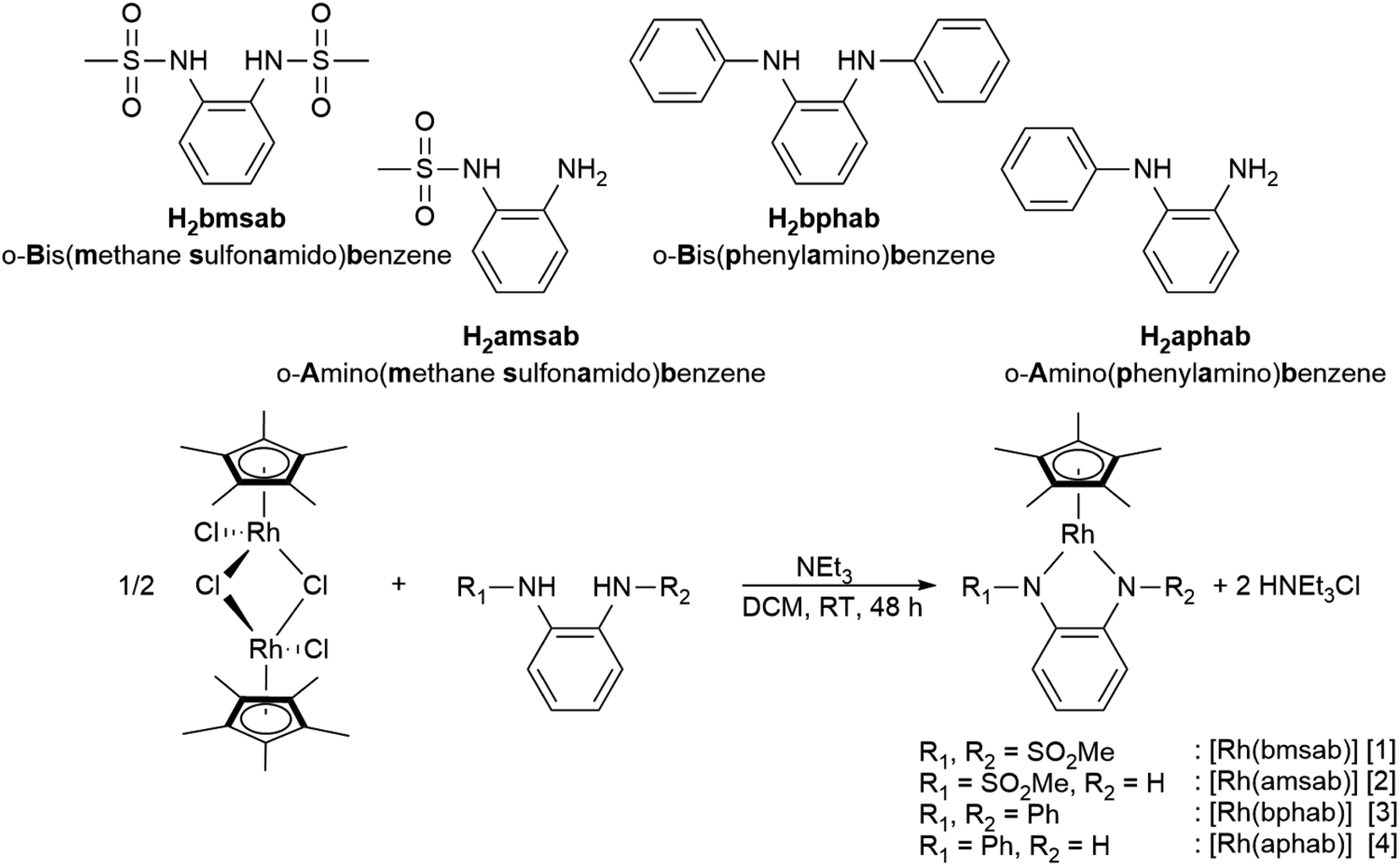
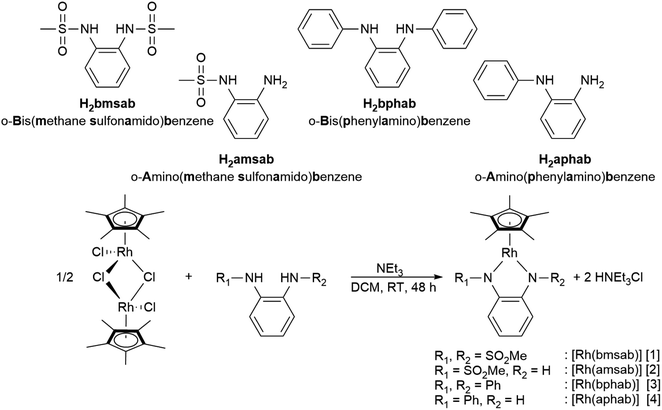
We set out to investigate the influence of both symmetry and donor capacity of substituted diamidobenzene ligands on the electrochemical behavior of mononuclear Cp*Rh(III) complexes. Thus, we chose a set of four ligands (Scheme 2) with substituents on either only one or both N donor atoms. As substituents we compared the strongly electron withdrawing sulfonamido moiety (FSO2Me = 0.53) and the mildly electron-withdrawing phenyl group (FPh = 0.12), F being the modified Swain–Lupton constant according to Hansch, Leo and Taft.61 All four complexes were synthesized by treatment of [Cp*RhCl2]2 with two equivalents of ligand and an excess of NEt3 in DCM (Scheme 2). Following work-up and purification, all compounds were obtained as microcrystalline solids in moderate to good yields. 1H- and 13C-NMR, EA, HR-MS as well as molecular structures from single crystal X-ray diffraction confirm the expected constitution of all compounds. Furthermore, the 1H-NMR spectra in DCM-d2 indicate diamagnetic compounds, as no paramagnetic contribution to the chemical shift is discernible (Fig. S1–S8†). In case of [1] and [3], the number of signals confirms the expected C2 symmetry in solution (Fig. S1 and S5†).
 |
| | Scheme 2 The differently substituted ligands used in this study and the synthetic route to their respective complexes. | |
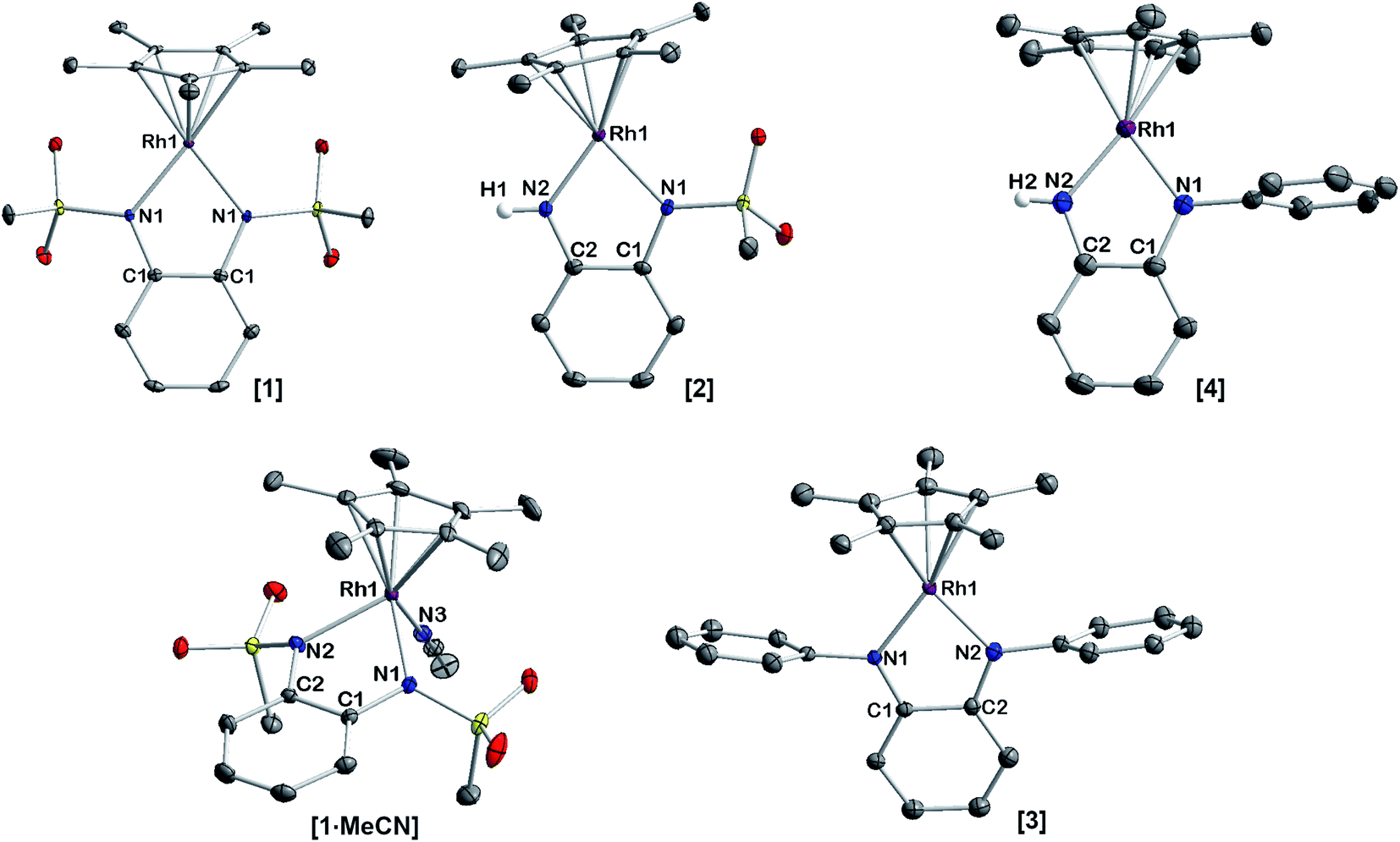
A striking feature of all complexes is their intense color in solution with extinction coefficients ranging from 7 × 103 to 15 × 103 L mol−1 cm−1 in DCM (Fig. S25†), indicating charge-transfer (CT) transitions and a sizable orbital mixing between ligand and metal. While complexes [2], [3] and [4] display qualitatively similar spectra in different solvents, [1] shows a distinctively different behavior in acetonitrile solution (Fig. S25†), indicating strong interaction with the coordinating solvent. This is revealing of the strongly electron-withdrawing character of the sulfonyl groups on the bmsab ligand, increasing the electrophilicity of the RhIII center and thus its affinity for MeCN. An inspection of the molecular structures obtained from DCM solutions sheds light on the bonding in complexes [1] to [4] (Fig. 1). All compounds crystallize as coordinatively unsaturated, formally five-coordinate complexes from the non-coordinating solvent. However, dissolution of [1] in acetonitrile and subsequent crystallization yields the coordinatively saturated complex [1·MeCN]. The bond lengths in the diamidobenzene ligand indicate the presence of a fully reduced, dianionic ligand bound to a RhIII center (Tables 1 and S10†): C–N bond lengths between 1.359(2) and 1.414(2) Å together with C–C bond lengths around 1.40 Å inside the aromatic ring fall into the previously described values for a dianionic diamidobenzene donor.57,62 In case of the coordinatively unsaturated compounds, the N,N-donor is arranged perpendicularly to the plane of the Cp* ligand, with complex [1] showing a slight deviation from perpendicularity. The perpendicular orientation allows for optimal π overlap between the Rh center and the lone pairs of the N donor, leading to electronic saturation and the ligand–metal orbital mixing prerequisite for CT transitions.
 |
| | Fig. 1 Molecular structures of the neutral complexes [1] to [4] in the crystal. Ellipsoids drawn at 50% probability. Solvent moelcules and H atoms omitted for clarity. | |
Table 1 Selected bond lengths in neutral complexes [1]–[4]. All values given in Å
|
|
[1]
|
[1·MeCN]
|
[2]
|
[3]
|
[4]
|
|
Complex [3] crystallizes with two different molecules in the asymmetric unit which display slightly different bond lengths. We have given values for the more symmetric molecule, as NMR data indicate C2 symmetry in solution.
|
| C1–N1 |
1.414(2) |
1.414(2) |
1.417(3) |
1.382(6) |
1.371(2) |
| C2–N2 |
1.414(2) |
1.424(2) |
1.366(3) |
1.373(7) |
1.359(2) |
| Rh–N1 |
2.051(2) |
2.098(2) |
1.937(2) |
1.985(4) |
1.961(2) |
| Rh–N2 |
2.051(2) |
2.113(2) |
2.075(2) |
1.982(5) |
1.983(2) |
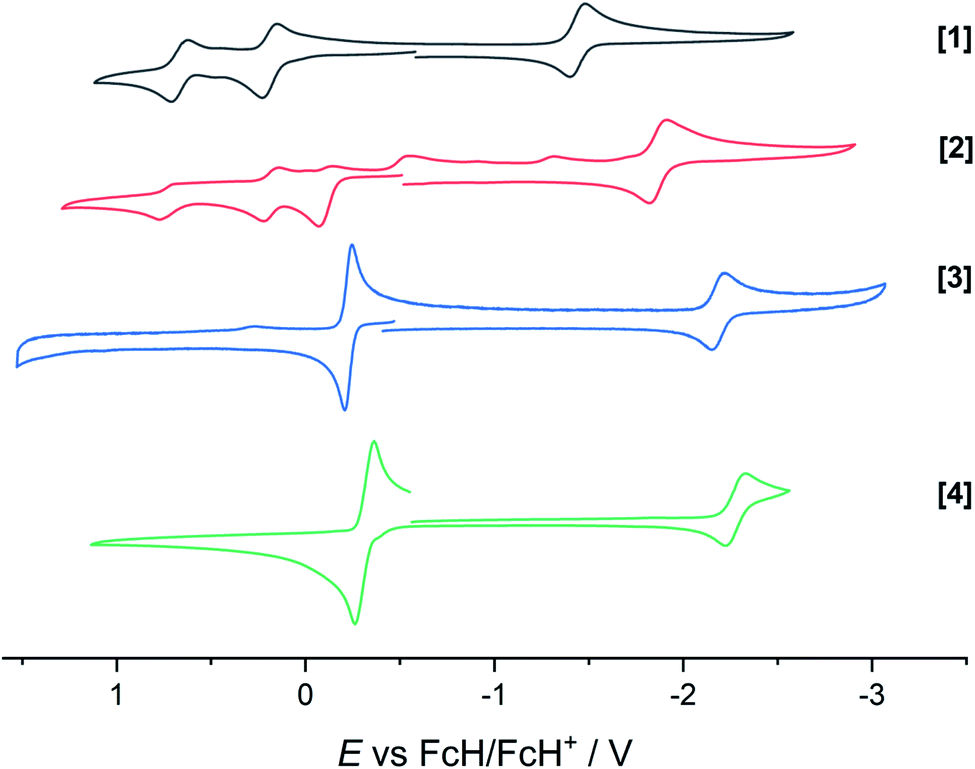
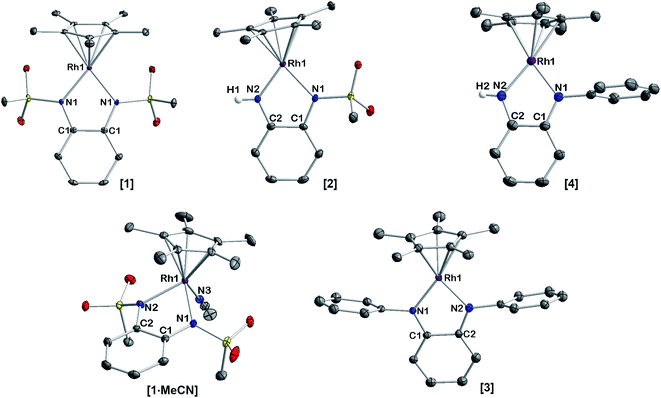
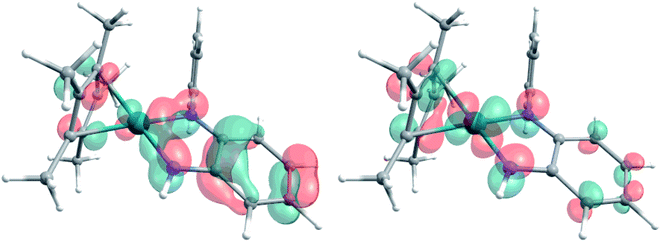
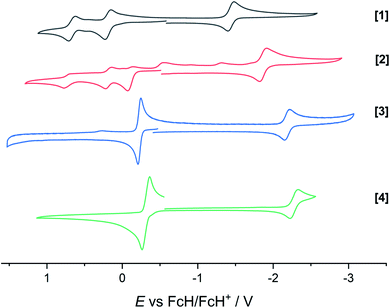
The extent of π donation is strongly influenced by the nature of the N-bound substituents. Evidently, in case of the bis(methane sulfonamide)benzene in [1], the lower limit of π saturation is almost reached, as even a relatively weak donor such as acetonitrile is sufficient to convert [1] into the coordinatively saturated form [1·MeCN]. The N–Rh bond lengths may serve as a metric to gain further insights into the relative donor strength of the other ligands. As expected, the Rh–N bond lengths contract with decreasing electron-withdrawing effect of the N-substituents. In all cases, the NH donor displays the shortest bond length. The expected increase in (π-)donor strength in the series [1] to [4] is thus confirmed by the molecular structures. π-Bonding and antibonding interactions also contribute to the chemically relevant frontier orbitals of the complexes. DFT calculations show qualitatively similar HOMOs and LUMOs for complexes [1] to [4]: while the HOMO has a π-bonding character regarding the metal-diamide interaction with major contributions from the chelating NCCN-moiety, the LUMO is π-antibonding and has a more pronounced 4d character. Fig. 2 shows the frontier orbitals of [4] as a representative example; the corresponding orbitals of [1]–[3] are shown in Fig. S28.† As the HOMO is mostly ligand-centered, one can expect a strong influence of the substitution pattern on the oxidation processes. Indeed, as can be seen from the cyclic voltammograms recorded in acetonitrile (Fig. 3), the four complexes show both qualitatively and quantitatively distinctive electrochemical features. While all complexes display a reversible reduction (vide infra), the behavior during the anodic scan depends on the nature of the substituents. Half-wave potentials are listed in Table 2; further details on the electrochemical properties can be found in the ESI, Section 4.† The symmetric sulfonamide complex [1.MeCN] shows two well-separated, reversible one-electron oxidations. While asymmetric [2] also shows one-electron processes, these are not reversible, indicating at least one EC mechanism and a complex follow-up chemistry, likely involving the N–H group. Contrasting this behavior, both [3] and [4] only show one feature in the anodic scan: each complex displays a reversible two-electron oxidation. This is in line with results we recently published on the Ir(III) analogue of [4].56 The increasing donor strength, i.e. higher electron density on the chelating NCCN moiety, is also reflected in the cathodic shift of the oxidation potentials on going from [1] to [4]. As the DFT calculations indicate, the diamidobenzene moieties are likely the loci of the oxidations.25 To verify this assumption, we performed spectro-electrochemical (SEC) measurements on complexes [1·MeCN], [3] and [4] in acetonitrile, as [2] only displayed irreversible oxidations.
 |
| | Fig. 2 Representative HOMO (left) and LUMO (right) of complex [4], calculated at the TPSSh/def2-TZVP level of theory. Contour value 0.05 A. | |
 |
| | Fig. 3 Cyclic voltammograms of complexes [1] to [4]. Measured in dry and degassed MeCN/TBAPF6. GC WE, Ag RE, Pt CE. Scan rate 100 mV s−1. Current is normalized. | |
Table 2 Half-wave potentials as observed in cyclic voltammetry in MeCN/TBAPF6. GC WE, Ag RE, Pt CE. All potentials given against the FcH/FcH+ redox couple. The third oxidation in [2] is omitted as a follow-up process
|
|
[1]
|
[2]
|
[3]
|
[4]
|
|
E
1/2 (1st ox.)/V |
0.20 |
−0.11 |
−0.23 |
−0.31 |
|
E
1/2 (2nd ox.)/V |
0.67 |
0.18 |
— |
— |
|
E
1/2 (red.)/V |
−1.44 |
−1.86 |
−2.18 |
−2.27 |
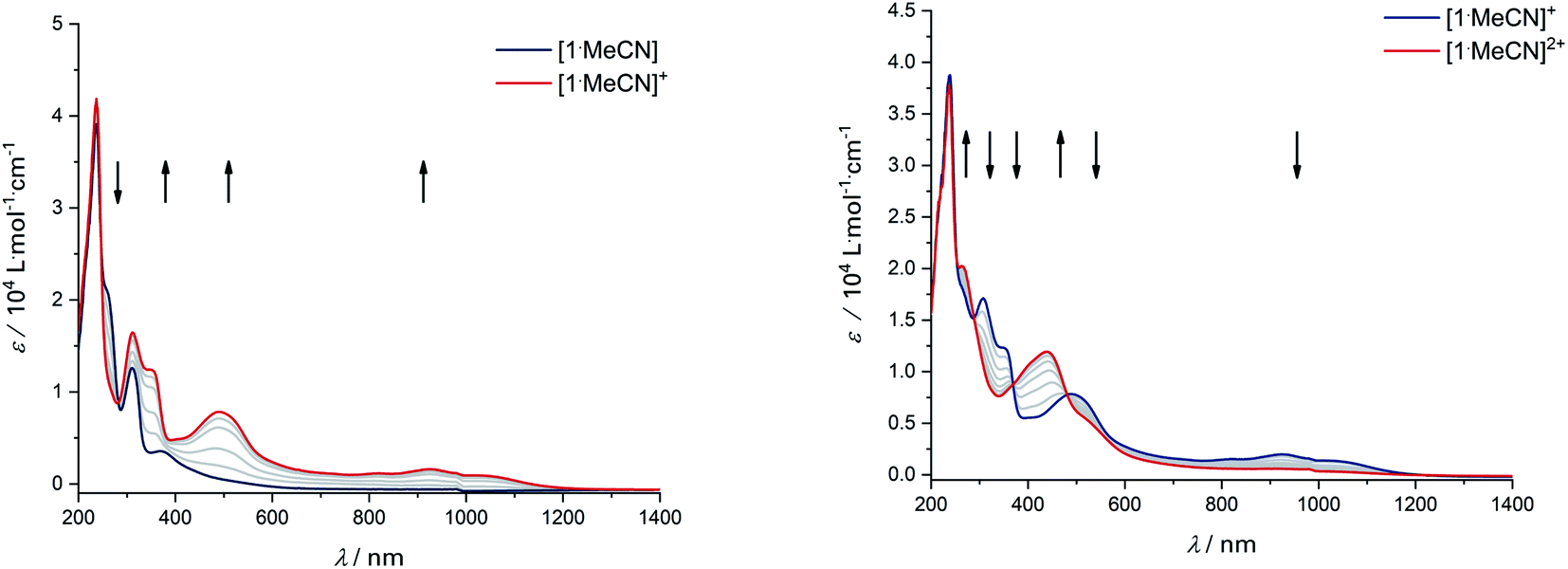
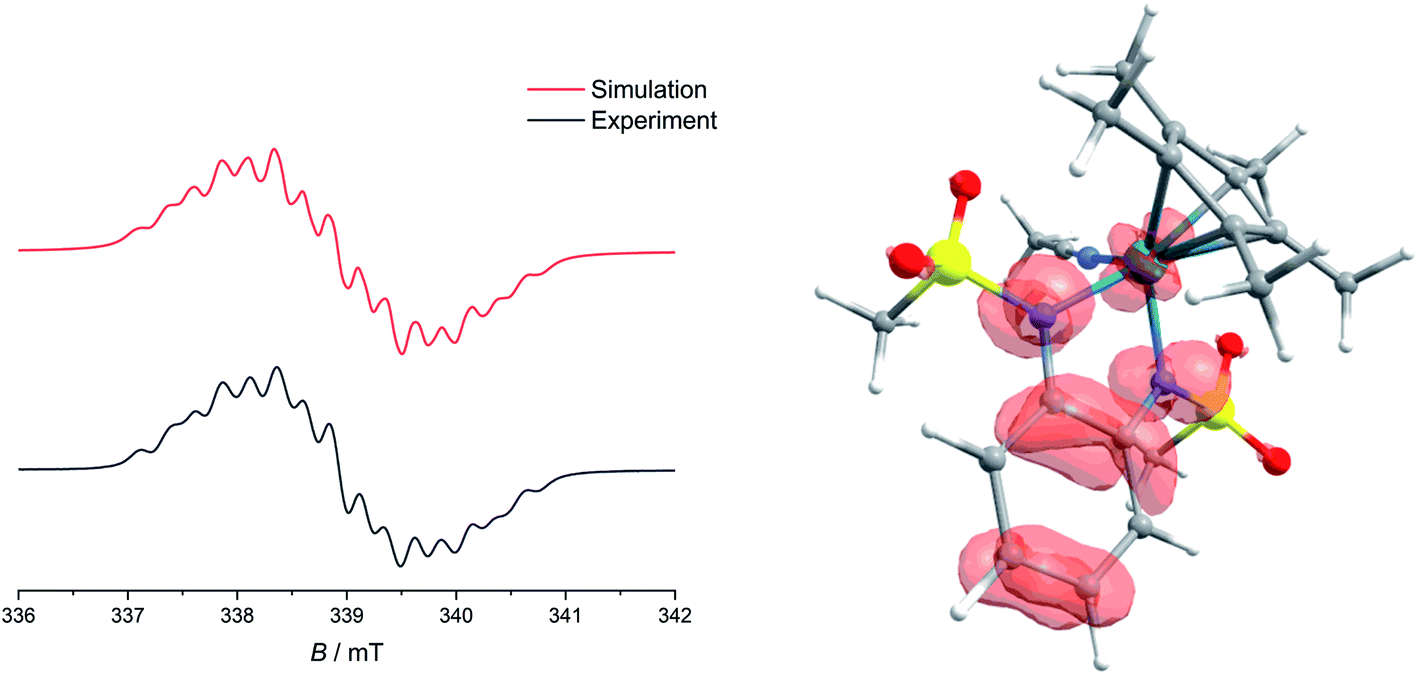
Upon the first oxidation, broad, long wavelength bands appear in the spectrum of [1·MeCN]+, concomitant with the appearance of a band at around 500 nm (Fig. 4, left). These features are typical for metal-bound radical ligands.63,64 The radical nature of [1·MeCN]+ was unequivocally shown by EPR-SEC (Fig. 5, left): oxidation leads to a line-rich spectrum centered around g = 1.99, which can be simulated by taking into account hyperfine coupling to two equivalent N nuclei (AN1 = 14.1 MHz), an additional N nucleus (AN2 = 9.4 MHz), the 103Rh nucleus (ARh = 20.1 MHz) and two sets of two equivalent protons (AH1 = 12.4 MHz, AH2 = 7 MHz).
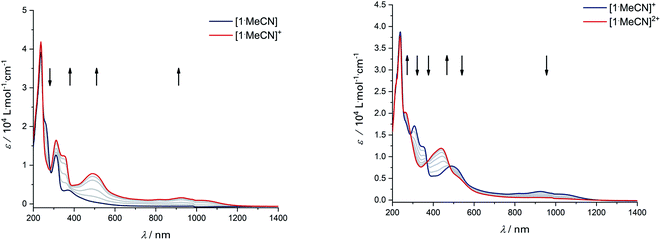 |
| | Fig. 4 UV/Vis-SEC of complex [1·MeCN] in MeCN/n-Bu4PF6 at 293 K. Left: Spectral changes during first oxidation. Right: Spectral changes during second oxidation. | |
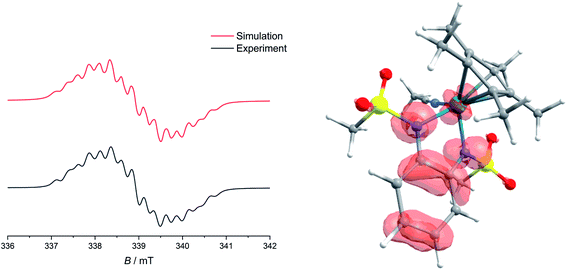 |
| | Fig. 5 Left: Experimental and simulated EPR spectra of [1.MeCN]+, obtained from electrolysis in MeCN/n-Bu4PF6 at 293 K. Right: Spin-density of [1.MeCN]+, calculated at the TPSSh/def2-TZVP level of theory. | |
This is in line with an unpaired electron delocalized over the chelating CNNC moiety, with additional contribution from the metal center and the aromatic backbone to the SOMO. DFT calculations reproduce these findings (Fig. 5, right). No signs of decomposition were observed for [1·MeCN]+ on the spectroelectrochemical timescale (i.e. several minutes). Removal of a second electron leads to a disappearance of the bands in the NIR region and new bands between 400 and 600 nm (Fig. 4, right). Such spectral signatures indicate MLCT and LL’CT transitions to an iminoquinonoid ligand;57 however, within seconds the doubly oxidized complex begins to decompose.
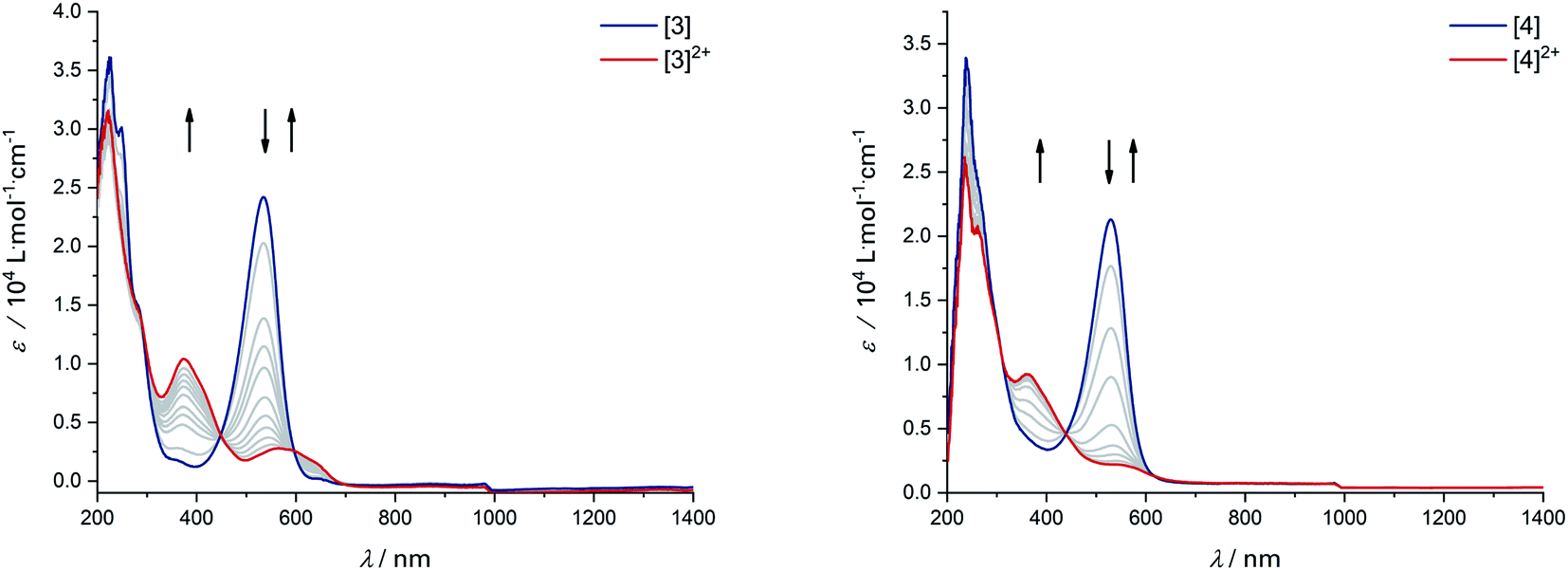
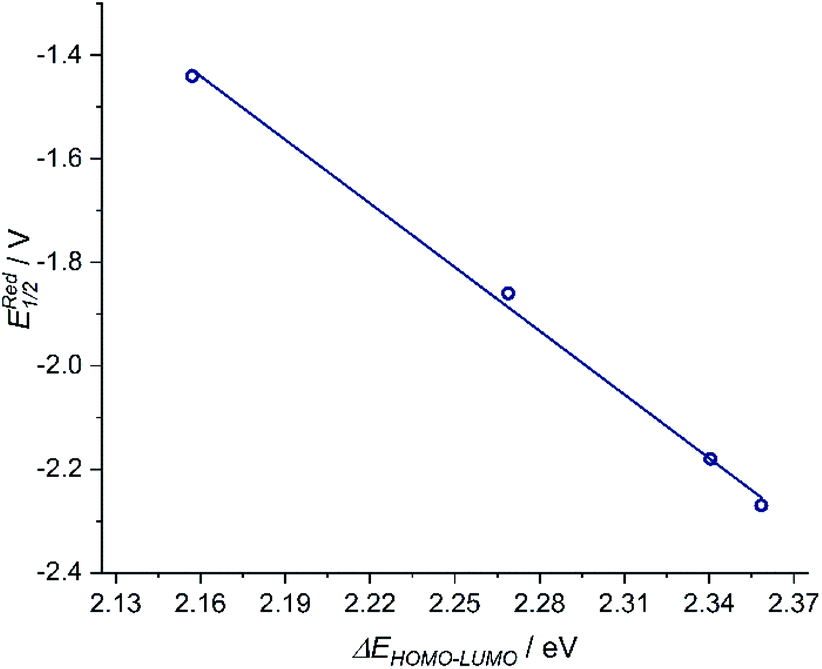
The donor strength of an imine with strongly electron-withdrawing substituents is probably so low that dissociation of the oxidized ligand occurs. The two-electron oxidations of [3] and [4] directly lead to spectral features that indicate an o-iminoquinonoid ligand structure (Fig. 6), but in these cases, the oxidized species are stable on the spectroelectrochemical timescale. The electrochemical and spectroscopic results indicate that modifications on the [N–R] handle have a strong influence on the potential and separation of oxidation events in diamidobenzene ligands. By comparing structurally related systems, these results point to a design strategy for ligands that may serve as either one- or two-electron reservoirs: the electron-poor sulfonamide moiety seems to preferentially stabilize ligand-centered radicals, while the [N–H] group appears to induce follow-up reactions of such radicals. This is likely due to radical H atom abstraction from the NH handle after single-electron oxidation. In contrast, both [N–H] and [N–Ph] substituents support the concerted two-electron oxidation to a stable iminoquinoid ligand. Ligand-centered one- and two-electron oxidations have been described previously for analogous systems;9,56,57 however, the reversible reductions at potentials between −1.44 V ([1]) and −2.27 V ([4]) against FcH/FcH+ are a novel feature. The cathodic shifts of the potentials follow the trend in donor strength described above and show a linear correlation with the calculated HOMO–LUMO gaps of complexes [1]–[4] (see Fig. 7), which indicates a similar electronic structure in the reduced species and suggests the possibility of predictively designing complexes with a desired reduction potential. At the TPSSh/def2-TZVP level of theory, the correlation is described by the following formula:
| ERed1/2 [V] = 7.43 V − 4.10 × ΔEHOMO–LUMO [eV] |
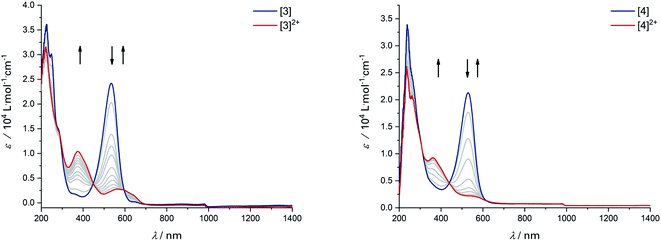 |
| | Fig. 6 UV/Vis-SEC of complexes [3] and [4] in MeCN/n-Bu4PF6 at 293 K. Left: Spectral changes during two-electron oxidation of [3]. Right: Spectral changes during two-electron oxidation of [4]. | |
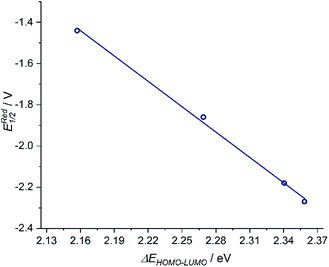 |
| | Fig. 7 Left: Correlation of EReduction1/2 and ΔEHOMO–LUMO with a goodness-of-fit R2 = 0.997. | |
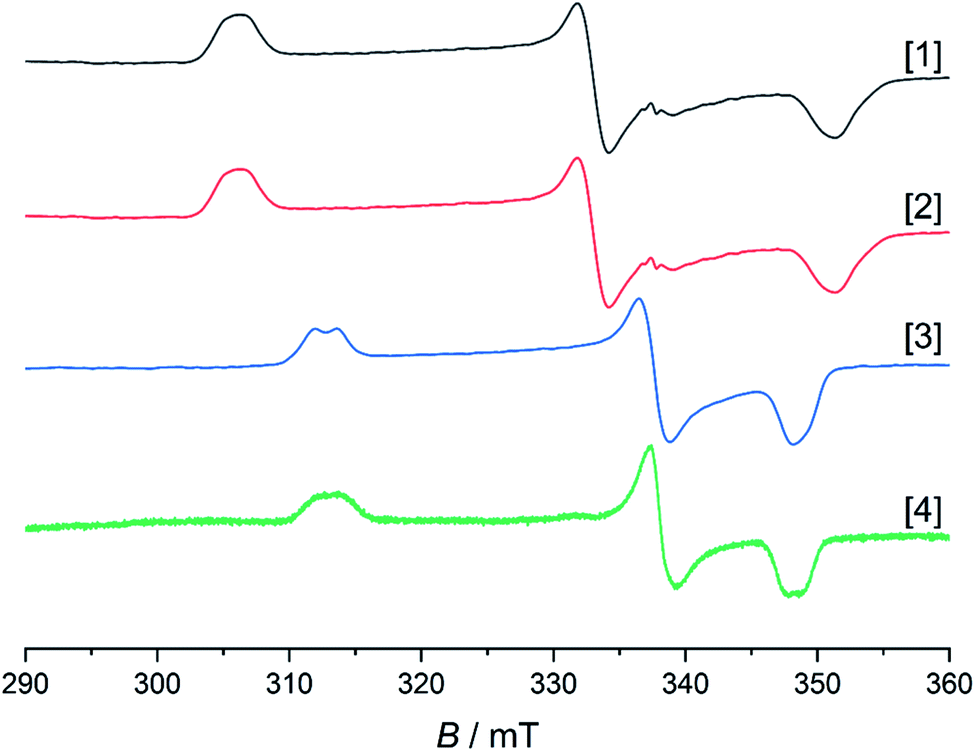
As mentioned above, all compounds show qualitatively similar LUMO shapes with a sizable contribution of a metal-centered orbital, which points to a reduction to formally RhII. EPR-SEC studies verified the assumption of metal-centered processes: the (electro-)chemically reduced species show rhombic, anisotropic spectra indicative of metalloradicals (Fig. 8 and S17†). The g-values are in line with previously described RhII complexes65,66 and particularly similar to a [Cp*RhII] moiety bound to a doubly reduced azobispyridine system.59 In case of electrogenerated [3]− and [4]−, hyperfine coupling to the 103Rh nucleus is partially resolved in g1 and g3. UV/Vis-SEC measurements confirm the reversibility of the processes and the stability of the reduced species in solution (Fig. S21–S24†). Unrestricted DFT calculations on the reduced species show a major contribution (55–64%) of Rh to the spin population (Fig. S29†). The calculated g-values are in qualitative agreement with the experimental values (see Table 3).
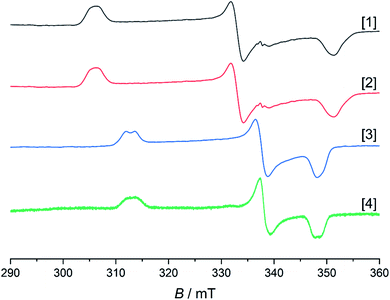 |
| | Fig. 8 EPR spectra of electrochemically generated [1]−, [2]−, [4]− in frozen MeCN and chemically reduced [3]− in frozen THF. Spectra recorded at 103 K. Intensities are normalized. Due to lower concentration in the SEC experiment, the signal to noise ratio is also lower for [4]−. | |
Table 3 Experimental g-values and calculated values at the UKS-TPSSh/def2-TZVP level of theory
|
|
[1]−
|
[2]−
|
[3]−
|
[4]−
|
|
g-Values exp., (calc.) |
1.92, (1.94) |
1.93, (1.96) |
1.94, (1.96) |
1.94, (1.96) |
| 2.03, (2.03) |
2.03, (2.02) |
2.00, (2.00) |
2.00, (2.00) |
| 2.21, (2.16) |
2.20, (2.13) |
2.16, (2.12) |
2.16, (2.12) |
| Spin population Rh (Loewdin) |
64% |
59% |
55% |
55% |
The EPR- and UV/Vis-SEC results clearly indicate that the reduction of the neutral Rh(III) complexes leads to the formation of stable, mononuclear, anionic Rh(II) compounds. As is apparent from Fig. 4 and Table 3, the g-anisotropy decreases with increasing donor strength. This trend can be qualitatively rationalized by inspecting the relevant orbitals as calculated on the TPSSh/def2-TZVP level of theory: the SOMO corresponds to the LUMO of the neutral complexes (Fig. 2) and shows π* symmetry regarding the metal diamide interaction with a large contribution from the metal dyz orbital. Stronger π donors will engender a larger energy splitting between the bonding and antibonding π orbital, effectively pushing up the SOMO in energy. In a first approximation, the deviation from gel = 2.0023 depends inversely on the energy difference between the ground state and excited states, in which electrons are promoted from the doubly occupied 4d orbitals into the SOMO. Energetically pushing up the SOMO thus increases this energy difference and consequently reduces the g anisotropy. An additional factor is the increasing covalency of the metal–ligand bonds, which is also reflected in the decrease of Rh-centered spin population (Table 3).
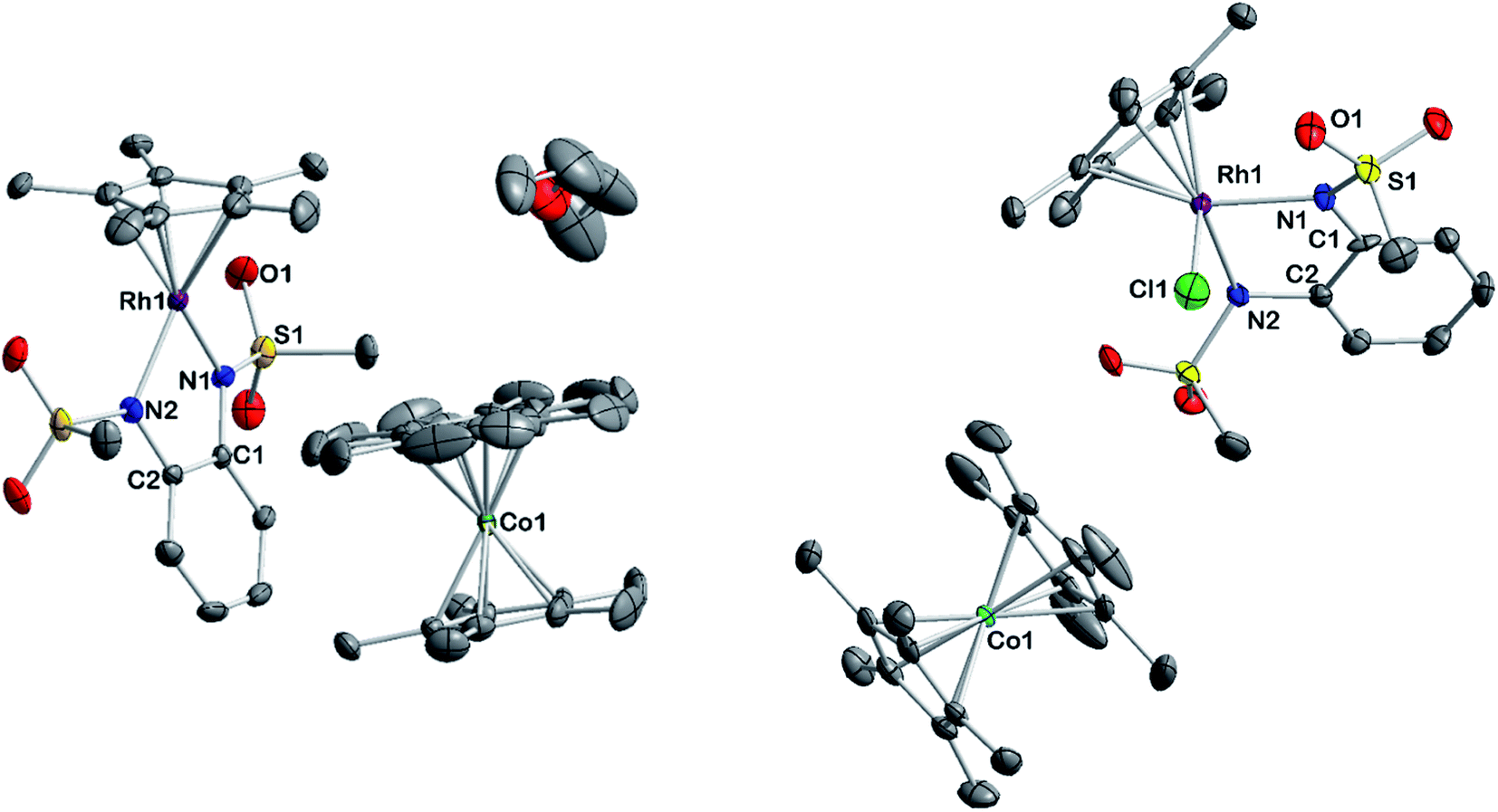
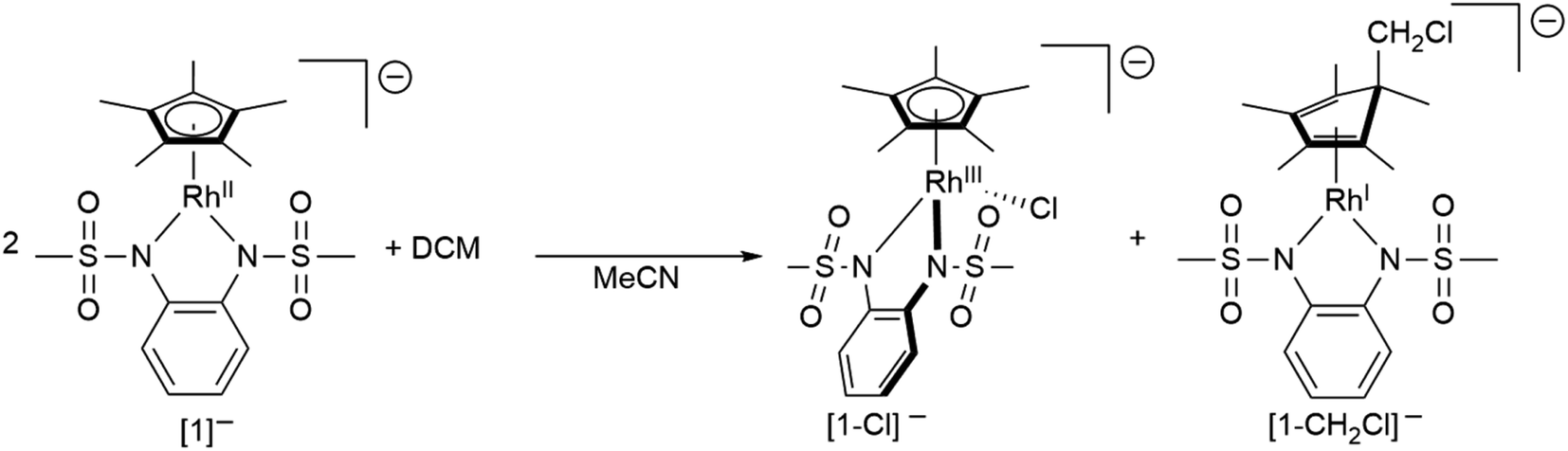
To gain insight into the structure and reactivity of the reduced species, we conducted a preliminary case study on [1]− as a representative example. A symmetrically substituted species was chosen as we expected this to be advantageous regarding both crystallization and data analysis in reactivity studies. Treatment of [1.MeCN] with [CoCp*2] in acetonitrile gave a very air-sensitive green solution, from which [CoCp*2][1] was isolated as crystalline material upon layering with diethyl ether and storage at −30 °C. [CoCp*2][1] crystallizes in the space group P21 with one molecule of diethyl ether. An inspection of the molecular structure given in Fig. 9 on the left, shows that upon reduction, the bound acetonitrile dissociates and [1]− adopts a coordinatively unsaturated geometry due to the increased electron density at the central metal. This leaves a free coordination site open for further reactivity. In comparison with the neutral species, the Rh–N bond lengths are elongated by ∼0.05 Å. Characteristic bond lengths and angles are given in Table S11.† Initial reactivity studies attest to the metalloradical behavior of [1]−: EPR spectra show the formation of a thermally unstable rhodium superoxide upon contact with O2 (Fig. S26†). The formation of superoxido species has been previously described for other RhII complexes.67–69 We also investigated the reactivity of [1]− towards the geminal dihalocarbon dichloromethane, as d7 metalloradicals are known to activate RX bonds.70 Indeed, upon treating an acetonitrile solution of [1]− with dichloromethane, more than 70% conversion is observed within 4 h (yields spectroscopically determined). Two products are formed, which can be identified by their 1H- and 13C-NMR spectra along with mass spectrometry. The homolytic splitting of the C–Cl bond leads to the formation of an anionic RhIII chloride complex and subsequently to the addition of the ·CH2Cl fragment to the Cp* unit in a second molecule of [1]− (see Scheme 3). A detailed interpretation of the spectra is given in the ESI.† The net reaction in Scheme 3 presents a disproportionation of two RhII molecules into a RhIII and a RhI complex – such reactions have been previously described for different RhII species.67,71,72 The reactivity towards DCM is very similar to the ‘RX’ reaction originally described by Wilkinson73 and later Kölle74 for cobaltocene and decamethylcobaltocene, respectively. While we haven't been able to quantitatively separate the two reaction products so far, we succeeded in crystallizing the chlorido adduct. The molecular structure is shown in Fig. 9 on the right. These results show that the metalloradical reactivity of decamethylcobaltocene is transferred onto the RhII metalloradical [1]−. Considering the substantially more negative potentials in [2]−, [3]− and [4]−, the activation of very stable carbon-halogen bonds seems likely. Additional reactivity studies towards that end are currently being undertaken in our laboratories.
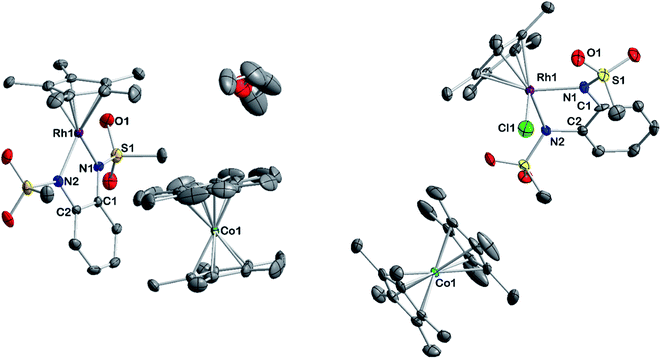 |
| | Fig. 9 Molecular structure of reduced complex [CoCp*2][1] (left) and one product of its reaction with DCM [CoCp*2][1-Cl] (right). Ellipsoids drawn at 50% probability. H atoms omitted for clarity. | |
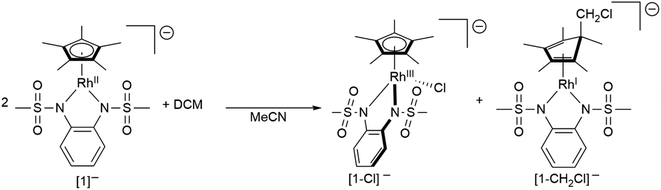 |
| | Scheme 3 The reaction of [1]− with DCM. | |
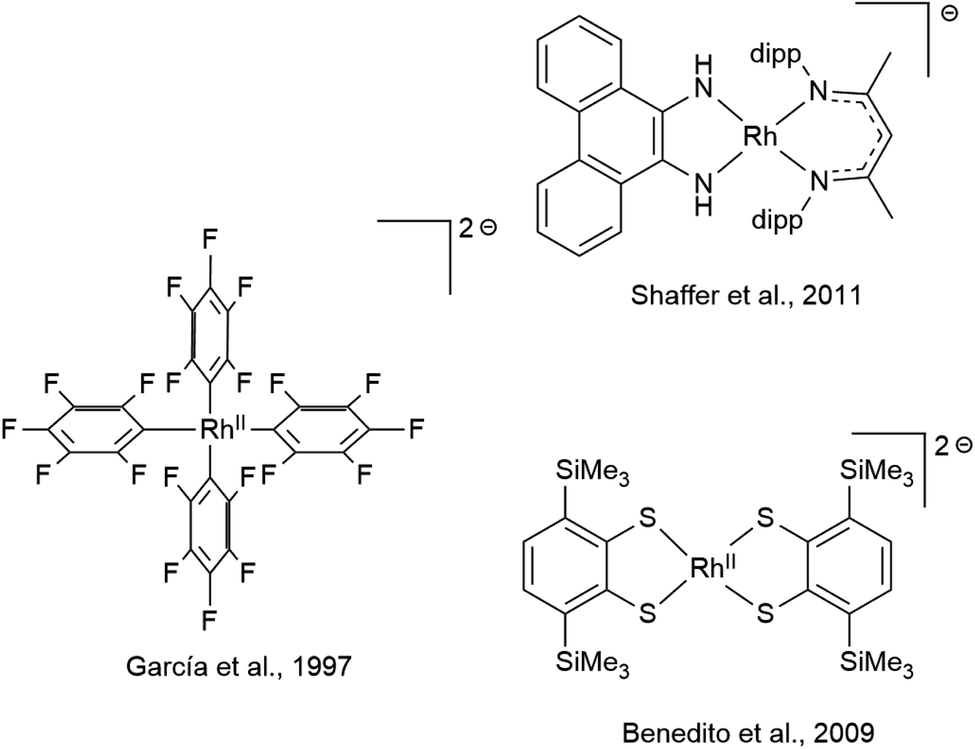
While mononuclear RhII complexes are still rather uncommon and often rely on sophisticated ligand design,75–78 anionic RhII complexes are particularly rare.79 Only very few have been structurally characterized, three of which are shown in Scheme 4.80–82 RhII complexes have been shown, inter alia, to activate C–C83 and C–H84 bonds as well as dihydrogen85,86 and to engage in H atom transfer.87 The reactivity of RhII-ate complexes however has been scarcely explored, even though they were recently proposed as intermediates in the photo-induced ortho-C–H borylation of arenes.88 The diamidobenzene scaffold allows access to a new class of electron-rich RhII-ate complexes with tunable potential. Note that the complexes [1] to [4] span a potential range of 830 mV regarding the RhIII/RhII couple. Considering the plethora of additional substitution patterns of diamidobenzene accessible via facile functionalization of either o-dibromobenzene or o-phenylenediamine, this potential range could be extended to over 1 V. The linear correlation of the reduction potential with the HOMO–LUMO gap possibly allows a predictive design strategy. Therefore, diamidobenzene complexes of [Cp*Rh] constitute a promising compound class to study the possibilities of RhII-ate complexes in chemical transformations.
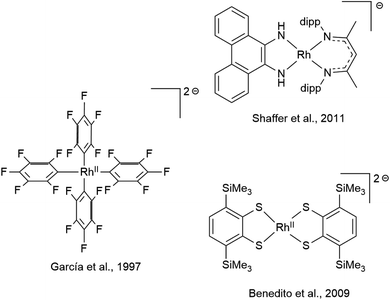 |
| | Scheme 4 Previously reported and structurally characterized anionic RhII complexes. | |
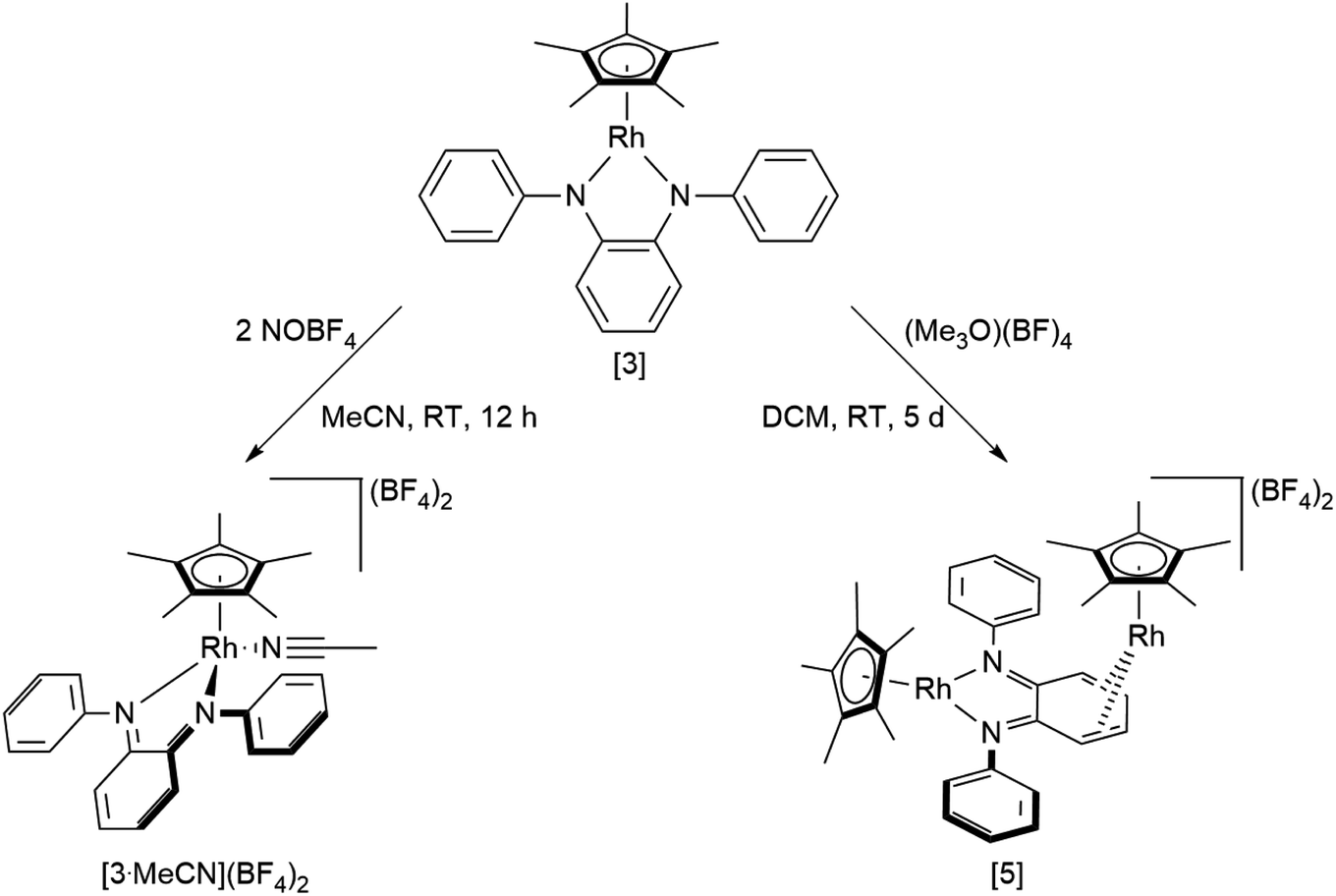
An investigation into the oxidative reactivity (Scheme 5) offered novel insights as well. As a case study, we focused on the symmetrically substituted species [3]. Isolation and crystallographic characterization (Fig. 10, left) of the chemically oxidized complex [3.MeCN]2+ confirmed the ligand-centered oxidation: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond lengths of 1.301(3) Å, a contraction of the C3–C4 and C5–C6 bond distances to around 1.34 Å together with an elongation of the residual C–C bonds in the backbone are all in line with an o-iminoquinonoid structure. The reduced donor strength of the oxidized ligand leads to additional coordination of a solvent molecule, similar to structures observed in other RhIII diimine complexes.89 The dianionic ligand in [3] can therefore act as an electron reservoir, analogous to the diamidobenzene ligand which enables the oxidative addition of [CH3]+ groups to an IrIII center.56 Thus, we treated symmetrical, phenyl-substituted complex [3] with one equivalent of Meerwein's salt (Me3O)(BF4) to see whether a similar reactivity would be observed (Scheme 5).
N bond lengths of 1.301(3) Å, a contraction of the C3–C4 and C5–C6 bond distances to around 1.34 Å together with an elongation of the residual C–C bonds in the backbone are all in line with an o-iminoquinonoid structure. The reduced donor strength of the oxidized ligand leads to additional coordination of a solvent molecule, similar to structures observed in other RhIII diimine complexes.89 The dianionic ligand in [3] can therefore act as an electron reservoir, analogous to the diamidobenzene ligand which enables the oxidative addition of [CH3]+ groups to an IrIII center.56 Thus, we treated symmetrical, phenyl-substituted complex [3] with one equivalent of Meerwein's salt (Me3O)(BF4) to see whether a similar reactivity would be observed (Scheme 5).
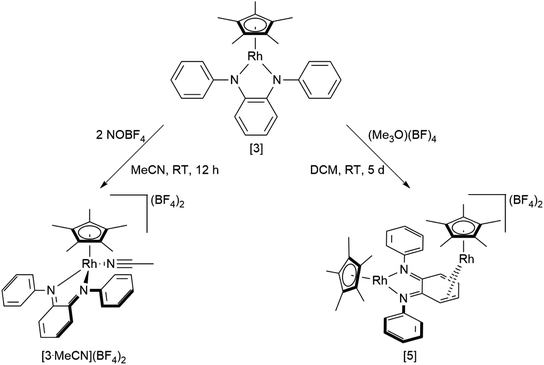 |
| | Scheme 5 Reactivity of [3] towards oxidizing substrates NOBF4 and Meerwein's salt. | |
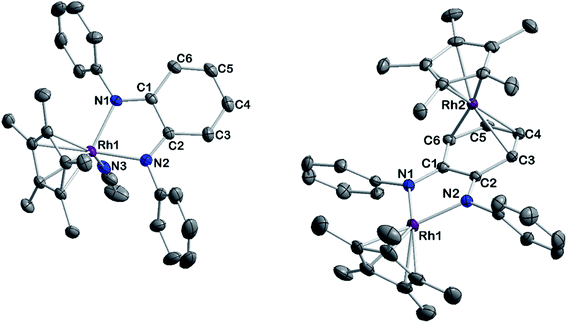 |
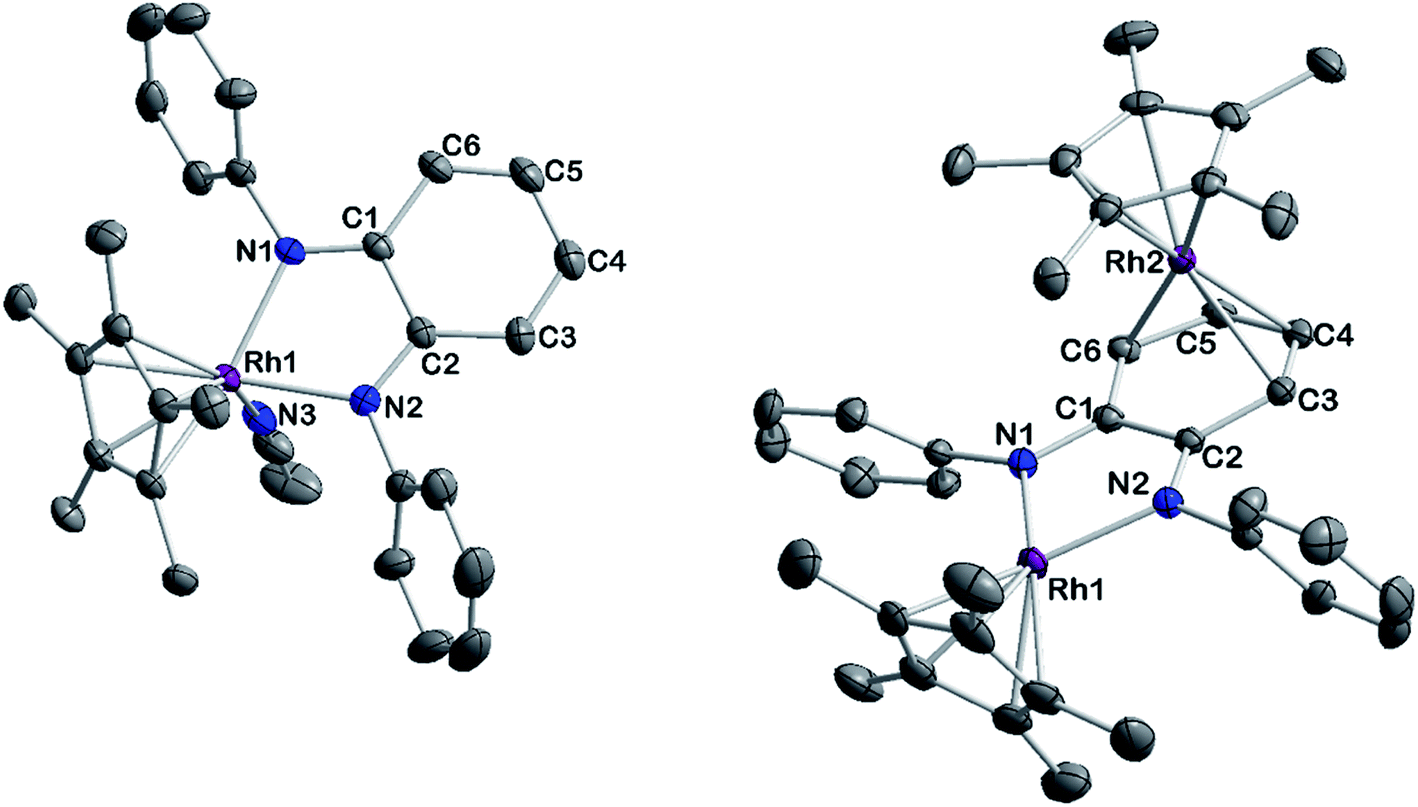
| | Fig. 10 ORTEP representation of the molecular structures of [3.MeCN]2+ (left) and [5]2+ (right). Ellipsoids drawn at 50% probability. H atoms, anions and solvent molecules have been omitted for clarity. | |
However, upon work-up and crystallization a completely different product was obtained: we isolated the dicationic, dinuclear complex [5](BF4)2 in 46% yield, in which a second [Cp*Rh] moiety is coordinated to the backbone of the o-diiminoquinone (Fig. 10, right). 1H- and 13C-NMR-spectra as well as ESI mass spectra confirm that the same constitution is present in solution: characteristic doublet splittings of the 13C resonances in the ligand backbone are proof of bonding to the 103Rh nucleus. Similar structures were described by Amouri and co-workers for o-benzoquinones in 2009:90 they developed a synthetic access to π-bonded rhodio-o-benzoquinones, in which a [Cp*RhI] fragment is coordinated in η4-fashion to the ligand backbone. These compounds were also shown to act as organometallic linkers via the chelating benzoquinone moiety (Scheme 5).65
The unexpected formation of a heterovalent, dinuclear rhodium complex with a certain amount of redox ambiguity is reminiscent of results described by Tejel, de Bruin and co-workers in 2008.91 They used a combination of structural and NMR data to describe the valency of two distinct Rh sites in a dinuclear species. Following that approach, the 1JRh,C couplings in [5](BF4)2 offer a first hint at an appropriate description of the bonding and valency in this compound: only C3/C6 and C4/C5 show coupling to a Rh nucleus (Fig. S12†). This is in line with shorter bond lengths between these carbon atoms and Rh2. A side-view of the molecular structure (Fig. S36†) shows a loss of planarity (and hence, aromaticity) in the ligand backbone. Both the spectral and structural features are comparable to the results obtained by Amouri and co-workers.65,90 Taken together, these findings imply the presence of a neutral iminoquinone ligand, which binds to a RhI moiety through its diolefinic backbone. Both the Rh–C bond distances of around 2.2 Å and the CC bond lengths of around 1.4 Å correspond to reported values for diolefins coordinated to RhI.92,93 The bond lengths between C3–C4 and C5–C6 are elongated by approximately 0.06 Å compared to the iminoquinonoid structure in [3.MeCN]2+ (Table 4). While the group of Amouri has shown that π-bonded metallo-o-benzoquinones and the heavier S- and Se-congeners may serve as organometallic ligands, our results extend this concept to o-iminoquinones, and this to the best of our knowledge is the first example of such a structural motif with o-diiminoquinones. Considering the possible photochemical applications of such ligand systems, an extension to N-donor analogues offers interesting new possibilities. Despite its structural similarity to previously reported complexes mentioned above, the mechanism of the formation of [5](BF4)2 is fundamentally different. We followed the reaction by 1H-NMR spectroscopy (Fig. S33†) and were able to identify the initial formation of the methylated Rh complex [3-CH3]+ by the appearance of a doublet in the high-field region (δ = 0.75 ppm, 2JRh,H = 2.4 Hz), which shows a chemical shift and coupling constant in the range of previously reported Rh–CH3 groups.94–96 A weak resonance at δ = 3.02 ppm may be tentatively assigned to chloromethane.97 This might indicate that the metal-bound methyl group abstracts chloride from the solvent. The related Ir–CH3 complex we reported on was stable in DCM but showed reactivity towards haloforms.56 In light of the frequently observed higher reactivity of Rh species, it seems plausible that the Rh–CH3 complex is reactive towards DCM. Additionally, signals at δ = 3.20 ppm and δ = 6.26 ppm in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio are similar to resonances described for N,N′-diphenyl-N-methylphenylenediamine.98 Hence, a methylation of the diamidobenzene ligand cannot be ruled out as a possible mechanistic step. These findings show an important difference between the isoelectronic complexes [3] and [Cp*Rh(bpy)]: the group of Blakemore isolated and characterized the stable methylated half-sandwich compound [Cp*Rh(bpy)Me] in 2017;96 in contrast, the diolefinic backbone in our system appears to favor the rearrangement to a dinuclear complex. While the isolation of [3-CH3]+ from solvents other than DCM has so far been unsuccessful, its preparation and the transformation into the homobimetallic, heterovalent, complex [5](BF4)2 are matters of on-going research in our group. Whether this reactivity can also be extended to differently substituted diamidobenzenes is also currently under investigation.
:1 ratio are similar to resonances described for N,N′-diphenyl-N-methylphenylenediamine.98 Hence, a methylation of the diamidobenzene ligand cannot be ruled out as a possible mechanistic step. These findings show an important difference between the isoelectronic complexes [3] and [Cp*Rh(bpy)]: the group of Blakemore isolated and characterized the stable methylated half-sandwich compound [Cp*Rh(bpy)Me] in 2017;96 in contrast, the diolefinic backbone in our system appears to favor the rearrangement to a dinuclear complex. While the isolation of [3-CH3]+ from solvents other than DCM has so far been unsuccessful, its preparation and the transformation into the homobimetallic, heterovalent, complex [5](BF4)2 are matters of on-going research in our group. Whether this reactivity can also be extended to differently substituted diamidobenzenes is also currently under investigation.
Table 4 Bond lengths in complex [3]2+ and [5]2+. All values given in Å
|
|
[3]2+
|
[5]2+
|
| Rh–N1/N2 |
2.088(2)/2.099(2) |
2.018(3)/2.012(3) |
| N1–C1/N2–C2 |
1.301(2)/1.301(2) |
1.348(4)/1.339(4) |
| C1–C2 |
1.476(3) |
1.451(4) |
| C2–C3/C1–C6 |
1.411(2)/1.411(2) |
1.426(4)/1.418(4) |
| C3–C4/C5–C6 |
1.346(3)/1.343(2) |
1.400(5)/1.410(4) |
| C4–C5 |
1.437(2) |
1.412(5) |
| Rh2–C4/C5 |
— |
2.183(3)/2.200(3) |
| Rh2–C3/C6 |
— |
2.232(3)/2.232(3) |
Conclusion
We have shown the synthesis and comprehensive (spectro-)elecotrchemical characterization of four different diamidobenzene complexes of RhIII. The substituents on the N-atoms of the diamidobenzene ligands have a very strong influence on the bonding, donor, electrochemical and spectroscopic properties of the Rh complexes. Depending on the substitution pattern, the redox-active ligands display very different donor properties and prefer either one- or two-electron oxidations. All complexes can be reversibly reduced to electron-rich, mononuclear RhII metalates at potentials that correlate with the electron-releasing character of the ligand. To the best of our knowledge, this is the first time that isolable mononuclear RhII metallates with such extreme tuning of redox potentials have been reported. An isolated example was shown to undergo typical metalloradical reactivity such as O2 activation and C–Cl bond homolysis. Furthermore, oxidation-mediated dinucleation via a transient Rh–CH3 species allowed the first characterization of a π-bonded rhodio-o-diiminoquinone. Such structures are completely unprecedented in the chemistry of metal complexes of o-diiminoquinones. In summary, our research confirms the potential of diamidobenzene complexes of Rh(III) in organometallic chemistry, as they combine salient features of metal- and ligand-centered reactivity in one molecular entity. As we have shown, such features can lead to the generation of completely unexpected properties, as well as the generation of totally unprecedented structures.
Data availability
The data that support the findings of this study are available in the ESI† of this article.
Author contributions
SS and BS designed the project. SS carried out all the synthesis, characterization, reactivity studies, spectroscopic, electrochemical and spectroelectrochemical measurements, and DFT calculations. RW, JB and UA were responsible for solving the crystal structures. SS and BS jointly wrote the manuscript with help from the other authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster). Financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, SA 1840/9-1) is kindly acknowledged.
References
-
Rhodium Organometallics, ed., D. M. P. Mingos and R. H. Crabtree, Elsevier, Amsterdam, 2007, vol. 7 Search PubMed.
- H. Werner, Electron-Rich Half-Sandwich Complexes: Metal Bases par excellence, Angew. Chem., Int. Ed. Engl., 1983, 22, 927–949 CrossRef.
- U. Kölle and M. Grätzel, Organometallic Rhodium(III) Complexes as Catalysts for the Photoreduction of Protons to Hydrogen on Colloidal TiO2, Angew. Chem., Int. Ed. Engl., 1987, 26, 567–570 CrossRef.
- S. Chardon-Noblat, S. Cosnier, A. Deronzier and N. Vlachopoulos, Electrochemical Properties of [(C5Me5)RhIII(L)Cl]+ Complexes (L = 2,2′-Bipyridine or 1,10-Phenanthroline Derivatives) in Solution in Related Polypyrrolic Films. Application to Electrocatalytic Hydrogen Generation, J. Electroanal. Chem., 1993, 352, 213–228 CrossRef CAS.
- W. C. Henke, D. Lionetti, W. N. G. Moore, J. A. Hopkins, V. W. Day and J. D. Blakemore, Ligand Substituents Govern the Efficiency and Mechanistic Path of Hydrogen Production with Cp*Rh Catalysts, ChemSusChem, 2017, 10, 4589–4598 CrossRef CAS PubMed.
- Y. Hu, L. Li, A. P. Shaw, J. R. Norton, W. Sattler and Y. Rong, Synthesis, Electrochemistry, and Reactivity of New Iridium(III) and Rhodium(III) Hydrides, Organometallics, 2012, 31, 5058–5064 CrossRef CAS.
- D. Lionetti, V. W. Day and J. D. Blakemore, Structural and Chemical Properties of Half-Sandwich Rhodium Complexes Supported by the Bis(2-pyridyl)methane Ligand, Dalton Trans., 2019, 48, 12396–12406 RSC.
- P. M. Maitlis, (Pentamethylcyclopentadienyl)rhodium and -Iridium Complexes: Approaches to New Types of Homogeneous Catalysts, Acc. Chem. Res., 1978, 11, 301–307 CrossRef CAS.
- D. Fujita, A. Kaga, H. Sugimoto, Y. Morimoto and S. Itoh, Controlling Coordination Number of Rhodium(III) Complex by Ligand-Based Redox for Catalytic C–H Amination, Bull. Chem. Soc. Jpn., 2020, 93, 279–286 CrossRef CAS.
- A. John Blacker, E. Clot, S. B. Duckett, O. Eisenstein, J. Grace, A. Nova, R. N. Perutz, D. J. Taylor and A. C. Whitwood, Synthesis and Structure of “16-electron” Rhodium(III) Catalysts for Transfer Hydrogenation of a Cyclic Imine: Mechanistic Implications, Chem. Commun., 2009, 6801–6803 RSC.
- A. Nova, D. J. Taylor, A. J. Blacker, S. B. Duckett, R. N. Perutz and O. Eisenstein, Computational Studies Explain the Importance of Two Different Substituents on the Chelating Bis(amido) Ligand for Transfer Hydrogenation by Bifunctional Cp*Rh(III) Catalysts, Organometallics, 2014, 33, 3433–3442 CrossRef CAS.
- A. Rajput, A. K. Sharma, S. K. Barman, A. Saha and R. Mukherjee, Valence Tautomerism and Delocalization in Transition Metal Complexes of o-Amidophenolates and Other Redox-active Ligands. Some Recent Results, Coord. Chem. Rev., 2020, 414, 213240 CrossRef CAS.
- C. G. Pierpont, Studies on Charge Distribution and Valence Tautomerism in Transition Metal Complexes of Catecholate and Semiquinonate Ligands, Coord. Chem. Rev., 2001, 216–217, 99–125 CrossRef CAS.
- M. D. Ward and J. A. McCleverty, Non-innocent Behaviour in Mononuclear and Polynuclear Complexes: Consequences for Redox and Electronic Spectroscopic Properties, J. Chem. Soc., Dalton Trans., 2002, 275–288 RSC.
- N. Deibel, D. Schweinfurth, J. Fiedler, S. Záliš and B. Sarkar, Isomeric Separation in Donor-Acceptor Systems of Pd(II) and Pt(II) and a Combined Structural, Electrochemical and Spectroelectrochemical Study, Dalton Trans., 2011, 40, 9925–9934 RSC.
-
P. J. Chirik, Forum Issue on Redox-Active Ligands, Inorg. Chem., 2011, 50, 9737–9914 Search PubMed.
-
T. Storr and R. N. Mukherjee, Forum Issue on Applications of Metal Complexes with Ligand-Centered Radicals, Inorg. Chem., 2018, 57, 9577–10480 Search PubMed.
- K. Chłopek, E. Bill, T. Weyhermüller and K. Wieghardt, Molecular and Electronic Structure of Five-coordinate Complexes of Iron(II/III) Containing o-Diiminobenzosemiquinonate(1-) Pi Radical Ligands, Inorg. Chem., 2005, 44, 7087–7098 CrossRef PubMed.
- D. Herebian, E. Bothe, F. Neese, T. Weyhermüller and K. Wieghardt, Molecular and Electronic Structures of Bis-(o-diiminobenzosemiquinonato)metal(II) Complexes (Ni, Pd, Pt), Their Monocations and -Anions, and of Dimeric Dications Containing Weak Metal-Metal Bonds, J. Am. Chem. Soc., 2003, 125, 9116–9128 CrossRef CAS PubMed.
- A. L. Balch and R. H. Holm, Complete Electron-Transfer Series of the [M-N 4 ] Type, J. Am. Chem. Soc., 1966, 88, 5201–5209 CrossRef CAS.
- S. Pascal and O. Siri, Benzoquinonediimine Ligands: Synthesis, Coordination Chemistry and Properties, Coord. Chem. Rev., 2017, 350, 178–195 CrossRef CAS.
- E. Bill, E. Bothe, P. Chaudhuri, K. Chlopek, D. Herebian, S. Kokatam, K. Ray, T. Weyhermüller, F. Neese and K. Wieghardt, Molecular and Electronic Structure of Four- and Five-coordinate Cobalt Complexes Containing Two o-Phenylenediamine- or Two o-Aminophenol-type Ligands at Various Oxidation Levels: An Experimental, Density Functional, and Correlated Ab initio Study, Chem.–Eur. J., 2004, 11, 204–224 CrossRef CAS PubMed.
- D. Herebian, K. E. Wieghardt and F. Neese, Analysis and Interpretation of Metal-radical Coupling in a Series of Square Planar Nickel Complexes: Correlated Ab initio and Density Functional Investigation of Ni(L(ISQ))(2) (L(ISQ)=3,5-di-tert-butyl-o-diiminobenzosemiquinonate(1-)), J. Am. Chem. Soc., 2003, 125, 10997–11005 CrossRef CAS PubMed.
- M. M. Khusniyarov, T. Weyhermüller, E. Bill and K. Wieghardt, Tuning the Oxidation Level, the Spin State, and the Degree of Electron Delocalization in Homo- and Heteroleptic Bis(alpha-diimine)iron Complexes, J. Am. Chem. Soc., 2009, 131, 1208–1221 CrossRef CAS PubMed.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, New Avenues for Ligand-Mediated Processes--Expanding Metal Reactivity by the Use of Redox-active Catechol, o-Aminophenol and o-Phenylenediamine Ligands, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- J. I. van der Vlugt, Radical-Type Reactivity and Catalysis by Single-Electron Transfer to or from Redox-Active Ligands, Chem.–Eur. J., 2019, 25, 2651–2662 CrossRef CAS PubMed.
- D. Kalinina, C. Dares, H. Kaluarachchi, P. G. Potvin and A. B. P. Lever, Spectroscopic, Electrochemical, and Computational Aspects of the Charge Distribution in Ru(acac)2(R-o-benzoquinonediimine) Complexes, Inorg. Chem., 2008, 47, 10110–10126 CrossRef CAS PubMed.
- J. Rusanova, E. Rusanov, S. I. Gorelsky, D. Christendat, R. Popescu, A. A. Farah, R. Beaulac, C. Reber and A. B. P. Lever, The Very Covalent Diammino(o-benzoquinonediimine) Dichlororuthenium(II). An Example of Very Strong Pi-back-Donation, Inorg. Chem., 2006, 45, 6246–6262 CrossRef CAS PubMed.
- A. B. P. Lever and S. I. Gorelsky, Comparison of o-Benzoquinonediimine with Bipyridine and Bipyrazine in Electronic Coupling to Ruthenium(II), as a Function of Spectator Ligand, Coord. Chem. Rev., 2000, 208, 153–167 CrossRef CAS.
- N. Leconte, J. Moutet, K. Herasymchuk, R. M. Clarke, C. Philouze, D. Luneau, T. Storr and F. Thomas, Mn(IV) and Mn(V)-Radical Species Supported by the Redox Non-innocent Bis(2-amino-3,5-di-tert-butylphenyl)amine Pincer Ligand, Chem. Commun., 2017, 53, 2764–2767 RSC.
- N. Leconte, J. Moutet, T. Constantin, F. Molton, C. Philouze and F. Thomas, Coordination Chemistry of the Redox Non-Innocent Ligand Bis(2-amino-3,5-di- tert -butylphenyl)amine with Group 10 Metal Ions (Ni, Pd, Pt), Eur. J. Inorg. Chem., 2018, 1752–1761 CrossRef CAS.
- N. Leconte, S. Gentil, F. Molton, C. Philouze, A. Le Goff and F. Thomas, Complexes of the Bis(di- tert -butyl-aniline)amine Pincer Ligand: The Case of Copper, Eur. J. Inorg. Chem., 2020, 2691–2699 CrossRef CAS.
- A. I. Nguyen, R. A. Zarkesh, D. C. Lacy, M. K. Thorson and A. F. Heyduk, Catalytic Nitrene Transfer by a Zirconium( iv ) Redox-active Ligand Complex, Chem. Sci., 2011, 2, 166–169 RSC.
- A. I. Nguyen, K. J. Blackmore, S. M. Carter, R. A. Zarkesh and A. F. Heyduk, One- and Two-electron Reactivity of a Tantalum(V) Complex with a Redox-active Tris(amido) Ligand, J. Am. Chem. Soc., 2009, 131, 3307–3316 CrossRef CAS PubMed.
- J. Ciccione, N. Leconte, D. Luneau, C. Philouze and F. Thomas, Geometric and Electronic Structures of Nickel(II) Complexes of Redox Noninnocent Tetradentate Phenylenediamine Ligands, Inorg. Chem., 2016, 55, 649–665 CrossRef CAS PubMed.
- N. Leconte, A. Du Moulinet d'Hardemare, C. Philouze and F. Thomas, A Highly Active Diradical Cobalt(iii) Catalyst for the Cycloisomerization of Alkynoic Acids, Chem. Commun., 2018, 54, 8241–8244 RSC.
- N. Leconte, J. Ciccione, G. Gellon, C. Philouze and F. Thomas, Unprecedented Redox-driven Ligand Ejection in Nickel(II)-diiminosemiquinonate Radical Complexes, Chem. Commun., 2014, 50, 1918–1920 RSC.
- N. Leconte, F. Berthiol, C. Philouze and F. Thomas, Copper Complexes of the Tetradentate N,N′ -Bis(2-amino-3,5-di- tert -butylphenyl)-2,2′-diaminobiphenyl Ligand, Eur. J. Inorg. Chem., 2021, 1481–1489 CrossRef CAS.
- T. Janes, M. Xu and D. Song, Synthesis and Reactivity of Li and TaMe3 Complexes Supported by N,N'-Bis(2,6-diisopropylphenyl)-o-phenylenediamido Ligands, Dalton Trans., 2016, 45, 10672–10680 RSC.
- M. M. Khusniyarov, T. Weyhermüller, E. Bill and K. Wieghardt, Reversible Electron Transfer Coupled to Spin Crossover in an Iron Coordination Salt in the Solid State, Angew. Chem., Int. Ed., 2008, 47, 1228–1231 CrossRef CAS PubMed.
- M. M. Khusniyarov, E. Bill, T. Weyhermüller, E. Bothe, K. Harms, J. Sundermeyer and K. Wieghardt, Characterization of Three Members of the Electron-transfer Series Fe(pda)2n (n=2-, 1-, 0) by Spectroscopy and Density Functional Theoretical Calculations pda=redox Non-innocent Derivatives of N,N'-Bis(pentafluorophenyl)-o-phenylenediamide(2-, 1-, 0), Chem.–Eur. J., 2008, 14, 7608–7622 CrossRef CAS PubMed.
- T. Janes, J. M. Rawson and D. Song, Syntheses and Structures of Li, Fe, and Mo Derivatives of N,N'-Bis(2,6-diisopropylphenyl)-o-phenylenediamine, Dalton Trans., 2013, 42, 10640–10648 RSC.
- S. Krupski, R. Pöttgen, I. Schellenberg and F. E. Hahn, Benzannulated N-Heterocyclic Germylenes and Stannylenes with Sterically Demanding N,N'-Substituents, Dalton Trans., 2014, 43, 173–181 RSC.
- S. Sobottka, M. Nößler, A. L. Ostericher, G. Hermann, N. Z. Subat, J. Beerhues, M. Behr-van der Meer, L. Suntrup, U. Albold, S. Hohloch, J. C. Tremblay and B. Sarkar, Tuning PtII -Based Donor-Acceptor Systems through Ligand Design: Effects on Frontier Orbitals, Redox Potentials, UV/Vis/NIR Absorptions, Electrochromism, and Photocatalysis, Chem.–Eur. J., 2020, 26, 1314–1327 CrossRef CAS PubMed.
- T. Matsumoto, H.-C. Chang, M. Wakizaka, S. Ueno, A. Kobayashi, A. Nakayama, T. Taketsugu and M. Kato, Nonprecious-metal-assisted Photochemical Hydrogen Production from Ortho-phenylenediamine, J. Am. Chem. Soc., 2013, 135, 8646–8654 CrossRef CAS PubMed.
- B. Weber and F. A. Walker, Solution NMR Studies of Iron(II) Spin-crossover Complexes, Inorg. Chem., 2007, 46, 6794–6803 CrossRef CAS PubMed.
- H. Bamberger, U. Albold, J. Dubnická Midlíková, C.-Y. Su, N. Deibel, D. Hunger, P. P. Hallmen, P. Neugebauer, J. Beerhues, S. Demeshko, F. Meyer, B. Sarkar and J. van Slageren, Iron(II), Cobalt(II), and Nickel(II) Complexes of Bis(sulfonamido)benzenes: Redox Properties, Large Zero-Field Splittings, and Single-Ion Magnets, Inorg. Chem., 2021, 60, 2953–2963 CrossRef CAS PubMed.
- Y. Rechkemmer, F. D. Breitgoff, M. van der Meer, M. Atanasov, M. Hakl, M. Orlita, P. Neugebauer, F. Neese, B. Sarkar and J. van Slageren, A Four-coordinate Cobalt(II) Single-ion Magnet with Coercivity and a Very High Energy Barrier, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- K. Y. Monakhov, J. van Leusen, P. Kögerler, E.-L. Zins, M. E. Alikhani, M. Tromp, A. A. Danopoulos and P. Braunstein, Linear, Trinuclear Cobalt Complexes with o-Phenylene-bis-Silylamido Ligands, Chem.–Eur. J., 2017, 23, 6504–6508 CrossRef CAS PubMed.
- M. Bera, K. Keshari, A. Bhardwaj, G. Gupta, B. Mondal and S. Paria, Electrocatalytic Water Oxidation Activity of Molecular Copper Complexes: Effect of Redox-Active Ligands, Inorg. Chem., 2022, 61, 3152–3165 CrossRef CAS PubMed.
- M. van der Meer, Y. Rechkemmer, I. Peremykin, S. Hohloch, J. van Slageren and B. Sarkar, (Electro)catalytic C-C Bond Formation Reaction with a Redox-active Cobalt Complex, Chem. Commun., 2014, 50, 11104–11106 RSC.
- J. Jacquet, E. Salanouve, M. Orio, H. Vezin, S. Blanchard, E. Derat, M. Desage-El Murr and L. Fensterbank, Iminosemiquinone Radical Ligands Enable Access to a Well-defined Redox-active Cu(II)-CF3 Complex, Chem. Commun., 2014, 50, 10394–10397 RSC.
- N. Deibel, D. Schweinfurth, S. Hohloch, J. Fiedler and B. Sarkar, Donor-acceptor Systems of Pt(II) and Redox-induced Reactivity towards Small Molecules, Chem. Commun., 2012, 48, 2388–2390 RSC.
- Q. Liang, J. C. DeMuth, A. Radović, N. J. Wolford, M. L. Neidig and D. Song, 2Fe-2S Cluster Supported by Redox-Active o-Phenylenediamide Ligands and Its Application toward Dinitrogen Reduction, Inorg. Chem., 2021, 60, 13811–13820 CrossRef CAS PubMed.
- W. Zhou, B. O. Patrick and K. M. Smith, Influence of Redox Non-innocent Phenylenediamido Ligands on Chromium Imido Hydrogen-atom Abstraction Reactivity, Chem. Commun., 2014, 50, 9958–9960 RSC.
- S. Sobottka, M. B. van der Meer, E. Glais, U. Albold, S. Suhr, C.-Y. Su and B. Sarkar, A Coordinatively Unsaturated Iridium Complex with an Unsymmetrical Redox-active Ligand: (Spectro)electrochemical and Reactivity Studies, Dalton Trans., 2019, 48, 13931–13942 RSC.
- M. van der Meer, S. Manck, S. Sobottka, S. Plebst and B. Sarkar, Redox Activity and Bond Activation in Iridium–Diamidobenzene Complexes: A Combined Structural, (Spectro)electrochemical, and DFT Investigation, Organometallics, 2015, 34, 5393–5400 CrossRef CAS.
- E. T. Ouellette, J. S. Magdalenski, R. G. Bergman and J. Arnold, Applications of Low-Valent Transition Metalates: Development of a Reactive Noncarbonyl Rhenium(I) Anion, Acc. Chem. Res., 2022, 55, 783–793 CrossRef CAS PubMed.
- W. Kaim, R. Reinhardt, S. Greulich and J. Fiedler, Resolving the Two-Electron Process for the Couple [(C5Me5)M(N∧N)Cl]+/[(C5Me5)M(N∧N)] (M = Rh, Ir) into Two One-Electron Steps Using the 2,2‘-Azobis(pyridine) N∧N Ligand, Fast Scan Cyclovoltammetry, and Spectroelectrochemistry: Detection of Radicals instead of MII Intermediates, Organometallics, 2003, 22, 2240–2244 CrossRef CAS.
- M. Ladwig and W. Kaim, Spectroscopic and Electrochemical Properties of the Isomeric Bidiazine Complexes [(C5Me5)CiRh(bdz)]+ and (C5Me5)Rh(bdz) and Their Relevance to the Catalysis of the 2 H+ → H2 Reaction by 2,2′-Bipyridine Analogues, J. Organomet. Chem., 1991, 419, 233–243 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- S. Bhattacharya, P. Gupta, F. Basuli and C. G. Pierpont, Structural Systematics for o-C(6)H(4)XY Ligands with X,Y= O, NH, and S Donor Atoms. o-Iminoquinone and o-Iminothioquinone Complexes of Ruthenium and Osmium, Inorg. Chem., 2002, 41, 5810–5816 CrossRef CAS PubMed.
- W. Kaim, Concepts for Metal Complex Chromophores Absorbing in the Near Infrared, Coord. Chem. Rev., 2011, 255, 2503–2513 CrossRef CAS.
- W. Kaim, The Transition Metal Coordination Chemistry of Anion Radicals, Coord. Chem. Rev., 1987, 76, 187–235 CrossRef CAS.
- A. Damas, B. Ventura, J. Moussa, A. Degli Esposti, L.-M. Chamoreau, A. Barbieri and H. Amouri, Turning on Red and Near-infrared Phosphorescence in Octahedral Complexes with Metalated Quinones, Inorg. Chem., 2012, 51, 1739–1750 CrossRef CAS PubMed.
- M. J. Shaw, W. E. Geiger, J. Hyde and C. White, 16-Electron through 19-Electron Complexes of the 1,5-COD and 1,3-COD Isomers of (η 5 -C 5 Ph 5 )Rh(η 4 -C 8 H 12 ): Electrochemical Evidence for an Oxidatively Induced Agostic Interaction, Organometallics, 1998, 17, 5486–5491 CrossRef CAS.
- D. G. H. Hetterscheid, B. de Bruin, J. M. M. Smits and A. W. Gal, Disproportionation of Rh II (cod) to Rh I (cod) and Rh III (cycloocta-2,5-dien-1-yl): Hydrogen Atom Transfer vs. Electron and Proton Transfer, Organometallics, 2003, 22, 3022–3024 CrossRef CAS.
- A. Bakac and L. M. Thomas, Macrocyclic Rhodium(III) Hydrides and a Monomeric Rhodium(II) Complex, Inorg. Chem., 1996, 35, 5880–5884 CrossRef CAS.
- X. X. Zhang and B. B. Wayland, Sterically Demanding Diporphyrin Ligands and Rhodium(II) Porphyrin Bimetalloradical Complexes, Inorg. Chem., 2000, 39, 5318–5325 CrossRef CAS PubMed.
-
Comprehensive Organometallic Chemistry, in The Synthesis, Reactions and Structures of Organometallic Compounds, ed., G. Wilkinson, F. G. A. Stone, and E. W. Abe, Pergamon Press Ltd., Oxford, 1st edn, 1982, vol. 5 Search PubMed.
- D. G. DeWit, Reactivity of Mononuclear Rhodium(II) Compounds, Coord. Chem. Rev., 1996, 147, 209–246 CrossRef.
- D. G. H. Hetterscheid, J. M. M. Smits and B. de Bruin, Chloride-Triggered Disproportionation of a Mononuclear Rh II (nbd) Species to Rh I (nbd) and Rh III (η 1-norbornenyl) Complexes: Possibilities for Wacker Type Mono-oxygenation of Norbornadiene to Norbornenone, Organometallics, 2004, 23, 4236–4246 CrossRef CAS.
- M. L. H. Green, L. Pratt and G. Wilkinson, A New Type of Transition Metal–Cyclopentadiene Compound, J. Chem. Soc., 1959, 3753–3767 RSC.
- U. Kölle and F. Khouzami, Permethylmetallocene, II. Decamethylcobaltocen: Synthese und Umwandlung in Methylierte (Aren)(cyclopentadienyl)cobalt-Kationen, Chem. Ber., 1981, 114, 2929–2937 CrossRef.
- F. M. Dixon, J. R. Farrell, P. E. Doan, A. Williamson, D. A. Weinberger, C. A. Mirkin, C. Stern, C. D. Incarvito, L. M. Liable-Sands, L. N. Zakharov and A. L. Rheingold, Rational Design of a Novel Mononuclear Rhodium(II) Complex, Organometallics, 2002, 21, 3091–3093 CrossRef CAS.
- A. Takaoka and J. C. Peters, A Homologous Series of Cobalt, Rhodium, and Iridium Metalloradicals, Inorg. Chem., 2012, 51, 16–18 CrossRef CAS PubMed.
- K. Fuchigami, N. P. Rath and L. M. Mirica, Mononuclear Rhodium(II) and Iridium(II) Complexes Supported by Tetradentate Pyridinophane Ligands, Inorg. Chem., 2017, 56, 9404–9408 CrossRef CAS PubMed.
- W. I. Dzik, L. Fuente Arruga, M. A. Siegler, A. L. Spek, J. N. H. Reek and B. de Bruin, Open-Shell Organometallic [M II (dbcot(bislutidylamine))] 2+ Complexes (M = Rh, Ir): Unexpected Base-Assisted Reduction of the Metal Instead of Amine Ligand Deprotonation, Organometallics, 2011, 30, 1902–1913 CrossRef CAS.
-
B. de Bruin, D. G. H. Hetterscheid, A. J. J. Koekkoek and H. Grützmacher, in Progress in Inorganic Chemistry, ed. K. D. Karlin, Wiley; John Wiley [distributor], Hoboken, N.J., Chichester, 2007, pp. 247–354 Search PubMed.
- D. W. Shaffer, S. A. Ryken, R. A. Zarkesh and A. F. Heyduk, Redox Behavior of Rhodium 9,10-Phenanthrenediimine Complexes, Inorg. Chem., 2011, 50, 13–21 CrossRef CAS PubMed.
- F. L. Benedito, T. Petrenko, E. Bill, T. Weyhermüller and K. Wieghardt, Square Planar Bis{3,6-bis(trimethylsilyl)benzene-1,2-dithiolato}metal Complexes of Cr(II), Co(III), and Rh(II): An Experimental and Density Functional Theoretical Study, Inorg. Chem., 2009, 48, 10913–10925 CrossRef CAS PubMed.
- M. P. García, M. V. Jiménez, A. Cuesta, C. Siurana, L. A. Oro, F. J. Lahoz, J. A. López, M. P. Catalán, A. Tiripicchio and M. Lanfranchi, Synthesis and Reactivity of Mononuclear (Pentachlorophenyl)rhodium(II) Complexes. Structural Relevance of Rhodium− o -Chlorine Secondary Bonding, Organometallics, 1997, 16, 1026–1036 CrossRef.
- K. S. Chan, X. Z. Li, W. I. Dzik and B. de Bruin, Carbon-carbon Bond Activation of 2,2,6,6-Tetramethyl-piperidine-1-oxyl by a Rh(II) Metalloradical: A Combined Experimental and Theoretical Study, J. Am. Chem. Soc., 2008, 130, 2051–2061 CrossRef CAS PubMed.
- B. B. Wayland, S. Ba and A. E. Sherry, Activation of Methane and Toluene by Rhodium(II) Porphyrin Complexes, J. Am. Chem. Soc., 1991, 113, 5305–5311 CrossRef CAS.
- A. G. Bunn, M. Wei and B. B. Wayland, Reactivity Patterns of H2 and CO with a Rhodium(II) Salen Derivative: Formation of Hydride, Formyl, and Dimetal Ketone Complexes and Rhodium Reduction, Organometallics, 1994, 13, 3390–3392 CrossRef CAS.
- J. Wassenaar, B. de Bruin, M. A. Siegler, A. L. Spek, J. N. H. Reek and J. I. van der Vlugt, Activation of H2 by a Highly Distorted Rh(II) Complex with A New C3-Symmetric Tripodal Tetraphosphine Ligand, Chem. Commun., 2010, 46, 1232–1234 RSC.
- D. G. H. Hetterscheid, M. Klop, R. J. N. A. M. Kicken, J. M. M. Smits, E. J. Reijerse and B. de Bruin, Hydrogen-atom Transfer in Open-shell Organometallic Chemistry: The Reactivity of Rh(II)(cod) and Ir(II)(cod) Radicals, Chem.–Eur. J., 2007, 13, 3386–3405 CrossRef CAS PubMed.
- J. Tanaka, Y. Nagashima, A. J. Araujo Dias and K. Tanaka, Photo-Induced ortho-C-H Borylation of Arenes through In Situ Generation of Rhodium(II) Ate Complexes, J. Am. Chem. Soc., 2021, 143, 11325–11331 CrossRef CAS PubMed.
- J. D. Blakemore, E. S. Hernandez, W. Sattler, B. M. Hunter, L. M. Henling, B. S. Brunschwig and H. B. Gray, Pentamethylcyclopentadienyl Rhodium Complexes, Polyhedron, 2014, 84, 14–18 CrossRef CAS.
- J. Moussa, M. N. Rager, L. M. Chamoreau, L. Ricard and H. Amouri, Unprecedented π-Bonded Rhodio- and Iridio- o -Benzoquinones as Organometallic Linkers for the Design of Chiral Octahedral Bimetallic Assemblies, Organometallics, 2009, 28, 397–404 CrossRef CAS.
- C. Tejel, M. A. Ciriano, M. P. del Río, F. J. van den Bruele, D. G. H. Hetterscheid, N. Tsichlis i Spithas and B. de Bruin, Deprotonation Induced Ligand-to-Metal Electron Transfer: Synthesis of a Mixed-Valence Rh(−I,I) Dinuclear Compound and Its Reaction with Dioxygen, J. Am. Chem. Soc., 2008, 130, 5844–5845 CrossRef CAS PubMed.
- A. J. Blake, R. O. Gould, M. A. Halcrow and M. Schröder, Synthesis of Cationic Half-sandwich Rhodium( I ) Complexes of 1,4,7-Trithiacyclononane ([9]aneS 3 ). The Single-crystal Structures of [Rh([9]aneS 3 )(C 2 H 4 ) 2 ]PF 6 [Rh([9]aneS 3 )(C 8 H 12 )]BF 4 and [Rh([9]aneS 3 )(C 4 H 6 )]PF 6 ·0.25OEt 2, J. Chem. Soc., Dalton Trans., 1994, 2197–2208 RSC.
- S. S. D. Brown, S. N. Heaton, M. H. Moore, R. N. Perutz and G. Wilson, Synthesis and Photoisomerization of Divinyltetramethyldisiloxane and Divinyltetramethyldisilazane Complexes of (η 5 -C 5 R 5 )Rh (R = H, Me). Crystal and Molecular Structure of (η 5 -C 5 Me 5 )Rh[η 4 -(CH 2 CH)Me 2 SiOSiMe 2 (CHCH 2 )], Organometallics, 1996, 15, 1392–1404 CrossRef CAS.
- W. J. Hoogervorst, K. Goubitz, J. Fraanje, M. Lutz, A. L. Spek, J. M. Ernsting and C. J. Elsevier, (Bis(imino)aryl)rhodium(III) Halide and Methyl Compounds, Organometallics, 2004, 23, 4550–4563 CrossRef CAS.
- L. Gonsalvi, J. A. Gaunt, H. Adams, A. Castro, G. J. Sunley and A. Haynes, Quantifying Steric Effects of α-Diimine Ligands. Oxidative Addition of MeI to Rhodium(I) and Migratory Insertion in Rhodium(III) Complexes, Organometallics, 2003, 22, 1047–1054 CrossRef CAS.
- D. Lionetti, V. W. Day and J. D. Blakemore, Synthesis and Electrochemical Properties of Half-Sandwich Rhodium and Iridium Methyl Complexes, Organometallics, 2017, 36, 1897–1905 CrossRef CAS.
- G. Barany, A. L. Schroll, A. W. Mott and D. A. Halsrud, A General Strategy for Elaboration of the Dithiocarbonyl Functionality, -(C:O)SS-: Application to the Synthesis of Bis(chlorocarbonyl)disulfane and Related Derivatives of Thiocarbonic Acids, J. Org. Chem., 1983, 48, 4750–4761 CrossRef CAS.
- M. Chakrabarty, A. Batabyal and S. Khasnobis, On Attempted Oxidative Cyclisation of Isomeric N,N'-Diphenylphenylenediamines and Their N,N '-Dimethyl Derivatives by Palladium(II)
Acetate and Uv Light, Synth. Commun., 2000, 30, 3651–3668 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a